VYTORIN®
ORGANON BRASIL
ezetimiba + sinvastatina
Apresentações.
VYTORIN® é apresentado na forma de comprimidos disponíveis em quatro dosagens, em caixas com:
- VYTORIN®10/10 (ezetimiba 10 mg/sinvastatina 10 mg): 28 comprimidos.
- VYTORIN®10/20 (ezetimiba 10 mg/sinvastatina 20 mg): 14 ou 28 comprimidos.
- VYTORIN®10/40 (ezetimiba 10 mg/sinvastatina 40 mg): 14 ou 28 comprimidos.
- VYTORIN®10/80 (ezetimiba 10 mg/sinvastatina 80 mg): 28 comprimidos.
Composição.
Os ingredientes ativos de VYTORIN® são ezetimiba e sinvastatina.
USO ORAL
USO ADULTO E PEDIÁTRICO (10-17 ANOS DE IDADE):
- VYTORIN®10/10 (ezetimiba 10 mg/sinvastatina 10 mg)
- VYTORIN®10/20 (ezetimiba 10 mg/sinvastatina 20 mg)
- VYTORIN®10/40 (ezetimiba 10 mg/sinvastatina 40 mg)
USO ADULTO:
- VYTORIN®10/80 (ezetimiba 10 mg/sinvastatina 80 mg)
Ingredientes inativos:
Hidroxianisol butilado, ácido cítrico monoidratado, croscarmelose sódica, hidroxipropilmetilcelulose, lactose monoidratada, estearato de magnésio, celulose microcristalina e propilgalato.
Indicações.
Hipercolesterolemia Primária
VYTORIN® é indicado como terapia adjuvante à dieta para reduzir níveis elevados de colesterol total, colesterol ligado a lipoproteína de baixa densidade (LDL-C), apolipoproteína B (apo B), triglicérides (TG) e colesterol não ligado a lipoproteína de alta densidade (não-HDL-C) e para aumentar os níveis de colesterol ligado à lipoproteína de alta densidade (HDL-C) em pacientes adultos e adolescentes (10 a 17 anos de idade) com hipercolesterolemia primária (heterozigótica familiar e não familiar) ou hiperlipidemia mista.
A administração concomitante de fenofibrato e VYTORIN® pode ser feita para pacientes adultos com hiperlipidemia mista que necessitem de redução de TG e não-HDL-C e aumento de HDL-C adicionais.
Hipercolesterolemia Familiar Homozigótica (HFHo)
VYTORIN® é indicado para reduzir os níveis elevados de colesterol total e de LDL-C em pacientes adultos e adolescentes (10 a 17 anos de idade) com HFHo. Os pacientes também podem receber tratamentos adjuvantes (p. ex., aférese de LDL).
Resultados de eficácia.
Em estudos clínicos controlados, VYTORIN® reduziu significativamente o colesterol total, o colesterol de lipoproteína de baixa densidade (LDL-C), a apolipoproteína B (apo B), os triglicérides (TG) e o colesterol não ligado à lipoproteína de alta densidade (não-HDL-C) e aumentou o colesterol ligado a lipoproteína de alta densidade (HDL-C) em pacientes com hipercolesterolemia.
Hipercolesterolemia Primária
VYTORIN®
São relatados cinco estudos multicêntricos e duplo-cegos conduzidos com VYTORIN® em pacientes com hipercolesterolemia primária: dois foram comparativos com sinvastatina, dois foram comparativos com atorvastatina e um foi comparativo com rosuvastatina.
Em um estudo multicêntrico, duplo-cego, controlado com placebo e com 12 semanas de duração, 887 pacientes hipercolesterolêmicos foram distribuídos de modo randômico em dez grupos de tratamento: placebo, ezetimiba (10 mg), sinvastatina (10 mg, 20 mg, 40 mg, ou 80 mg) ou a administração concomitante de ezetimiba e sinvastatina equivalente a VYTORIN®10/10, 10/20, 10/40 e 10/80. Quando os pacientes que receberam VYTORIN® foram comparados aos que receberam todas as doses de sinvastatina, VYTORIN® reduziu significativamente o colesterol total, o LDL-C, a apo B, os TG, o colesterol não-HDL-C e a proteína C-reativa. Os efeitos de VYTORIN® no HDL-C foram semelhantes aos observados com a sinvastatina. Uma análise adicional mostrou que VYTORIN® aumentou significativamente o HDL-C em comparação com o placebo (veja Tabela 1).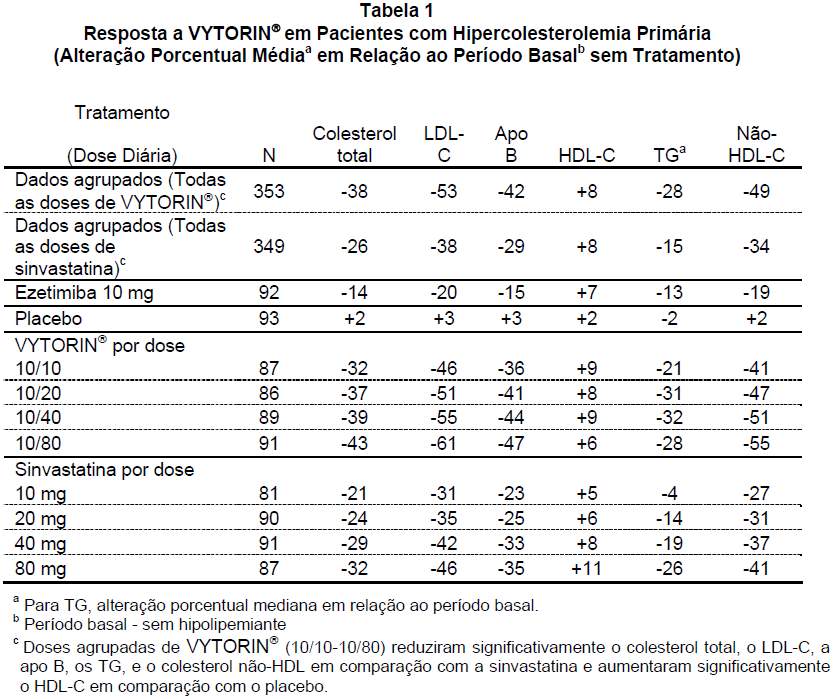
Em um estudo com desenho semelhante, os resultados para todos os parâmetros lipídicos foram, em geral, consistentes. Em uma análise agrupada desses dois estudos, a resposta dos lípides a VYTORIN® foi semelhante em pacientes com níveis de TG maiores ou menores do que 200 mg/dL.
