EXHER
NATCOFARMA
everolimo
Antineoplásico.
MEDICAMENTO SIMILAR EQUIVALENTE AO MEDICAMENTO REFERÊNCIA
Apresentações.
Exher® comprimidos de 5mg e 10 mg em embalagem contendo 28 comprimidos (4 blisters com 7 comprimidos cada).
USO ORAL
USO ADULTO E PEDIÁTRICO ACIMA DE 3 ANOS (uso pediátrico apenas para tratamento de SEGA associado ao TSC)
Composição.
Cada comprimido de Exher® 5 mg e 10 mg contém respectivamente 5 mg e 10 mg de everolimo. Excipientes: butilidroxitolueno, hipromelose, lactose, acetona, crospovidona e estearato de magnésio.
Informações técnicas.
1. INDICAÇÕES
Exher® é indicado para o tratamento de:
- Mulheres na pós-menopausa com câncer de mama avançado, receptor hormonal positivo, em combinação com um inibidor da aromatase, após terapia endócrina prévia.
- Pacientes com tumores neuroendócrinos avançados (NET) localizados no estômago e intestino, pulmão ou pâncreas;
- Pacientes com câncer avançado do(s) rim(ns) (Carcinoma avançado de Células Renais (CCR)) cuja doença tenha progredido durante ou após o tratamento com VEGFR - TKI, quimioterápicos ou imunoterápicos;
- Pacientes com angiomiolipoma renal associado à complexo de esclerose tuberosa (TSC) não necessitando de cirurgia imediata (em pacientes acima de 18 anos);
- Pacientes com astrocitoma subependimário de células gigantes (SEGA, um tumor cerebral específico) associado à complexo de esclerose tuberosa (TSC).
2. RESULTADOS DE EFICÁCIA
Câncer de mama avançado receptor hormonal positivo
O estudo BOLERO-2 (Estudo CRAD001Y2301), de Fase III, multicêntrico, duplo-cego, randomizado de everolimo + exemestano versus placebo + exemestano foi realizado com mulheres na pós-menopausa com câncer de mama avançado, receptor de estrógeno positivo e HER 2-neu/não amplificado, com recorrência ou progressão após terapia prévia com letrozol ou anastrozol. As pacientes foram randomizadas em uma proporção de 2:1 para receber everolimo (10 mg ao dia) ou placebo, além de exemestano em regime aberto (25 mg ao dia). A randomização foi estratificada com base na sensibilidade documentada à terapia hormonal prévia ou não e pela presença de metástase visceral ou não. A sensibilidade à terapia hormonal prévia foi definida como (1) benefício clínico documentado (resposta completa [RC], resposta parcial [RP], doença estável [SD] ≥ 24 semanas) a pelo menos uma terapia hormonal prévia no contexto da doença avançada, ou (2) pelo menos um histórico de 24 meses de terapia hormonal adjuvante antes da recorrência16.
O desfecho primário do estudo foi a sobrevida livre de progressão (SLP) avaliada pelos Critérios de Avaliação de Resposta em Tumores Sólidos (RECIST), com base na avaliação do investigador (radiologia local). As análises corroborativas de SLP foram baseadas em uma revisão radiológica central independente.
Os desfechos secundários incluíram sobrevida global (SG), Taxa de Resposta Global (TRG), Taxa de Benefício Clínico (TBC), Segurança, mudança na Qualidade de vida (QoL) e tempo até deterioração da Capacidade Funcional por ECOG. Os desfechos adicionais incluíram alterações nos marcadores de remodelamento ósseo nas semanas 6 e 12.
