TERROSA
GEDEON RICHTER
teriparatida
Antiosteoporótico.
Apresentações.
Terrosa® pack: embalagem com 1 refil de 3 mL, contendo 2,4 mL de solução injetável e 1 caneta aplicadora de uso exclusivo.
Terrosa® refil: embalagem com 1 ou 3 refis de 3 mL, contendo cada um 2,4 mL de solução injetável de uso exclusivo com a caneta aplicadora do Terrosa®
USO SUBCUTÂNEO
USO ADULTO ACIMA DE 18 ANOS
Composição.
Cada mL da solução contém: teriparatida* 250 mcg
Excipientes: ácido acético glacial, acetato de sódio tri-hidratado, manitol, metacresol e água para injetáveis. Ácido clorídrico e/ou hidróxido de sódio podem ter sido adicionados durante a fabricação para ajuste do pH.
*A teriparatida, rhPTH (1-34), produzida na E. Coli, utilizando a tecnologia de DNA recombinante, é idêntica à sequência de aminoácidos 34-N-terminal do hormônio da paratireoide humano endógeno.
Informações técnicas.
1. INDICAÇÕES
Terrosa® é indicado para o tratamento da osteoporose com alto risco para fraturas tanto em mulheres na pós- menopausa como em homens. O alto risco para fraturas inclui uma história de fratura osteoporótica, ou a presença de múltiplos fatores de risco para fraturas, ou falha ao tratamento prévio para osteoporose conforme decisão médica.
Terrosa® também é indicado para o tratamento da osteoporose associada à terapia sistêmica com glicocorticoides, tanto em homens quanto em mulheres.
2. RESULTADOS DE EFICÁCIA
Terrosa® é um medicamento biológico desenvolvido pela via da comparabilidade (biossimilar). O programa de desenvolvimento do produto foi projetado para demonstrar a comparabilidade entre Terrosa® e o medicamento comparador FORTÉO®.
Fatores de risco
Fatores de risco independentes, por exemplo, baixa DMO (Densidade Mineral Óssea), idade, existência de fratura anterior, histórico familiar de fraturas de quadril, alta renovação óssea e baixo índice de massa corporal devem ser considerados para identificar mulheres e homens com risco aumentado de fraturas osteoporóticas que poderiam beneficiar do tratamento.
Mulheres na pré-menopausa com osteoporose induzida por glicocorticoides devem ser consideradas de alto risco de fratura se tiverem uma fratura prevalente ou uma combinação de fatores de risco que as colocam em alto risco de fratura (por exemplo, baixa densidade óssea [por exemplo, T-score ≤ - 2 ], terapia sustentada com altas doses de glicocorticoides [por exemplo, ≥7,5 mg / dia por pelo menos 6 meses], alta atividade da doença subjacente, baixos níveis de esteróides sexuais).
Tratamento da osteoporose em mulheres na pós-menopausa
A segurança e eficácia de teriparatida, uma vez ao dia, por um período de exposição mediano de 19 meses, foram demonstradas em um estudo clínico, duplo-cego, multicêntrico, placebo-controlado em 1.637 mulheres (idade média 69,5 anos) na pós-menopausa com osteoporose. Todas as pacientes receberam 1.000 mg de cálcio e, no mínimo 400 UI de vitamina D por dia. Noventa por cento das pacientes no estudo tinham uma ou mais fraturas vertebrais no início, a DMO vertebral média foi de 0,82 g/cm2 (equivalente a um índice T = -2,6). As radiografias da coluna no início e no final do tratamento foram avaliadas em todas as pacientes. A avaliação final de eficácia primária foi a ocorrência de novas fraturas vertebrais.
Um estudo clínico, fase 4, randomizado, duplo-cego, controlado por com risedronato, de 24 meses incluiu 1.360 mulheres na pós-menopausa com osteoporose estabelecida. Dessas mulheres, 680 foram randomizadas para o grupo teriparatida e 680 foram randomizadas para o grupo risedronato oral, 35 mg semanal. No período basal, a média de idade das participantes era de 72,1 anos com média de 2 fraturas vertebrais prevalentes; 57,9% das pacientes receberam terapia prévia com bisfosfonato e 18,8% administraram glicocorticoides concomitantemente durante o estudo. O estudo de seguimento de 24 meses foi completado por 1.013 (74,5%) pacientes. A dose cumulativa média (mediana) de glicocorticoide foi de 474,3 (66,2) mg no braço de teriparatida e de 898,0 (100,0) mg no braço de risedronato. A ingestão média (mediana) de vitamina D, foi de 1.433 UI/dia (1.400 UI/dia) no braço de teriparatida e de 1.191 UI/dia (900 UI/dia) no braço de risedronato. A incidência de novas fraturas vertebrais em pacientes que tiveram radiografias da coluna vertebral no período basal e durante o acompanhamento, foi de 28/516 (5,4%) no braço de teriparatida, e de 64/533 (12,0%) no braço de risedronato, com risco relativo (IC de 95%) = 0,44 (0,29 - 0,68), P < 0,0001. A incidência cumulativa de fraturas clínicas agrupadas (fraturas clínicas vertebrais e não vertebrais) foi de 4,8% no grupo de teriparatida e de 9,8% no grupo risedronato, com taxa de risco (IC de 95%) = 0,48 (0,32 - 0,74), P = 0,0009.
