TASIGNA
NOVARTIS
nilotinibe
Antineoplásico.
Apresentações.
Cápsulas - via oral. Embalagem com 112 cápsulas de 200 mg.
USO ADULTO
Composição.
Cada cápsula de Tasigna® contém 220,60 mg de cloridrato de nilotinibe monoidratado, equivalente a 200 mg de nilotinibe. Excipientes: lactose monoidratada, crospovidona, poloxaleno 188, dióxido de silício, estearato de magnésio. Componentes da cápsula: gelatina, dióxido de titânio, óxido de ferro amarelo.
Indicações.
Tasigna® é indicado para o tratamento de pacientes adultos com leucemia mieloide crônica (LMC) cromossomo Philadelphia positivo em fase crônica ou em fase acelerada após falha ou intolerância a pelo menos uma terapia prévia, incluindo imatinibe.
Resultados de eficácia.
LMC Ph+ após falha ou intolerância: Um estudo aberto, multicêntrico, de fase II foi conduzido para determinar a eficácia de Tasigna® em pacientes com LMC após falha ou intolerância ao imatinibe, com braços separados de tratamento para as fases crônica (FC) e acelerada (FA) da doença. O estudo está em andamento. A eficácia foi baseada em 321 pacientes envolvidos em FC e em 137 pacientes envolvidos em FA. A duração mediana do tratamento foi de 561 dias e 264 dias, respectivamente (vide Tabela 2). Tasigna® foi administrado continuamente (duas vezes ao dia, 2 horas após a refeição e sem alimentação adicional após pelo menos uma hora) a menos que houvesse uma evidência de resposta inadequada ou de progressão da doença. O escalonamento da dose para 600 mg duas vezes ao dia foi permitido.
Os critérios considerados na resistência ou falha ao imatinibe incluíram falha para obter resposta hematológica completa (após 3 meses), resposta citogenética (após 6 meses) ou resposta citogenética maior (após 12 meses) ou progressão da doença após respostas prévias citogenética ou hematológica. Os critérios considerados na intolerância ao imatinibe incluíram pacientes que descontinuaram imatinibe devido a toxicidade e por não apresentarem respostas citogenéticas maior na ocasião em que ingressaram no estudo.
Em geral, 73% dos pacientes eram resistentes e 27% eram intolerantes ao imatinibe. A maioria dos pacientes tinha um longo histórico de LMC, que incluía extensos tratamentos anteriores com outros agentes antineoplásicos tais como imatinibe, hidroxiureia, interferona e alguns destes pacientes também apresentavam transplante de medula óssea prévio mal sucedido. A média da dose anterior mais alta de imatinibe foi de 600 mg/dia para pacientes em FC e em FA, e a dose anterior mais alta de imatinibe foi > 600 mg/dia em 74% de todos pacientes, sendo 40% dos pacientes com doses prévias de imatinibe ≥ 800 mg/dia.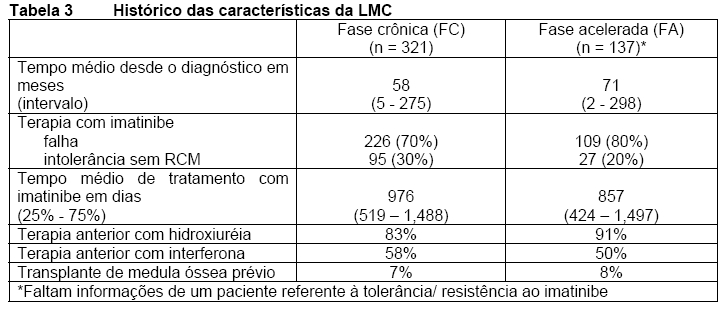
O objetivo principal nos pacientes em FC foi a resposta citogenética maior (RCM), definida como eliminação (RCC, resposta citogenética completa) ou redução significante para < 35% das metáfases Ph+ (resposta citogenética parcial) das células hematopoiéticas Ph+. A resposta hematológica completa (RHC) em pacientes na FC foi avaliada como um objetivo secundário. O objetivo principal nos pacientes em FA foi a resposta hematológica (RH) global confirmada, definida também como uma resposta hematológica completa, nenhuma evidência de leucemia ou retorno à fase crônica.
Fase crônica: A taxa de RCM nos 321 pacientes em FC foi 59%. A maioria dos respondedores atingiu rapidamente a RCM, dentro de 3 meses (média de 2,8 meses) a partir do início do tratamento com Tasigna®, e estas respostas foram mantidas. A taxa de RCC foi de 44%. O tempo médio para atingir RCM foi de 3 meses (média de 3,3 meses). Dos pacientes que atingiram RCM, 77% (IC 95%: 71%-84%) manteve a resposta aos 24 meses. A duração média da RCM não foi atingida. Dos pacientes que atingiram RCC, 84% (IC 95%: 77%-91%) mantiveram a resposta aos 24 meses. A duração média da RCC não foi atingida. Pacientes com RHC ao início atingiram RCM mais rápido (1,4 vs 2,8 meses). Dos pacientes em FC sem RHC ao início, 76% atingiram RHC, e o tempo médio para a RHC foi de 1 mês, com a duração média ainda não atingida. A taxa de sobrevida global estimada aos 24 meses para pacientes com LMC - FC foi de 87%.
Fase acelerada: A taxa global de RH confirmada em 137 pacientes em FA foi de 55%. A maioria dos respondedores atingiu precocemente a RH no tratamento com Tasigna® (média de 1 mês) e isto foi duradouro (duração média de RH confirmada foi de 21,5 meses). Dos pacientes que atingiram RH, 49% (IC 95%: 35%-62%) mantiveram resposta aos 24 meses. A taxa de RCM foi de 32% com tempo médio para resposta de 2,8 meses. Dos pacientes que atingiram RCM, 66% (IC 95%: 50%-82%) mantiveram resposta aos 24 meses. A duração média da RCM não foi atingida. As taxas de resposta para os dois braços de tratamentos estão relatadas na Tabela 4.
