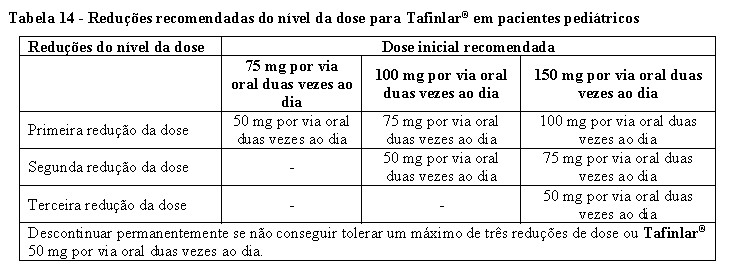
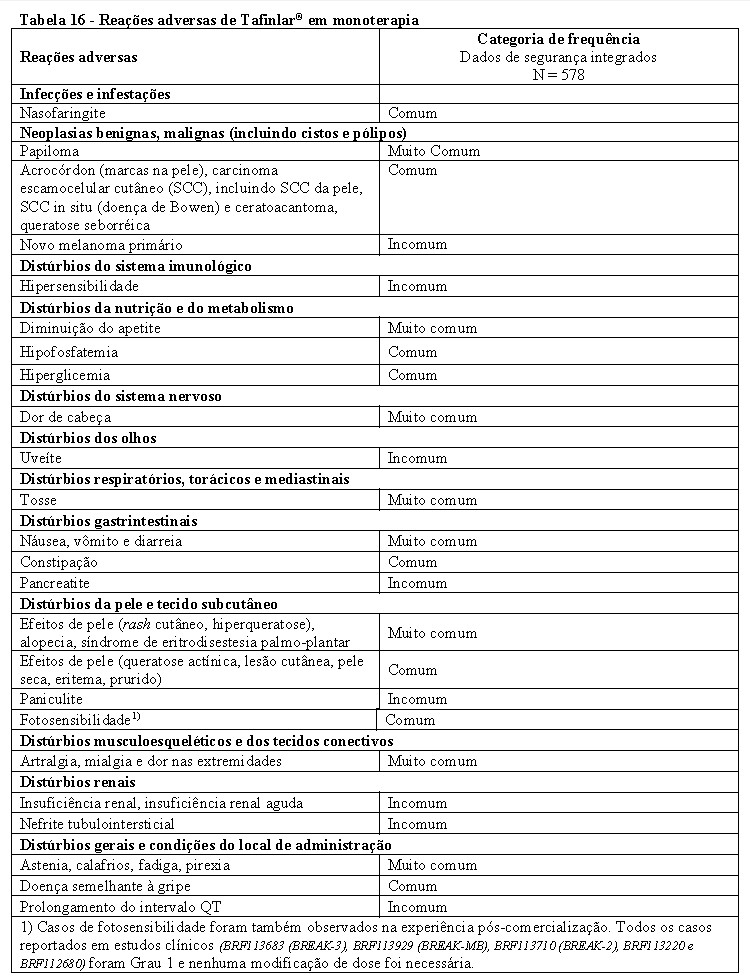
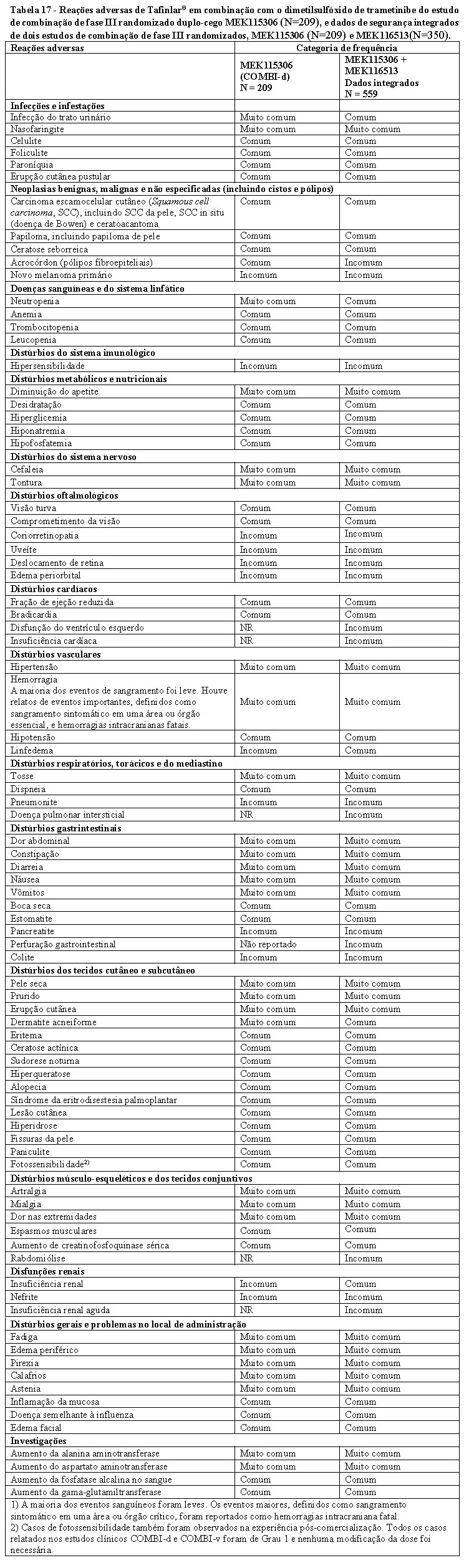
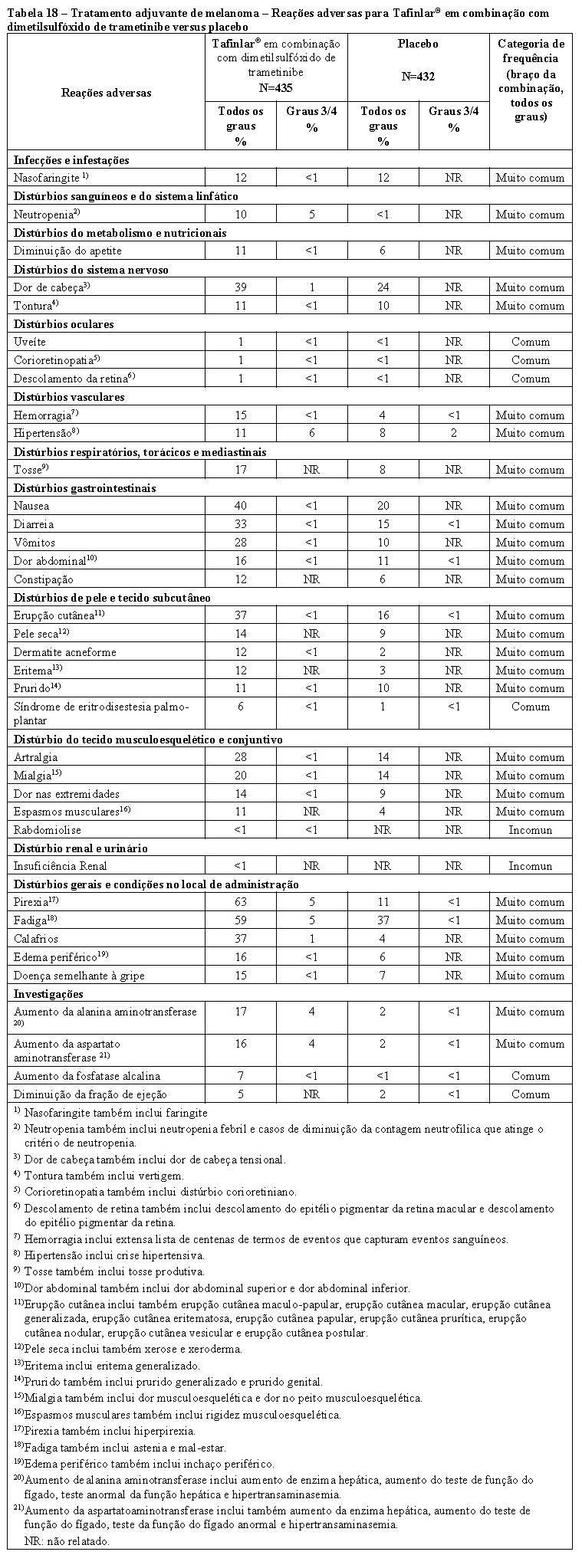
TAFINLAR
NOVARTIS
dabrafenibe
Antineoplásico.
Apresentações.
Cápsulas duras de 50 ou 75 mg em cartuchos com 120 cápsulas.
VIA ORAL
USO ADULTO E PEDIÁTRICO ACIMA DE 6 ANOS (conforme indicação abaixo)
Composição.
Tafinlar® 50 mg: cada cápsula dura contém 50 mg de dabrafenibe equivalente a 59,25 mg de mesilato de dabrafenibe.
Excipientes: celulose microcristalina, estearato de magnésio, dióxido de silício, óxido de ferro vermelho, dióxido de titânio, hipromelose, óxido de ferro preto, goma laca, álcool butílico, álcool isopropílico, propilenoglicol e hidróxido de amônio.
Tafinlar® 75 mg: cada cápsula dura contém 75 mg de dabrafenibe equivalente a 88,88 mg de mesilato de dabrafenibe.
Excipientes: celulose microcristalina, estearato de magnésio, dióxido de silício, óxido de ferro vermelho, dióxido de titânio, hipromelose, óxido de ferro preto, goma laca, álcool butílico, álcool isopropílico, propilenoglicol e hidróxido de amônio.
Informações técnicas.
1. INDICAÇÕES
Melanoma metastático ou irressecável
Tafinlar® como monoterapia ou em combinação com dimetilsulfóxido de trametinibe é indicado para o tratamento de pacientes adultos com melanoma metastático ou irressecável com mutação de BRAF V600.