No total, 724 pacientes foram randomizadas em uma proporção de 2:1 para a combinação de everolimo (10 mg ao dia) + exemestano (25 mg ao dia) (n = 485) ou placebo + exemestano (25 mg ao dia) (n = 239). Os dois grupos de tratamento foram em geral, equilibrados no que se refere às características demográficas basais da doença e histórico de uso prévio de antineoplásicos. A idade média das pacientes era de 61 anos (faixa de 28 a 93), sendo 75% caucasianas16. A duração média do tratamento cego foi de 24,0 semanas para as pacientes recebendo everolimo + exemestano e de 13,4 semanas para aquelas recebendo placebo + exemestano.
Os resultados de eficácia foram obtidos a partir da observação da análise final de 510 eventos de SLP locais e 320 eventos de SLP centralmente avaliados. As pacientes no braço de placebo+exemestano não foram alocadas para o braço com everolimo no momento da progressão.16
O estudo demonstrou um benefício clínico estatisticamente significativo de everolimo + exemestano quando comparado a placebo + exemestano através de um prolongamento de 2,45 vezes da mediana de SLP (mediana: 7,82 meses versus 3,19 meses), resultando em uma redução de risco de 55% na progressão ou morte (SLP HR 0,45; IC 95%: 0,38, 0,54; teste de log-rank unilateral com valor p < 0,0001 de acordo com a avaliação do investigador local (vide Tabela 1-1 e Figura 1-1 BOLERO-2). 16
A análise de SLP com base em uma avaliação radiológica central independente foi favorável e mostrou um prolongamento de 2,7 vezes da mediana de sobrevida livre de progressão (11,01 meses versus 4,14 meses), resultando em uma redução de risco de 62% na progressão ou morte (SLP HR 0,38; IC 95%: 0,31, 0,48; teste log-rank unilateral com valor p < 0,0001) (vide Tabela 1-1 e Figura 1-2 BOLERO-2 - Curvas de sobrevida livre de progressão de Kaplan-Meier (revisão radiológica independente)). 16
A resposta objetiva de acordo com a avaliação do investigador baseada no RECIST foi observada em 12,6% dos pacientes (IC 95%: 9,8, 15,9) no braço de everolimo + exemestano vs. 1,7% (IC 95%: 0,5-4,2) no braço de placebo + exemestano (p < 0,0001 para comparação entre os braços). A taxa de benefício clínico para everolimo + exemestano foi 51,3% vs. 26,4% no braço de controle; p < 0,0001 (vide Tabela 1-1). 16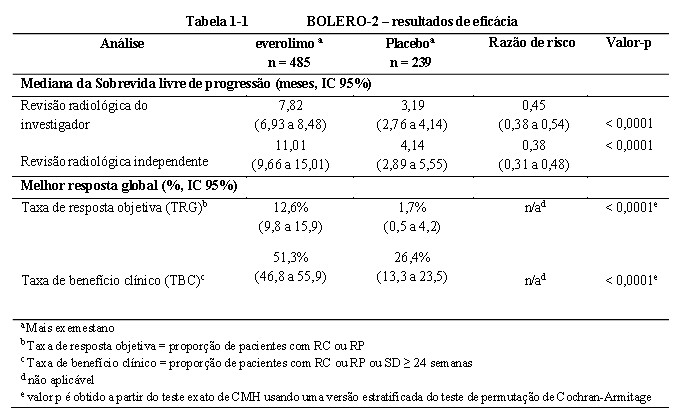
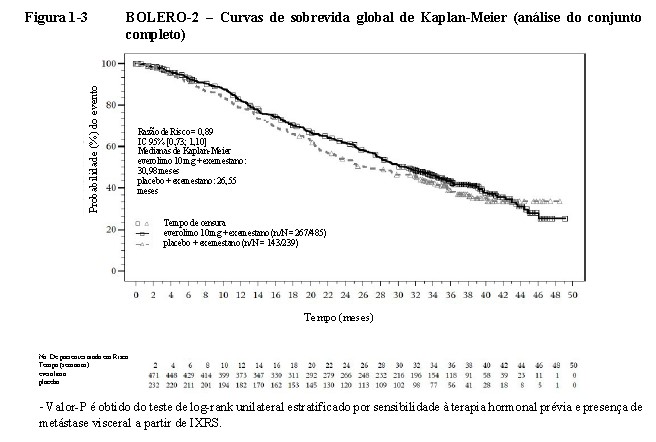
No final da análise de Sobrevida Global (SG), a duração média da SG foi de 31 meses vs 26,6 meses para o braço everolimo + exemestano vs o braço placebo + exemestano, respectivamente [Razão de risco = 0,89 (IC 95%: 0,73 a 1,10; p=0,1426)] (vide Figura 1- 3).