A taxa de sobrevida total estimada aos 24 meses para pacientes com LMC - FA foi de 70%.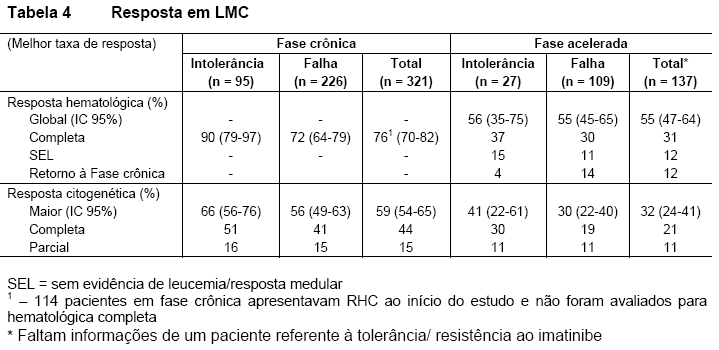
Os braços separados de tratamento também foram incluídos no estudo de fase 2 para avaliar Tasigna® em um grupo de pacientes em FC e FA, que foram extensivamente pré-tratados com múltiplas terapias incluindo agentes inibidores da tirosinoquinase em adição ao imatinibe. Destes pacientes, 30/36 (83%) eram resistentes ao tratamento e não apresentavam intolerância. Em 22 pacientes em FC avaliados para eficácia, Tasigna® induziu uma taxa de RCM em 32% e uma taxa de RHC de 50%. Em 11 pacientes em FA avaliados para eficácia, o tratamento induziu uma taxa de RH global de 36%.
Após falha do imatinibe, 24 mutações diferentes do BCR-ABL foram observadas em 42% dos pacientes com LMC em fase crônica e 54% dos pacientes com LMC em fase acelerada que foram avaliados para mutações. Tasigna® demonstrou eficácia em pacientes com presença de uma variedade de mutações do BCR-ABL associadas com resistência ao imatinibe, exceto T315I.
Adicionalmente, com o objetivo de confirmar o perfil de segurança do nilotinibe em uma população maior de pacientes, o estudo 2109, um estudo fase 3, aberto, multicêntrico, internacional, em pacientes adultos com LMC em fase crônica, fase acelerada e crise blástica, resistentes ou intolerantes ao imatinibe foi conduzido. Em comparação com o estudo fase 2 anteriormente descrito, o número total de pacientes do estudo 2109 com LMC em fase crônica e em fase acelerada foi de 1.219 e 157 pacientes respectivamente. Na população em FC, 566 foram avaliados para resposta hematológica e 532 foram avaliados para resposta citogénetica. No grupo em FA, 90 pacientes foram avaliados para resposta hematológica e 84 pacientes para resposta citogenética, respectivamente.
Nos pacientes com LMC em FC, foi observada uma taxa de resposta hematológica de 62%, sendo 41% resposta hematológica completa. Em relação a resposta citogenética, obteve-se uma taxa de resposta citogenética maior de 42%, sendo 26% resposta citogenética completa e 15% parcial. A duração mediana de exposição ao nilotinibe nos pacientes em FC foi de 184 dias.
Nos pacientes com LMC em FA, a taxa de resposta hematológica foi de 44%, sendo 22% em resposta hematológica completa, e uma taxa de resposta citogenética maior de 12%, sendo 6% às custas de resposta citogenética completa e 6% parcial. Retornaram à fase crônica 10% dos pacientes e 12% não tinham evidência de leucemia ao exame da medula óssea. A duração mediana de exposição ao nilotinibe nos pacientes em FA foi de 137 dias.
Caract. farmacológicas.
Propriedades farmacodinâmicas
Grupo farmacoterapêutico: agentes antineoplásicos - inibidor da proteína tirosinoquinase. Código ATC: L01XE08.
Tasigna® é um potente e seletivo inibidor da atividade -tirosinoquinase-ABL da oncoproteína BCR-ABL em linhagens celulares e principalmente em células leucêmicas cromossomo Philadelphia positivo. O medicamento se liga fortemente ao sítio de ligação do ATP de tal forma que se torna um potente inibidor do tipo selvagem da BCR-ABL e mantém atividade contra 32 das 33 formas mutantes da BCR-ABL resistentes ao imatinibe. Como consequência de sua atividade bioquímica, o nilotinibe inibe seletivamente a proliferação e induz a apoptose em linhagens celulares e, principalmente, em células leucêmicas cromossomo Philadelphia positivo de pacientes com LMC. Em modelos murinos de LMC, em monoterapia com administração oral, nilotinibe reduz a carga tumoral e prolonga a sobrevida.
Tasigna® tem pouco ou nenhum efeito sobre a maioria das outras proteínas-quinase avaliadas, incluindo Src, com exceção das seguintes quinases: PDGF, Kit CSF-1R, DDR e efrina, as quais são inibidas nas concentrações médias atingidas após administração oral nas doses terapêuticas recomendadas para o tratamento da LMC (vide Tabela 1).