Tratamento adjuvante de melanoma
Tafinlar® em combinação com dimetilsulfóxido de trametinibe é indicado para o tratamento adjuvante de pacientes adultos com melanoma de estágio III com mutação BRAF V600, após ressecção completa.
Câncer de pulmão avançado de células não pequenas
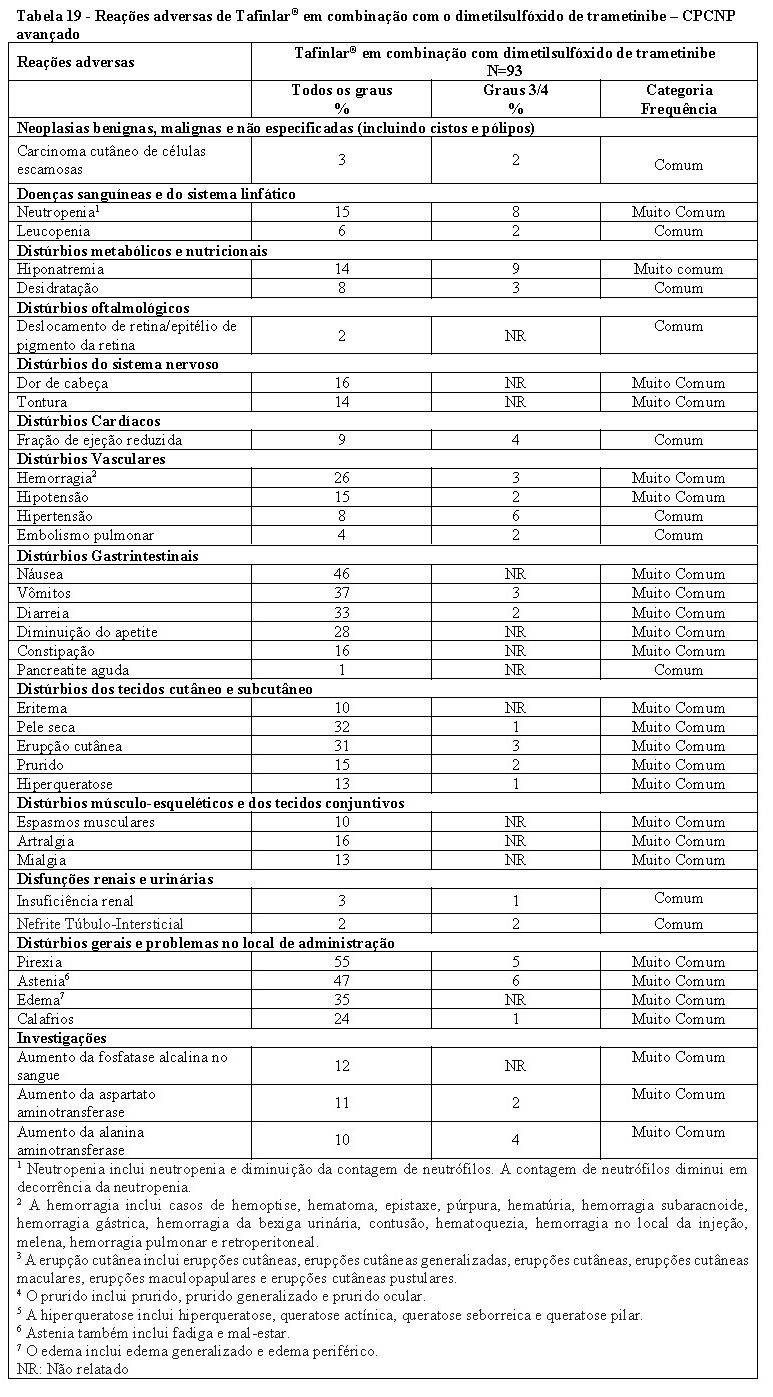
Tafinlar® em combinação com dimetilsulfóxido de trametinibe é indicado para o tratamento de pacientes com câncer de pulmão metastático de células não pequenas (CPCNP) com mutação de BRAF V600E.
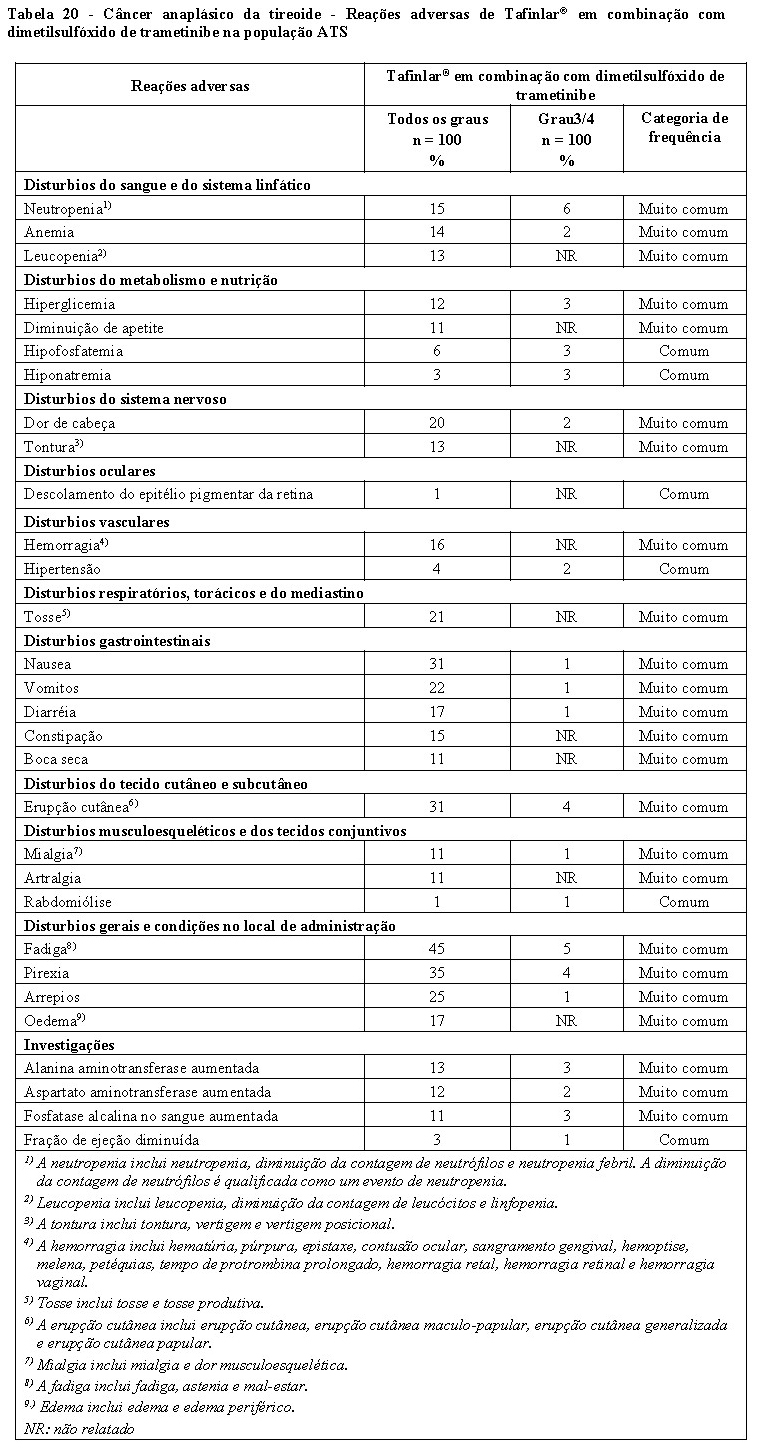
Câncer anaplásico de tireoide localmente avançado ou metastático
Tafinlar® em combinação com dimetilsulfóxido de trametinibe é indicado para o tratamento de pacientes com câncer anaplásico de tireoide (CAT) localmente avançado ou metastático com mutação de BRAF V600E.
Tumores sólidos irressecáveis ou metastáticos
Tafinlar® em combinação com dimetilsulfóxido de trametinibe é indicado para o tratamento de pacientes adultos e pediátricos acima de 6 anos de idade com tumores sólidos irressecáveis ou metastáticos com mutação BRAF V600E que progrediram após tratamento prévio e que não têm opções de tratamento alternativas satisfatórias.
2. RESULTADOS DE EFICÁCIA
Estudos clínicos
Melanoma irressecável ou mestastático
Tafinlar® em monoterapia
A eficácia de Tafinlar® no tratamento de pacientes adultos com melanoma irressecável ou metastático positivo para mutação BRAF V600 foi sido avaliada em 3 estudos (BRF113683 [BREAK-3], BRF113929 [BREAK-MB], e BRF113710 [BREAK-2]) incluindo pacientes com BRAF V600E e/ou mutações do V600K.
Pacientes não tratados previamente
A segurança e eficácia de Tafinlar® foram avaliadas em um estudo fase III, randomizado, aberto, [BREAK-3] comparando Tafinlar® a dacarbazina (DTIC) em pacientes com melanoma positivo para mutação BRAF V600E avançado (irressecável Estágio III) ou metastático (Estágio IV) não tratados previamente. A triagem incluiu teste central da mutação de BRAF V600E usando um ensaio de mutação de BRAF realizado na amostra do tumor mais recente disponível.
O estudo incluiu 250 pacientes randomizados 3:1 para receber ou Tafinlar® 150 mg duas vezes ao dia ou DTIC intravenoso 1000 mg/m2 a cada 3 semanas. O objetivo primário para este estudo era avaliar a eficácia do Tafinlar® comparado ao DTIC com relação à sobrevida livre de progressão (SLP) para pacientes com melanoma irressecável ou metastático com mutação positiva para BRAF V600E. Aos pacientes no braço do DTIC foi permitido receber Tafinlar® independente após confirmação radiográfica de progressão inicial. As características do período basal foram balanceadas entre os grupos de tratamento. Sessenta por cento dos pacientes eram homens e 99,6% eram caucasianos, a idade mediana era de 52 anos com 21% dos pacientes sendo ≥ 65 anos, 98,4% tinham condição de ECOG (Eastern Cooperative Oncology Group) de 0 ou 1, e 97% dos pacientes tinham doença metastática.
