SOLIRIS
ALEXION
eculizumabe
Trat. da hemoglobinúria paroxística noturna.
Apresentações.
Soliris® (eculizumabe) 300 mg (10 mg/mL): embalagem com um frasco-ampola contendo 30 ml de solução estéril para diluição para infusão intravenosa.
Solução para Diluição para Infusão Via Intravenosa
USO ADULTO E PEDIÁTRICO
Composição.
Cada 1 mL de Soliris® (eculizumabe) contém: eculizumabe 10 mg. Excipientes: fosfato de sódio monobásico, fosfato de sódio dibásico, cloreto de sódio, polissorbato 80 e água para injetáveis.
Informações técnicas.
Infecção menigocócica
Devido ao seu mecanismo de ação, a utilização de Soliris® aumenta a suscetibilidade dos pacientes a infecção meningocócica (Neisseria meningitidis). Estes pacientes podem estar em risco de contrair doença meningocócica devido a qualquer um dos sorotipos. No sentido de reduzir o risco de infecção, todos os pacientes devem ser vacinados pelo menos 2 semanas antes de receber Soliris®, a menos que o risco de atrasar a terapia com Soliris®ultrapasse os riscos de desenvolver uma infecção meningocócica. Os pacientes que iniciaram o tratamento com Soliris® em menos de 2 semanas após receberem a vacina meningocócica devem receber tratamento com antibióticos profiláticos apropriados até 2 semanas após a vacinação. Recomenda-se a vacinação contra os sorotipos A, C, Y, W135 e B onde disponível, na prevenção dos sorotipos meningocócicos patogênicos mais comuns. Os pacientes devem receber a vacina de acordo com as diretrizes clínicas de vacinação atuais.
A vacinação pode ainda ativar o complemento. Como resultado, pacientes com doenças mediadas pelo complemento, incluindo HPN e SHUa, podem experimentar um aumento nos sinais e sintomas de suas doenças de base, tais quais hemólise (HPN) e MAT (SHUa). Desse modo, os pacientes devem ser monitorados rigorosamente para identificação de sintomas da doença após a vacinação recomendada.
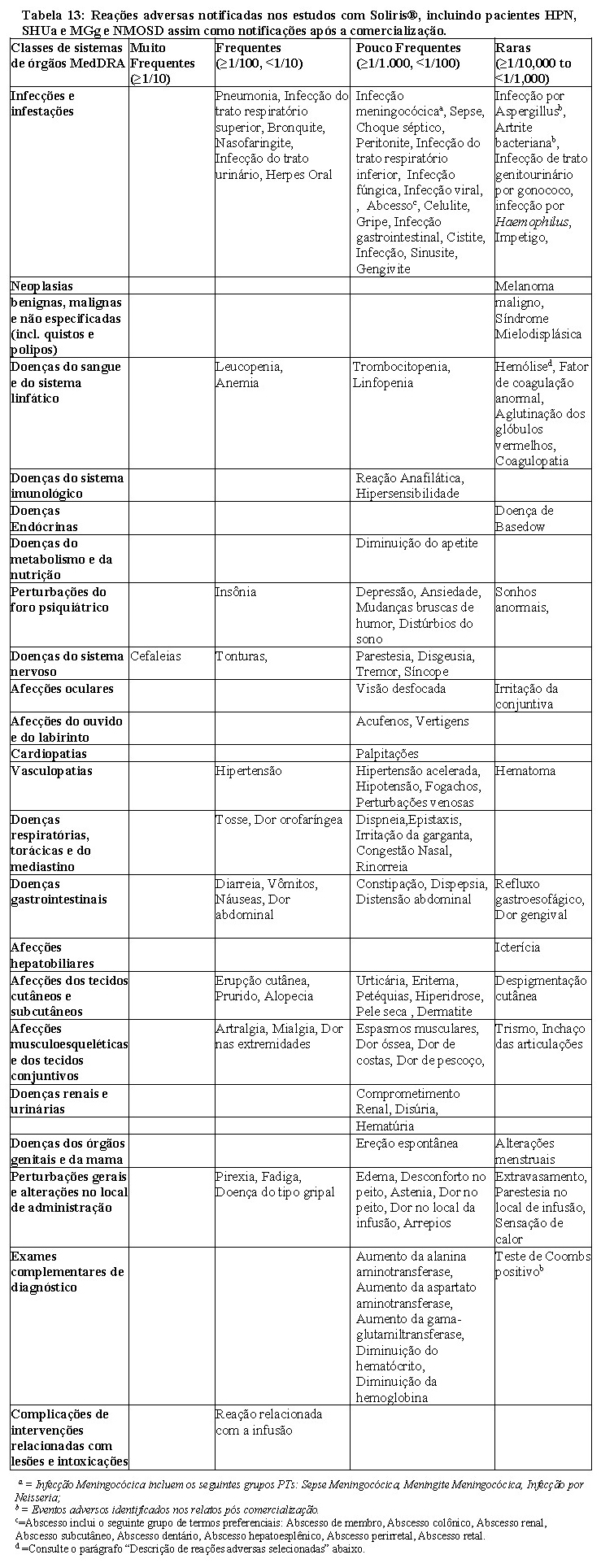
A vacinação pode não ser suficiente para prevenir a infecção meningocócica. Devem ponderar-se as orientações oficiais sobre o uso adequado de agentes antibacterianos. Foram notificados casos graves ou fatais de infecção meningocócica em pacientes tratados com Soliris®. Sepse é a apresentação comum das infecções meningocócicas em pacientes tratados com Soliris (vide seção 9 Reações Adversas). Todos os pacientes devem ser monitorizados para a detecção de sinais precoces de infecção meningocócica, examinados de imediato no caso de suspeita de infecção e, se necessário, tratados com antibióticos apropriados. Os pacientes devem ser informados sobre estes sinais e sintomas e as medidas a tomar para procurar cuidados médicos imediatos. Os médicos devem discutir os benefícios e os riscos da terapia com Soliris® com os pacientes e fornecer-lhes um cartão de segurança do paciente.
1. INDICAÇÕES
Soliris® (eculizumabe) é indicado em adultos e crianças para o tratamento de pacientes com:
-Hemoglobinúria paroxística noturna (HPN).
A evidência do benefício clínico de Soliris® (eculizumabe) foi demonstrada no tratamento de pacientes com hemólise e sintoma(s) clínico(s) indicativo(s) de alta atividade da doença, independente do histórico de transfusões.
-Síndrome hemolítico urêmica atípica (SHUa).
Soliris® não é indicado para pacientes com síndrome hemolítico urêmica relacionada a toxina Shiga de Escherichia coli.
2. RESULTADOS DE EFICÁCIA
Eculizumabe é um anticorpo (IgG2/4k) monoclonal humanizado, produzido numa linha celular de NS0 por tecnologia de DNA recombinante.