Em um estudo multicêntrico, duplo-cego, controlado, com duração de 23 semanas, 710 pacientes com doença arterial coronariana (DAC) ou equivalente de risco de DAC pelos critérios estabelecidos nas diretrizes do NCEP ATP III e LDL-C ≥130 mg/dL foram distribuídos de modo randômico em quatro grupos de tratamento: administração concomitante de ezetimiba e sinvastatina equivalente a VYTORIN® (10/10, 10/20 e 10/40) ou 20 mg de sinvastatina. A dose de sinvastatina dos pacientes que não atingiram LDL-C < 100 mg/dL foi titulada a intervalos de 6 semanas para a dose máxima de 80 mg. Na 5ª semana, as reduções de LDL-C com VYTORIN® 10/10, 10/20 ou 10/40 foram significativamente maiores do que as obtidas com 20 mg de sinvastatina. Além disso, na 5ª semana, significativamente mais pacientes que receberam VYTORIN® 10/10, 10/20 ou 10/40 atingiram a meta de LDL-C em comparação aos que receberam 20 mg de sinvastatina (veja Tabela 2). Os resultados na 5ª semana em termos de redução do LDL-C e de porcentagem de pacientes que atingiram a meta de LDL-C foram consistentes com os resultados obtidos no final do estudo (23ª semana).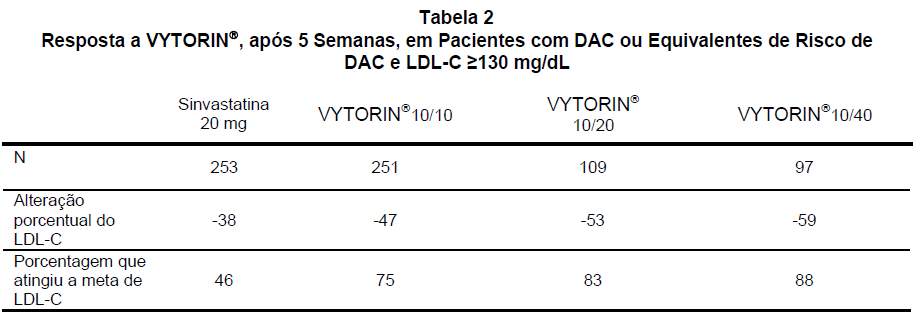
Em um estudo multicêntrico, duplo-cego, de 6 semanas de duração, 1.902 pacientes com hipercolesterolemia primária que não atingiram a meta de LDL-C estabelecida pelo Programa Nacional de Educação em Colesterol (NCEP) ATP III foram distribuídos de modo randômico para um de oito grupos de tratamento: VYTORIN® (10/10, 10/20, 10/40 ou 10/80) ou atorvastatina (10 mg, 20 mg, 40 mg ou 80 mg). Quando os pacientes que receberam todas as doses de VYTORIN® foram comparados àqueles que receberam todas as doses de atorvastatina, VYTORIN® reduziu os níveis de colesterol total, LDL-C, apo B e não HDL-C e aumentou os níveis de HDL-C significativamente mais do que a atorvastatina. Os efeitos de VYTORIN® nos TG foram semelhantes aos efeitos observados com a atorvastatina (veja Tabela 3).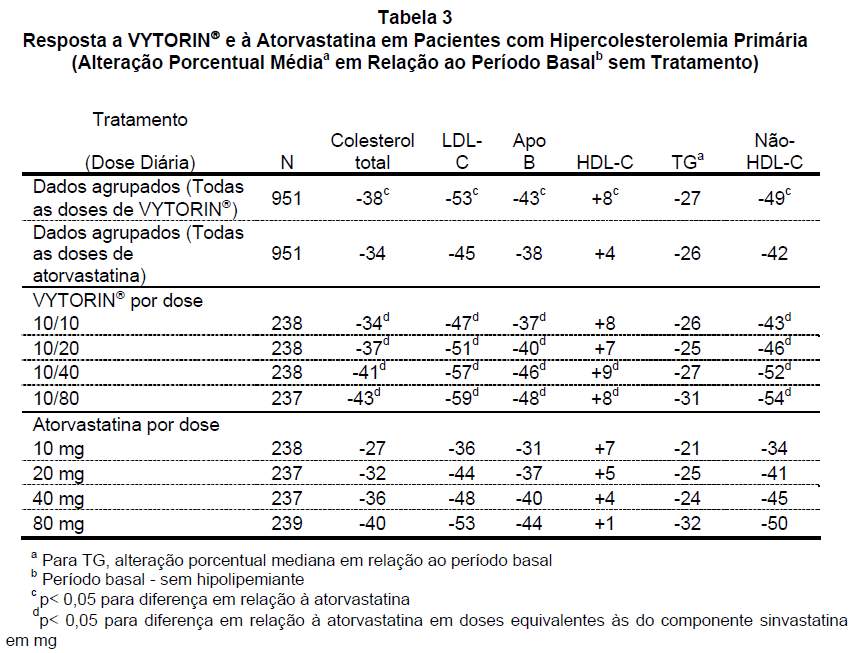
Em um estudo de titulação forçada, multicêntrico, duplo-cego, com 24 semanas de duração, 788 pacientes com hipercolesterolemia primária que não haviam atingido as metas de LDL-C do NCEP ATP III foram distribuídos de modo randômico para receber a administração concomitante de ezetimiba e sinvastatina equivalente a VYTORIN® (10/10 e 10/20) ou 10 mg de atorvastatina. Nos três grupos de tratamento, a dose da vastatina foi titulada a intervalos de 6 semanas até 80 mg. A cada comparação de dose pré-especificada, VYTORIN® proporcionou reduções maiores de LDL-C em comparação com a atorvastatina (veja Tabela 4).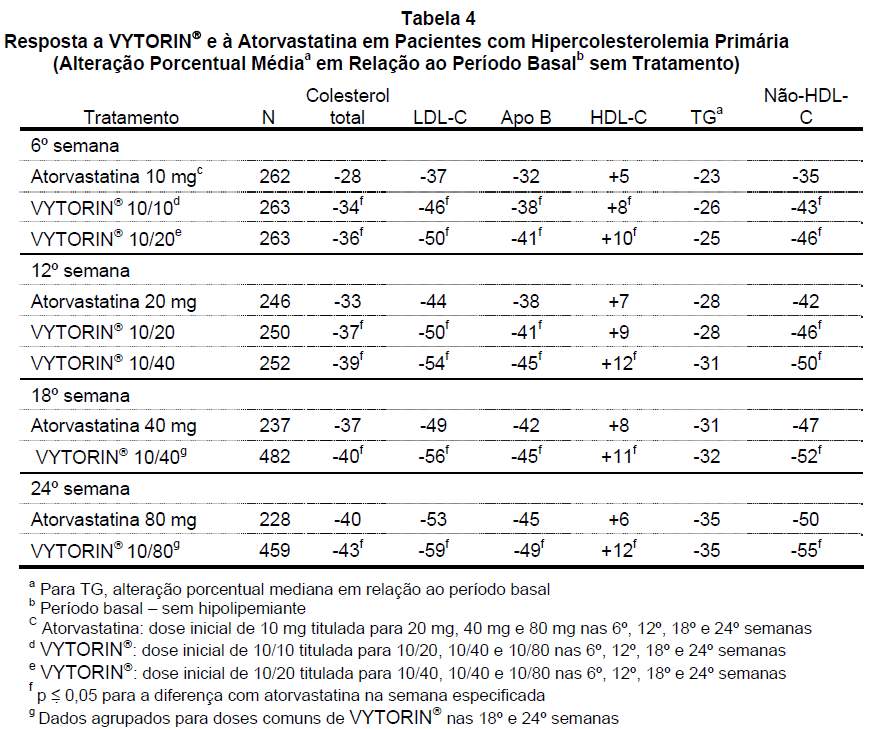