A análise primária foi baseada em 118 eventos no momento de corte de dados. A avaliação do investigador para os dados de eficácia estão resumidos na Tabela 1 e Figura 1.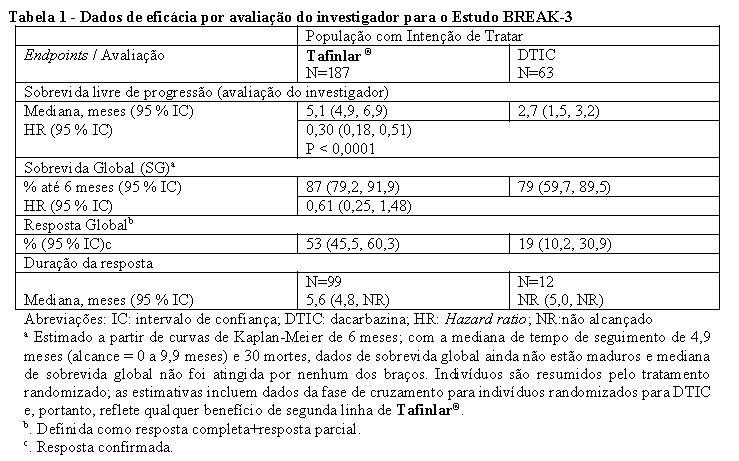
Vinte e oito indivíduos (44 %) randomizados para DTIC cruzaram para o Tafinlar® seguindo a progressão da doença verificada de forma independente. O tempo mediano em Tafinlar® após o cruzamento foi de 2,8 meses e a taxa de resposta global (TRG) não confirmada foi de 46%.
Pacientes com metástases cerebrais
BREAK-MB foi um estudo multicêntrico, aberto, de duas coortes, de Fase II desenhado para avaliar a resposta intracranial de Tafinlar® em indivíduos com confirmação histológica (Estágio IV) de melanoma com mutação positiva BRAF (V600E ou V600K) metastático para o cérebro. Os indivíduos foram incluídos na Coorte A (indivíduos sem tratamento local prévio para metástases cerebrais) ou Coorte B (indivíduos que receberam tratamento local prévio para metástases cerebrais). Os resultados estão resumidos na Tabela 2.
Pacientes que progrediram de um inibidor BRAF:
Existem dados limitados em pacientes utilizando a combinação de dimetilsulfóxido de trametinibe com dabrafenibe que progrediram previamente de um inibidor BRAF. Esses dados demonstram que a eficácia da combinação será menor nesses pacientes. Portanto, outras opções de tratamento devem ser consideradas antes do tratamento com a combinação dimetilsulfóxido de trametinibe e dabrafenibe na população tratada anteriormente com outro inibidor de BRAF.
Pacientes que não foram tratados previamente ou falharam em pelo menos uma terapia sistêmica prévia
BRF113710 (BREAK-2) foi um estudo multicêntrico, global, aberto, braço único, de Fase II que incluiu 92 indivíduos de pesquisa com melanoma metastático confirmado histologicamente (Estágio IV) com melanoma positivo para mutação BRAF V 600E ou V600K confirmada. Os indivíduos eram virgens de tratamento (n=15) ou receberam tratamento prévio (n=77) na presença de metástases (por exemplo, quimioterapia, imunoterapia, terapia alvo prévia, etc.).
O investigador avaliou a taxa de resposta confirmada na eficácia primária em uma população de pacientes com melanoma metastático BRAF V600E (n=76) foi de 59 % (95% IC: 48,2; 70,3) incluindo 7% de resposta completa. A mediana de SLP foi de 6,3 meses (95% IC: 4,6; 7,7) e a duração mediana da resposta foi de 5,2 meses (95 % IC: 3,9, não calculado). A terapia sistêmica prévia não pareceu afetar significativamente a resposta. O investigador avaliou a taxa de resposta confirmada na eficácia secundária em uma população de pacientes com melanoma metastático com mutação positiva BRAF V600K (n=16) foi de 13% (95% IC: 0,0; 28,7) com duração mediana da resposta de 5,3 meses (95 % IC: 3,7; 6,8). Não houve resposta completa na população de pacientes V600K.
Tafinlar® em combinação com dimetilsulfóxido de trametinibe:
A eficácia e a segurança da dose recomendada de dimetilsulfóxido de trametinibe (2 mg uma vez ao dia) em combinação com Tafinlar® (150 mg duas vezes ao dia) para o tratamento de pacientes adultos com melanoma metastático ou irressecável com mutação de BRAF V600 foram estudadas em dois estudos pivotais de fase III.
MEK115306 (COMBI-d)
O MEK115306 (COMBI-d) foi um estudo de fase III, randomizado, duplo-cego, comparando a combinação de Tafinlar® e dimetilsulfóxido de trametinibe versus Tafinlar® e placebo como terapia de primeira linha em indivíduos com melanoma cutâneo irresecável (estágio IIIC) ou metastático (estágio IV) com mutação BRAF V600E/K positiva. O desfecho primário do estudo foi a sobrevida livre de progressão (SLP) avaliada pelo investigador com um desfecho secundário de sobrevida global (SG). Os participantes foram estratificados por nível de lactato desidrogenase (DHL) ( > o limite superior da normalidade (ULN) versus ≤ ULN) e mutação BRAF (V600E versus V600K).
Um total de 423 participantes foi randomizado 1:1 para o braço de terapia combinada (dimetilsulfóxido de trametinibe 2 mg uma vez ao dia e Tafinlar® 150 mg duas vezes ao dia) (N = 211) ou braço de monoterapia com Tafinlar® (150 mg duas vezes ao dia) (N = 212). As características basais estavam equilibradas entre os grupos de tratamento. O sexo masculino constituiu 53% dos pacientes e a idade média foi de 56 anos. A maioria dos pacientes obteve uma pontuação de desempenho do ECOG [Eastern Cooperative Oncology Group (Grupo Oncológico Cooperativo do Leste)] de 0 (72%) e tinha doença estágio IVM1c (66%). A maioria dos pacientes teve mutação BRAF V600E (85%). Os 15% de pacientes restantes tiveram mutação BRAF V600K. Indivíduos com metástase cerebral não foram incluídos neste estudo.