Propriedades farmacocinéticas
Absorção
Os picos de concentração de nilotinibe são alcançados 3 horas após a administração oral. A absorção de nilotinibe após administração oral foi aproximadamente de 30%. A biodisponibilidade absoluta de nilotinibe não foi determinada. Em comparação com uma solução oral (pH de 1,2 a 1,3), a biodisponibilidade relativa das cápsulas de nilotinibe é aproximadamente 50%. Em voluntários sadios, a Cmáx e a área sob a curva (AUC) da concentração sérica x tempo do nilotinibe aumentam em 112% e 82%, respectivamente, quando Tasigna® é administrado com alimento comparados com jejum. A administração de Tasigna® 30 minutos ou 2 horas após a refeição aumenta a biodisponibilidade de nilotinibe para 29% ou 15%, respectivamente (vide "Posologia", "Precauções", "Advertências" e "Interações medicamentosas e outras formas de interação"). A absorção de nilotinibe (biodisponibilidade relativa) pode ser reduzida em aproximadamente 48% e 22% em pacientes com gastrectomia total e parcial, respectivamente.
Doses únicas de 400 mg de nilotinibe, administradas usando 2 cápsulas de 200 mg em que o conteúdo de cada cápsula foi disperso em uma colher de sopa de suco de maçã, mostraram ser bioequivalentes em relação a administração única de 2 cápsulas intactas de 200 mg.
Distribuição
A razão sangue/plasma do nilotinibe é 0,68. A ligação às proteínas plasmáticas foi de aproximadamente 98% com base em estudos in vitro.
Biotransformação
As principais vias metabólicas identificadas em indivíduos sadios foram a oxidação e a hidroxilação. O principal componente sérico circulante foi o nilotinibe. Nenhum dos metabólitos contribuiu significantemente para a atividade farmacológica do nilotinibe.
Eliminação
Após uma dose única de nilotinibe radiomarcado em indivíduos sadios, mais de 90% da dose foi eliminada dentro de 7 dias, principalmente nas fezes. A droga-mãe respondeu por 69% da dose.
Linearidade/não-linearidade
A exposição de nilotinibe no estado de equilíbrio foi dose-dependente, com aumentos menos proporcionais à dose na exposição sistêmica com doses maiores de 400 mg administradas uma vez ao dia. A exposição sérica diária de 400 mg de nilotinibe duas vezes ao dia foi 35% maior no estado de equilíbrio do que com a administração de 800 mg uma vez ao dia. Não houve aumento relevante na exposição ao nilotinibe quando a dose foi aumentada de 400 mg duas vezes ao dia para 600 mg duas vezes ao dia.
Características nos pacientes
As condições de estado de equilíbrio foram alcançadas no oitavo dia. Um aumento na exposição sérica de nilotinibe entre a primeira dose e o estado de equilíbrio foi de, aproximadamente, 2 vezes na dose de uma vez ao dia e de 3,8 vezes na dose de duas vezes ao dia. A meia-vida de eliminação aparente estimada da farmacocinética de dose múltipla com doses diárias foi de aproximadamente 17 horas. A variabilidade da farmacocinética do nilotinibe interpaciente, foi considerada de moderada a alta.
Dados de segurança pré-clínica
O nilotinibe foi avaliado quanto à segurança farmacológica, a toxicidade de dose repetida, a genotoxicidade, em estudos de toxicidade reprodutiva, estudos de fototoxicidade e um estudo de carcinogenicidade em ratos.
O nilotinibe não teve efeitos no SNC e nas funções respiratórias. Estudos de segurança cardíaca in vitro demonstraram um sinal pré-clínico de prolongamento do intervalo QT. Nenhum efeito foi observado em ECG de cães e macacos tratados até 39 semanas ou em estudos telemétricos especiais em cães.
Estudos de toxicidade de dose repetida em cães com duração de até 4 semanas e em macacos Cynomolgus com duração de até 9 meses, revelaram que o fígado é o principal órgão afetado pela toxicidade do nilotinibe. Alterações incluindo aumento de atividade da alanina aminotransferase e da fosfatase alcalina, e achados histopatológicos (principalmente hiperplasia/hipertrofia das células sinusoidais ou nas células de Kupffer, hiperplasia do canal da bile e fibrose periportal). Em geral, as alterações clínicas e bioquímicas foram totalmente reversíveis após um período de recuperação de quatro semanas, as alterações histológicas apresentaram apenas reversibilidade parcial. Exposições às menores doses que mostraram efeitos hepáticos foram menores que a exposição em humanos na dose de 800 mg/dia. Foram observadas apenas alterações hepáticas pouco importantes em camundongos ou ratos tratados por até 26 semanas. Em ratos, cães e macacos foram observados aumentos geralmente reversíveis nos níveis de colesterol. No estudo de 2 anos de carcinogenicidade em ratos, o maior órgão afetado para lesões não-neoplásicas foi o útero (dilatação, ectasia vascular, hiperplasia de células endoteliais, inflamação e/ou hiperplasia epitelial).
Estudos de genotoxicidade em sistemas bacterianos in vitro e em sistemas de mamíferos in vitro e in vivo com e sem ativação metabólica não revelaram nenhuma evidência de um potencial mutagênico para o nilotinibe.
Nenhuma evidência de carcinogenicidade em um estudo de 2 anos de carcinogenicidade com ratos sobre a administração de nilotinibe a 5, 15 e 40 mg/kg/dia. Exposições (em termos de AUC) na dose mais elevada foram representadas aproximadamente duas vezes a três vezes a exposição diária humana (baseada na AUC) ao nilotinibe no estado de equilíbrio na dose de 800 mg/dia.
O nilotinibe não induziu teratogenicidade, mas demonstrou embrio e fetotoxicidade em doses nas quais também apresentou toxicidade materna. Um aumento de abortos pós-implantação foi observado em ambos estudos de fertilidade, tanto no tratamento de machos como de fêmeas, e no estudo de embriotoxicidade com o tratamento de fêmeas. Embrioletalidade e efeitos fetais (principalmente pesos fetais diminuídos, variações viscerais e esqueléticas) em ratos e reabsorção aumentada de fetos e variações esqueléticas em coelhos foram detectadas nos estudos de embriotoxicidade. Fêmeas expostas ao nilotinibe em doses menores ou iguais às observadas em humanos nas doses de 800 mg/dia não apresentaram efeitos adversos.