Efeito sobre a incidência de fraturas
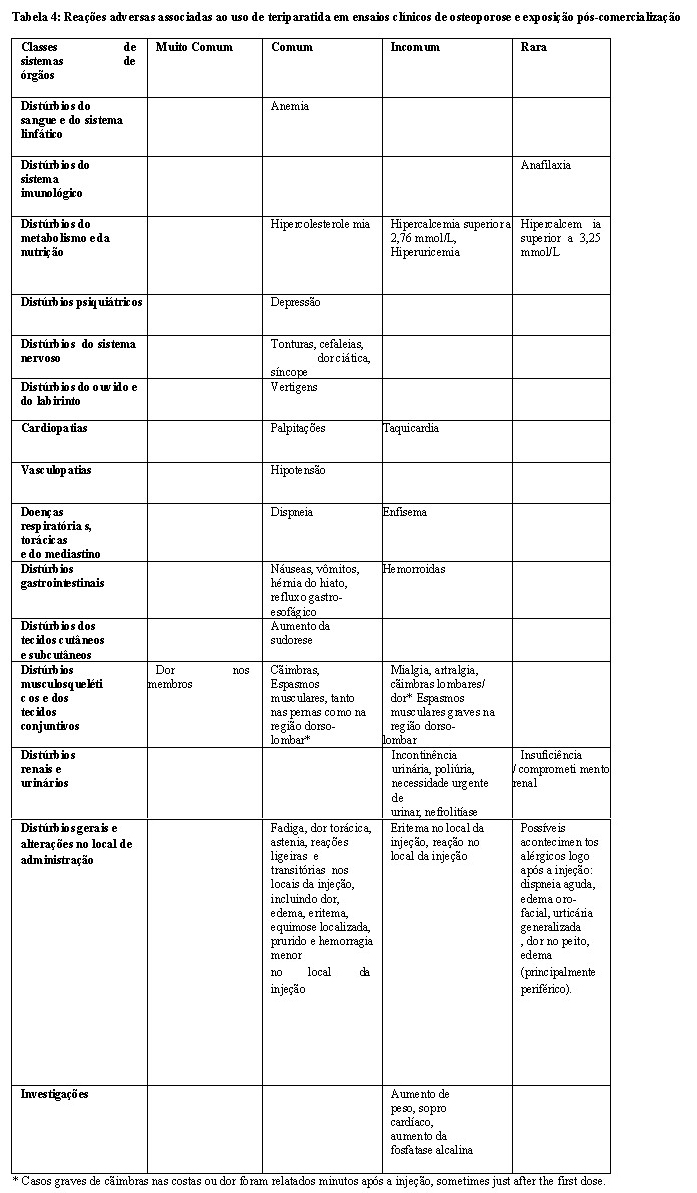
Novas fraturas vertebrais: A teriparatida, quando administrada com cálcio e vitamina D e comparado ao cálcio e vitamina D isolados, reduziu o
Fraturas osteoporóticas não vertebrais: A teriparatida reduziu significativamente a incidência de qualquer fratura não vertebral de 5,5% no
Efeito sobre a DMO: A teriparatida aumentou a DMO da coluna lombar das mulheres na pós-menopausa em 96%. O aumento estatisticamente significativo foi observado em 3 meses e persistiu durante o período de tratamento. O aumento da DMO neste grupo de mulheres foi estatisticamente significante quando comparado com o valor inicial da DMO.
Histologia óssea: os efeitos da teriparatida sobre a histologia óssea foram avaliados em biópsias da crista ilíaca de 35 mulheres na pós-menopausa tratadas por até 2 anos com placebo ou teriparatida 20 mcg ou 40 mcg por dia, cálcio e vitamina D. Os aumentos na DMO e na resistência óssea à fratura alcançados com teriparatida ocorreram sem evidência de toxicidade celular ou efeitos adversos sobre a arquitetura ou mineralização óssea.
Os resultados de um período de tratamento até 24 meses (mediana de 19 meses) com teriparatida, demonstram uma redução de fraturas estatisticamente significativa (Tabela 1). Para prevenir uma ou mais novas fraturas vertebrais, 11 mulheres tiveram que ser tratadas por 19 meses em média.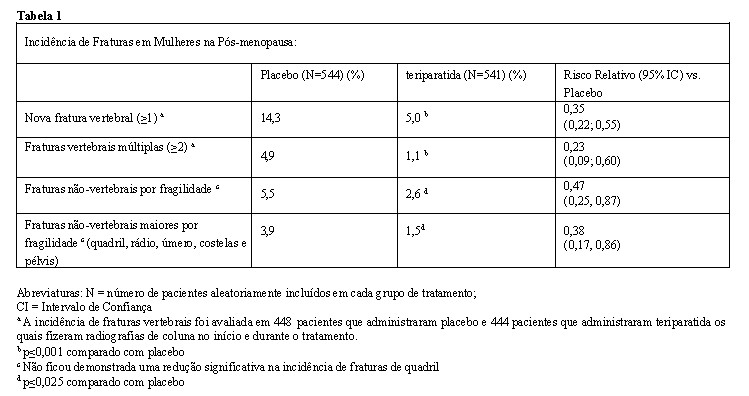
Após 19 meses de tratamento (mediana), a DMO, aumentou na zona da coluna lombar e no total do quadril, em cerca de 9% e 4%, respectivamente, quando comparado com placebo (p < 0,001).
Eficácia pós-tratamento: após tratamento com teriparatida, 1.262 mulheres na pós-menopausa do estudo pivotal, participaram num estudo de seguimento, pós-tratamento. O objetivo principal do estudo foi o de recolher dados de segurança da teriparatida. Durante este período observacional, foram permitidos outros tratamentos para a osteoporose e foi efetuada uma avaliação adicional das fraturas vertebrais.
Durante uma mediana de 18 meses após a interrupção do tratamento com teriparatida, verificou- se a redução de 41% (p = 0,004) comparativamente com placebo no número de pacientes com um mínimo de uma nova fratura vertebral.