Em um estudo multicêntrico, duplo-cego, de 6 semanas de duração, 2.959 pacientes com hipercolesterolemia primária que não haviam atingido as metas de LDL-C do NCEP ATP III foram distribuídos de modo randômico para um de seis grupos de tratamento: VYTORIN® (10/20, 10/40 ou 10/80) ou rosuvastatina (10 mg, 20 mg ou 40 mg). Quando os pacientes que receberam todas as doses de VYTORIN® foram comparados àqueles que receberam todas as doses de rosuvastatina, VYTORIN® reduziu significativamente mais os níveis de colesterol total, LDL-C, apo B e não HDL-C do que a rosuvastatina. Os efeitos de VYTORIN® em relação ao HDL-C foram semelhantes aos efeitos observados com a rosuvastatina (veja Tabela 5).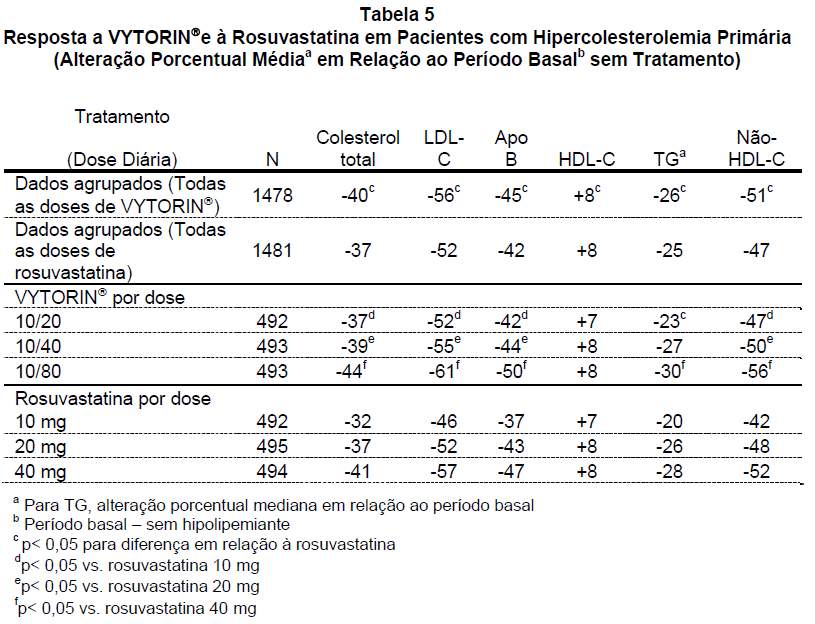
Em um estudo duplo-cego, controlado com placebo, com 8 semanas de duração, 240 pacientes com hipercolesterolemia que já usavam sinvastatina em monoterapia e que não haviam atingido as metas de LDL-C do Programa Nacional de Educação Sobre Colesterol (NCEP) [100 a 160 mg/dL], dependendo das características no período basal foram distribuídos de modo randômico para receber 10 mg de ezetimiba ou placebo, além da terapia com sinvastatina já em andamento. Entre os pacientes em uso de sinvastatina e que não haviam atingido as metas de LDL-C no período basal (~80%), significativamente mais pacientes distribuídos de modo randômico para a ezetimiba coadministrada com a sinvastatina atingiram as metas de LDL-C no final do estudo em comparação com os pacientes distribuídos de modo randômico para o placebo: 76% e 21,5%, respectivamente. As reduções de LDL-C correspondentes para ezetimiba ou placebo administrados concomitantemente à sinvastatina também foram significativamente diferentes (27% ou 3%, respectivamente). Além disso, a administração concomitante de ezetimiba e sinvastatina diminuiu significativamente o colesterol total, a apo B e os TG em relação à administração concomitante de placebo e sinvastatina.
Em um estudo multicêntrico, duplo-cego, com duração de 24 semanas, 214 pacientes com diabetes mellitus tipo 2 que receberam tiazolidinedionas (rosiglitazona ou pioglitazona) durante 3 meses, no mínimo, e 20 mg de sinvastatina durante 6 semanas, no mínimo, com LDL-C médio de 93 mg/dL, foram distribuídos de modo randômico para receber 40 mg de sinvastatina ou os princípios ativos equivalentes a VYTORIN® 10/20 administrados concomitantemente.
VYTORIN® 10/20 foi significativamente mais eficaz do que a duplicação da dose de sinvastatina para 40 mg na redução adicional do LDL-C (-21% e 0%, respectivamente), do colesterol total (-14% e -1%, respectivamente), da apo B (-14% e -2%, respectivamente) e do colesterol não-HDL (-20% e -2%, respectivamente), além das reduções observadas com 20 mg de sinvastatina. Os resultados para o HDL-C e os TG não foram significativamente diferentes entre os dois grupos de tratamento. Os resultados não foram afetados pelo tipo de tiazolidinediona utilizada.
Administração Concomitante com Fenofibrato
Em um estudo clínico multicêntrico, duplo-cego, controlado com placebo, com duração de até 12 semanas, 611 pacientes com hiperlipidemia mista foram distribuídos de modo randômico para receber placebo, VYTORIN® 10/20 apenas, 160 mg de fenofibrato apenas, ou VYTORIN® 10/20 e 160 mg de fenofibrato.