Em um estudo pré e pós-natal, a administração oral de nilotinibe para ratas fêmeas desde o 6° dia de gestação ao 21° dia ou 22° após o parto, resultou em efeitos maternos (consumo reduzido de alimentos e menores ganhos de peso) e em períodos de gestação mais longos na dose de 60 mg/kg. A dose materna de 60 mg/kg foi associada com peso diminuído do filhote e mudanças em alguns parâmetros físicos de desenvolvimento (o dia médio para desdobramento da orelha, erupção dos dentes e abertura dos olhos foi mais precoce). A dose de 20 mg/kg não apresentou efeitos adversos na fêmea e na cria.
Em um estudo de desenvolvimento juvenil, nilotinibe foi administrado via oral por gavage a ratos a partir da primeira semana após o parto até a idade adulta (70 dias após o parto) em doses de 2, 6 e 20 mg/kg/dia. Os efeitos foram limitados a dose de 20 mg/kg/dia e consistiram em reduções nos parâmetros de peso e consumo de alimento com recuperação após o fim da administração. A dose de 6 mg/kg/dia em ratos jovens não apresentou efeitos adversos. Em geral, o perfil de toxicidade nos ratos jovens foi comparável ao observado em ratos adultos.
O nilotinibe mostrou ter absorção de luz nas frequências UV-A e UV-B, e ser distribuído na pele, apresentando potencial fototóxico in vitro. Entretanto, nenhuma fototoxicidade foi observada in vivo. Assim sendo, o risco do nilotinibe causar fotossensibilização em pacientes é considerado muito baixo.
Contraindicações.
Hipersensibilidade conhecida ao nilotinibe ou a qualquer excipiente do produto.
Advertências e precauções.
Advertências
Uso em idosos, crianças e outros grupos de risco
Crianças e Adolescentes
Dados de segurança e eficácia em crianças e adolescentes abaixo de 18 anos de idade não foram estabelecidos.
Pacientes Idosos
Aproximadamente 30% dos pacientes dos estudos clínicos tinham 65 anos ou mais. Não foram observadas grandes diferenças na segurança e eficácia em pacientes com idade ≥ 65 anos, comparados com pacientes adultos de 18 a 65 anos de idade.
Pacientes com insuficiência renal
Não foram realizados estudos clínicos envolvendo pacientes com insuficiência na função renal. Os estudos clínicos excluíram pacientes com concentração de creatinina sérica > 1,5 vezes o limite superior da normalidade. Como o nilotinibe e seus metabólitos não são excretados por via renal, não é esperada uma diminuição no clearance (depuração) corpóreo total em pacientes com insuficiência renal.
Pacientes com insuficiência hepática
A insuficiência hepática tem um efeito modesto na farmacocinética do nilotinibe. O ajuste de dose não é considerado necessário em pacientes com insuficiência hepática, mas estes devem ser tratados com cautela (vide "Precauções" e "Advertências").
Distúrbios cardíacos
Nos estudos clínicos, pacientes com doenças cardíacas não controladas ou significantes, incluindo infarto recente do miocárdio, insuficiência cardíaca congestiva, angina instável, ou bradicardia clinicamente significantes foram excluídos.
Deve-se ter cuidado com pacientes com insuficiência cardíaca clinicamente relevante (vide "Precauções" e "Advertências").
Gravidez
Tasigna® enquadra-se na categoria D de risco na gravidez.
Não há dados adequados sobre o uso de Tasigna® em mulheres grávidas. Estudos em animais não mostraram teratogenicidade, mas embrio e fetotoxicidade foram observadas em doses que também apresentaram toxicidade materna (vide "Dados de segurança pré-clínica"). Tasigna® não deve ser utilizado durante a gravidez, a menos que seja necessário. Se o medicamento for utilizado durante a gravidez, a paciente deve ser informada sobre os riscos potenciais para o feto.
Mulheres com potencial de engravidar
Mulheres com potencial de engravidar devem ser aconselhadas a utilizar um método contraceptivo altamente eficaz durante o tratamento com Tasigna® .
Lactação
Não é conhecido se o nilotinibe é excretado no leite humano. Estudos em animais demonstram que o nilotinibe é excretado no leite. As mulheres não devem amamentar enquanto estiverem tomando Tasigna®, pois um risco para o recém-nascido não pode ser excluído.
Fertilidade
Nenhum efeito na contagem/motilidade dos espermatozoides e na fertilidade foi observado em ratos machos e fêmeas até a dose mais alta testada de aproximadamente 5 vezes maior do que a dosagem recomendada para humanos (vide "Dados de segurança pré-clínica").
Efeitos na habilidade de dirigir e utilizar máquinas
Nenhum estudo sobre os efeitos do nilotinibe na habilidade de dirigir e operar máquinas foi realizado. Pacientes que sentirem tontura, distúrbio visual ou outras reações adversas com um impacto potencial na habilidade de dirigir ou de operar máquinas com segurança, não devem realizar estas atividades enquanto persistam as reações adversas (vide "Reações adversas").
Precauções
Mielossupressão
O tratamento com Tasigna® está frequentemente associado à trombocitopenia, neutropenia e anemia (NCI CTC - National Cancer Institute Common Toxicity Criteria -Graus 3/4). Esta ocorrência é mais frequente em pacientes com LMC após falha ou intolerância ao imatinibe e em particular em pacientes com LMC-FA. Contagem sanguínea completa deve ser realizada a cada duas semanas nos dois primeiros meses e depois, mensalmente, ou quando indicado clinicamente. Mielossupressão foi geralmente reversível e comumente controlada com interrupção temporária do tratamento com Tasigna® ou diminuição da dose (vide "Posologia").