Eficácia e segurança clínicas
Hemoglobinúria Paroxística Noturna
A segurança e eficácia de Soliris® em pacientes hemolíticos com HPN foram avaliadas num estudo randomizado, duplo cego, controlado por placebo, com duração de 26 semanas (C04-001). Os pacientes com HPN também foram tratados com Soliris® num estudo com um grupo único de 52 semanas (C04-002) e num estudo de extensão de longa duração (E05-001). Os pacientes receberam vacinação meningocócica antes de receberem Soliris®. Em todos os estudos, a dose de eculizumabe foi de 600 mg a cada 7 ± 2 dias durante 4 semanas, a que se seguiram 900 mg 7±2 dias mais tarde e, depois, 900 mg a cada 14 ± 2 dias até ao final do estudo. Soliris® foi administrado por infusão intravenosa com uma duração de aproximadamente 25 a 45 minutos. Iniciou-se também um registro observacional, não intervencional, em pacientes com HPN (M07-001), para caracterizar a história natural da doença em pacientes não tratados e os resultados clínicos durante o tratamento com Soliris®.
No estudo C04-001, foram incluídos pacientes com HPN que receberam pelo menos 4 transfusões nos 12 meses precedentes, com confirmação de pelo menos 10% de células HPN por citometria de fluxo e com contagens de plaquetas correspondentes a, pelo menos, 100.000/mL. Os pacientes foram distribuídos aleatoriamente pelo braço Soliris® (n = 43) ou placebo (n = 44). Antes da randomização, todos os pacientes foram sujeitos a um período inicial de observação para confirmar a necessidade de transfusão de eritrócitos e para identificar a concentração de hemoglobina (o "valor de referência") que iria definir a estabilização dos valores de hemoglobina e os resultados de transfusões. O valor de referência de hemoglobina para indicação de transfusão era igual ou menor que 9 g/dl em pacientes sintomáticos e igual ou menor que 7 g/dl em pacientes assintomáticos. Os parâmetros primários de avaliação da eficácia foram a estabilização da hemoglobina (pacientes que mantiveram uma concentração de hemoglobina acima do valor de referência de hemoglobina para gatilho transfusional, evitando a necessidade de transfusão de eritrócitos durante o período total de 26 semanas) e a necessidade de transfusão de sangue. A fadiga e a qualidade de vida relacionada com aspectos de saúde foram parâmetros de avaliação secundários relevantes. A hemólise foi monitorizada principalmente pela medição de níveis séricos de desidrogenase láctica (DHL) e a proporção de eritrócitos HPN foi monitorizada por citometria de fluxo. Os pacientes que recebiam anticoagulantes e corticosteróides sistêmicos no início do tratamento continuaram estas medicações. As principais características no início do estudo eram equilibradas (ver Tabela 1).
No estudo não controlado C04-002, pacientes com HPN que receberam pelo menos uma transfusão nos 24 meses precedentes e apresentando, pelo menos, 30.000 plaquetas/mL, receberam Soliris® durante um período de 52 semanas. Medicamentos concomitantes incluíram agentes antitrombóticos em 63% dos pacientes e corticosteróides sistêmicos em 40% dos pacientes. As características no início do estudo são apresentadas na Tabela 1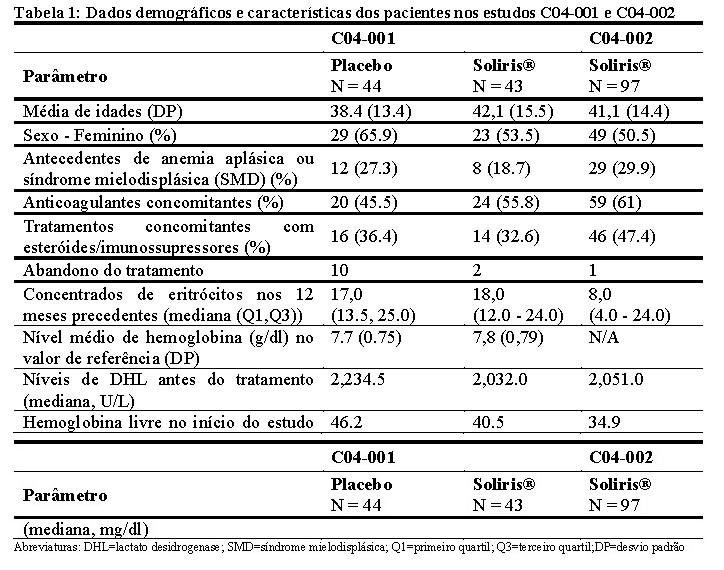
No estudo C04-001, os pacientes tratados com Soliris® apresentaram uma redução significativa da hemólise (p < 0,001), resultando em melhora da anemia, indicada por um aumento da estabilização da hemoglobina e pela redução da necessidade de transfusões de eritrócitos, em comparação com os pacientes tratados com placebo (ver Tabela 2). Estes efeitos foram observados nos pacientes de cada um dos 3 níveis de transfusão de eritrócitos antes do estudo (4 à 14 unidades; 15 à 25 unidades; > 25 unidades).
Após 3 semanas de tratamento com Soliris®, os pacientes referiram menor fadiga e melhora na qualidade de vida relacionada com aspectos de saúde. Devido ao tamanho da amostra e à duração do estudo, os efeitos de Soliris® sobre acontecimentos trombóticos não puderam ser avaliados. No estudo C04-002, 96 dos 97 pacientes incluídos completaram o estudo (1 paciente morreu após um evento trombótico). Durante o período do tratamento, manteve-se uma redução na hemólise intravascular, avaliada pelos níveis séricos de DHL, a qual resultou no aumento da capacidade de evitar transfusões, na redução da necessidade de transfusões de eritrócitos e na redução da fadiga. (ver Tabela 2).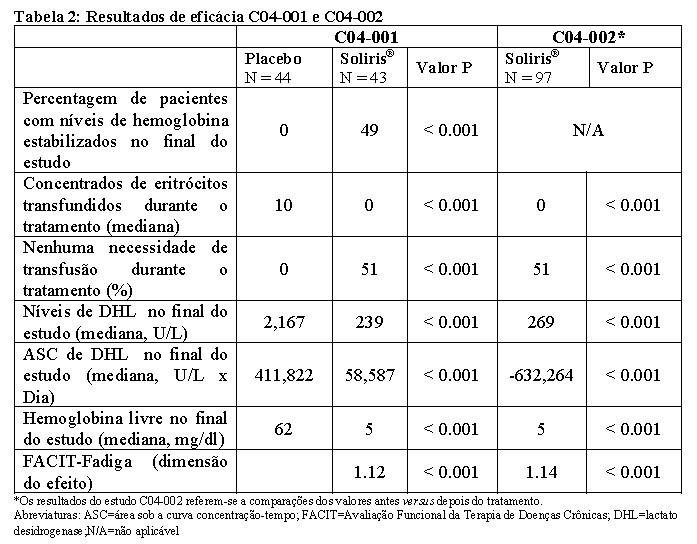
Dos 195 pacientes originários dos estudos C04-001, C04-002 e outros estudos iniciais, os pacientes com HPN tratados com Soliris® foram incluídos num estudo de extensão de longo prazo (E05-001). Todos os pacientes mantiveram uma redução da hemólise intravascular durante o período total de exposição ao Soliris®, que variou entre 10 e 54 meses. Observaram-se menos acontecimentos trombóticos com o tratamento com Soliris® do que no mesmo período de tempo anterior ao tratamento. No entanto, esta observação foi demonstrada em estudos clínicos não controlados.