A taxa de SLP de doze meses foi de 33% para os pacientes recebendo everolimo + exemestano comparado a 11% do braço recebendo placebo + exemestano17.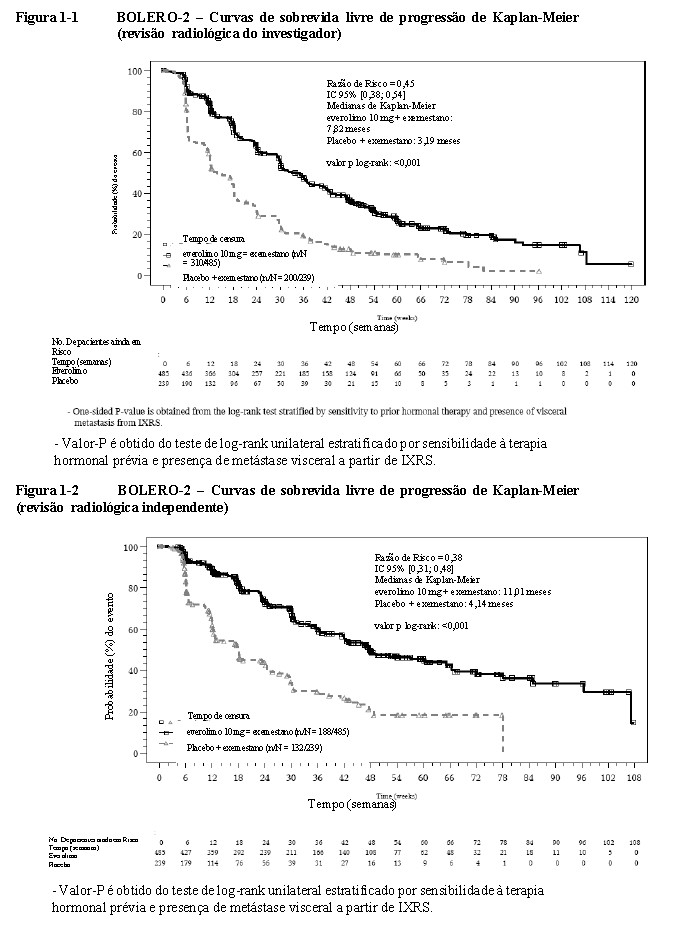
O efeito do tratamento estimado pela SLP foi corroborado por uma análise de subgrupos planejada referente a SLP de acordo com a avaliação do investigador. Para todos os subgrupos analisados, um efeito de tratamento positivo foi observado com everolimo + exemestano com uma razão de risco estimada vs. placebo + exemestano variando de 0,25 a 0,62 (vide Tabela 1-2)16. As análises de subgrupos demonstraram um efeito de tratamento homogêneo e consistente, independentemente da sensibilidade à terapia hormonal prévia e presença de metástase visceral e ao longo dos principais subgrupos demográficos e de prognósticos16.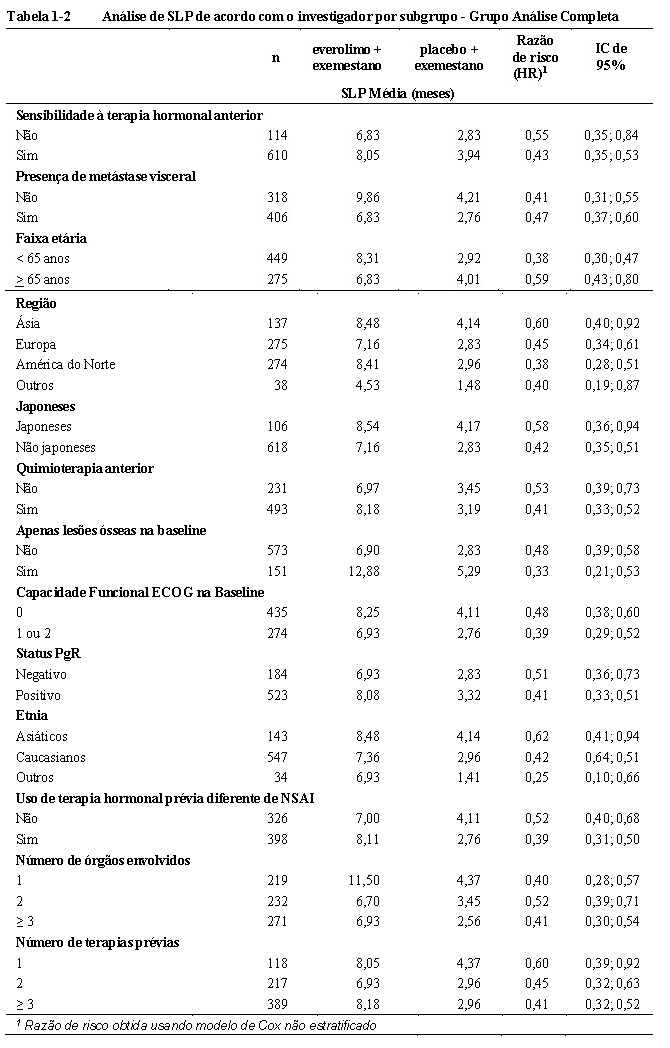