A SG mediana e as taxas de sobrevida estimadas em 1 ano, 2 anos, 3 anos, 4 anos e 5 anos são apresentadas na Tabela 3. Uma análise de SG em 5 anos demonstrou benefício contínuo para a combinação de Tafinlar® e dimetilsulfóxido de trametinibe em comparação à monoterapia com Tafinlar®; a SG mediana para o braço de terapia combinada foi aproximadamente 7 meses mais longa do que para monoterapia com Tafinlar® (25,8 meses versus 18,7 meses), com taxas de sobrevida em 5 anos de 32% para a combinação versus 27% para monoterapia com Tafinlar® (Tabela 3, Figura 2). A curva de SG de Kaplan-Meier parece estabilizar de 3 a 5 anos (vide Figura 2). A taxa de sobrevida global em 5 anos foi de 40% (IC de 95%: 31,2, 48,4) no braço de terapia combinada versus 33% (IC de 95%: 25,0, 41,0) no braço de monoterapia com Tafinlar® para pacientes que tiveram um nível basal normal de lactato desidrogenase, e 16% (IC de 95%: 8.4, 26.0) no braço de terapia combinada versus 14% (IC de 95%: 6.8, 23.1) no braço de monoterapia com Tafinlar® para pacientes com um nível basal elevado de lactato desidrogenase.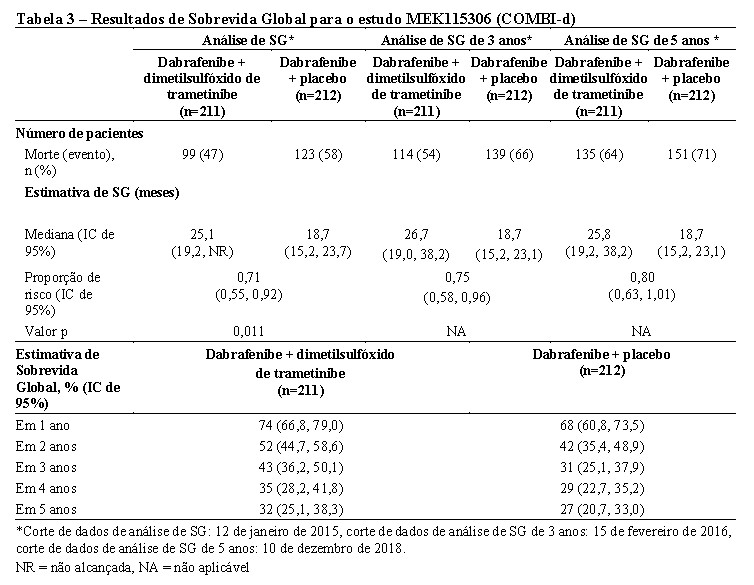
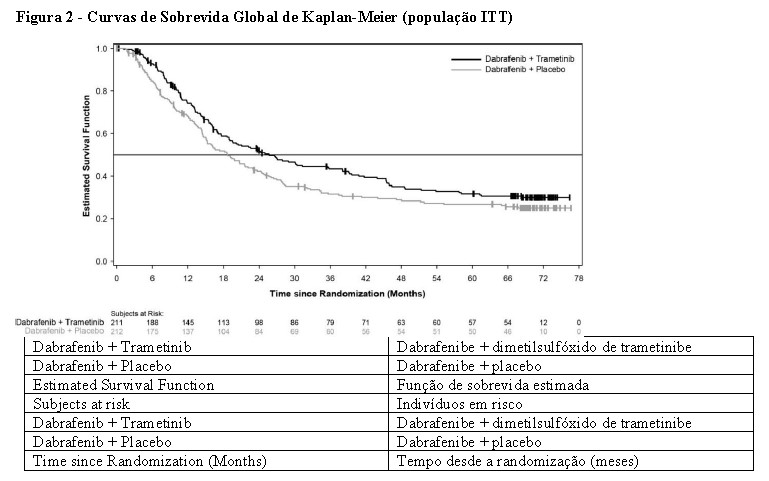
As melhoras clinicamente significativas para o desfecho primário da SLP foram mantidas ao longo de um período de 5 anos no braço de terapia combinada em comparação à monoterapia com Tafinlar®. Também foram observadas melhoras clinicamente significativas para a taxa de resposta global (TRG) e uma maior duração da resposta no braço de terapia combinada em comparação com a monoterapia com Tafinlar® (Tabela 4).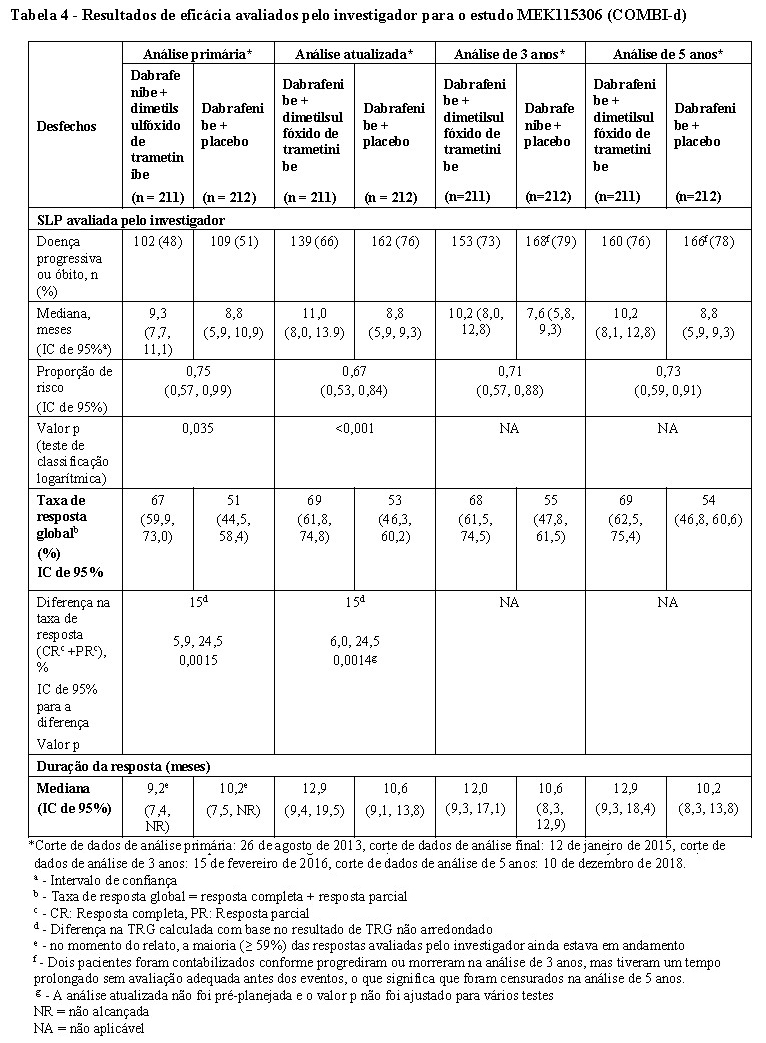
MEK116513 (COMBI-v):
O estudo MEK116513 foi um estudo de fase III de dois braços, aberto, randomizado, que comparou a terapia combinada de Tafinlar® e dimetilsulfóxido de trametinibe à monoterapia com vemurafenibe em melanoma metastático com mutação BRAF V600 positiva. O desfecho primário do estudo foi sobrevida global. Os indivíduos foram estratificados por nível de lactato desidrogenase (DHL) ( > o limite superior da normalidade (ULN) versus ≤ ULN) e mutação BRAF (V600E versus V600K).
Um total de 704 indivíduos foi randomizado 1:1 para o braço de terapia combinada (dimetilsulfóxido de trametinibe 2 mg uma vez ao dia e Tafinlar® 150 mg duas vezes ao dia) ou braço de monoterapia com vemurafenibe (960 mg duas vezes ao dia). A maioria dos indivíduos era branca ( > 96%) e do sexo masculino (55%), com uma idade média de 55 anos (24% tinham ≥ 65 anos). A maioria dos indivíduos tinha doença M1c estágio IV (61%). A maioria dos indivíduos tinha DHL ≤ ULN (67%), situação de desempenho ECOG de 0 (70%), e doença visceral (78%) na avaliação inicial. No geral, 54% dos indivíduos tinham < 3 locais da doença na avaliação inicial. A maioria dos indivíduos tinha mutação BRAF V600E (89%). Indivíduos com metástase cerebral não foram incluídos neste estudo.
Uma análise de SG de 5 anos demonstrou benefício contínuo para a combinação de Tafinlar® e dimetilsulfóxido de trametinibe comparado à monoterapia com vemurafenibe; a SG mediana para o braço de terapia combinada foi aproximadamente 8 meses mais longa do que a SG mediana para monoterapia com vemurafenibe (26,0 meses versus 17,8 meses), com taxas de sobrevida em 5 anos de 36% para a combinação versus 23% para monoterapia com vemurafenibe (Tabela 5, Figura 3) A curva de SG de Kaplan-Meier parece estabilizar de 3 a 5 anos (vide Figura 3). A taxa de sobrevida global em 5 anos foi de 46% (IC de 95%: 38,8, 52,0) no braço de terapia combinada versus 28% (IC de 95%: 22,5, 34,6) no braço de monoterapia com vemurafenibe para pacientes que tiveram um nível basal normal de lactato desidrogenase, e 16% (IC de 95%: 9.3, 23.3) no braço de terapia combinada versus 10% (IC de 95%: 5.1, 17.4) no braço de monoterapia com vemurafenibe para pacientes com um nível basal elevado de lactato desidrogenase.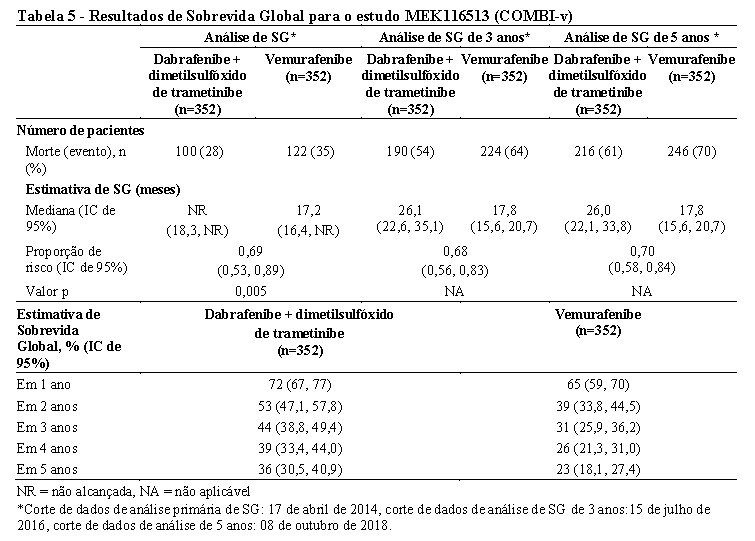
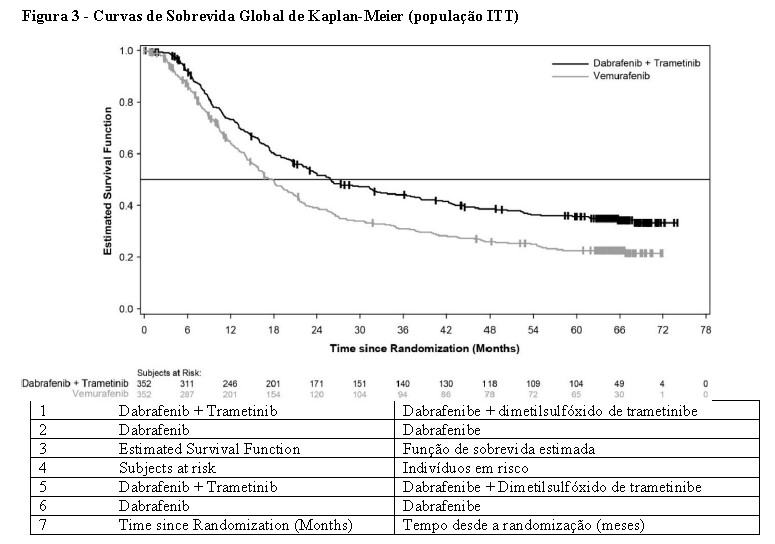
As melhoras clinicamente significativas para o desfecho secundário da SLP foram mantidas ao longo de um período de 5 anos no braço de terapia combinada em comparação à monoterapia com vemurafenibe. Também foram observadas melhoras clinicamente significativas para a taxa de resposta global (TRG) e uma maior duração da resposta no braço de terapia combinada em comparação à monoterapia com vemurafenibe (Tabela 6).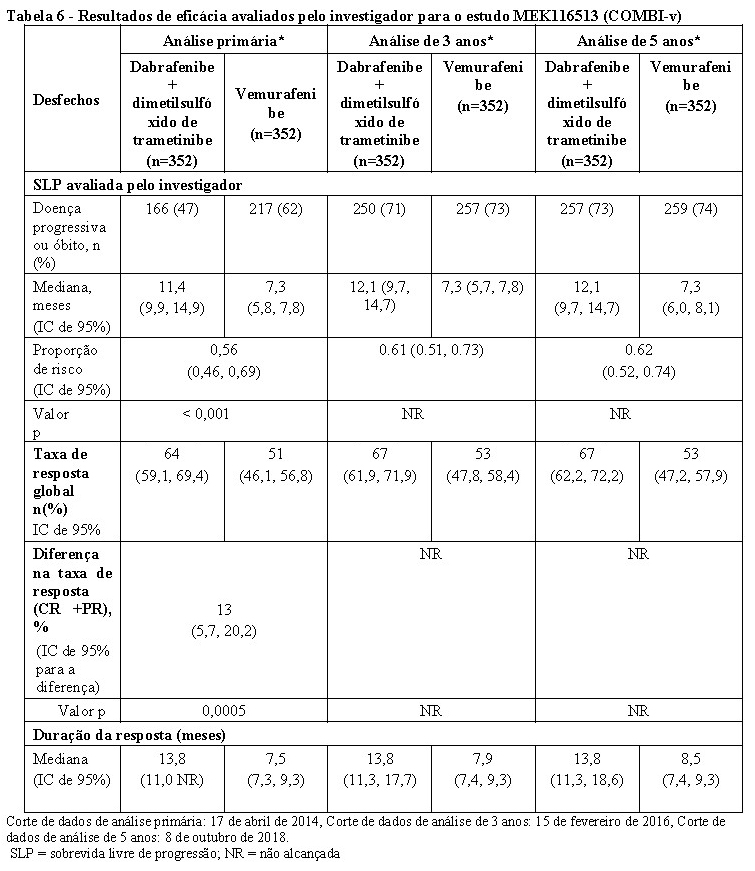
Corte de dados de análise primária: 17 de abril de 2014, Corte de dados de análise de 3 anos: 15 de fevereiro de 2016, Corte de dados de análise de 5 anos: 8 de outubro de 2018.