VYTORIN® administrado concomitantemente ao fenofibrato diminuiu significativamente o colesterol total, o LDL-C, a apo B, o colesterol não-HDL e os TG em comparação com o fenofibrato administrado isoladamente e reduziu significativamente os níveis de apo B, colesterol não-HDL e TG e aumentou os níveis de HDL-C em comparação com VYTORIN® administrado isoladamente (veja Tabela 6).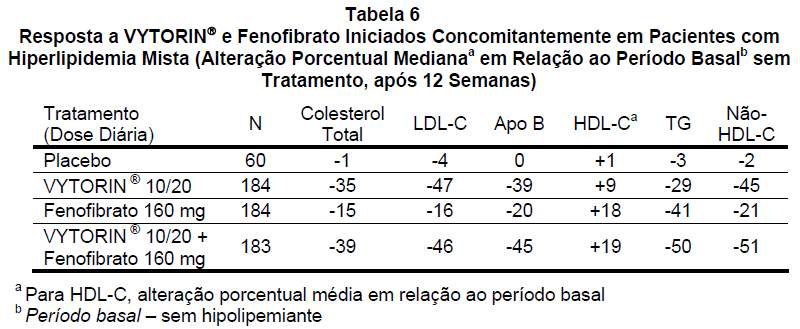
Estudos Clínicos em Pacientes Pediátricos (10 a 17 Anos de Idade)
Em um estudo multicêntrico, duplo-cego, controlado, 142 meninos e 106 meninas pós-menarca, de 10 a 17 anos de idade (média de idade de 14,2 anos) com hipercolesterolemia familiar heterozigótica (HFHe) foram distribuídos de modo randômico para receber a administração concomitante de ezetimiba e sinvastatina equivalentes a VYTORIN® ou sinvastatina apenas. Os critérios de inclusão nesse estudo foram: 1) níveis de LDL-C no período basal situados entre 160 e 400 mg/dL e 2) histórico médico e apresentação clínica compatíveis com HFHe. Os pacientes receberam VYTORIN® (10/10, 10/20 ou 10/40) ou sinvastatina (10 mg, 20 mg ou 40 mg) durante 6 semanas, VYTORIN® 10/40 ou sinvastatina 40 mg nas 27 semanas seguintes e VYTORIN® 10/10, 10/20, ou 10/40 em esquema aberto, durante 20 semanas, subseqüentemente.
Na 6ª semana VYTORIN® (todas as doses) reduziu os níveis de colesterol total, LDL-C, Apo B, e colesterol não-HDL-significativamente mais que a sinvastatina (todas as doses). Os resultados para TG e HDL-C foram semelhantes nos dois grupos de tratamento (veja Tabela 7). Na 33ª semana, VYTORIN® reduziu os níveis de colesterol total, LDL-C, Apo B, TG e colesterol não-HDL significativamente mais que a sinvastatina. Os aumentos de HDL-C foram semelhantes nos dois grupos de tratamento. Além disso, na 33ª semana, significativamente mais pacientes que receberam VYTORIN® 10/40 (63%) atingiram a meta ideal da American Academy of Pediatrics (AAP) para LDLC ( < 110 mg/dL) em comparação com os que receberam sinvastatina 40 mg (27%). Na 53ª semana, as alterações porcentuais médias em relação ao período basal para todas as doses de VYTORIN® foram: -39% (colesterol total); -49% (LDL-C); -23% (apo B); +3% (HDL-C); -17% (TG) e -46% (não-HDL-C).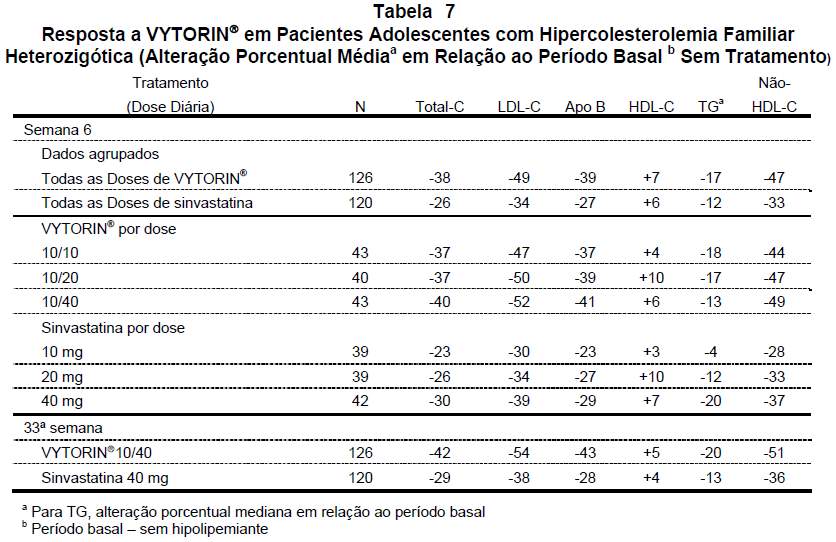
A segurança e a eficácia das doses acima de 10/40 mg/dia não foram estudadas em crianças. A eficácia a longo prazo da terapia com VYTORIN® na infância para reduzir a morbidade e a mortalidade na idade adulta não foi estudada.
Ezetimiba
Em dois estudos multicêntricos, duplo-cegos, controlados com placebo, com duração de 12 semanas, envolvendo 1.719 pacientes com hipercolesterolemia primária, a ezetimiba diminuiu significativamente o colesterol total (13%), o LDL-C (19%), a apo B (14%) e os TG (8%) e aumentou o HDL-C (3%) em comparação com o placebo. A redução de LDL-C foi consistente em relação à idade, sexo, raça e LDL-C no período basal. Além disso, a ezetimiba não exerceu efeito nas concentrações plasmáticas das vitaminas lipossolúveis A, D e E e no tempo de protrombina e não afetou a produção de adrenocorticóides pelas supra-renais.
Sinvastatina
VYTORIN® contém sinvastatina. Em dois estudos clínicos de grande porte, controlados com placebo, o estudo 4S - Estudo Escandinavo de Sobrevida com a Sinvastatina (n= 4.444 pacientes) e o estudo HPS - Estudo de Proteção do Coração (n= 20.536 pacientes), os efeitos do tratamento com a sinvastatina foram avaliados em pacientes sob alto risco de eventos coronarianos por doença coronariana preexistente, diabetes, doença vascular periférica e histórico de AVC ou de outra doença vascular cerebral. A sinvastatina comprovou reduzir o risco de mortalidade por todas as causas (total) ao reduzir as mortes por DAC, o risco de infarto do miocárdio não-fatal e de AVC e a necessidade de procedimentos de revascularização coronariana e não-coronariana. O incremento do benefício na morbimortalidade cardiovascular com VYTORIN®, além do já demonstrado com a sinvastatina, não foi estabelecido.