Prolongamento do intervalo QT
Dados in vitro sugerem que nilotinibe tem o potencial de prolongar a repolarização ventricular cardíaca (intervalo QT).
No estudo de fase II com pacientes com LMC após falha ou intolerância ao imatinibe em fase crônica e acelerada, tratados com nilotinibe 400 mg duas vezes ao dia, a mudança do tempo médio do intervalo QTcF, a partir do nível basal, no estado de equilíbrio foi 5 e 8 msec, respectivamente. QTcF de > 500 msec foi observado em 4 pacientes ( < 1 % destes pacientes). Em estudos com voluntários sadios com exposições que foram comparáveis às exposições observadas em pacientes, a mudança no tempo médio do QTcF com placebo a partir do nível basal foi 7 msec (IC ± 4 msec). Nenhum indivíduo teve QTcF > 450 msec. Além disso, nenhuma arritmia clinicamente relevante foi observada durante a condução dos estudos clínicos. Em particular, nenhum episódio de torsade de pointes (transitórios ou mantidos) foi observado. O prolongamento significativo do intervalo QT pode ocorrer quando Tasigna® é tomado inapropriadamente com alimento e/ou inibidores fortes de CYP3A4 e/ou produtos medicinais com conhecido potencial de prolongar o intervalo QT. Por isso, a coadministração com alimentos deve ser evitada e o uso concomitante com inibidores fortes de CYP3A4 e/ou produtos medicinais com conhecido potencial de prolongar o intervalo QT devem ser evitados (vide "Precauções", "Advertências", "Interação com alimentos" e "Interações medicamentosas e outras formas de interação"). A presença de hipocalemia e hipomagnesemia pode aumentar ainda mais este efeito (vide "Posologia").
Tasigna® deve ser utilizado com cautela em pacientes que têm, ou com risco significativo de desenvolver, o prolongamento do intervalo QTc, tais como:
• síndrome do QT longo;
• doenças cardíacas não controladas ou significativas, incluindo infarto recente do miocárdio, insuficiência cardíaca congestiva, angina instável, ou bradicardia clinicamente significativa.
Morte súbita
Nos estudos clínicos, casos incomuns (0,1 a 1%) de morte súbita foram relatados em pacientes recebendo Tasigna® com um histórico médico anterior de doença cardíaca ou fatores de risco cardíaco significativos. Comorbidades em adição à malignidade subjacente também estavam frequentemente presentes, bem como medicações concomitantes. Anomalias de repolarização ventricular podem ter sido fatores de contribuição. Com base na exposição de pós-lançamento em pacientes/ano, a taxa de mortes súbitas como relatos espontâneos é de 0,02% por paciente ao ano.
Interações medicamentosas.
A administração de Tasigna® com agentes que são fortes inibidores da CYP3A4 e medicamentos que podem prolongar o intervalo QT tais como antiarrítmicos deve ser evitada (vide "Posologia" e "Interações medicamentosas e outras formas de interação"). Se o tratamento com qualquer um desses agentes for requerido, recomenda-se, se possível, que a terapia com Tasigna® seja interrompida (vide "Interações medicamentosas e outras formas de interação"). Se a interrupção temporária do tratamento com Tasigna® não for possível, indica-se o monitoramento cuidadoso do indivíduo quanto ao prolongamento do intervalo QT (vide "Posologia", "Interações medicamentosas e outras formas de interação" e "Propriedades farmacocinéticas").
O uso concomitante de Tasigna® com produtos medicinais que são indutores potentes de CYP3A4 parece reduzir a exposição ao nilotinibe a uma extensão clinicamente relevante. Portanto, em pacientes recebendo Tasigna® , o uso concomitante de agentes terapêuticos alternativos com menor potencial para indução de CYP3A4 deve ser selecionado (vide "Interações medicamentosas e outras formas de interação").
Interação com alimentos
A biodisponibilidade do nilotinibe é aumentada pelos alimentos. Tasigna® não deve ser tomado junto com alimentos (vide "Posologia", "Interações medicamentosas e outras formas de interação") e deve ser ingerido duas horas após a refeição. Nenhum alimento deve ser consumido por pelo menos uma hora após a ingestão da dose.
Para pacientes com dificuldade de deglutição, o conteúdo da cada cápsula deve ser disperso em uma colher de sopa de suco de maçã e deve ser ingerido imediatamente. Não mais do que uma colher de sopa de suco de maçã e nenhum outro alimento deve ser usado (vide "Posologia"). Suco de grapefruit e outros alimentos que sejam conhecidos como inibidores da CYP3A4 devem ser evitados em qualquer momento.
Insuficiência hepática
A insuficiência hepática tem um efeito modesto na farmacocinética de nilotinibe. A administração de uma dose única de nilotinibe resultou em aumentos na AUC de 35%, 35% e 19% em pacientes com insuficiência hepática leve, moderada e grave respectivamente, comparado ao grupo controle de indivíduos com função hepática normal. A Cmáx prevista no estado de equilíbrio de nilotinibe mostrou aumentos de 29%, 18% e 22%, respectivamente. Os estudos clínicos excluíram pacientes com ALT e/ou AST > 2,5 (ou > 5, se relacionada à doença) vezes o limite superior da normalidade e/ou bilirrubina total > 1,5 vezes o limite superior da normalidade. O metabolismo do nilotinibe é principalmente hepático. Recomenda-se cautela em pacientes com insuficiência hepática (vide "Posologia - Recomendações de monitoramento").
Lipase sérica
Foi observada elevação na lipase sérica. Recomenda-se cautela em pacientes com história prévia de pancreatite. Caso as elevações de lipase sejam acompanhadas por sintomas abdominais, a dose deve ser interrompida e um diagnóstico apropriado deve ser considerado para excluir a pancreatite. (vide "Posologia").