O Registro HPN (M07-001) foi usado para avaliar a eficácia de Soliris® em pacientes com HPN sem história de transfusão de eritrócitos. Estes pacientes tinham alta atividade da doença, definida por hemólise elevada (DHL ≥1,5x LSN - Limite Superior da Normalidade) e a presença do(s) seguinte(s) sintoma(s) clínicos relacionado(s): fadiga, hemoglobinúria, dor abdominal, falta de ar (dispneia), anemia (hemoglobina < 10,0 g/dL), eventos adversos vasculares graves (incluindo trombose), disfagia ou disfunção erétil.
No Registro HPN, observou-se nos pacientes tratados com Soliris® uma redução na hemólise e sintomas associados. Aos 6 meses, os pacientes tratados com Soliris® sem história de transfusão de eritrócitos tiveram uma redução significativa (p < 0.001) dos níveis de DHL (mediana de DHL de 305 U/L; Tabela 3). Além disso, 74% dos pacientes sem histórico de transfusão e tratados com Soliris® tiveram efeitos clinicamente significativos na Avaliação Funcional da Terapia de Doenças Crônicas (FACIT) - Pontuação de Fadiga (i.e., aumento em 4 pontos ou mais) e 84% na escala (EORTC) de fadiga (i.e., diminuição em 10 pontos ou mais).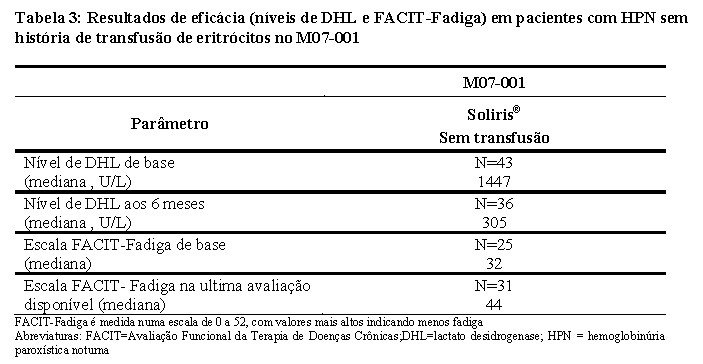
Síndrome Hemolítico Urêmica Atípica
Foram usados os dados de 100 pacientes em 4 estudos prospectivos controlados, 3 em pacientes adultos e adolescentes (C08-002A/B, C08-003A/B, C10-004) 1 em pacientes pediátricos e adolescentes (C10-003) e trinta pacientes num estudo retrospectivo (C09-001r) para avaliar a eficácia de Soliris® no tratamento do SHUa.
O C08-002A/B foi um estudo prospectivo, controlado, aberto, realizado em pacientes na fase inicial da SHUa com evidência de manifestações clínicas de microangiopatia trombótica (MAT), com uma contagem de plaquetas ≤ 150 x 109/L apesar da terapia com plasmaférese ou infusão de plasma, e com valores de DHL e de creatinina sérica acima dos limites superiores normais. O C08-003A/B foi um estudo prospectivo, controlado, aberto, que decorreu em pacientes numa fase mais tardia da SHUa, sem evidência aparente de manifestações clínicas de MAT e a receberem terapia crônica com plasmaférese ou infusão de plasma fresco congelado (≥1 tratamento de plasmaférese ou infusão de plasma fresco congelado a cada 2 semanas e não mais que 3 tratamentos de plasmaférese ou infusão de plasma fresco congelado/semana durante pelo menos 8 semanas antes da primeira dose). Em ambos os estudos prospectivos os pacientes foram tratados com Soliris® durante 26 semanas e a maioria dos pacientes foram incluídos em um estudo de extensão de longa duração, aberto. Todos os pacientes incluídos em ambos os estudos prospectivos tinham um nível de ADAMTS-13 (uma desintegrina e metaloproteinase com trombospondina tipo 1 motivo 1 membro 13) acima de 5%.
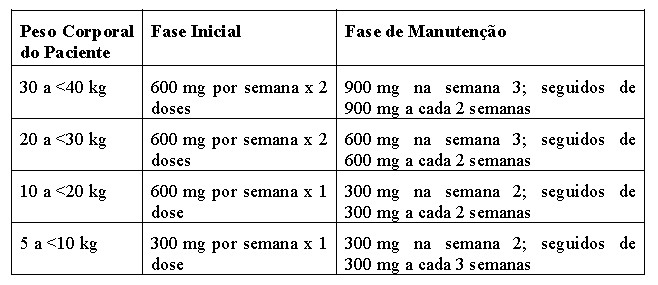
Os pacientes receberam vacinação meningocócica antes do tratamento com Soliris® ou receberam tratamento profilático com os antibióticos apropriados até 2 semanas após a vacinação. Em todos os estudos, a dose de Soliris® (eculizumabe) em pacientes adultos e adolescentes com SHUa foi de 900 mg a cada 7 ± 2 dias durante 4 semanas, em seguida de 1.200 mg 7 ± 2 dias mais tarde, e posteriormente 1.200 mg a cada 14 ± 2 dias durante a duração do estudo. Soliris® foi administrado por infusão intravenosa durante 35 minutos. O regime posológico nos pacientes pediátricos e nos adolescentes com peso inferior a 40 kg foi definido com base numa simulação farmacocinética (PK) que identificou a dose e o calendário recomendado com base no peso corporal (vide seção 8 "Posologia e Modo de Usar").
Os parâmetros de avaliação primários incluíram a alteração da contagem de plaquetas relativamente ao valor no início do estudo no estudo C08-002A/B e o estado livre de eventos da MAT no estudo C08-003A/B. Os parâmetros de avaliação adicionais incluíram a taxa de intervenções associadas à MAT, normalização hematológica, resposta completa da MAT, alterações na DHL, função renal e qualidade de vida. O estado livre de eventos da MAT foi definido como a ausência durante pelo menos 12 semanas dos seguintes: diminuição na contagem de plaquetas > 25% comparada ao valor no início do estudo, plasmaférese ou infusão de plasma, e nova diálise. As intervenções associadas à MAT foram definidas como plasmaférese ou infusão de plasma ou nova diálise. A normalização hematológica foi definida como a normalização da contagem de plaquetas e dos níveis de DHL mantidos durante ≥ 2 avaliações consecutivas por ≥ 4 semanas. A resposta completa da MAT foi definida como a normalização hematológica e uma redução ≥ 25% nos níveis séricos de creatinina mantidos durante ≥ 2 avaliações consecutivas por ≥ 4 semanas.
As características no início do estudo são apresentadas na Tabela 4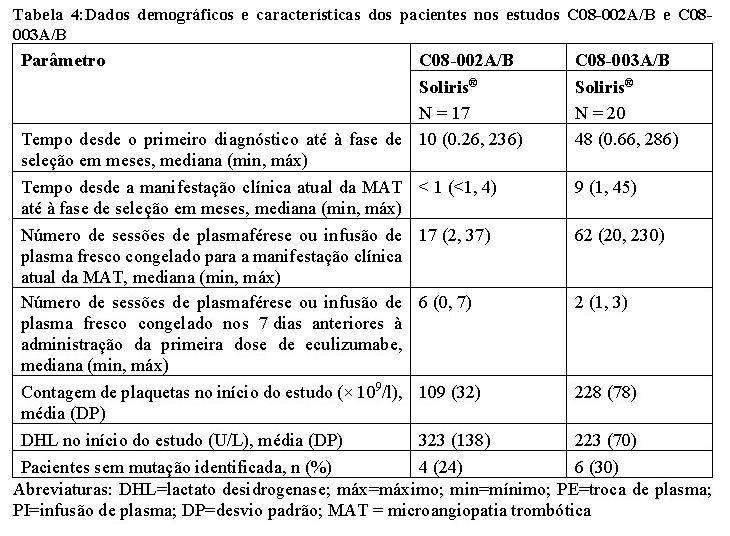
Os pacientes no estudo em SHUa C08-002 A/B receberam Soliris® por um período mínimo de 26 semanas. Após completarem o período inicial de tratamento de 26 semanas, a maioria dos pacientes continuou a receber Soliris® (eculizumabe) através da inclusão num estudo de extensão. No estudo em SHUa C08-002A/B, a duração mediana da terapia com Soliris® foi de aproximadamente 100 semanas (intervalo: 2 semanas a 145 semanas).