SLP = sobrevida livre de progressão; NR = não alcançada
Pacientes com melanoma metastático com metástases cerebrais
Estudo BRF117277 / DRB436B2204 (COMBI-MB)
A eficácia e segurança de Tafinlar® em combinação com dimetilsulfóxido de trametinibe em pacientes com melanoma BRAF mutado positivo que se metastizou para o cérebro foi estudada em um estudo multicêntrico, aberto, não ranzomizado, de Fase II (estudo COMBI-MB).
Um total de 125 pacientes foram inscritos em quatro coortes:
• Coorte A: pacientes com melanoma mutado BRAFV600E com metástases cerebrais assintomáticas sem terapia local prévia dirigida ao cérebro e estado de desempenho ECOG de 0 ou 1.
• Coorte B: pacientes com melanoma mutado BRAFV600E com metástases cerebrais assintomáticas com terapia local prévia dirigida ao cérebro e estado de desempenho ECOG de 0 ou 1.
• Coorte C: pacientes com melanoma mutado BRAFV600D/K/R com metástases cerebrais assintomáticas, com ou sem terapia local prévia dirigida ao cérebro e estado de desempenho ECOG de 0 ou 1.
• Coorte D: pacientes com melanoma mutado BRAFV600D/E/K/R com metástases cerebrais sintomáticas, com ou sem terapia local prévia dirigida ao cérebro e estado de desempenho ECOG de 0 ou 1 ou 2.
O desfecho primário do estudo foi a resposta intracraniana na Coorte A, definida como a porcentagem de pacientes com uma resposta intracraniana confirmada, avaliada pelo investigador usando o Critério de Avaliação de Resposta modificado em Tumores Sólidos (RECIST) versão 1.1. Os resultados de eficácia estão resumidos na Tabela 7. Os desfechos secundários foram a duração da resposta intracraniana, taxa de resposta global (TRG), sobrevida livre de progressão (SLP) e sobrevida global (SG). Os resultados de eficácia estão resumidos na Tabela 7.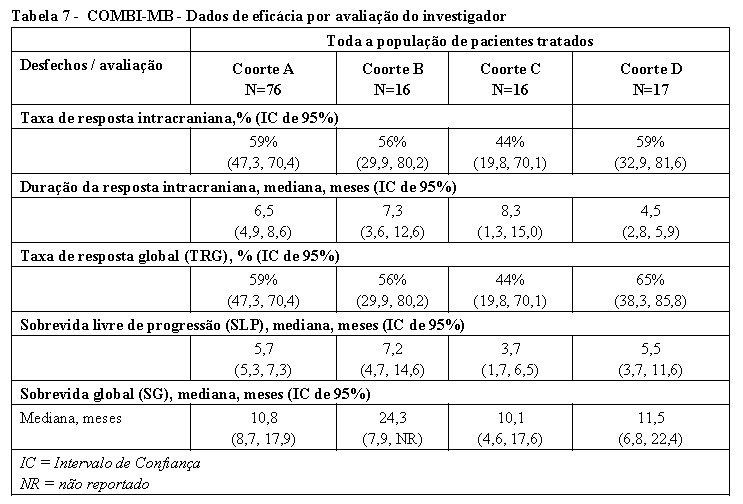
Tratamento adjuvante de melanoma
Estudo BRF115532 / DRB436F2301 (COMBI-AD)
A eficácia e segurança de Tafinlar® em combinação com dimetilsulfóxido de trametinibe foi estudado em um estudo fase III, multicêntrico, randomizado, duplo-cego, controlado por placebo, em pacientes com melanoma de estágio III com mutação BRAF V600, após ressecção completa.
Os pacientes foram randomizados 1: 1 para receber a terapia combinada com dabrafenibe e dimetilsulfóxido de trametinibe (Tafinlar® 150 mg duas vezes ao dia e dimetilsulfóxido de trametinibe 2 mg uma vez ao dia) ou dois placebos por um período de 12 meses. A inclusão exigiu ressecção completa do melanoma com linfadenectomia completa dentro de 12 semanas antes da randomização. Qualquer tratamento anticâncer sistêmico prévio, incluindo radioterapia, não foi permitido. Pacientes com história prévia de malignidade, se livre da doença por pelo menos 5 anos, eram foram elegíveis. Os pacientes que apresentaram neoplasias malignas com mutações RAS ativadas confirmadas não foram elegíveis. Os pacientes foram estratificados pelo estado de mutação BRAF (V600E ou V600K) e estágio de doença antes da cirurgia (por sub-estágio do estágio III, indicando diferentes níveis de envolvimento dos linfonodos e tamanho do tumor primário e ulceração). O desfecho primário foi a sobrevida livre de recidiva (SLR) avaliada pelo investigador, definida como o tempo desde a randomização até a recidiva da doença ou a morte por qualquer causa. A avaliação radiológica do tumor foi realizada a cada 3 meses nos primeiros dois anos e, a cada 6 meses a partir disso, até a primeira recaída ser observada. Os desfechos secundários incluem a sobrevida global (SG, desfecho secundário) e a sobrevida livre de metástase distante (SLMD).
Um total de 870 pacientes foram randomizados para os braços de terapia em combinação (n = 438) e placebo (n = 432). A maioria dos pacientes eram caucasianos (99%) e do sexo masculino (55%), com idade mediana de 51 anos (18% tinham ≥65 anos). O estudo incluiu pacientes com todos os sub-estágios do estágio III da doença antes da ressecção; 18% destes pacientes apresentavam comprometimento dos linfonodos identificáveis somente por microscópio e sem ulceração tumoral primária. A maioria dos pacientes apresentava uma mutação BRAF V600E (91%).
A duração mediana do seguimento no momento da análise primária foi de 2,83 anos no braço da combinação de dabrafenibe e dimetilsulfóxido de trametinibe e 2,75 anos no braço placebo.