Num estudo clínico aberto, 503 mulheres na pós-menopausa com osteoporose grave e uma fratura por fragilidade nos 3 anos anteriores (83%) que tinham recebido terapia prévia para a osteoporose, foram tratadas com teriparatida durante um período máximo de 24 meses, Aos 24 meses, o aumento médio desde o início do estudo da DMO na coluna lombar, quadril total e colo do fêmur foi de 10,5%, 2,6% e 3,9%, respetivamente. O aumento médio da DMO entre 18 e 24 meses foi de 1,4%, 1,2% e 1,6% na coluna lombar, quadril total e colo do fêmur, respectivamente.
Tratamento para aumentar massa óssea em homens com osteoporose primária ou hipogonadal
A eficácia e segurança de teriparatida uma vez ao dia, com exposição média de 10 meses, foi demonstrada em um estudo clínico duplo-cego, multicêntrico, placebo-controlado em 437 homens com osteoporose idiopática ou secundária ao hipogonadismo (definida por uma baixa testosterona livre matinal ou FSH ou LH elevados). No basal, os índices -T da densidade mineral óssea da coluna e do colo do fêmur, foram -2.2 e -2.1, respetivamente. No início do estudo, 35% dos doentes tinham uma fratura vertebral e 59% tinham uma fratura não vertebral. Todos os pacientes receberam 1.000 mg de cálcio e, no mínimo, 400 UI de vitamina D por dia. O objetivo primário de eficácia era uma alteração na DMO da coluna lombar.
Homens tratados com teriparatida tiveram aumentos significantes na DMO da coluna lombar, colo do fêmur, quadril total e corpo inteiro. A teriparatida aumentou a DMO da coluna lombar em homens, com aumentos significativos já nos primeiros 3 meses e que continuaram por todo o período de tratamento. Após 12 meses, a DMO, aumentou na coluna lombar e no quadril total em cerca de 5% e 1%, respetivamente, comparativamente com placebo. No entanto, não ficou demonstrado um efeito significativo nos índices de fraturas
A teriparatida foi eficaz independentemente da idade, taxa inicial de remodelação óssea e DMO inicial.O tratamento com teriparatida aumentou a DMO da coluna lombar desde o início do tratamento, em 94% dos homens tratados.
Tratamento da osteoporose induzida por glicocorticoide
A eficácia da teriparatida para o tratamento da osteoporose induzida por glicocorticoide foi demonstrada em estudo de 18 meses de fase primaria a 36 meses, randomizado, duplo-cego, com comparador controlado (alendronato 10 mg/dia), em 428 pacientes recebendo terapia com glicocorticoide (equivalente a 5 mg ou mais de prednisona por pelo menos 3 meses). Vinte e oito por cento dos pacientes tiveram uma ou mais fraturas vertebrais radiografadas no início do estudo clínico. Todos os pacientes receberam 1.000 mg de cálcio e, no mínimo, 800 UI de vitamina D por dia.
Este estudo incluiu mulheres na pós-menopausa (N = 277), mulheres na pré-menopausa (N = 67) e homens (N = 83). No início do estudo, as mulheres na pós-menopausa tinham uma idade média de 61 anos, escore T médio de DMO da coluna lombar de -2,7, dose equivalente de prednisona mediana de 7,5 mg/dia e 34% tinham uma ou mais fraturas vertebrais radiográficas; mulheres na pré-menopausa tinham idade média de 37 anos, escore T médio de DMO da coluna lombar de -2,5, dose equivalente de prednisona mediana de 10 mg/dia e 9% tinham uma ou mais fraturas vertebrais radiográficas; e os homens tinham idade média de 57 anos, índice T médio de DMO da coluna lombar de - 2,2, dose equivalente de prednisona mediana de 10 mg/dia e 24% tinham uma ou mais fraturas vertebrais radiográficas.
Sessenta e nove porcento dos pacientes concluíram a fase inicial de 18 meses. No desfecho de 18 meses, a teriparatida aumentou significativamente a DMO da coluna vertebral (7,2%) em comparação com alendronato (3,4%) (p < 0,001). A teriparatida aumentou a DMO do quadril total (3,6%) comparado com alendronato (2,2%) (p < 0,01), assim como o colo do fêmur (3,7%) comparado com alendronato (2,1%) (p < 0,05). Em pacientes tratados com teriparatida a DMO da coluna vertebral, quadril total e colo do fêmur aumentou entre 18 e 24 meses um incremento de 1,7%, 0,9% e 0,4%, respectivamente.
Aos 36 meses, a análise de raios-X da coluna vertebral de 169 pacientes com alendronato e 173 pacientes com teriparatida mostrou que 13 pacientes no grupo de alendronato (7,7%) experimentaram uma nova fratura vertebral em comparação com 3 pacientes no grupo de teriparatida (1,7%) (p = 0,01). Além disso, 15 de 214 pacientes no grupo de alendronato (7,0%) experimentaram uma fratura não vertebral em comparação com 16 de 214 pacientes no grupo de teriparatida (7,5%) (p = 0,84).
Em mulheres na pré-menopausa, o aumento na DMO desde o início até o ponto final de 18 meses foi significativamente maior no grupo de teriparatida em comparação com o grupo de alendronato na coluna lombar (4,2% versus -1,9%; p < 0,001) e quadril total (3,8% versus 0,9 %; p = 0,005). No entanto, nenhum efeito significativo nas taxas de fratura foi demonstrado.