Foram observados uma redução na atividade do complemento terminal e um aumento na contagem de plaquetas relativamente aos valores no início do estudo após o início de Soliris®. Observou-se uma redução na atividade do complemento terminal em todos os pacientes após o início de Soliris®. A Tabela 5 resume os resultados de eficácia do estudo em SHUa C08-002A/B. Todas as taxas dos parâmetros de eficácia melhoraram ou se mantiveram, durante 2 anos de tratamento. A resposta completa da MAT foi mantida por todos os respondedores. Quando o tratamento foi mantido por mais de 26 semanas, 2 pacientes adicionais adquiriram e mantiveram a resposta completa da MAT devido à normalização do DHL (um paciente) e uma diminuição da creatinina sérica (dois pacientes).
A função renal, avaliada pela taxa de filtração glomerular estimada (TFGe), melhorou e manteve-se durante a terapia com Soliris®. Quatro dos 5 pacientes que necessitaram de diálise quando da entrada no estudo, descontinuaram a diálise durante a duração do tratamento com Soliris®, e 1 paciente voltou a necessitar de nova diálise. Os pacientes notificaram melhora da qualidade de vida (QoL) relacionada com a saúde.
No estudo em SHUa C08-002A/B, as respostas ao Soliris® foram semelhantes nos pacientes com e sem mutações identificadas nos genes que codificam as proteínas reguladoras do complemento.
Os pacientes no estudo em SHUa C08-003A/B receberam Soliris® por um período mínimo de 26 semanas. Após completarem o período inicial de tratamento de 26 semanas, a maioria dos pacientes continuou a receber Soliris® através da inclusão num estudo de extensão. No estudo em SHUa C08-003A/B, a duração mediana da terapia com Soliris® foi de aproximadamente 114 semanas (intervalo: 26 a 129 semanas). A Tabela 5 resume os resultados de eficácia do estudo em SHUa C08-003A/B.
No estudo em SHUa C08-003A/B, as respostas ao Soliris® foram semelhantes nos pacientes com e sem mutações identificadas nos genes que codificam as proteínas reguladoras do complemento. Observou-se uma redução na atividade do complemento terminal em todos os pacientes após o início de Soliris®. Todas as taxas dos parâmetros de eficácia melhoraram ou se mantiveram durante 2 anos de tratamento. A resposta completa da MAT foi mantida por todos os respondedores. Quando o tratamento foi mantido por mais de 26 semanas, 6 pacientes adicionais adquiriram e mantiveram a resposta completa da MAT devido a uma diminuição da creatinina sérica. Nenhum paciente necessitou de nova diálise com Soliris®. A função renal, avaliada pela TFGe, aumentou durante a terapia com Soliris®.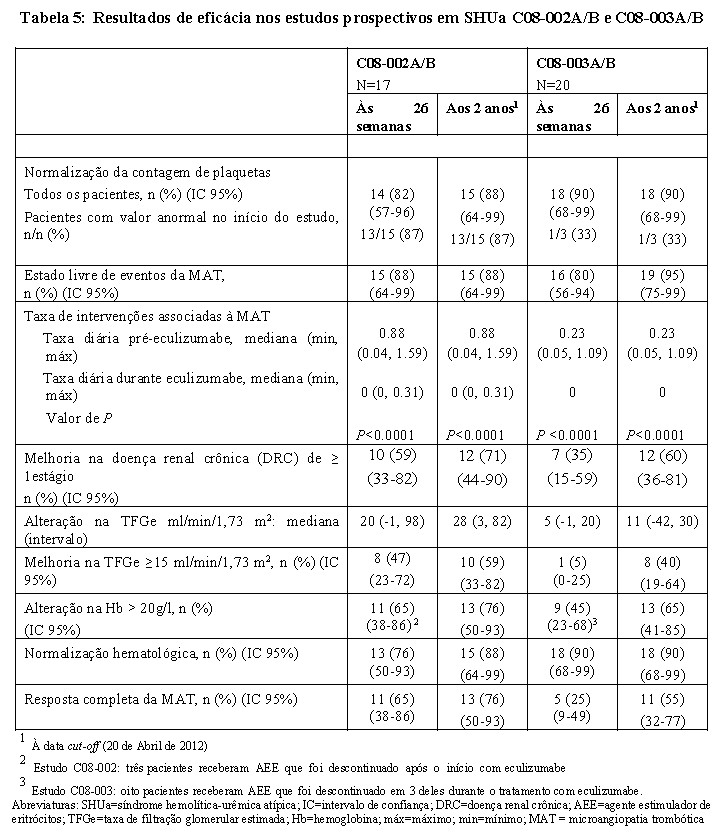
O estudo C10-004 em SHUa incluiu quarenta e um pacientes que apresentaram sinais de MAT. Para estarem aptos para a inclusão no estudo, foi requerido que os pacientes tivessem uma contagem de plaquetas inferior ao limite mais baixo do intervalo normal (LLN), evidência de hemólise como um aumento do DHL sérico, e a creatinina sérica acima dos limites superiores do normal, sem necessidade de diálise crônica. A mediana da idade do paciente foi de 35 anos (intervalo: 18 a 80 anos). Todos os pacientes incluídos no estudo C10-004 em SHUa apresentaram um nível de ADAMTS-13 acima dos 5%. Uma mutação identificável das proteínas reguladoras do complemento ou autoanticorpo foi reportada em 51% dos pacientes. Um total de 35 pacientes receberam plasmaférese ou infusão de plasma fresco congelado antes do eculizumabe. A Tabela 6 resume as principais características clínicas iniciais e relacionadas com a doença dos pacientes incluídos no estudo C10-004 em SHUa.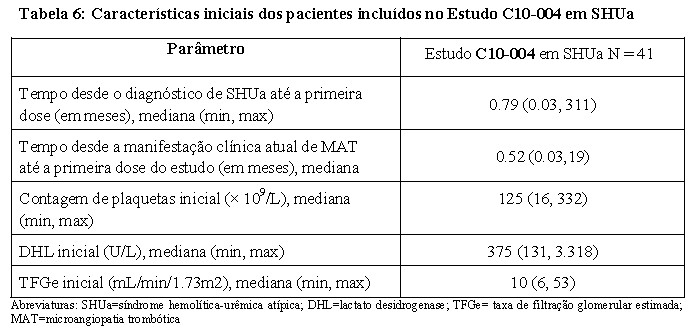
Os pacientes no estudo C10-004 em SHUa receberam Soliris® no mínimo durante 26 semanas. Após o período de tratamento inicial de 26 semanas, a maioria dos pacientes foram escolhidos para continuar em dosagem crônica.