Hipercolesterolemia Familiar Homozigótica (HFHo)
Foi conduzido um estudo duplo-cego, randômico, com duração de 12 semanas, envolvendo pacientes com diagnóstico clínico e/ou genotípico de HFHo. Foram analisados os dados de um subgrupo de pacientes (n= 14) que recebeu 40 mg de sinvastatina no período basal. O aumento da dose da sinvastatina de 40 mg para 80 mg (n= 5) reduziu o LDL-C em 13% a partir do período basal com 40 mg de sinvastatina. A administração concomitante de ezetimiba e sinvastatina equivalente a VYTORIN® (10/40 e 10/80 agrupados, n= 9) reduziu o LDL-C em 23% em relação ao período basal com 40 mg de sinvastatina. Entre os pacientes que receberam a administração concomitante de ezetimiba e sinvastatina equivalente a VYTORIN® (10/80, n= 5), houve redução de 29% do LDL-C em relação ao período basal com 40 mg de sinvastatina.
Caract. farmacológicas.
VYTORIN® é um hipolipemiante que inibe seletivamente a absorção intestinal de colesterol e de fitosteróis relacionados e a síntese endógena de colesterol.
FARMACOLOGIA CLÍNICA
Mecanismo de Ação
VYTORIN®
O colesterol plasmático é derivado da absorção intestinal e da síntese endógena. VYTORIN® contém ezetimiba e sinvastatina, dois compostos com mecanismos de ação complementares sobre os lípides. VYTORIN® reduz o colesterol total, o colesterol de lipoproteína de baixa densidade (LDL-C), a apolipoproteína B (apo B), os triglicérides (TG) e o colesterol não ligado à lipoproteína de alta densidade (não-HDL-C) elevados e aumenta o colesterol ligado a lipoproteína de alta densidade (HDL-C) por meio da dupla inibição da síntese e da absorção do colesterol.
Ezetimiba
A ezetimiba inibe a absorção intestinal do colesterol; é ativa por via oral e seu mecanismo de ação difere do de outras classes de compostos redutores do colesterol (por exemplo, das vastatinas, dos seqüestrantes de ácidos biliares [resinas], dos derivados do ácido fíbrico e dos fitostanóis). O alvo molecular da ezetimiba é o transportador de esterol, Niemann-Pick_C1-Like (NPC1L1), o qual é responsável pela captação intestinal do colesterol e dos fitosteróis.
A ezetimiba localiza-se na borda em escova dos enterócitos do intestino delgado, onde inibe a absorção do colesterol, diminuindo assim a oferta de colesterol do intestino para o fígado; as vastatinas reduzem a síntese hepática do colesterol e juntos, esses mecanismos distintos propiciam redução complementar do colesterol.
Em um estudo clínico com duração de 2 semanas que envolveu 18 pacientes hipercolesterolêmicos, VYTORIN® inibiu a absorção intestinal de colesterol em 54% quando comparado ao placebo.
Inúmeros estudos pré-clínicos foram realizados para determinar a seletividade da ezetimiba em relação à inibição da absorção do colesterol. A ezetimiba inibiu a absorção do [14C]-colesterol sem afetar a absorção dos TG, dos ácidos graxos, dos ácidos biliares, da progesterona, do etinilestradiol ou das vitaminas lipossolúveis A e D.
Sinvastatina
Após ingestão, a sinvastatina, que é uma lactona inativa, é hidrolisada no fígado ao beta-hidroxiácido ativo correspondente, que tem potente atividade inibitória sobre a HMG-CoA redutase (3 hidróxi-3 metilglutaril CoA redutase). Essa enzima catalisa a conversão da HMG-CoA em mevalonato, uma etapa inicial e limitante da velocidade de biossíntese do colesterol.
A sinvastatina mostrou reduzir concentrações normal e elevada de LDL-C. O LDL é formado a partir da lipoproteína de densidade muito baixa (VLDL) e seu catabolismo ocorre predominantemente pelo receptor de LDL de alta afinidade. O mecanismo do efeito redutor de LDL da sinvastatina pode envolver a redução da concentração de colesterol VLDL (VLDL-C) e a indução do receptor de LDL, o que leva à redução da produção e ao aumento do catabolismo do LDL-C. A apolipoproteína B também baixa consideravelmente durante o tratamento com sinvastatina. Além disso, a sinvastatina aumenta moderadamente o HDL-C e reduz os TG plasmáticos. Como resultado dessas alterações, as razões de colesterol total para HDL-C e de LDL-C para HDL-C são reduzidas.
Farmacocinética
Absorção
Ezetimiba: após administração oral, a ezetimiba é rapidamente absorvida e extensivamente conjugada a um glicuronídeo fenólico farmacologicamente ativo (glicuronídeo de ezetimiba), cujas concentrações plasmáticas máximas (Cmáx) médias ocorrem em 1 a 2 horas; já para a ezetimiba, estas concentrações são atingidas em 4 a 12 horas. A biodisponibilidade absoluta da ezetimiba não pode ser determinada, uma vez que o composto é praticamente insolúvel em meios aquosos apropriados para injeção.
A administração concomitante de alimentos (com altos teores de gorduras ou sem gordura) não exerceu efeito na biodisponibilidade oral da ezetimiba administrada em comprimidos de 10 mg.
Sinvastatina: demonstrou-se que a biodisponibilidade do beta-hidroxiácido para a circulação sistêmica após uma dose oral de sinvastatina foi menor do que 5% da dose, o que é compatível com a ampla extração hepática de primeira passagem. Os principais metabólitos da sinvastatina presentes no plasma humano são o beta-hidroxiácido e quatro metabólitos ativos adicionais.
Em jejum, os perfis plasmáticos dos inibidores total e ativo não foram afetados quando a sinvastatina foi administrada imediatamente antes de uma refeição-teste.
Distribuição
Ezetimiba: a ezetimiba e o glicuronídeo de ezetimiba ligam-se às proteínas plasmáticas humanas em 99,7% e 88% a 92%, respectivamente.
Sinvastatina: a sinvastatina e o beta-hidroxiácido ligam-se às proteínas plasmáticas humanas (95%).