Dados Comparativos de Eficácia e Segurança (Terrosa® e Fortéo®)
Estudo RGB-10-00: Estudo Comparativo Farmacocinético/Farmacodinâmico
Este foi um estudo farmacocinético comparativo em mulheres adultas saudáveis, randomizado, controlado por ativo, duplo-cego, centro único e dose fixa única de 20 mg/ 80 mL. Os indivíduos foram randomizados para as sequências de tratamento AB ou BA, onde o Tratamento A foi a administração de dose única de Terrosa® e o Tratamento B foi a administração de dose única de Fortéo®.
Um total de 54 mulheres saudáveis na pré-menopausa entraram no estudo e foram randomizadas para qualquer um dos 2 braços de tratamento. Das pacientes inscritas, 53 completaram o estudo e o conjunto final de análise de farmacocinética consistia em 51 de 54 pacientes.
As comparações estatísticas dos parâmetros de farmacocinética da teriparatida plasmática após a administração de uma dose única subcutânea de Terrosa® 20 mg/80 mL versus uma dose única subcutânea de Fortéo® 20 mg/80 estão resumidas na Tabela 2 abaixo: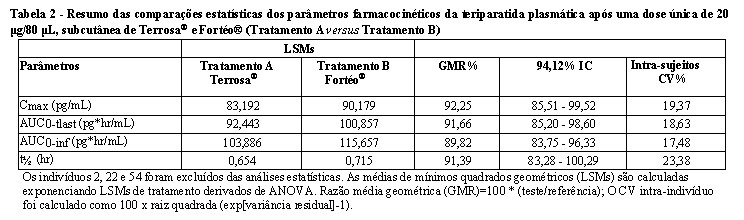
Abreviaturas: AUC0-inf: área sob a curva concentração-tempo do tempo zero ao infinito; AUC0-tlast: área sob a curva concentração-tempo desde o tempo zero até a última concentração quantificável; IC: intervalo de confiança; Cmax: concentração plasmática máxima; t½: meia-vida terminal; tlast: tempo na última concentração quantificável.
Com base nesses resultados, o perfil farmacocinético/farmacodinâmico de Terrosa® é considerado equivalente ao Forsteo®, quando administrado em dose única subcutânea de 20 mg/80 mL.
Estudo RGB1023O31: Estudo Clínico Comparativo de Fase III
O estudo RGB1023O31 foi um estudo comparativo de eficácia/segurança de Fase III, multicêntrico, randomizado, controlado por ativo, de grupos paralelos. O objetivo do estudo foi demonstrar eficácia e segurança comparáveis, incluindo imunogenicidade, de Terrosa® e Fortéo® (teriparatida 20 mg/80 mL solução injetável), dose diária subcutânea administrada por 52 semanas em pacientes japoneses com osteoporose com alto risco de fratura.
O estudo RGB1023O31 foi bem sucedido e demonstrou eficácia comparável de Terrosa® e Fortéo®. Para o desfecho primário, o intervalo de confiança (IC) de 95% bilateral estava dentro do intervalo da margem de equivalência pré- especificada (± 2,8%), confirmando a equivalência terapêutica de Terrosa® para Fortéo®.
2.1 Referências bibliográficas
1. Neer RM, Arnaud CD, Zanchetta JR, Prince R, Gaich GA, Reginster JY, Hodsman AB, Eriksen EF, Ish-Shalom S, Genant HK, Wang O, Mitlak BH. Effect of parathyroid hormone (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med 2001; 344: 1434-41.
2. Lindsay R, Scheele WH, Neer R, Pohl G, Adami S, Mautalen C, Reginster JY, Stepan JJ, Myers SL, Mitlak BH. Sustained vertebral fracture risk reduction after withdrawal of teriparatide in postmenopausal women with osteoporosis. Arch Intern Med 2004; 164(18): 2024-30.
3. Obermayer-Pietsch BM, Marin F, McCloskey EV, Hadji P, Farrerons J, Boonen S, Audran M, Barker C, Anastasilakis AD, Fraser WD, Nickelsen T, EUROFORS Investigators. Effects of two years of daily teriparatide treatment on BMD in postmenopausal women with severe osteoporosis with and without prior antiresorptive treatment. J Bone Miner Res 2008; 23(10): 1591-600.
4. Kendler DL, Marin F, Zerbini CAF, Russo LA, Greenspan SL, Zikan V, Bagur A, Malouf-Sierra J, Lakatos P, Fahrleitner-Pammer A, Lespessailles E, Minisola S, Body JJ, Geusens P, Möricke R, López-Romero P. Effects of teriparatide and risedronate on new fractures in post-menopausal women with severe osteoporosis (VERO): a multicentre, double-blind, double-dummy, randomised controlled trial. Lancet 2018; 391: 230-40.
5. Orwoll ES, Scheele WH, Paul S, Adami S, Syversen U, Diez-Perez A, Kaufman JM, Clancy AD, Gaich GA. The effect of teriparatide [human parathyroid hormone (1-34)] therapy on bone density in men with osteoporosis. J Bone Miner Res. 2003; 18(1): 9-17.
6. Saag KG, Shane E, Boonen S, Marín F, Donley DW, Taylor KA, Dalsky GP, Marcus R. Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N Engl J Med 2007; 357: 2028-39.
7. Saag KG, Zanchetta JR, Devogelaer JP, Adler RA, Eastell R, See K, Krege JH, Krohn K, Warner MR. Effects of teriparatide versus alendronate for treating glucocorticoid-induced osteoporosis thirty-six-month results of a randomized, double-blind, controlled trial. Arthritis Rheum 2009; 60(11): 3346-55.
3 CARACTERÍSTICAS FARMACOLÓGICAS
Grupo farmacoterapêutico: homeostase do cálcio, hormônio da paratireóide e análogos, código ATC: H05AA02
Terrosa® é um medicamento biossimilar que demonstrou semelhança em qualidade, segurança e eficácia ao medicamento comparador FORTÉO Colter Pen.