Os resultados para a análise primária de SLR são apresentados na Tabela 8. O estudo mostrou uma diferença estatisticamente significativa para o desfecho primário de SLR avaliado pelo investigador entre os braços tratados, com uma estimativa de redução de risco de 53% no braço da combinação de dabrafenibe e dimetilsulfóxido de trametinibe em comparação com o braço grupo placebo (HR = 0,47; IC 95%: 0,39, 0,58; p = 1,53 × 10-14).Os resultados foram consistentes através dos subgrupos, incluindo fatores de estratificação para o estágio da doença e o tipo de mutação BRAF V600. A SLR mediana foi de 16,6 meses para o braço placebo e não foi alcançado para o braço da combinação no momento da análise primária.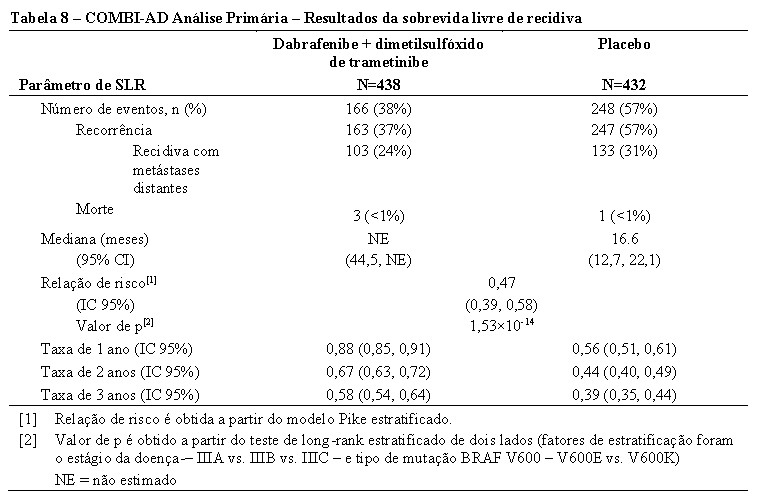
Com base em dados atualizados com um adicional de 29 meses de acompanhamento em comparação com a análise primária (acompanhamento mínimo de 59 meses), o benefício de SLR foi mantido com uma RR estimada de 0,51 (IC de 95%: 0,42, 0,61) (Figura 4). A taxa de SLR de 5 anos foi de 52% (IC de 95%: 48, 58) no braço da combinação em comparação com 36% (IC de 95%: 32, 41) no braço placebo.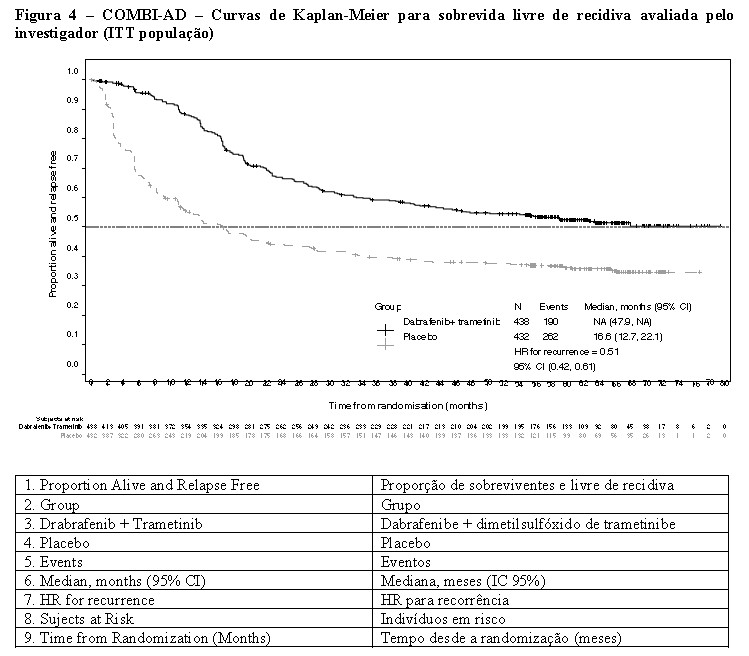
A duração mediana do acompanhamento no momento da análise final de sobrevida global foi de 8,3 anos no braço de combinação e 6,9 anos no braço de placebo. A taxa de risco estimada para sobrevida global foi de 0,80 (IC de 95%: 0,62, 1,01; p=0,063) com 125 eventos (29%) no braço de combinação e 136 eventos (31%) no braço de placebo. As taxas estimadas de sobrevida global de 5 anos foram de 79% no braço de combinação e 70% no braço de placebo, e as taxas estimadas de sobrevida global de 10 anos foram de 66% no braço de combinação e 63% no braço de placebo. Em pacientes que passaram a receber terapias anticâncer subsequentes após o tratamento do estudo, as terapias incluíram terapia direcionada em 21% no braço de combinação e 37% no braço de placebo, e imunoterapia em 29% no braço de combinação e 29% no braço de placebo. As curvas de Kaplan-Meier para a análise final de sobrevida global são mostradas na Figura 5. 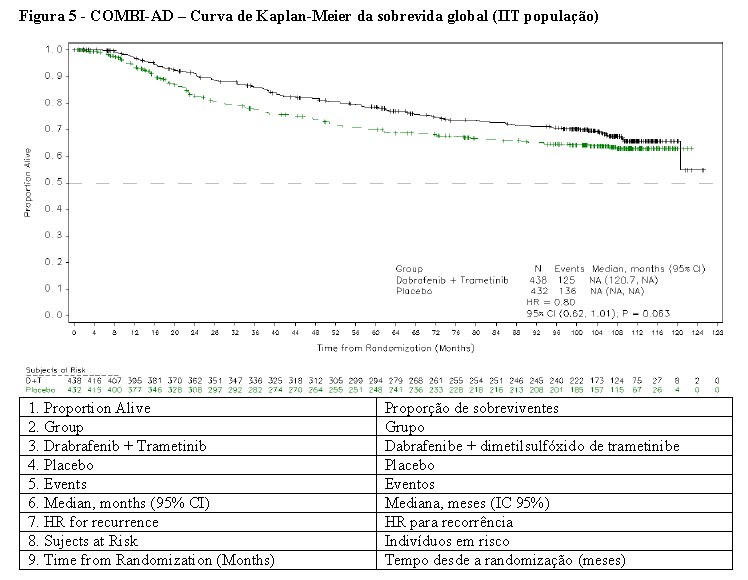
CPCNP Avançado
Estudo E2201 (BRF113928)
A eficácia e a segurança de Tafinlar® em combinação com dimetilsulfóxido de trametinibe foram estudadas em um estudo aberto de Fase II, Coorte 3, multicêntrico, não randomizado, que incluíram pacientes com CPCNP em estágio IV com mutação de BRAF V600E.
Os critérios de elegibilidade principais foram CPCNP metastático, positivo para mutação BRAF V600E, confirmado por um laboratório local certificado; pacientes sem tratamento prévio com inibidores de BRAF ou inibidor de MEK e ausência de mutação EGFR ou rearranjo ALK (a não ser que os pacientes tenham progredido na terapia prévia com inibidor de tirosina quinase). Para as coortes A e B, os critérios de inclusão também incluíram progressão tumoral documentada após o recebimento de, no mínimo, um regime quimioterápico à base de platina prévio aprovado para o CPCNP de estágio avançado/metastático, mas não mais que três tratamentos sistêmicos prévios. Para a coorte C, os pacientes não poderiam ter recebido tratamento sistêmico prévio para o CPCNP de estágio avançado/metastático.
O desfecho primário foi a taxa de resposta global avaliada pelo investigador (TRG) usando os "Critérios de avaliação de resposta em tumores sólidos" (RECIST 1.1). Os pontos finais secundários incluíram a duração da resposta (DoR), a sobrevida livre de progressão (SLP), a sobrevida global (SG), a segurança e a farmacocinética populacional. TRG, DoR e SLP também foram avaliados por um Comitê de Revisão Independente (IRC) como uma análise de sensibilidade.
Os Coortes foram incluídos sequencialmente:
• Coorte B (n = 57): terapia combinada (Tafinlar® 150 mg duas vezes ao dia e dimetilsulfóxido de trametinibe 2 mg uma vez por dia): 59 pacientes incluídos. 57 pacientes receberam previamente de uma a três linhas de tratamento sistêmico para sua doença metastática. Dois pacientes não apresentaram nenhum tratamento sistêmico prévio e foram incluídos na análise para pacientes incluídos no coorte C.
• Coorte C (n = 36): terapia combinada (Tafinlar® 150 mg duas vezes ao dia e dimetilsulfóxido de trametinibe 2 mg uma vez por dia): 34 pacientes incluídos (nota: os dois pacientes da coorte B que não apresentaram nenhum tratamento sistêmico prévio foram incluídos na análise para pacientes incluídos no coorte C para um total de 36 pacientes). Todos os pacientes receberam medicação em estudo como tratamento de primeira linha para doença metastática.
Entre o total de 93 pacientes que foram incluídos na terapia combinada em Coortes B e C, a maioria dos pacientes era caucasiana (n = 79, 85%). Houve discreta superioridade de mulheres em relação a homens (54% vs 46%). A idade média foi de 64 anos em pacientes que tiveram pelo menos uma terapia prévia e 68 anos em pacientes que não receberam tratamento para a doença avançada. A maioria dos pacientes (n = 87, 94%) incluída nos Coortes de terapia combinada tinha estado de desempenho pelo ECOG de 0 ou 1. Vinte e seis (26) pacientes (28%) nunca haviam fumado. Noventa e um (91) pacientes (97,8%) tiveram uma histologia não escamosa. Na população pré-tratada, 38 pacientes (67%) haviam recebido uma linha de terapia sistêmica contra o câncer metastático. No momento da análise primária, para o desfecho primário, a TRG avaliada pelo investigador foi de 61,1% (IC 95%, 43,5, 76,9) na população de primeira linha e 66,7% (IC 95%, 52,9%, 78,6%) na população previamente tratada. Estes resultados atingiram a significância estatística para rejeitar a hipótese nula de que a TRG de dimetilsulfóxido de trametinibe em combinação com Tafinlar® para ambas as populações de CPCNP era menor ou igual a 30%.
Os resultados TRG avaliados pelo IRC foram consistentes com a avaliação do investigador (Tabela 9).
A análise final da eficácia realizada 5 anos após a primeira dose do último indivíduo é apresentada na Tabela 9.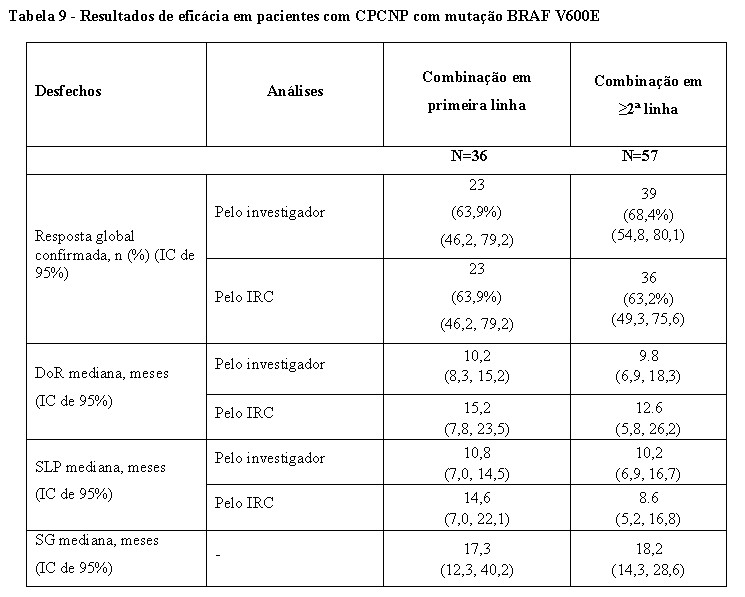
Câncer anaplásico de tireoide localmente avançado ou metastático
Estudo BRF117019 / CDRB436X2201
A eficácia e segurança de Tafinlar® em combinação com dimetilsulfóxido de trametinibe foram estudadas em um estudo de Fase II, nove coortes, multicêntrico, não randomizado e aberto em pacientes com cânceres raros com a mutação BRAF V600E, incluindo câncer anaplásico de tireoide (CAT) localmente avançado ou metastático.