Gastrectomia total
A biodisponibilidade de nilotinibe pode ser reduzida em pacientes com gastrectomia total (vide "Propriedades farmacocinéticas"). Deve-se considerar acompanhamento mais frequente destes pacientes.
Síndrome de Lise Tumoral
Casos de Síndrome de Lise Tumoral (SLT) foram relatados em pacientes tratados com Tasigna® . Para as recomendações de monitoramento vide "Posologia".
Lactose
Como as cápsulas contêm lactose, Tasigna® não é recomendado para pacientes com problemas hereditários de intolerância à galactose, deficiência grave de lactase, deficiência ou má absorção de glucose-galactose.
Interações medicamentosas e outras formas de interação
Medicamentos que podem aumentar as concentrações plasmáticas de nilotinibe
O nilotinibe é metabolizado principalmente pelo fígado e também é um substrato para a bomba de efluxo multimedicamentoso da P-glicoproteína (Pgp). Portanto, a absorção e a subsequente eliminação do nilotinibe absorvido via sistêmica podem ser influenciadas por sustâncias que afetam a CYP3A4 e/ou a Pgp.
Em um estudo de Fase I de nilotinibe administrado em combinação com imatinibe (um substrato e moderador de Pgp e CYP3A4), ambos os medicamentos tiveram um efeito inibitório leve na CYP3A4 e/ou PgP. Quando os dois medicamentos foram administrados concomitantemente, a AUC de imatinibe foi aumentada em 18% a 39%, e a AUC de nilotinibe foi aumentada em 18% a 40%.
A biodisponibilidade do nilotinibe em indivíduos sadios foi aumentada em 3 vezes quando coadministrado com um forte inibidor da CYP3A4, o cetoconazol. Portanto, o tratamento concomitante com fortes inibidores da CYP3A4 deve ser evitado (incluindo, mas não limitado a cetoconazol, itraconazol, voriconazol, ritonavir, claritromicina, e telitromicina) (vide "Posologia", "Precauções -Prolongamento do intervalo QT"). Medicações concomitantes alternativas, com nenhuma ou com uma mínima inibição da CYP3A4, devem ser consideradas.
Medicamentos que podem diminuir as concentrações plasmáticas de nilotinibe
Indutores de atividade da CYP3A4 podem aumentar o metabolismo do nilotinibe e, com isso, diminuir as concentrações plasmáticas de nilotinibe. A administração concomitante de medicamentos que induzem a CYP3A4 (por ex.: fenitoína, rifampicina, carbamazepina, fenobarbital, e erva de São João) pode reduzir a exposição ao nilotinibe. Em pacientes para os quais indutores da CYP3A4 são indicados, agentes alternativos com um menor potencial de indução da enzima devem ser considerados.
Em voluntários sadios recebendo um indutor de CYP3A4, a rifampicina, na dose de 600 mg por dia por 12 dias, a exposição sistêmica (AUC) ao nilotinibe foi diminuída aproximadamente em 80%.
O nilotinibe tem uma solubilidade pH dependente, com solubilidade mais baixa em pH mais alto. Em indivíduos sadios recebendo 40 mg de esomeprazol uma vez ao dia por 5 dias, o pH gástrico foi aumentado notavelmente, mas a absorção de nilotinibe foi diminuída modestamente (diminuição de 27% na Cmáx e 34% na AUC0-∞). Tasigna® pode ser usado concomitantemente com esomeprazol ou outros inibidores da bomba de próton, se necessário.
Medicamentos cuja concentração sistêmica pode ser alterada pelo nilotinibe
O nilotinibe é identificado como um inibidor competitivo da CYP3A4, CYP2C8, CYP2C9, CYP2D6 e da UGT1A1 in vitro, com o valor de Ki sendo mais baixo para CYP2C9 (Ki = 0,13 microM). Em indivíduos sadios, nas concentrações clinicamente relevantes de nilotinibe, a farmacocinética ou farmacodinâmica da varfarina, um substrato sensível da CYP2C9, não foi alterada. Tasigna® pode ser usado concomitantemente com varfarina sem aumentar o efeito anticoagulante. Além disso, a administração de uma dose única de Tasigna® com midazolam em indivíduos sadios aumentou a exposição ao midazolam em 30%, contudo a taxa metabólica de 1-hidróxi-midazolam para midazolam não foi alterada.
Medicamentos antiarrítmicos e outras drogas que podem prolongar o intervalo QT
O uso concomitante de medicamentos antiarrítmicos (incluindo, mas não limitado a amiodarona, disopiramida, procainamida, quinidina, e sotalol) e outras drogas que podem prolongar o intervalo QT (incluindo, mas não limitado a cloroquina, halofantrina, claritromicina, haloperidol, metadona, moxifloxacino, bepridil e pimozida) deve ser evitado (vide "Precauções").
Outras interações que podem afetar as concentrações séricas
A absorção de nilotinibe é aumentada quando administrado com alimentos, resultando em concentração sérica mais alta (vide "Posologia", "Precauções", "Advertências" e "Propriedades farmacocinéticas").
Suco de grapefruit e outros alimentos conhecidos por inibir a CYP3A4 devem ser evitados a qualquer momento.
Cuidados de armazenamento.
Este medicamento deve ser armazenado em temperatura ambiente (entre 15 e 30°C), e depois de aberto mantido na embalagem original.
Posologia e modo de usar.
Modo de usar e cuidados de conservação depois de aberto
Via oral. Conservar em temperatura ambiente (entre 15 e 30 °C), e depois de aberto manter na embalagem original.