Descrição: teriparatida, derivada de DNA recombinante, contém hormônio paratireoideano humano recombinante (1- 34) e também é chamado PTHrh (1-34). Ele é o primeiro medicamento de uma nova classe de agentes formadores de osso. A teriparatida tem peso molecular de 4.117,8 daltons e é idêntica ao hormônio paratireoideano humano natural na sequência dos primeiros 34 aminoácidos da porção N-terminal.
3.1 Propriedades farmacológicas
Mecanismo de ação: o hormônio paratireoideano endógeno (PTH) constituído por 84 aminoácidos é o regulador primário do metabolismo de cálcio e fosfato no osso e no rim. A teriparatida (rhPTH(1-34)) é um fragmento ativo (1- 34) do hormônio paratireoideano endógeno humano. As ações fisiológicas do PTH abrangem a estimulação de formação óssea por efeitos diretos nas células formadoras de osso (osteoblastos), e indiretos no aumento da reabsorção tubular renal de cálcio, na excreção do fosfato e no aumento da absorção intestinal de cálcio. As ações biológicas do PTH são mediadas através da ligação aos receptores PTH-específicos na superfície da célula. A teriparatida liga-se a esses receptores com a mesma afinidade do PTH e com as mesmas ações no osso e no rim. Como o PTH endógeno, a teriparatida não se acumula nos ossos ou em outros tecidos.
A teriparatida é um agente de formação óssea para o tratamento da osteoporose. Os efeitos esqueléticos da teriparatida dependem do parâmetro de exposição sistêmica. A administração de teriparatida uma vez ao dia aumenta a aposição de osso novo nas superfícies trabecular e cortical (endósteo e periósteo) do osso pela estimulação preferencial da atividade osteoblástica sobre a atividade osteoclástica. Estes efeitos únicos da teriparatida são manifestados por aumentos rápidos na massa óssea e dos marcadores de remodelação óssea. Ao contrário, o excesso constante do PTH endógeno, como ocorre no hiperparatireoidismo, pode ser prejudicial ao esqueleto, pois a reabsorção óssea pode ser mais estimulada do que a formação óssea.
3.2 Propriedades farmacodinâmicas
Efeitos sobre o metabolismo mineral
A teriparatida afeta o metabolismo do cálcio e do fósforo em um padrão consistente com as ações conhecidas do PTH
endógeno (por exemplo: aumento no cálcio sérico e diminuição de fósforo sérico).
Concentrações séricas de cálcio
Quando 20 mcg de teriparatida são administradas uma vez ao dia, a concentração sérica de cálcio aumenta transitoriamente, começando aproximadamente 2 horas após a administração e atingindo a concentração máxima 4 a 6 horas após a administração (aumento mediano de 0,4 mg/dL). A concentração sérica de cálcio começa a decair aproximadamente 6 horas após a administração e retorna às concentrações iniciais em 16 a 24 horas após cada dose. Não foi observada hipercalcemia mantida em qualquer dose estudada.
Em um estudo clínico de homens com osteoporose primária ou secundária ao hipogonadismo, os efeitos sobre o cálcio sérico foram similares àqueles observados em mulheres na pós-menopausa. A concentração máxima mediana de cálcio sérico medida 4 a 6 horas após a administração de teriparatida foi 9,44 mg/dL nos 12 meses.
Excreção urinária de cálcio
Em um estudo com mulheres na pós-menopausa com osteoporose, que receberam suplementação de cálcio e vitamina D, teriparatida aumentou a excreção urinária de cálcio. A excreção urinária mediana de cálcio foi de 190 mg/dia nos 6 meses e 170 mg/dia nos 12 meses. Os valores medianos nos meses 6 e 12 foram 30 mg/dia e 12 mg/dia mais altos, respectivamente, do que aqueles de pacientes tratadas com placebo. A incidência de hipercalciúria ( > 300 mg/dia) foi semelhante em pacientes tratadas com teriparatida e placebo.
Fósforo e vitamina D
Em estudos de dose única, teriparatida provocou fosfatúria transitória e reduções leves transitórias na concentração de fósforo sérico. Entretanto, não foi observada hipofosfatemia em estudos clínicos com teriparatida.
Em estudos clínicos com a administração diária de teriparatida, o aumento mediano na concentração sérica de 1,25- dihidroxivitamina D aos 12 meses foi de 19% em mulheres, e de 14% em homens. A concentração sérica de 25- hidroxivitamina D em 12 meses reduziu 19% nas mulheres e 10% nos homens.
Efeitos sobre os marcadores de remodelação óssea
A administração diária de teriparatida a mulheres na pós-menopausa e a homens, ambos com osteoporose, estimulou a formação óssea, como mostrado pelos rápidos aumentos dos marcadores no soro, fosfatase alcalina ósseo-específica (FAOS) e peptídeo pró-colágeno tipo I carboxi-terminal (PICP). Dados sobre marcadores bioquímicos de remodelação óssea foram disponibilizados nos primeiros 12 meses de tratamento. Foram observadas em 1 mês de tratamento concentrações máximas de PICP aproximadamente 41% acima das iniciais, seguidas por um declínio a valores próximos dos iniciais após 12 meses. As concentrações de FAOS aumentaram em 1 mês de tratamento e continuaram aumentando mais lentamente de 6 a 12 meses. Os aumentos máximos de FAOS atingidos foram de 45% acima dos valores iniciais em mulheres e de 23% em homens. Após a suspensão da terapia, as concentrações de FAOS voltaram ao valor inicial. Os aumentos nos marcadores de formação foram acompanhados por aumentos secundários nos marcadores de reabsorção óssea: N-telopeptídeo (NTX) e deoxipiridinolina (DPD) urinário, consistentes com o processo de acoplamento fisiológico de formação e reabsorção óssea na remodelação óssea. As alterações de FAOS, NTX e DPD foram um pouco menores em homens que em mulheres, possivelmente devido à exposição sistêmica mais baixa a teriparatida em homens.