Após o início da terapia com Soliris®, observou-se uma redução na atividade do complemento terminal e um aumento na contagem das plaquetas em relação ao início do tratamento. Soliris® reduziu os sinais da atividade da MAT mediada pelo complemento, como demonstrado por um aumento na média da contagem de plaquetas desde o início do estudo até às 26 semanas.
No estudo C10-004 em SHUa, a média (±DP) da contagem de plaquetas aumentou de 119 ± 66 x 109/L no início do estudo para 200 ± 84 x 109/L ao fim de 1 semana; este efeito foi mantido durante as 26 semanas (média (±DP) da contagem e plaquetas à semana 26: 252 ± 70 x109/L). A função renal, avaliada pela TFGe, melhorou durante a terapêutica com Soliris®. Vinte dos 24 pacientes que precisaram de diálise no início do estudo foram capazes de interromper a diálise durante o tratamento com Soliris®. A Tabela 7 resume os resultados de eficácia no estudo C10-004 em SHUa.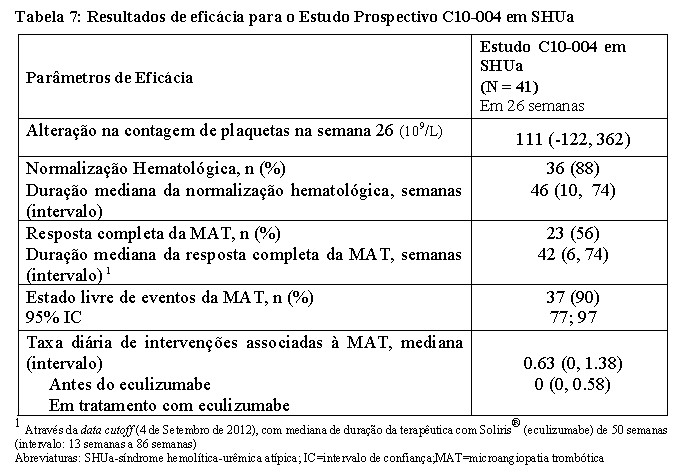
O tratamento mais prolongado com Soliris® (mediana de 52 semanas, com intervalo de 15 a 126 semanas) foi associado a um aumento da taxa de melhora clinicamente significativa em pacientes adultos com SHUa. Quando o tratamento com Soliris® foi continuado por mais do que 26 semanas, três pacientes adicionais (63% dos pacientes no total) atingiram resposta completa da MAT e quatro pacientes adicionais (98% dos pacientes no total) atingiram a normalização hematológica. Na última avaliação, vinte e cinco dos quarenta e um pacientes (61%) atingiram uma melhora da TFGe ≥ 15 mL/min/1.73 m2 a partir do basal.
População pediátrica
Hemoglobinúria Paroxística Noturna
Um total de sete pacientes pediátricos com HPN, com um peso mediano de 57,2 kg (intervalo de 48,6 a 69,8 Kg) e idade entre os 11 e 17 anos (idade mediana: 15,6 anos), receberam Soliris® no estudo M07-005.
O tratamento com eculizumabe no regime posológico proposto na população pediátrica foi associado a uma redução da hemólise intravascular avaliada através dos níveis séricos de DHL. Também resultou numa diminuição marcada ou eliminação de transfusões sanguíneas, e uma tendência de melhora global nas funções gerais. A eficácia do tratamento com eculizumabe em pacientes pediátricos com HPN mostrou-se consistente com o observado em pacientes adultos com HPN incluídos nos estudos principais em HPN (C04-001 e C04-002) (Tabelas 2 e 8).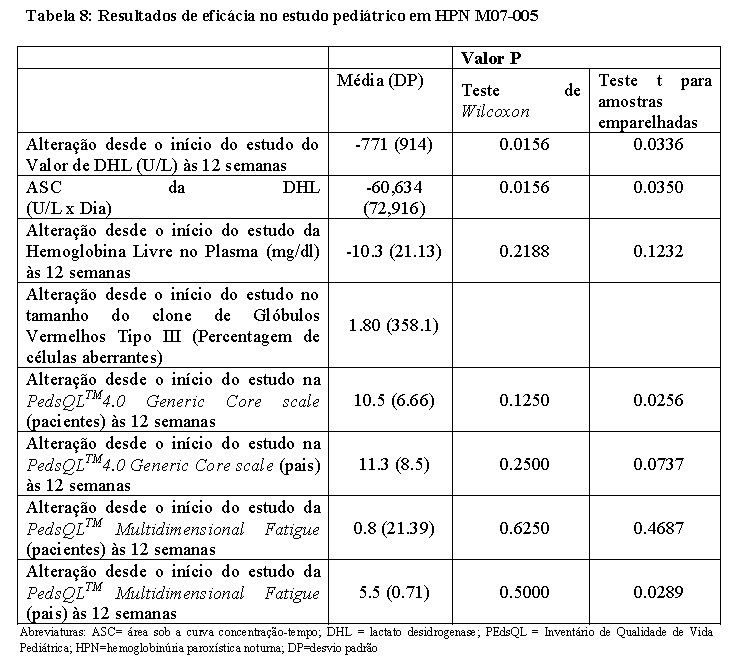
Síndrome Hemolítico Urémico Atípica
Um total de 15 pacientes pediátricos (com idades entre os 2 meses e os 12 anos) recebeu Soliris® (eculizumabe) no estudo em SHUa C009-001r. Quarenta e sete por cento dos pacientes tinham uma mutação identificada nas proteínas reguladoras do complemento ou anticorpo. O tempo mediano desde o diagnóstico de SHUa até à primeira dose de Soliris® foi de 14 meses (intervalo < 1, 110 meses). O tempo mediano desde a manifestação atual de MAT até à primeira dose de Soliris® foi de 1 mês (intervalo < 1 a 16 meses). A duração mediana da terapia com Soliris® foi de 16 semanas (intervalo 4 a 70 semanas) para crianças com idade < 2 anos (n=5) e 31 semanas (intervalo 19 a 63 semanas) para crianças dos 2 a < 12 anos de idade (n=10).
Em geral, os resultados de eficácia para estes pacientes pediátricos pareceram consistentes com o que foi observado nos pacientes incluídos nos estudos principais em SHUa C08-002 e C08-003 (Tabela 5). Nenhum paciente pediátrico necessitou de nova diálise durante o tratamento com Soliris®.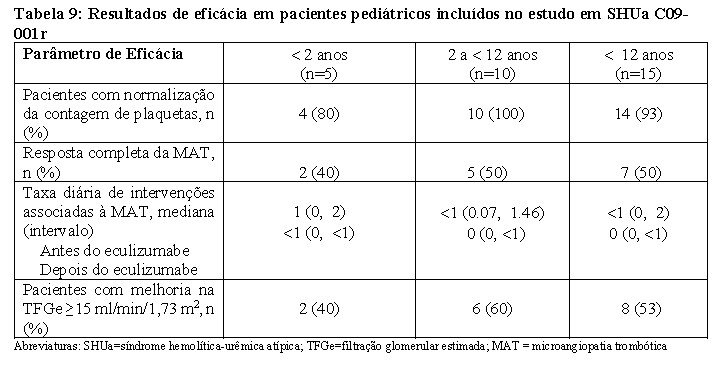
Em pacientes pediátricos com uma duração mais curta da atual manifestação clínica grave da MAT antes do eculizumabe, houve controle da MAT e melhoria da função renal com o tratamento com eculizumabe (Tabela 9).