A farmacocinética de doses única e múltipla de sinvastatina não mostrou acúmulo do medicamento após administração múltipla. Em todos esses estudos de farmacocinética, a concentração plasmática máxima dos inibidores ocorreu 1,3 a 2,4 horas após a dose.
Metabolismo
Ezetimiba: a ezetimiba é metabolizada principalmente no intestino delgado e no fígado, por conjugação do glicuronídeo (uma reação de fase II) e excreção biliar subseqüente. Observou-se metabolismo oxidativo mínimo (uma reação de fase I) em todas as espécies avaliadas. A ezetimiba e o glicuronídeo de ezetimiba são os principais derivados do fármaco detectados no plasma, constituindo aproximadamente 10% a 20% e 80% a 90% do total, respectivamente. Tanto a ezetimiba quanto o glicuronídeo de ezetimiba são eliminados lentamente do plasma, com evidência de recirculação êntero-hepática significativa. A meia-vida da ezetimiba e do glicuronídeo de ezetimiba é de aproximadamente 22 horas.
Sinvastatina: a sinvastatina é uma lactona inativa que é rapidamente hidrolisada in vivo para o betahidroxiácido correspondente, um potente inibidor da HMG-CoA redutase. A hidrólise ocorre principalmente no fígado; a velocidade de hidrólise no plasma humano é muito lenta.
A sinvastatina é bem absorvida em humanos e passa por ampla extração hepática de primeira passagem. A extração no fígado depende do fluxo sangüíneo hepático. O fígado é o principal local de ação, com excreção posterior dos equivalentes do fármaco na bile. Conseqüentemente, a disponibilidade do fármaco ativo na circulação sistêmica é baixa.
A meia-vida do metabólito beta-hidroxiácido após uma injeção intravenosa é de 1,9 horas, em média.
Eliminação
Ezetimiba: após administração oral de 20 mg de [14C]-ezetimiba a seres humanos, a ezetimiba total respondeu por cerca de 93% da radioatividade plasmática total. Aproximadamente 78% e 11% da carga radioativa administrada foram recuperados nas fezes e na urina, respectivamente, ao longo de um período de coleta de 10 dias. Após 48 horas, os níveis plasmáticos de radioatividade eram indetectáveis.
Sinvastatina: após uma dose oral de sinvastatina radioativa em humanos, 13% da radioatividade foi excretada na urina e 60% nas fezes em 96 horas. A quantidade recuperada nas fezes representa os equivalentes do fármaco absorvido excretados na bile, assim como o fármaco não absorvido. Após uma injeção intravenosa do metabólito beta-hidroxiácido, apenas 0,3% da dose IV, em média, foi excretada na urina como inibidor.
Contraindicações.
• Hipersensibilidade aos princípios ativos ou a qualquer dos excipientes.
• Hepatopatia ativa ou elevações persistentes e inexplicadas das transaminases séricas.
• Gravidez e lactação (veja ADVERTÊNCIAS, Gravidez e Lactação).
• Quando houver necessidade de administrar VYTORIN® com fenofibrato, consulte a Circular aos Médicos (bula) de fenofibrato.
Advertências e precauções.
Quando houver necessidade de administrar VYTORIN® com fenofibrato, consulte a Circular aos Médicos (bula) de fenofibrato.
Miopatia/Rabdomiólise
A sinvastatina, a exemplo de outros inibidores da HMG-CoA redutase, ocasionalmente provoca miopatia que se manifesta como dor, dolorimento ou fraqueza musculares e creatina quinase (CK) acima de 10 vezes o limite superior da normalidade (LSN). Algumas vezes a miopatia apresenta-se como rabdomiólise, com ou sem insuficiência renal aguda secundária à mioglobinúria e, raramente, pode ser fatal. O risco de miopatia é aumentado por níveis elevados de atividade inibitória da HMG-CoA redutase no plasma.
• VYTORIN® contém sinvastatina, portanto o risco de miopatia/rabdomiólise aumenta com o uso concomitante de VYTORIN® e:
- inibidores potentes do CYP3A4: itraconazol, cetoconazol, eritromicina, claritromicina, telitromicina, inibidores da protease do HIV ou nefazodona, particularmente com doses mais altas de VYTORIN® (veja INTERAÇÕES MEDICAMENTOSAS).
- outros medicamentos: genfibrozila e outros fibratos particularmente com doses mais altas de VYTORIN® . Em um estudo que envolveu 184 pacientes e no qual VYTORIN® 10/20 mg/dia e fenofibrato 160 mg/dia foram administrados concomitantemente durante até 12 semanas, não houve relatos de miopatia. (Veja INTERAÇÕES MEDICAMENTOSAS).
-ciclosporina ou danazol, particularmente com doses mais altas de VYTORIN® (veja INTERAÇÕES MEDICAMENTOSAS).
-amiodarona ou verapamil com doses mais altas de VYTORIN® (veja INTERAÇÕES MEDICAMENTOSAS). Foi relatada miopatia em 6% dos pacientes que receberam 80 mg de sinvastatina e amiodarona em um estudo clínico em andamento.
- diltiazem: pacientes em uso de diltiazem e VYTORIN® 10/80 concomitantemente apresentam risco ligeiramente aumentado de miopatia. Em estudos clínicos, o risco de miopatia em pacientes que tomaram 40 mg de sinvastatina e diltiazem foi semelhante ao dos pacientes que tomaram 40 mg de sinvastatina sem diltiazem (veja INTERAÇÕES MEDICAMENTOSAS).
Ácido Fusídico: pacientes em uso de ácido fusídico, e que passem a receber VYTORIN® concomitantemente podem apresentar risco aumentado de miopatia (veja INTERAÇÕES MEDICAMENTOSAS).
Niacina (≥1 g/dia) (veja INTERAÇÕES MEDICAMENTOSAS, Interações com medicamentos hipolipemiantes que podem causar miopatia quando administrados isoladamente).
• A exemplo do que ocorre com outros inibidores da HMG-CoA redutase, o risco de miopatia/rabdomiólise está relacionado à dose de sinvastatina. Em uma base de dados de estudos clínicos nos quais 41.050 pacientes receberam sinvastatina, dos quais 24.747 (aproximadamente 60%) durante pelo menos 4 anos, a incidência de miopatia foi de aproximadamente 0,02% com 20 mg, 0,08% com 40 mg e 0,53% com 80 mg. Nesses estudos, os pacientes foram cuidadosamente monitorados e alguns produtos medicinais causadores de interação medicamentosa foram excluídos.