Posologia
A terapia deve ser iniciada por um médico experiente no tratamento de pacientes com LMC. A dose recomendada de Tasigna® é de 400 mg duas vezes por dia (vide "Propriedades farmacocinéticas"). O tratamento deverá prosseguir enquanto o paciente continuar a ser beneficiado.
Tasigna® deve ser tomado duas vezes ao dia, em intervalos de 12 horas, aproximadamente, e não deve ser ingerido com alimentos. As cápsulas devem ser engolidas inteiras com água. Nenhum alimento deve ser consumido por, pelo menos, duas horas antes da dose ser ingerida e nenhum alimento deve ser consumido por, pelo menos, uma hora após a tomada da dose (vide "Precauções", "Advertências", "Interações medicamentosas e outras formas de interação" e "Propriedades farmacocinéticas").
Para pacientes com dificuldade de deglutição, o conteúdo da cada cápsula deve ser disperso em uma colher de sopa de suco de maçã e deve ser ingerido imediatamente. Não mais do que uma colher de sopa de suco de maçã e nenhum outro alimento deve ser usado (vide "Posologia" e "Propriedades farmacocinéticas").
Tasigna® pode ser administrado em combinação com fatores de crescimento hematopoiéticos, tais como eritropoetina ou G-CSF, se clinicamente indicados. Tasigna® pode ser administrado com hidroxiureia ou anagrelida, se clinicamente indicado.
Recomendações de monitoramento e ajustes de dose
O ECG é recomendado antes do início da terapia com Tasigna® e deve ser repetido após 7 dias e quando clinicamente indicado. Hipocalemia ou hipomagnesemia deve ser corrigido antes da administração de Tasigna® e os níveis sanguíneos de potássio e magnésio devem ser monitorados periodicamente durante a terapia, particularmente em pacientes com risco para anormalidades eletrolíticas (vide "Precauções" e "Advertências"). Devido a possível ocorrência de Síndrome de Lise Tumoral (SLT) são recomendadas correção de desidratação clinicamente significante e tratamento dos níveis elevados de ácido úrico antes do início da terapia com Tasigna® .
Tasigna® pode ser interrompido temporariamente e/ou sua dose pode ser reduzida em decorrência de toxicidades hematológicas (neutropenia, trombocitopenia) que não estejam relacionadas à leucemia subjacente (vide Tabela 5).
Se ocorrer toxicidade não-hematológica, moderada ou grave, clinicamente significante, o tratamento deve ser interrompido e poderá ser retomado com a dose de 400 mg uma vez ao dia, desde que a toxicidade esteja resolvida. Se clinicamente apropriado, o re-escalonamento da dose para 400 mg duas vezes ao dia deve ser considerado.
Elevações de lipase: para elevações de lipase de Graus 3 e 4, as doses devem ser reduzidas para 400 mg uma vez ao dia ou interrompidas. Os níveis de lipase devem ser testados mensalmente, ou quando clinicamente indicado (vide "Reações adversas"). Elevações de bilirrubina e transaminases hepáticas: para elevações de bilirrubina ou transaminases hepáticas de Graus 3 e 4, as doses devem ser reduzidas para 400 mg uma vez ao dia ou interrompidas. Os níveis de bilirrubina e de transaminases hepáticas devem ser testados mensalmente ou quando clinicamente indicado (vide "Reações adversas").
Se o paciente esquecer de tomar uma dose, ele não deve tomar uma dose adicional, mas deve tomar a próxima dose como de costume.
Reações adversas.
Os dados descritos abaixo refletem a exposição ao Tasigna® em 458 pacientes com LMC-FC Ph+ (n=321) e LMC-FA Ph+ (n=137) após falha ou intolerância a pelo menos uma terapia prévia, incluindo imatinibe em um estudo multicêntrico aberto tratado com a dose recomendada de 400 mg duas vezes ao dia.
As reações adversas não hematológicas relacionadas ao medicamento relatados como muitos comuns (≥ 10% nas populações de pacientes com LMC-FC e LMC-FA) foram: rash (erupção cutânea), prurido, náusea, fadiga, dor de cabeça, constipação, diarreia, vomito e mialgia. A maioria destas reações adversas foram de gravidade leve a moderada. Alopécia, espasmos musculares, diminuição de apetite, artralgia, dor óssea, dor abdominal, edema periférico e astenia foram menos frequentemente observados ( < 10% e ≥ 5%) e tiveram gravidade leve a moderada (Grau 1 ou 2).
Derrames pleurais e pericárdicos, assim como complicações devido a retenção de líquidos ocorreram em < 1% dos pacientes que receberam Tasigna®. Falha cardíaca foi observada em < 1% dos pacientes. Hemorragias gastrointestinais e de SNC foram relatados em 1% e < 1% dos pacientes respectivamente.
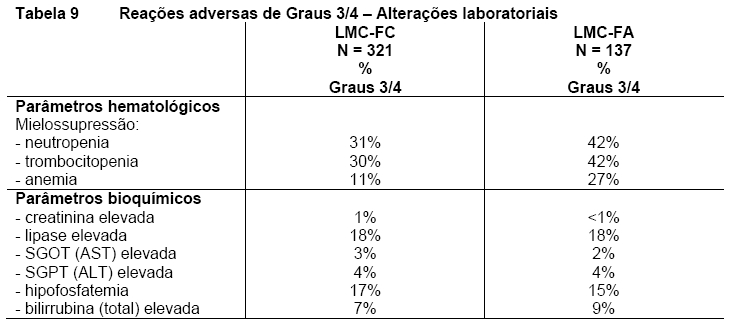
QTcF excedendo 500 msec foi observado nesse estudo em 4 pacientes ( < 1%). Nenhum episódio de Torsade de Pointes (transitório ou mantido) foi observado. Reações adversas hematológicas incluem mielossupressão: trombocitopenia (31%), neutropenia (17%) e anemia (14%). Vide Tabela 7 - Reações adversas de Graus 3/4 - Alterações laboratoriais. A descontinuação devido a reações adversas relacionadas a droga foi observada em 16% dos pacientes em FC e 10% dos pacientes em FA.