Em pacientes pediátricos com uma duração mais prolongada da atual manifestação clínica grave da MAT antes do eculizumabe, houve controle da MAT com o tratamento com eculizumabe. No entanto, a função renal não se alterou devido a dano renal irreversível prévio (Tabela 10).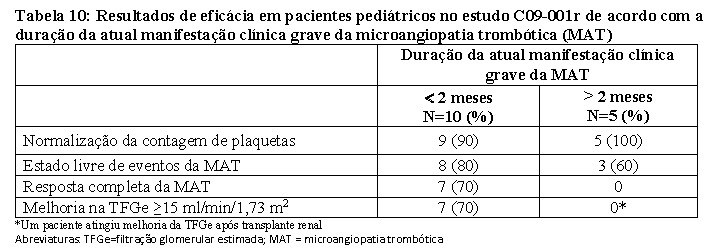
Um total de 22 pacientes pediátricos e adolescentes (com idades entre 5 meses e 17 anos) receberam Soliris® no Estudo C10-003 em SHUa.
No estudo C10-003, os pacientes deveriam apresentar contagem de plaquetas inferior ao limite mais baixo do intervalo normal (LLN), evidência de hemólise como um aumento da DHL sérica acima dos limites superiores do normal, nível de creatinina sérica ≥ percentil 97 da idade, sem necessidade de diálise crônica. A mediana da idade do paciente foi de 6,5 anos (intervalo: 5 meses a 17 anos). Os pacientes incluídos no estudo C10-003 em SHUa apresentaram um nível de ADAMTS-13 acima dos 5%. Cinquenta por cento dos pacientes tiveram uma mutação identificável nas proteínas reguladoras do complemento ou autoanticorpo. Um total de dez pacientes receberam plasmaférese ou infusão de plasma fresco congelado antes do eculizumabe. A Tabela 11 resume as principais características clínicas e relacionadas com a doença inicial dos pacientes incluídos no estudo C10-003 em SHUa.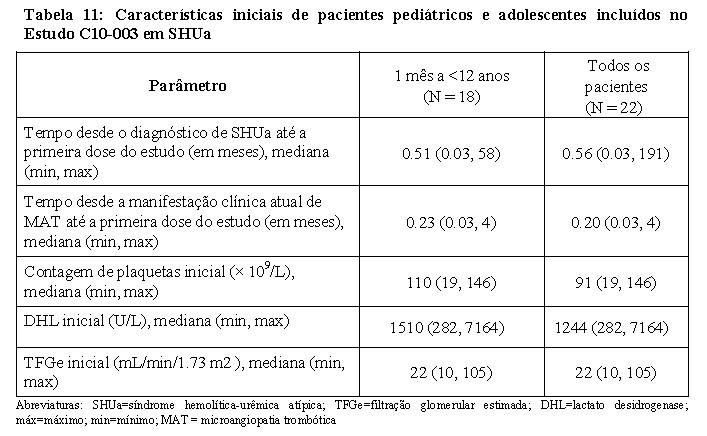
Os pacientes no estudo C10-003 em SHUa receberam Soliris® no mínimo durante 26 semanas. Após o período de tratamento inicial de 26 semanas, a maioria dos pacientes foram escolhidos para continuar em dosagem crônica.
Após o início da terapia com Soliris®, observou-se em todos os pacientes uma redução na atividade do complemento terminal. Soliris®reduziu os sinais da atividade MAT mediada pelo complemento, como demonstrado por um aumento na média da contagem de plaquetas desde o início do estudo até às 26 semanas. A média (±DP) da contagem de plaquetas aumentou de 88 ± 42 x 109/L no início do estudo para 281 ± 123 x 109/L ao fim de uma semana; este efeito foi mantido durante as 26 semanas (média (±DP) da contagem e plaquetas à semana 26: 293 ± 106 x109/L). A função renal, avaliada pela TFGe, melhorou durante a terapia com Soliris®. Nove dos onze pacientes em diálise no início do estudo não precisaram mais de diálise após 15 dias de tratamento com eculizumabe. As respostas foram similares em todas as idades desde os 5 meses aos 17 anos de idade. No estudo C10-003 em SHUa, as respostas ao Soliris® foram similares em pacientes com e sem mutações identificadas nos genes que codificam as proteínas reguladoras do complemento ou autoanticorpos antifator H.
A Tabela 12 resume os resultados de eficácia no estudo C10-003 em SHUa.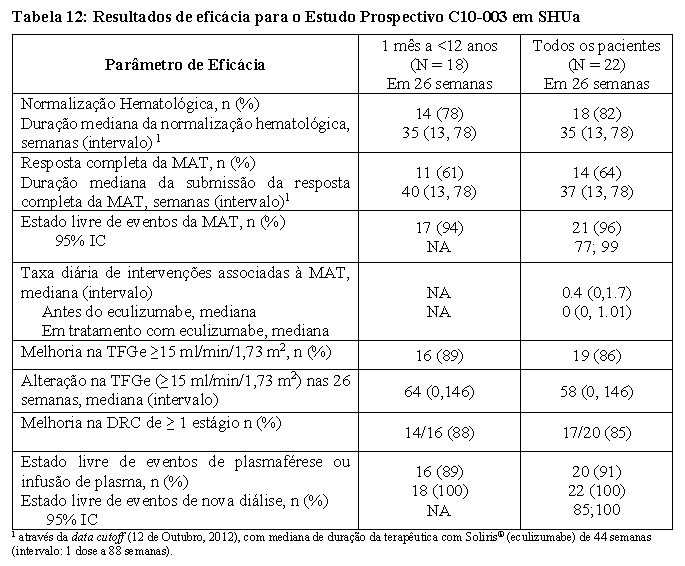
O tratamento mais prolongado com Soliris® (mediana de 55 semanas, com intervalo de 1 dia a 107 semanas) foi associado a um aumento da taxa de melhora clinicamente significativa em pacientes pediátricos e adolescentes com SHUa. Quando o tratamento com Soliris® foi continuado por mais do que 26 semanas, um paciente adicional (68% dos pacientes no total) atingiu resposta completa da MAT e dois pacientes adicionais (91% dos pacientes no total) atingiram a normalização hematológica.
Na última avaliação, dezenove dos vinte e dois pacientes (86%) atingiram uma melhoria da TFGe ≥ 15 mL/min/1.73 m2 a partir da linha de base. Nenhum paciente precisou de nova diálise com Soliris®.
Referências:
Hemoglobinúria Paroxística Noturna
ALMEIDA, Antonio M. et al. Clinical benefit of eculizumab in patients with no transfusion history in the International Paroxysmal Nocturnal Haemoglobinuria Registry. Intern Med J. 47(9):1026-1034, 2017
BRODSKY, Robert A. et al. Multicenter phase 3 study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Blood, v. 111, n. 4, p. 1840-1847, 2008.