3.3 Propriedades farmacocinéticas
A teriparatida é amplamente absorvida após injeção subcutânea e a biodisponibilidade absoluta é de 95%. As velocidades de absorção e eliminação são rápidas. O peptídeo atinge concentrações séricas máximas cerca de 30 minutos após injeção subcutânea de uma dose de 20 mcg e decai para concentrações não quantificáveis dentro de 3 horas. As concentrações molares máximas de teriparatida excedem ligeiramente o limite normal superior para o PTH endógeno em 4 a 5 vezes.
O volume de distribuição é de aproximadamente 1,7 L/kg por via subcutânea. Não foram efetuados estudos de excreção e de metabolismo com a teriparatida, mas acredita-se que o metabolismo periférico do hormônio paratireoidiano ocorre predominantemente no fígado e no rim. A depuração sistêmica da teriparatida (aproximadamente 62L/h em mulheres e 94 L/h em homens) excede a taxa plasmática hepática normal consistente tanto com a depuração hepática e extra-hepática.
A variabilidade entre indivíduos na depuração sistêmica e no volume de distribuição é de 25% a 50%. A meia-vida da teriparatida no plasma é de 5 minutos quando administrada por via intravenosa e aproximadamente 1 hora quando administrada por via subcutânea.
A meia-vida mais longa após a administração subcutânea reflete o tempo necessário para a absorção no local da injeção. De acordo com estudos de fase 4, os pacientes começam apresentar benefício na incidência de fraturas com 6 a 9 meses de tratamento com teriparatida.
Populações especiais
Sexo: a exposição sistêmica à teriparatida é aproximadamente 20% a 30% menor em homens do que em mulheres. Entretanto, nos estudos clínicos não houve diferenças quanto ao sexo com relação à segurança, tolerabilidade ou respostas farmacodinâmicas. Não é necessário ajuste de dose baseado no sexo.
Raça: as populações incluídas nas análises farmacocinéticas foram 98,5% de caucasianos. A influência da raça não pode ser determinada.
Geriátricos: não foram detectadas diferenças na farmacocinética da teriparatida com relação à idade (31 a 85 anos). Não é necessário o ajuste da dose baseada na idade.
Pediátricos: a farmacocinética da teriparatida não foi avaliada em populações pediátricas (ver ADVERTÊNCIAS E PRECAUÇÕES)
Insuficiência renal: não foram identificadas diferenças farmacocinéticas ou de segurança clinicamente relevantes em pacientes com insuficiência
Insuficiência cardíaca: não foram identificadas diferenças de segurança clinicamente relevantes em relação à farmacocinética, pressão sanguínea e
Insuficiência hepática: as células hepáticas de Kuppfer são possivelmente os locais principais para a clivagem do PTH (1-34) e do PTH (1-84) em fragmentos que são eliminados da circulação principalmente pelos rins. Entretanto, não foram realizados estudos em pacientes com insuficiência hepática.
Biodisponibilidade comparativa (bioequivalência)
A equivalência farmacocinética de Terrosa® com o medicamento comparador Fortéo® foi demonstrada em um estudo duplo-cego, randomizado e cruzado de Fase I, RGB-10-001 em 54 voluntários saudáveis. Os parâmetros farmacocinéticos estão resumidos na Tabela 3.
3.4 Dados pré-clínicos de segurança
A teriparatida não foi genotóxica em uma bateria padrão de testes. Não produziu efeitos teratogênicos em ratos, camundongos ou coelhos. Não foram observados efeitos importantes em ratas grávidas ou camundongos aos quais foi administrada teriparatida em doses diárias de 30 a 1000 mcg/kg. No entanto, a reabsorção fetal e a redução do tamanho da ninhada ocorreram em coelhas grávidas com doses diárias de 3 a 100 mcgs/kg. A embriotoxicidade observada em coelhos pode estar relacionada à sua sensibilidade muito maior aos efeitos do PTH no cálcio ionizado no sangue em comparação com roedores.
Ratos tratados com injeções diárias tiveram formação óssea exagerada dependente da dose e aumento da incidência de osteossarcoma, muito provavelmente devido a um mecanismo epigenético. A teriparatida não aumentou a incidência de nenhum outro tipo de neoplasia em ratos. Devido às diferenças na fisiologia óssea em ratos e humanos, a relevância clínica desses achados é provavelmente menor. Nenhum tumor ósseo foi observado em macacas ooforectomizadas tratadas por 18 meses ou durante um período de acompanhamento de 3 anos após a interrupção do tratamento. Além disso, nenhum osteossarcoma foi observado em ensaios clínicos ou durante o estudo de acompanhamento pós-tratamento.
Estudos em animais demonstraram que o fluxo sanguíneo hepático severamente reduzido diminui a exposição do PTH ao sistema de clivagem principal (células de Kupffer) e, consequentemente, a depuração do PTH (1-84).
4 CONTRAINDICAÇÕES
Hipersensibilidade à teriparatida ou a qualquer um dos seus excipientes mencionados na Composição;
Gravidez e amamentação (vide Item 5 "Advertências e Precauções");
Hipercalcemia preexistente (vide Item 5 "Advertências e Precauções");
Insuficiëncia renal grave;
Doenças ósseas metabólicas (incluindo hiperparatiroidismo e a Doença óssea de Paget) exceto osteoporose primária ou osteoporose induzida por glicocorticoide.