O estudo teve análises provisórias pré-especificadas que foram realizadas aproximadamente a cada 12 semanas. Pacientes receberam Tafinlar® 150 mg duas vezes ao dia e dimetilsulfóxido de trametinibe 2 mg uma vez ao dia. O desfecho primário foi a taxa de resposta global avaliada pelo investigador (TRG) usando os 'Critérios de Avaliação de Resposta em Tumores Sólidos' (RECIST 1.1 avaliado pelo investigador). Os desfechos secundários incluíram a duração da resposta (DoR), sobrevida livre de progressão (SLP), sobrevida global (SG) e segurança. TRG, DoR e SLP também foram avaliados por um Comitê de Revisão Independente (IRC).
Trinta e seis pacientes foram incluídos e avaliados quanto à resposta na coorte CAT. A idade mediana foi de 71 anos (variação: 47 a 85); 44% eram homens, 50% brancos, 44% asiáticos; e 94% tinham status de desempenho ECOG de 0 ou 1. Os tratamentos anticâncer anteriores incluíram cirurgia (n = 30, 83%), radioterapia de feixe externo (n = 30, 83%) e terapia sistêmica (n = 24, 67%) para CAT. Os testes de laboratório central confirmaram a mutação BRAF V600E em 33 pacientes (92%).
Para o desfecho primário, a TRG avaliada pelo investigador foi de 56% (IC 95%: 38,1, 72,1) na coorte CAT. Os resultados da TRG avaliados pelo IRC e pela avaliação do investigador foram consistentes (Tabela 10).
As respostas foram duráveis com uma DoR mediana na coorte CAT de 14,4 meses (IC 95%: 7,4, 43,6) pela avaliação do investigador e uma SLP mediana de 6,7 meses (IC 95%: 4,7, 13,8).
Para pacientes com CAT, a SG mediana foi 14,5 meses (IC 95%: 6,8, 23,2). A estimativa de Kaplan-Meier da sobrevida global em 12 meses para pacientes com CAT foi de 51,7% (IC 95%: 33,6, 67,1).
Tumores sólidos irressecáveis ou metastáticos
A segurança e eficácia de Tafinlar® em combinação com dimetilsulfóxido de trametinibe para o tratamento de tumores sólidos irressecáveis ou metastáticos positivos para mutação BRAF V600E foram avaliadas nos estudos clínicos BRF117019, NCI-MATCH e CTMT212X2101, e são suportadas por resultados em COMBI-d, COMBI-v, e BRF113928.
Em estudos com adultos, os pacientes receberam Tafinlar® 150 mg duas vezes ao dia e dimetilsulfóxido de trametinibe 2 mg uma vez ao dia. As principais medidas de resultados de eficácia foram TRG por RECIST v1.1, critérios RANO [GAG] ou RANO modificados [GBG] e duração da resposta (DoR).
Estudo BRF117019 e Estudo NCI-MATCH
O estudo BRF117019 é um estudo aberto, não randomizado, multicêntrico, multicoorte em pacientes adultos com tumores selecionados com a mutação BRAF V600E, incluindo glioma de alto grau (GAG) (n = 45), câncer do trato biliar (CTB) (n = 43), glioma de baixo grau (GBG) (n = 13), adenocarcinoma de intestino delgado (AID) (n = 3), tumor estromal gastrointestinal (TEG) (n = 1) e câncer anaplásico de tireoide. Os pacientes foram incluídos com base em avaliações locais do status da mutação BRAF V600E; um laboratório central confirmou a mutação BRAF em 93 de 105 pacientes.
O braço H (EAY131-H) do estudo NCI-MATCH é um estudo aberto de braço único que incluiu pacientes com mutação BRAF V600E. Pacientes com melanoma, câncer de tireoide ou CCR foram excluídos. O status da mutação BRAF V600E para inclusão no estudo foi determinado por teste laboratorial central ou local. O estudo incluiu pacientes adultos com tumores sólidos, incluindo tumores gastrointestinais (n = 14), tumores pulmonares (n = 7), tumores ginecológicos ou peritoneais (n = 6), tumores do SNC (n = 4) e ameloblastoma de mandíbula (n = 1).
Entre os 131 pacientes incluídos no BRF117019 e NCI-MATCH com os tipos de tumor mostrados na Tabela 11, as características basais foram: idade mediana de 51 anos com 20% com 65 anos ou mais; 56% do sexo feminino; 85% brancos, 9% asiáticos, 3% negros, 3% outros; e 37% ECOG 0, 56% ECOG 1 e 6% ECOG 2. Dos 131 pacientes, 90% receberam terapia sistêmica prévia.
Os resultados de eficácia em pacientes com tumores sólidos estão resumidos na Tabela 11.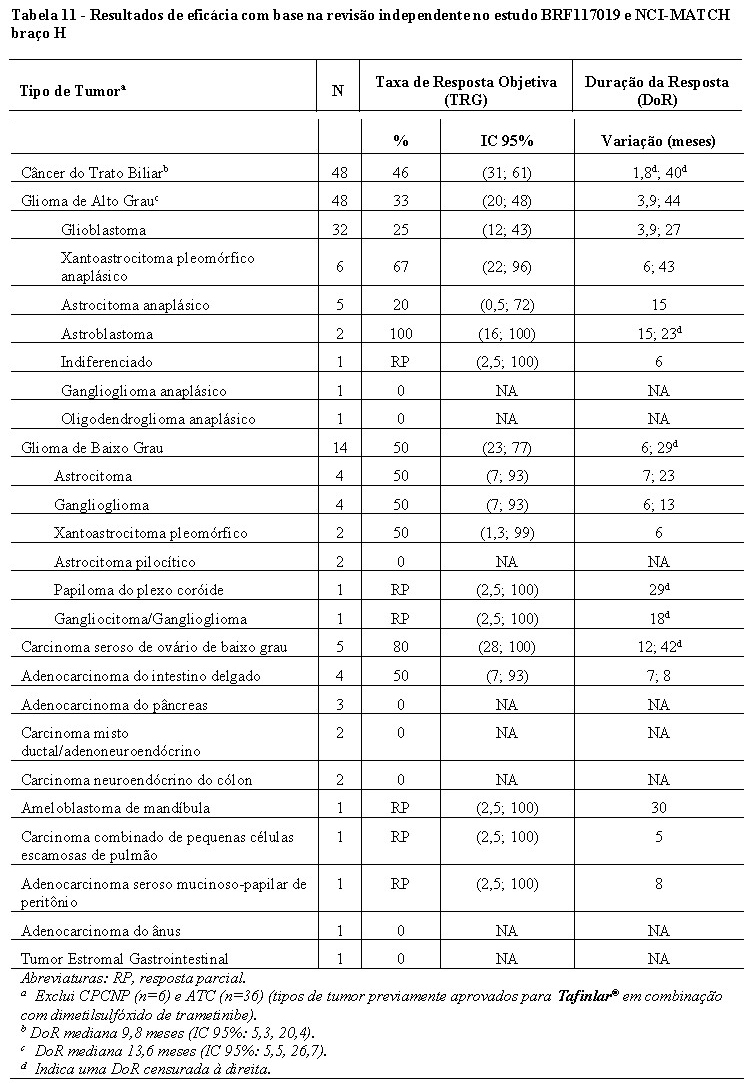
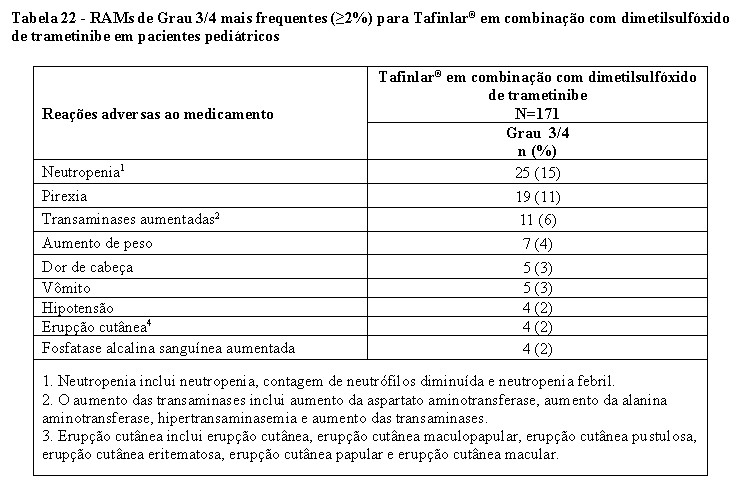
Estudo CTMT212X2101 (X2101)
O estudo X2101 foi um estudo de coorte múltipla, aberto e multicêntrico em pacientes pediátricos com tumores sólidos refratários ou recorrentes. A parte C foi um escalonamento de dose de Tafinlar® em combinação com dimetilsulfóxido de trametinibe em pacientes com mutação BRAF V600E. A parte D foi uma fase de expansão de coorte de Tafinlar® em combinação com dimetilsulfóxido de trametinibe em pacientes com GBG com mutação BRAF V600E. A principal medida de resultado de eficácia foi TRG conforme avaliado pelo comitê de revisão independente de acordo com os critérios RANO.
A eficácia de Tafinlar® em combinação com dimetilsulfóxido de trametinibe foi avaliada em 48 pacientes pediátricos, incluindo 34 pacientes com GBG e 2 pacientes com GAG.