HILL, Anita et al. Sustained response and long-term safety of eculizumab in paroxysmal nocturnal hemoglobinuria. Blood, v. 106, n. 7, p. 2559-2565, 2005.
HILL, Anita; ROTHER, Russell P.; HILLMEN, Peter. Improvement in the symptoms of smooth muscle dystonia during eculizumab therapy in paroxysmal nocturnal hemoglobinuria. Haematologica, v. 90, n. 12 Suppl, p. ECR40-ECR40, 2005.
HILL, Anita et al. Effect of eculizumab on haemolysis?associated nitric oxide depletion, dyspnoea, and measures of pulmonary hypertension in patients with paroxysmal nocturnal haemoglobinuria. British journal of haematology, v. 149, n. 3, p. 414-425, 2010.
HILLMEN, Peter et al. Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. New England Journal of Medicine, v. 350, n. 6, p. 552-559, 2004.
HILLMEN, Peter et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. New England Journal of Medicine, v. 355, n. 12, p. 1233-1243, 2006.
HILLMEN, Peter et al. Effect of the complement inhibitor eculizumab on thromboembolism in patients with paroxysmal nocturnal hemoglobinuria. Blood, v. 110, n. 12, p. 4123-4128, 2007.
HILLMEN, Peter et al. Long?term effect of the complement inhibitor eculizumab on kidney function in patients with paroxysmal nocturnal hemoglobinuria. American journal of hematology, v. 85, n. 8, p. 553-559, 2010.
HILLMEN, Peter et al. Long?term safety and efficacy of sustained eculizumab treatment in patients with paroxysmal nocturnal haemoglobinuria. British journal of haematology, v. 162, n. 1, p. 62-73, 2013.
KELLY, Richard J. et al. Long-term treatment with eculizumab in paroxysmal nocturnal hemoglobinuria: sustained efficacy and improved survival. Blood, v. 117, n. 25, p. 6786-6792, 2011.
KELLY, Richard J. Et al. Eculizumab in Pregnant Patients with Paroxysmal Nocturnal Hemoglobinuria. N Engl J Med. 10;373(11):1032-9. 2015
REISS, Ulrike M. et al. Efficacy and safety of eculizumab in children and adolescents with paroxysmal nocturnal hemoglobinuria. Pediatric blood & cancer, v. 61, n. 9, p. 1544-1550, 2014.
RICHARDS, Stephen J. et al. The Effect of Eculizumab Therapy on Red Cell Response Kinetics in Patients with Paroxysmal Nocturnal Hemoglobinuria. Blood, v. 106, n. 11, p. 1047-1047, 2005.
Síndrome Hemolítico Urémico Atípica
FAKHOURI, Fadi et al. Terminal complement inhibitor eculizumab in adult patients with atypical hemolytic uremic syndrome: a single-Arm, Open-Label Trial. American Journal of Kidney Diseases, v. 68, n. 1, p. 84-93, 2016.
GREENBAUM, Larry et al. Eculizumab inhibits thrombotic microangiopathy and improves renal function in pediatric patients with atypical hemolytic uremic syndrome. In: ASN Kidney Week 2013. American Society of Nephrology (ASN), 2013.
GREENBAUM, Larry A. et al. Eculizumab is a safe and effective treatment in pediatric patients with atypical hemolytic uremic syndrome. Kidney international, v. 89, n. 3, p. 701-711, 2016.
KEATING, Gillian M. Eculizumab: a review of its use in atypical haemolytic uraemic syndrome. Drugs, v. 73, n. 18, p. 2053-2066, 2013.
LEGENDRE, Christophe M. et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. New England Journal of Medicine, v. 368, n. 23, p. 2169-2181, 2013.
LICHT, Christoph et al. Hematologic and renal improvements in atypical hemolytic uremic syndrome patients with long disease duration and chronic kidney disease in response to eculizumab. In: PEDIATRIC NEPHROLOGY. 233 SPRING ST, NEW YORK, NY 10013 USA: SPRINGER, 2013. p. 1565-1566.
LICHT, Christoph et al. Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney international, v. 87, n. 5, p. 1061-1073, 2015.
WALLE, Johan Vande et al. Improved renal recovery in patients with atypical hemolytic uremic syndrome following rapid initiation of eculizumab treatment. Journal of nephrology, p. 1-8, 2016.
3. CARACTERISTICAS FARMACOLÓGICAS
3.1 Propriedades farmacodinâmicas
Grupo farmacoterapêutico: Imunossupressor seletivo, código ATC: L04AA25
Soliris® é um anticorpo IgG2/4k monoclonal humanizado recombinante, que se liga à proteína humana C5 do complemento e inibe a ativação do complemento terminal. O anticorpo Soliris® contém regiões constantes humanas e regiões murinas determinantes da complementaridade, enxertadas na estrutura de regiões variáveis de cadeia leve e pesada humana. Soliris® é composto por duas cadeias pesadas, com 448 aminoácidos, e duas cadeias leves, com 214 aminoácidos, tendo um peso molecular de aproximadamente 148 kDa.
Soliris® é produzido num sistema de expressão de mieloma murino (linha celular NS0) e purificado por afinidade e cromatografia de troca iônica. O processo de fabricação da substância ativa do medicamento inclui também inativação viral específica, bem como procedimentos de remoção.
Mecanismo de ação
O eculizumabe, a substância ativa de Soliris®, é um inibidor do complemento terminal que se liga de forma específica à proteína C5 do complemento, com alta afinidade, inibindo, deste modo, a sua clivagem em C5a e C5b e impedindo a geração do complexo de ataque à membrana (C5b-9) do complemento terminal. O eculizumabe preserva os componentes iniciais da ativação do complemento que são essenciais para a opsonização dos microrganismos e para a remoção dos imunocomplexos.
Em pacientes com HPN, a ativação não controlada do complemento terminal e a consequente hemólise intravascular mediada pelo complemento são bloqueadas com o tratamento com Soliris®.
Na maioria dos pacientes com HPN, concentrações séricas de eculizumabe correspondentes a aproximadamente 35 microgramas/mL são suficientes para a inibição completa da hemólise intravascular mediada pelo complemento terminal.
Na HPN, a administração crônica de Soliris® resultou em redução rápida e sustentada da atividade hemolítica mediada pelo complemento.
Em pacientes com SHUa, a ativação não controlada do complemento terminal e a consequente MAT mediada pelo complemento são bloqueadas com o tratamento com Soliris®.
Todos os pacientes tratados com Soliris®, quando administrado como recomendado, demonstraram uma redução rápida e sustentada na atividade do complemento terminal. Em todos os pacientes com SHUa, concentrações séricas de eculizumabe, correspondentes a aproximadamente 50-100 microgramas/mL são suficientes para a inibição completa da atividade do complemento terminal.
Na SHUa, a administração crônica de Soliris® resultou numa redução rápida e sustentada da MAT mediada pelo complemento.
3.2 Propriedades farmacocinéticas Farmacocinética e metabolismo farmacológico
Biotransformação
Os anticorpos humanos são sujeitos à digestão endocítica nas células do sistema reticuloendotelial. O eculizumabe contém apenas aminoácidos de ocorrência natural e não possui metabólitos ativos conhecidos. Os anticorpos humanos são predominantemente catabolizados por enzimas lisossomais em aminoácidos e peptídeos pequenos.