Aumentos inexplicáveis da fosfatase alcalina.
Antes da radioterapia externa ou por implante envolvendo os ossos devido ao aumento do risco para osteossarcoma (vide Item 5 "Advertências e Precauções "Carcinogênese e Mutagênese).
Pacientes com neoplasias ósseas ou metástases ósseas devem ser excluídos do tratamento com teriparatida.
Categoria de risco C. Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
5 ADVERTÊNCIAS E PRECAUÇÕES
A teriparatida não foi estudada em pacientes com hipercalcemia. Estes pacientes devem ser excluídos do tratamento com Terrosa® devido à possibilidade de exacerbação da hipercalcemia. A hipercalcemia deve ser excluída antes do tratamento com Terrosa®, mas não é necessária a monitoração de rotina do cálcio sérico durante o tratamento.
Os seguintes grupos de pacientes devem ser excluídos do tratamento com Terrosa®:
-pacientes com malignidades esqueléticas ou metástases ósseas;
-pacientes com outras doenças osteometabólicas diferentes da osteoporose (incluindo hiperparatireoidismo e Doença de Paget do osso) e aqueles pacientes com elevações inexplicadas da fosfatase alcalina sérica.
Terrosa® não deve ser usado em pacientes previamente submetidos a radioterapia externa ou radioterapia por implante envolvendo os ossos, uma vez que estes pacientes possuem um risco basal aumentado para osteossarcoma (ver Carcinogênese, mutagênese, danos à fertilidade).
Urolitíase
Terrosa® não foi estudado em pacientes com urolitíase ativa; contudo, nenhum aumento em urolitíase foi observado nos estudos clínicos. Se houver suspeita de urolitíase ativa ou hipercalciúria preexistente, deve ser considerada a medida da excreção de cálcio urinário. Terrosa® deve ser usado com cuidado em pacientes com urolitíase ativa ou recente devido ao potencial de exacerbação desta condição.
Hipotensão ortostática
Em estudos clínicos a curto prazo com teriparatida, foram observados episódios isolados de hipotensão ortostática transitória. Tipicamente, o evento iniciou nas 4 horas após a administração e desapareceu espontaneamente dentro de alguns minutos a poucas horas. Quando ocorreu hipotensão ortostática transitória, aconteceu nas primeiras doses, sendo aliviada pelo posicionamento dos pacientes em uma posição reclinada, e não impediu a continuação do tratamento.
Testes de laboratório Cálcio
sérico
A teriparatida pode induzir aumentos pequenos e transitórios do cálcio sérico com efeito máximo observado em aproximadamente 4 a 6 horas pós-dose. Caso o cálcio sérico seja avaliado, amostras de sangue devem ser coletadas pelo menos 16 horas após a administração de teriparatida para que haja tempo suficiente para ocorrer a diminuição dos efeitos da teriparatida.
Cálcio urinário
A teriparatida pode causar pequenos aumentos na excreção urinária de cálcio, mas a incidência de hipercalciúria não diferiu dos pacientes tratados com placebo em estudos clínicos (vide Item 3. Características Farmacológicas, Propriedades Farmacodinâmicas).
Ácido úrico sérico
A teriparatida pode causar pequenos aumentos nas concentrações séricas de ácido úrico. Em estudos clínicos, 3% dos pacientes tratados com teriparatida tiveram a concentração de ácido úrico elevada comparado a 1% dos pacientes tratados com placebo. Entretanto, a hiperuricemia não resultou em aumento de gota, urolitíase ou artralgia.
Função renal
Não foram observados eventos adversos renais significativos em estudos clínicos. As avaliações incluíram depuração de creatinina, medidas de ureia no sangue, creatinina e eletrólitos no soro, densidade e pH da urina e exame do sedimento urinário. Não foi realizada avaliação a longo prazo de pacientes com insuficiência renal grave, pacientes em diálise crônica ou pacientes com transplante renal. Precauções e cuidados devem ser tomados em pacientes com insuficiência renal moderada.
Imunogenicidade
No estudo clínico, os anticorpos com reação cruzada com a teriparatida foram detectados em 3,0% das pacientes recebendo teriparatida. Geralmente, anticorpos foram detectados primeiramente após 12 meses de tratamento e diminuíram após a retirada da terapia. Não houve evidências de reações de hipersensibilidade e de reações alérgicas entre estes pacientes. A formação de anticorpo não teve efeito aparente sobre o cálcio sérico ou sobre a resposta da DMO.
Carcinogênese, mutagênese, danos à fertilidade
Carcinogênese
Um estudo inicial de carcinogenicidade, em ratos com tempo de vida próximo a 2 anos, tratados com injeções diárias de teriparatida, apresentaram formação óssea exagerada e aumento na incidência de marcadores de osteossarcoma dependentes da dose administrada, muito provavelmente devido a mecanismo epigenético. A teriparatida não ocasionou aumento da incidência de neoplasia em outros tecidos.
O segundo estudo com ratos (com duração de 2 anos) foi realizado para avaliar se a ocorrência de osteossarcoma foi dependente da dose e duração do tratamento. O nível sem efeito observado (NOEL) foi identificado e é equivalente a 3 vezes a exposição humana a uma dose de 20 mcg, baseada na comparação da Área sob a Curva (AUC). A relevância desses achados para os humanos é incerta.
Em estudo de longo prazo em macacos e macacas maduras do ponto de vista esquelético foram tratados por 18 meses com teriparatida diária, em exposições de aproximadamente 6 vezes maior do que observados em humanos e então foram observados por um período adicional de 3 anos. Não foram detectados tumores ósseos.