Para pacientes com GBG com mutação BRAF V600E nas Partes C e D, a idade mediana foi de 10 anos (variação: 1 a 17); 50% eram homens, 75% brancos, 8% asiáticos, 3% negros; e 58% tinham status de desempenho de Karnofsky/Lansky de 100. Os tratamentos anticâncer anteriores incluíram cirurgia (83%), radioterapia externa (2,8%) e terapia sistêmica (92%). A TRG foi de 25% (IC 95%: 12%, 42%). Para os 9 pacientes que responderam, a DoR foi ≥6 meses para 78% dos pacientes, ≥12 meses para 56% dos pacientes e ≥24 meses para 44% dos pacientes.
Outros estudos
Análise de gerenciamento de pirexia
Pirexia é observada em pacientes tratados com a terapia combinada de Tafinlar® e dimetilsulfóxido de trametinibe. Os estudos de registro iniciais para a terapia de combinação no cenário de melanoma irressecável ou metastático (COMBI-d e COMBI-v; total N = 559) e no cenário de melanoma adjuvante (COMBI-AD, N = 435) recomendaram interromper apenas Tafinlar® em caso de pirexia. Em dois estudos subsequentes em melanoma irressecável ou metastático (braço de controle COMBI-i, N = 264) e na configuração de melanoma adjuvante (COMBI-Aplus, N = 552), interrupção de Tafinlar® e dimetilsulfóxido de trametinibe quando a temperatura do paciente era ≥ 38°C (COMBI-Aplus) ou ao primeiro sintoma de pirexia (COMBI-i; COMBI-Aplus para pirexia recorrente), resultou em melhores resultados relacionados à pirexia sem afetar a eficácia:
• Configuração de melanoma irressecável ou metastático (COMBI-d / v vs COMBI-i):
- grau 3/4 de pirexia reduzida de 6,6% para 3,4%
- hospitalização devido a pirexia reduzida de 12,3% para 6,1%
- pirexia com complicações (desidratação, hipotensão, disfunção renal, síncope, calafrios graves) reduzida de 6,4% para 1,9%
- as taxas de descontinuação do tratamento devido à pirexia foram comparáveis, 1,1% versus 1,9%
• Configuração de melanoma adjuvante (COMBI-AD vs COMBI-Aplus):
- grau 3/4 de pirexia reduzida de 5,7% para 4,3%
- hospitalização devido a pirexia reduzida de 11,0% para 5,1%
- pirexia com complicações (desidratação, hipotensão, disfunção renal, síncope, calafrios graves) reduzida de 6,0% para 2,2%
- descontinuação do tratamento devido à pirexia reduzida de 6,2% para 2,5%
Referências
1. [COMBI-d]: LONG G.V., Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma.
N ENGL J MED. November 11, 2014.
2. [COMBI-d]: SCHADENDORF, D. Health-related quality of life impact in a randomised phase III study of
the combination of dabrafenib and trametinib versus dabrafenib monotherapy in patients with BRAF V600
metastatic melanoma. Eur J Cancer (2015), http://dx.doi.org/10.1016/j.ejca.2015.03.004.
3. [COMBI-v]: ROBERT, C. Improved overall survival in melanoma with combined dabrafenib and trametinib.
N ENGL J MED. November 17, 2014.
4. [COMBI-v]: GROB, J.J. Comparison of dabrafenib and trametinib combination therapy with vemurafenib monotherapy on health-related quality of life in patients with unresectable or metastatic cutaneous BRAF Val600-mutation-positive melanoma (COMBI-v): results of a phase 3, open-label, randomised trial. Lancet Oncol 2015; 16: 1389-98. October, 2015.
5.Phase II Trial (BREAK-2) of the BRAF Inhibitor Dabrafenib (GSK2118436) in Patients With Metastatic Melanoma J Clin Oncol 31:3205-3211. © 2013.
6. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial Lancet Oncol 2012; 13: 1087-95.
7. Long GV, Hauschild A, Santinami M, et al. Adjuvant Dabrafenib Plus Trametinib for Stage III BRAF V600E/K-Mutant Melanoma. New England Journal of Medicine. 2017.
8. Planchard D. et al., Dabrafenib plus trametinib in patients with previously treated BRAFV600E-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol 2016; 17: 984-993.
9. Planchard D. et al., Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol 2017; 18(10):1307-1316.
10. Davies MA, Saiag P, Robert C, et al (2017) Dabrafenib plus trametinib in patients with BRAFV600-mutant melanoma brain metastases (COMBI-MB): a multicentre, multicohort, openlabel, phase 2 trial. Lancet Oncol; 18(7): 863-73.
11. Robert C, Grob JJ, Stroyakovskiy D, et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N Engl J Med 2019; 381:626-636.
12. Subbiah V, Kreitman RJ, Wainberg ZA, et al: Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAF V600-mutant anaplastic thyroid cancer. J Clin Oncol 2018; 36:7-13.
13. Tafinlar and Mekinist 2.5 Clinical Overview Adjuvant treatment of Stage III BRAF V600 mutation positive melanoma: 5-year efficacy update. Novartis. 19-Feb-2021.
14. Tafinlar and Mekinist 2.5 Clinical Overview in BRAF V600E Mutation-positive Non-small Cell Lung Cancer (NSCLC) - long term data. Novartis. 28-Jun-2021.
15. Tafinlar and Mekinist 2.5 Clinical Overview Rationale for changes to US Product Information - additional long-term efficacy in subjects with anaplastic thyroid cancer (ATC). Novartis. 17-Mar-2021.
16. Tafinlar and Mekinist. 2.5 Clinical Overview in rare BRAF V600E mutation-positive solid tumors. Novartis. 08-Mar-2022.
17. Tafinlar and Mekinist. 2.7.3 Summary of Clinical Efficacy in rare BRAF V600E mutation-positive solid tumors. Novartis. 24-Feb-2022.
18. Tafinlar and Mekinist. 2.5 Clinical Overview, Rationale for changes to Core Data Sheet (CDS) / Product Information - Adjuvant treatment of Stage III BRAF V600-mutant melanoma: Final results for Study CDRB436F2301 (COMBI-AD). Novartis. 24-Apr-2024.
19. Tafinlar and Mekinist. Clinical Study Report (BRF115532/DRB436F2301): COMBI-AD: A phase III randomized double blind study of dabrafenib (GSK2118436) in COMBInation with trametinib (GSK1120212) versus two placebos in the ADjuvant treatment of high-risk BRAF V600 mutation-positive melanoma after surgical resection. Novartis. 11-Jan-2024.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Grupo farmacoterapêutico: Inibidores da B-Raf serina-treonina quinase (BRAF). Código ATC: L01EC02
Propriedades Farmacodinâmicas
Mecanismo de ação
Tafinlar® em monoterapia
O dabrafenibe é um inibidor de RAF quinase ATP-competitivo, potente e seletivo, com valores de IC50 de 0,65, 0,5 e 1,84 nM para as enzimas BRAF V600E, BRAF V600K e BRAF V600D, respectivamente. As mutações oncogênicas em BRAF levam a ativação constitutiva da via RAS/RAF/MEK/ERK e estimulação do crescimento das células tumorais. As mutações de BRAF têm sido identificadas em uma alta frequência em cânceres específicos, incluindo aproximadamente 50% dos melanomas e 1-3% de CPCNP. A mutação de BRAF mais comumente observada, V600E, e a próxima mais comum, V600K, respondem por 95% das mutações de BRAF encontradas em todos os pacientes com câncer. Um número de substituições raras também ocorre incluindo V600D, V600G e V600R.
O dabrafenibe também inibe BRAF selvagem e enzimas CRAF com valores de IC50 de 3,2 E 5,0 nM, respectivamente em ensaios bioquímicos. O dabrafenibe inibe o crescimento celular de melanoma mutante BRAF V600 e o crescimento da linha celular de CPCNP e CAT in vitro e os modelos de xenoenxertos de melanoma in vivo.
Tafinlar® em combinação com dimetilsulfóxido de trametinibe
O dimetilsulfóxido de trametinibe é um inibidor reversível, altamente seletivo, alostérico da ativação da quinase regulada de sinal extracelular mitógeno-ativado 1 (MEK1) e inibidor da atividade da quinase MEK 2.
Proteínas MEK são componentes da via de sinalização da quinase relacionada ao sinal extracelular (ERK). O Tafinlar® e o dimetilsulfóxido de trametinibe inibem duas quinases nesta via, BRAF e MEK, e a combinação proporciona inibição concomitante da via. A combinação de Tafinlar® com dimetilsulfóxido de trametinibe é sinérgica nas linhagens de células do melanoma, CPCNP e CAT com mutação BRAF V600E positivo in vitro e retarda o surgimento de resistência in vivo em xenoenxertos de melanoma com mutação BRAF V600 positivo.
Efeitos Farmacodinâmicos
O Tafinlar® demonstrou supressão de um biomarcador farmacodinâmico à jusante (ERK fosforilado) em linhagens celulares de melanoma mutante BRAF V600, in vitro e em modelos animais.
Em indivíduos com melanoma mutante BRAF V600, a administração de Tafinlar® resultou em inibição de ERK fosforilado do tumor em relação ao período basal.
Eletrofisiologia cardíaca
O efeito potencial de Tafinlar® no prolongamento QT foi avaliado em um estudo QT com múltiplas doses. Uma dose supraterapêutica de 300 mg de Tafinlar® duas vezes ao dia foi administrada em 32 indivíduos com tumores com mutação BRAF-V600 positiva. Nenhum evento clinicamente relevante de Tafinlar® ou seus metabólitos no intervalo QTc foi observado.
Em ensaios clínicos, o prolongamento do QTc (QT corrigido pela frequência cardíaca) para ≥500 ms ocorreu em 0.8% (2/264) dos pacientes que receberam Tafinlar® mais trametinibe