Eliminação
Não foram realizados estudos específicos para avaliar as vias de excreção/eliminação hepática, renal, pulmonar ou gastrointestinal de Soliris®. Em rins normais, os anticorpos não são excretados, sendo excluídos da filtração devido ao seu tamanho.
Parâmetros farmacocinéticos
Em quarenta pacientes com HPN, utilizou-se um modelo de um compartimento para estimar os parâmetros farmacocinéticos após doses múltiplas. A depuração média foi de 0,31 ± 0,12 mL/hr/kg, o volume de distribuição médio foi de 110,3 ± 17,9 mL/kg, e a média da meia- vida de eliminação plasmática foi de 11,3 ± 3,4 dias. Com base nestes dados, prevê-se o estabelecimento de um estado de equilíbrio em aproximadamente 49 a 56 dias.
Em pacientes com HPN, a atividade farmacodinâmica está diretamente relacionada com as concentrações séricas de eculizumabe e a manutenção de níveis mínimos superiores a ≥ 35 microgramas/mL resulta num bloqueio completo da atividade hemolítica na maior parte dos pacientes com HPN.
Conduziu-se uma segunda análise PK na população com um modelo padrão de um compartimento, utilizando os dados PK de doses múltiplas de 37 pacientes com SHUa, que receberam o regime recomendado de Soliris® nos estudos C08-002A/B e C08-003A/B. Neste modelo, a depuração do Soliris® num paciente típico com SHUa com um peso de 70 kg foi de 0,0139 L/hr e o volume de distribuição foi 5,6 L. A meia- vida de eliminação foi de 297 h (aproximadamente 12,4 dias).
O segundo modelo PK na população foi aplicado à informação de PK de dose múltipla de 22 pacientes pediátricos com SHUa, recebendo o regime recomendado de Soliris® no estudo C10-003 em SHUa. A depuração e o volume de distribuição do Soliris® são dependentes do peso, o que constitui a base para um regime de doses categorizado pelo peso em pacientes pediátricos (vide seção 8 "Posologia e Modo de Usar"). Os valores de depuração de Soliris® em pacientes pediátricos com SHUa foram 10,4; 5,3 e 2,2 mL/h com peso corporal de 70, 30 e 10 Kg, respectivamente, e os valores correspondentes do volume de distribuição foram 5,23; 2,76 e 1,21 L, respectivamente. A meia-vida de eliminação correspondente permaneceu quase inalterada entre o intervalo de 349 a 378 h (aproximadamente 14,5 a 15,8 dias).
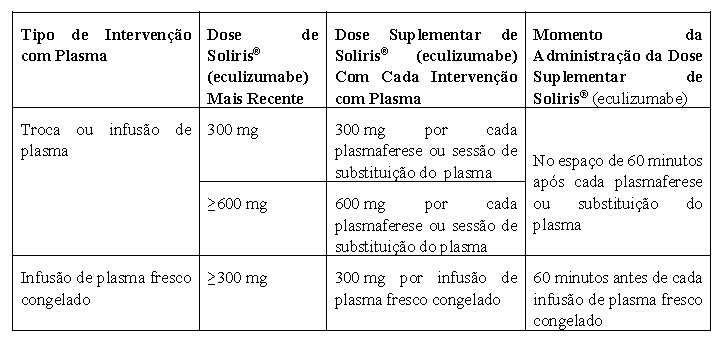
A depuração e a meia-vida do eculizumabe também foram avaliadas durante as intervenções de plasmaférese. A plasmaférese resultou numa diminuição de aproximadamente 50% nas concentrações de eculizumabe após uma intervenção de 1 hora e a meia-vida de eliminação do eculizumabe foi reduzida para 1,3 horas.
É recomendada uma posologia suplementar quando o Soliris® é administrado a pacientes com SHUa que estão em terapia de plasmaférese ou infusão de plasma (vide seção 8 "Posologia e Modo de Usar").
Todos os pacientes com SHUa tratados com Soliris®, quando administrado como recomendado, demonstraram uma redução rápida e sustentada na atividade do complemento terminal. Em pacientes com SHUa, a atividade farmacodinâmica está diretamente relacionada com as concentrações séricas de eculizumabe e a manutenção de níveis mínimos de aproximadamente 50-100 microgramas/mL resulta num bloqueio completo da atividade do complemento terminal em todos os pacientes com SHUa.
Populações especiais
HPN
Não foram efetuados estudos dedicados para avaliar a farmacocinética da administração de Soliris® em populações especiais de pacientes com HPN identificados pelo sexo, raça, idade (geriátricos) ou na presença do comprometimento renal ou hepático.
População pediátrica
A farmacocinética do eculizumabe foi avaliada no Estudo M07-005 incluindo sete pacientes pediátricos com HPN (idade entre os 11 e os 18 anos).
O peso foi uma covariável significativa, resultando em menor depuração do eculizumabe de 0,0105 L/h nos pacientes adolescentes. A posologia para pacientes pediátricos com peso < 40 kg é baseada nos pacientes pediátricos com SHUa.
SHUa
A farmacocinética de Soliris® foi estudada em pacientes com SHUa com diferentes graus de comprometimento renal e faixa etária. Não se observaram diferenças nos parâmetros farmacocinéticos nestas subpopulações de pacientes com SHUa.
4. CONTRA-INDICAÇÕES
Hipersensibilidade ao eculizumabe, às proteínas murinas ou a qualquer um dos excipientes da fórmula.
A terapêutica com Soliris® não deve ser iniciada em pacientes:
• com infecção por Neisseria meningitidis não resolvida.
• que não estejam vacinados contra Neisseria meningitidis (a menos que recebam tratamento profilático com antibióticos apropriados até 2 semanas após a vacinação).
5. ADVERTÊNCIAS E PRECAUÇÕES
Não se prevê que Soliris® afete o componente aplásico da anemia em pacientes com HPN associada à anemia aplásica.
Infecção meningocócica
Devido ao seu mecanismo de ação, a utilização de Soliris® aumenta a suscetibilidade dos pacientes a infecção meningocócica (Neisseria meningitidis). Pode ocorrer doença meningocócica devido a qualquer um dos sorotipos. No sentido de reduzir o risco de infecção, todos os pacientes devem ser vacinados pelo menos 2 semanas antes de receber Soliris®, a menos que o risco de atrasar a terapia com Soliris® ultrapasse os riscos de desenvolver uma infecção meningocócica. Os pacientes que iniciaram o tratamento com Soliris® em menos de 2 semanas após receberem a vacina meningocócica devem receber tratamento com antibióticos profiláticos apropriados até 2 semanas após a vacinação. A vacinação contra os sorotipos A, C, Y, W135 são recomendadas na prevenção dos sorogrupos meningocócicos comumente patogênicos. Vacinação contra o sorogrupo B onde disponível, é também recomendada. Os pacientes devem ser vacinados de acordo com as diretrizes clínicas de vacinação atuais.
A vacinação pode ainda ativar o complemento. Como resultado, pacientes com doenças mediadas pelo complemento, incluindo HPN e SHUa, podem experimentar um aumento nos sinais e sintomas de suas doenças de base, tais quais hemólise (HPN) e MAT (SHUa). Desse modo, os pacientes devem ser monitorados rigorosament