Conseqüentemente:
1. O uso concomitante de VYTORIN® com inibidores potentes do CYP3A4 (p. ex, itraconazol, cetoconazol, eritromicina, claritromicina, telitromicina, inibidores da protease do HIV ou nefazodona) deve ser evitado. Se o tratamento com itraconazol, cetoconazol, eritromicina, claritromicina ou telitromicina for inevitável, VYTORIN® deverá ser suspenso durante o curso do tratamento. Deve ser evitado o uso concomitante com outros medicamentos cujos efeitos inibitórios no citocromo CYP3A4 em doses terapêuticas são potentes, a menos que os benefícios do tratamento combinado superem o risco aumentado.
2. A segurança e a eficácia de VYTORIN® administrado com fibratos, exceto fenofibrato, não foram estabelecidas. Portanto, o uso concomitante de VYTORIN® e fibratos, exceto fenofibrato, deve ser evitado.
A administração concomitante de VYTORIN® em doses acima de 10/20 mg/dia e fenofibrato não foi estudada. Deve-se ter cuidado ao prescrever VYTORIN® e fenofibrato, uma vez que o fenofibrato pode causar miopatia quando administrado isoladamente. Em um estudo de 12 semanas no qual 184 pacientes receberam VYTORIN® 10/20 mg/dia + fenofibrato 160 mg/dia, a administração concomitante foi bem tolerada. Em outro estudo de 12 semanas de duração, no qual 411 pacientes receberam sinvastatina 20 mg/dia e fenofibrato 160 mg/dia, a administração concomitante também foi bem tolerada.
Há aumento do risco de miopatia quando a sinvastatina é utilizada concomitantemente com fibratos (especialmente a genfibrozila); o uso combinado de sinvastatina e genfibrozila deve ser evitado a menos que os benefícios superem o aumento do risco com essa combinação. Para pacientes recebendo genfibrozila concomitantemente, a dose de sinvastina não deve exceder 10 mg por dia.
Portanto, embora não recomendado, se VYTORIN® for utilizado em combinação com genfibrozila, a dose não deve exceder 10/10 mg por dia.
3. A dose de VYTORIN® não deve exceder 10/10 mg por dia para pacientes que estiverem recebendo concomitantemente ciclosporina ou danazol. Os benefícios do uso de VYTORIN® para pacientes que estejam recebendo ciclosporina ou danazol devem ser cuidadosamente avaliados em relação aos riscos dessas combinações de medicamentos e deve-se ter cautela ao iniciar a administração de VYTORIN® para pacientes que estejam recebendo ciclosporina (veja INTERAÇÕES MEDICAMENTOSAS).
4. A dose de VYTORIN® não deve exceder 10/20 mg/dia ao dia para pacientes que estejam recebendo concomitantemente amiodarona ou verapamil. O uso combinado de VYTORIN® em doses mais altas do que 10/20 mg ao dia com amiodarona ou verapamil deve ser evitado, a menos que o benefício clínico possa superar o risco aumentado de miopatia.
5. Pacientes recebendo ácido fusídico e VYTORIN® devem ser monitorados com atenção. A suspensão temporária de VYTORIN® pode ser considerada.
6. Deve-se ter cuidado ao se prescrever VYTORIN® e niacina (≥1 g/dia), uma vez que a niacina pode causar miopatia quando administrada isoladamente.
7. No início do tratamento com VYTORIN®, ou quando a dose de VYTORIN® for aumentada, todos os pacientes devem ser advertidos sobre o risco de miopatia e encorajados a relatar imediatamente qualquer dor, dolorimento ou fraqueza musculares inexplicados. O tratamento com VYTORIN® deve ser descontinuado imediatamente se houver suspeita de miopatia ou se esta for diagnosticada. A presença desses sintomas e/ou níveis de CK > 10 vezes o limite superior da normalidade indicam miopatia. Na maioria dos casos, quando os pacientes interrompem imediatamente o tratamento com sinvastatina, os sintomas musculares e o aumento de CK desaparecem. Deve-se considerar a dosagem periódica dos níveis de CK para pacientes que vão iniciar o tratamento com VYTORIN® ou para aqueles cuja dose está sendo aumentada, mas não há garantias de que esse monitoramento evitará miopatia.
8. Muitos dos pacientes que desenvolveram rabdomiólise durante o tratamento com sinvastatina apresentavam antecedentes clínicos complicados, incluindo insuficiência renal, geralmente como conseqüência de diabetes mellitus de longa duração. Esses pacientes requerem monitoração mais rigorosa se estiverem tomando VYTORIN®. O tratamento com VYTORIN® deve ser temporariamente interrompido alguns dias antes de uma cirurgia eletiva de vulto e diante de qualquer afecção clínica ou cirúrgica importante.
Enzimas Hepáticas
Em estudos clínicos controlados da administração concomitante de ezetimiba e sinvastatina foram observados aumentos consecutivos das transaminases séricas ≥ 3 vezes o limite superior da normalidade (veja REAÇÕES ADVERSAS).
Recomenda-se que sejam realizadas provas funcionais hepáticas antes do início do tratamento com VYTORIN® e a seguir, se for clinicamente indicado. Os pacientes titulados para a dose de 10/80 mg devem fazer um novo exame antes da titulação 3 meses após a titulação para a dose de 10/80 mg e periodicamente depois disso (p. ex., semestralmente), durante o primeiro ano do tratamento. Deve ser dada atenção especial aos pacientes que apresentarem aumento dos níveis de transaminases e, nesses pacientes, os exames devem ser repetidos imediatamente e realizados mais freqüentemente a seguir. Se os níveis de transaminase mostrarem evidências de progressão, particularmente se aumentarem para três vezes o limite superior da normalidade e forem persistentes, o medicamento deve ser descontinuado.
VYTORIN® deve ser usado com cautela em pacientes que consomem quantidades consideráveis de álcool e/ou com histórico de doença hepática. Hepatopatias ativas ou elevações inexplicadas e persistentes das transaminases são contra-indicações para o uso de VYTORIN®.
Insuficiência Hepática
Uma vez que os efeitos da exposição aumentada à ezetimiba em pacientes com insuficiência hepática moderada ou grave não são conhecidos, VYTORIN® não é recomendado para esses pacientes (veja USO EM IDOSOS, CRIANÇAS E OUTROS GRUPOS DE RISCO).
Fibratos
A segurança e a eficácia de VYTORIN® administrado com fibratos, exceto fenofibrato, não foram estudadas; portanto, a administração concomitante de VYTORIN® e fibratos, exceto fenofibrato, deve ser evitada (veja INTERAÇÕES MEDICAMENTOSAS).