Aumento do risco de osteossarcoma não foi observado em pacientes tratados com teriparatida em estudos observacionais. A ocorrência de osteossarcoma não foi observada nos estudos clínicos com teriparatida.
Até que mais dados clínicos estejam disponíveis, a duração do tratamento recomendada de 24 meses, não deve ser ultrapassada.
Mutagênese
A teriparatida não foi genotóxica em quaisquer dos seguintes sistemas de teste: teste de Ames para mutagênese bacteriana com e sem ativação metabólica, teste de linfoma de camundongo para mutação de células de mamíferos, teste de aberração cromossômica em células de ovário de hamster chinês e teste de micronúcleos in vivo em camundongos.
Danos à fertilidade
A teriparatida não teve efeitos sobre a fertilidade de ratos machos ou fêmeas em doses subcutâneas de até 300 mcg/Kg.
Estudos em coelhos demonstraram toxicidade reprodutiva (vide Item 3. Características Farmacológicas, Dados pré- clínicos de segurança). O efeito da teriparatida no desenvolvimento do feto não foi estudado. O risco potencial para o ser humano é desconhecido.
Gravidez
Categoria C - A teriparatida não produziu efeitos teratogênicos em fêmeas de ratos, camundongos ou coelhos. O efeito do tratamento com teriparatida sobre o desenvolvimento do feto humano não foi estudado. Teriparatida não deve ser administrada em mulheres grávidas.
As mulheres em risco de engravidar deverão utilizar um método contraceptivo eficaz durante a utilização de teriparatida. Se ocorrer gravidez, a teriparatida deve ser interrompida
Lactantes
A teriparatida é contraindicada em mulheres que estejam amamentando, pois não houve estudos clínicos para determinar se a teriparatida é secretada no leite materno.
Uso pediátrico e adolescentes
A segurança e eficácia da teriparatida em crianças e adolescentes com idade inferior a 18 anos não foi estabelecida. A teriparatida não deve ser administrada em pacientes pediátricos ou adolescentes (menores do que 18 anos) com epífase aberta.
A experiencia em população jovem, incluindo mulheres na pré menopausa, é limitada. O tratamento apenas deve ser iniciado se os benefícios forem claramente superiores aos riscos.
Uso geriátrico
Dos pacientes recebendo teriparatida no estudo clínico de tratamento da osteoporose de 1.637 mulheres na pós- menopausa, 75% tinham 65 anos ou mais e 23% tinham 75 anos ou mais. A segurança e eficácia da teriparatida foram similares independente da idade.
Dos pacientes recebendo teriparatida no estudo clínico do tratamento da osteoporose de 437 homens, 39% tinham 65 anos ou mais e 13% tinham 75 anos ou mais. A segurança e eficácia da teriparatida foram similares independente da idade.
Alguns pacientes podem sentir tontura após a administração de teriparatida. Caso o paciente sinta este sintoma, ele não deve dirigir ou operar máquinas até que se sinta melhor.
Rastreabilidade
A fim de melhorar a rastreabilidade dos medicamentos biológicos, o nome e o número do lote de cada refil administrado devem ser registados de forma clara pelo paciente no calendário.
Efeitos sobre a capacidade de dirigir veículos e operar máquinas
A teriparatida tem pouca ou nenhuma influência sobre a capacidade de conduzir e utilizar máquinas.
Alguns pacientes podem sentir hipotensão ortostática transitória ou tontura após a administração da teriparatida. Caso o paciente sinta estes sintomas, ele não deve dirigir ou operar máquinas até que se sinta melhor.
Teor de sódio
Terrosa®contém menos do que 1 mmol (23 mg) de sódio por dose, ou seja, é praticamente isento de sódio.
6 INTERAÇÕES MEDICAMENTOSAS
Não foram identificadas interações medicamentosas clinicamente significantes em estudos farmacodinâmicos agudos realizados entre teriparatida e os seguintes medicamentos: hidroclorotiazida, furosemida, atenolol e preparações de liberação prolongada de diltiazem, nifedipina, felodipina e nisoldipina.
Num estudo com 15 indivíduos saudáveis aos quais foi administrada digoxina diariamente até ao estado estacionário, uma dose única de teriparatida não alterou o efeito cardíaco da digoxina. No entanto, relatos de casos esporádicos sugeriram que a hipercalcemia pode predispor os pacientes à ãtoxicidade digitálica. Como a teriparatida aumenta transitoriamente o cálcio sérico, deve ser usada com cautela em pacientes que fazem uso de digitálicos.
A coadministração de raloxifeno ou terapia de reposição hormonal com teriparatida não alterou os efeitos sobre o cálcio sérico ou urinário ou sobre os eventos adversos.
Terrosa® pode ser administrado com alimento.
Não foram conduzidos estudos para avaliar a interação de teriparatida com plantas medicinais, álcool, nicotina e exames não laboratoriais.
7 CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Terrosa®deve ser armazenado sob refrigeração (2 a 8°C). Após a inserção do refil na caneta, o conjunto caneta-refil deve ser colocado no refrigerador imediatamente após a utilização.
Durante o período de uso, o tempo de exposição à temperatura ambiente deve ser minimizado e a dose deve ser administrada imediatamente
após a retirada de Terrosa®do refrigerador. Não congelar. Não usar Terrosa® se tiver sido congelado. Antes de inserir na caneta, manter o refil dentro da embalagem exterior para proteger da luz.
O prazo de validade do produto é de 24 meses quando mantido sob refrigeração, antes do primeiro uso. Após a primeira injeção, o produto deve ser usado em até 28 dias. Após esse período, o refil deve ser descartado, de acordo com as orientações locais, mesmo se ainda