Reações adversas mais frequentemente relatadas
As reações adversas não hematológicas (excluindo alterações laboratoriais), que foram relatadas em pelo menos 5% dos pacientes nos estudos clínicos com Tasigna®, estão apresentadas na Tabela 6. As reações adversas estão classificadas por ordem de frequência, a mais frequente primeiro, usando a seguinte convenção: muito comum (≥ 1/10) ou comum (≥ 1/100, < 1/10).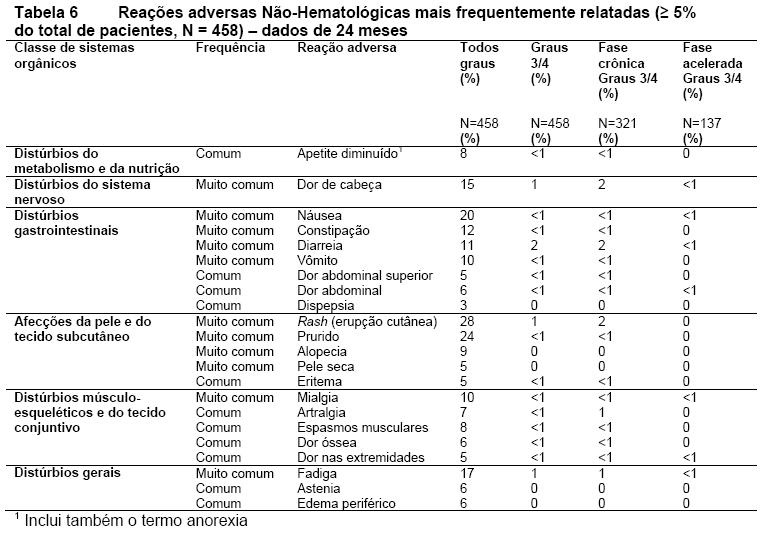
Adicionalmente, o estudo 2109, um estudo fase 3, aberto, multicêntrico, internacional, cujo objetivo primário foi o de confirmar o perfil de segurança do nilotinibe numa população maior de pacientes está relatado abaixo. O estudo 2109 envolveu pacientes adultos com LMC em fase crônica, fase acelerada e crise blástica, resistentes ou intolerantes ao imatinibe.
As medianas de duração de exposição ao nilotinibe foram de 184 dias para fase crônica, 17 dias para fase acelerada e 62 dias para pacientes em crise blástica. A dose mediana recebida foi de 792 mg, 787 mg e 783 mg diários respectivamente. A redução de dose do nilotinibe devido a eventos adversos ocorreu em 20% dos pacientes em fase crônica e em 22% dos pacientes em fase acelerada e em crise blástica. Entretanto a descontinuação do tratamento devido a eventos adversos foi observada em apenas 12% dos pacientes em fase crônica e em 13% e 11% dos pacientes em fase acelerada e crise blástica respectivamente.
A tabela 7 descreve os eventos adversos não hematológicos mais frequentemente observados neste estudo.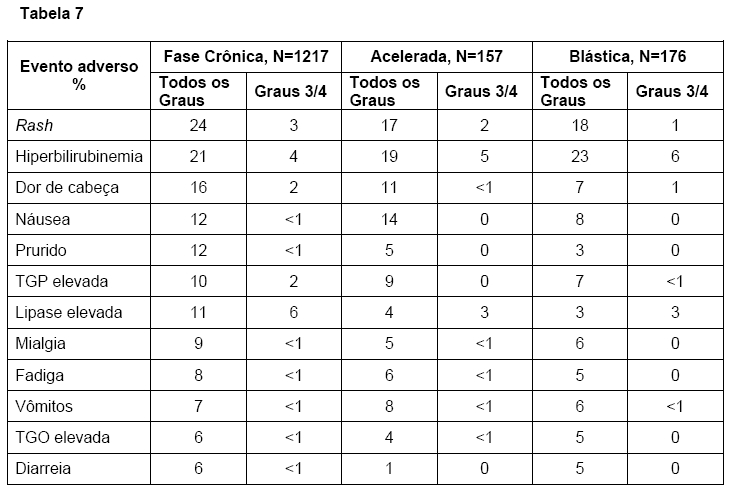
QTcF excedendo 500 msec foi observado em menos de 1% dos pacientes (em 3 pacientes com LMC em fase crônica e 1 paciente em fase acelerada e 1 em crise blástica).
A tabela 8 sumariza os eventos adversos hematológicos observados.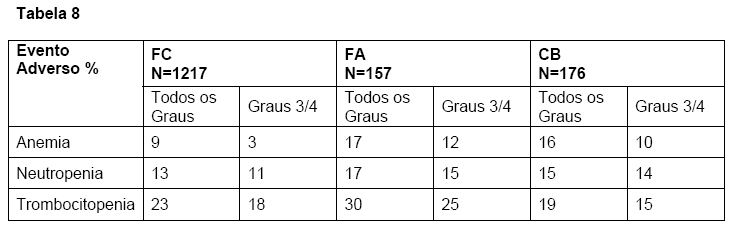
Dados adicionais dos estudos clínicos
As seguintes reações adversas foram relatadas em pacientes dos estudos clínicos com Tasigna® na dose recomendada com uma frequência menor que 5% (comuns ≥ 1/100 e < 1/10; incomuns ≥ 1/1000 e < 1/100); eventos únicos são capturados como de frequência desconhecida. Para anormalidades laboratoriais, eventos muitos comuns (≥ 1/10) não incluídos