Fenofibrato
Se houver suspeita de colelitíase em um paciente recebendo VYTORIN® e fenofibrato, deve-se investigar a vesícula biliar e considerar uma terapia hipolipemiante alternativa (veja EFEITOS ADVERSOS e a Circular aos Médicos (bula) de fenofibrato).
Ciclosporina
Para pacientes que estejam recebendo ciclosporina, deve-se ter cautela ao iniciar a terapia com VYTORIN® concomitantemente. As concentrações de ciclosporina devem ser monitoradas em pacientes que recebem VYTORIN® e ciclosporina (veja INTERAÇÕES MEDICAMENTOSAS).
Anticoagulantes
Se VYTORIN® for adicionado à varfarina, outro anticoagulante cumarínico ou fluindiona, a INR (International Normalized Ratio) deve ser apropriadamente monitorada.
Uso durante a Gravidez e Lactação Categoria de risco: X Este medicamento não deve ser utilizado por mulheres grávidas ou que possam ficar grávidas durante o tratamento.
Este medicamento causa malformação ao bebê durante a gravidez.
A aterosclerose é um processo crônico e, geralmente, a descontinuação dos medicamentos hipolipemiantes durante a gravidez deve ter pequeno impacto no risco a longo prazo associado à hipercolesterolemia primária.
VYTORIN®
VYTORIN® é contra-indicado durante a gravidez.
Sinvastatina
A segurança da sinvastatina em mulheres grávidas não foi estabelecida. Não foram conduzidos estudos clínicos controlados com sinvastatina envolvendo mulheres grávidas. Foram raros os relatos de anomalias congênitas após exposição intra-uterina a inibidores da HMG-CoA redutase. Entretanto, em uma análise de aproximadamente 200 gestações acompanhadas prospectivamente, em que houve exposição no primeiro trimestre à sinvastatina ou a outro inibidor da HMG-CoA redutase estreitamente relacionado, a incidência de anomalias congênitas foi comparável à observada na população geral. Esse número de gestações foi estatisticamente suficiente para excluir um aumento ≥ 2,5 vezes de anomalias congênitas em relação à incidência de base.
Embora não haja evidências de que a incidência de anomalias congênitas na prole de pacientes que tomaram sinvastatina - ou outro inibidor da HMG-CoA redutase estreitamente relacionado - seja diferente da observada na população geral, o tratamento da mãe com sinvastatina pode reduzir os níveis fetais de mevalonato, que é um precursor da biossíntese do colesterol. Por isso, VYTORIN® não deve ser usado por mulheres grávidas, que estão tentando engravidar ou com suspeita de gravidez. O tratamento com VYTORIN®deve ser suspenso durante toda a gravidez ou até que seja confirmado que a paciente não está grávida (veja CONTRA-INDICAÇÕES).
Ezetimiba
Não há dados clínicos disponíveis sobre exposição à ezetimiba durante a gestação.
Quando a ezetimiba foi administrada com sinvastatina, não foram observados efeitos teratogênicos em estudos de desenvolvimento embrio-fetal em ratas prenhes. Em coelhas prenhes, a incidência de malformações esqueléticas observada foi baixa.
Lactação
Estudos em ratos mostraram que a ezetimiba é excretada no leite. Não se sabe se os componentes ativos de VYTORIN® são excretados no leite materno humano; portanto, mulheres que estão amamentando não devem tomar VYTORIN®.
Uso Pediátrico
A segurança e a eficácia de VYTORIN® em pacientes de 10 a 17 anos de idade com hipercolesterolemia familiar heterozigótica foram avaliadas em um estudo clínico controlado em adolescentes (meninos e meninas com ao menos um ano pós-menarca). Os pacientes adolescentes que receberam VYTORIN® apresentaram perfil de eventos adversos semelhante ao dos pacientes adultos que receberam VYTORIN® . Doses maiores que 10/40 mg/dia não foram estudadas nessa população. Nesse estudo controlado, não houve efeito detectável sobre o crescimento ou a maturação sexual entre os adolescentes de ambos os sexos, ou qualquer efeito sobre a duração do ciclo menstrual entre as meninas [veja POSOLOGIA E ADMINISTRAÇÃO; REAÇÕES ADVERSAS; e RESULTADOS DE EFICÁCIA, Estudos Clínicos em Pacientes Pediátricos (10 a 17 Anos de Idade)]. VYTORIN® não foi estudado em pacientes com menos de 10 anos de idade ou em meninas na pré-menarca.
EFEITOS SOBRE A CAPACIDADE DE DIRIGIR E OPERAR MÁQUINAS
Não foram realizados estudos dos efeitos sobre a capacidade de dirigir e operar máquinas. No entanto, alguns efeitos adversos que foram relatados com VYTORIN® podem afetar em parte a capacidade de dirigir e operar máquinas. Podem ocorrer variações nas respostas individuais a VYTORIN® (veja EFEITOS ADVERSOS).
USO EM IDOSOS, CRIANÇAS E OUTROS GRUPOS DE RISCO
Uso Pediátrico
A absorção e o metabolismo da ezetimiba são semelhantes em crianças e adolescentes (10 a 18 anos de idade) e adultos. Com base na ezetimiba total, não há diferenças farmacocinéticas entre adolescentes e adultos. Não estão disponíveis dados de farmacocinética na população pediátrica < 10 anos de idade.
Uso em Idosos
As concentrações plasmáticas de ezetimiba total são aproximadamente 2 vezes mais elevadas nos indivíduos idosos (≥ 65 anos de idade) em relação aos jovens (18 a 45 anos de idade). A redução de LDL-C e o perfil de segurança são comparáveis em indivíduos idosos e jovens tratados com ezetimiba.
Insuficiência Hepática
Após dose única de 10 mg de ezetimiba, a área sob a curva (AUC) média para a ezetimiba total aumentou em aproximadamente 1,7 vez em pacientes com insuficiência hepática leve (escore de Child-Pugh de 5 ou 6) em comparação com indivíduos sadios. Em um estudo com duração de 14 dias no qual se administraram doses múltiplas (10 mg diariamente) a pacientes com insuficiência hepática moderada (escore de Child-Pugh de 7 a 9), a AUC média da ezetimiba total aumentou aproximadamente 4 vezes no 1° dia e no 14° dia, em comparação com o observado em indivíduos sadios. Não é necessário ajuste posológico para pacientes com insuficiência hepática leve. Uma vez que os efeitos da exposição aumentada à ezetimiba em pacientes com insuficiência hepática moderada ou grave (escore de Child-Pugh > 9)