A redução tumoral também foi evidente a partir do gráfico em cascata correspondente. Os resultados indicam que 70,8% dos pacientes no braço de everolimo + exemestano apresentaram diminuição tumoral versus 29,7% com placebo + exemestano (Figura 1-4)17.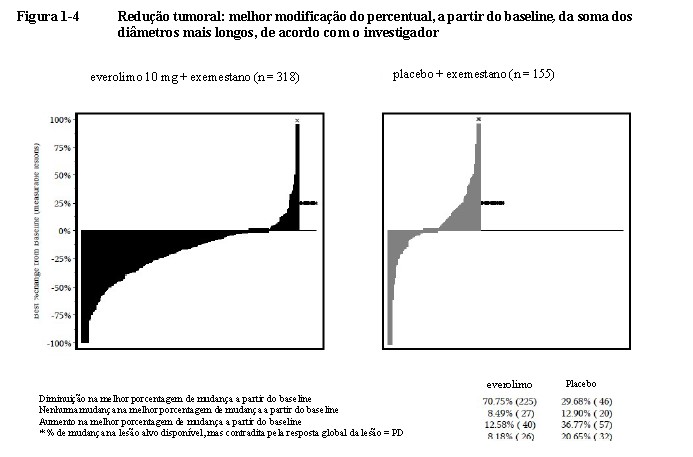
Não foram observadas diferenças estatisticamente significativas em relação aos parâmetros clínicos, entre os dois braços de tratamento, em termos de tempo até deterioração da capacidade funcional de ECOG (≥ 1 ponto) e mediana do tempo até deterioração (≥ 5%) das pontuações do domínio QLQ-C3016.
Efeito sobre os ossos
Não há dados a longo prazo sobre o efeito de everolimo sobre os ossos. Dados comparativos do estudo BOLERO-2 mostraram melhoria acentuada nos marcadores séricos de remodelamento ósseo durante as primeiras 12 semanas de terapia, sugerindo um efeito favorável no remodelamento ósseo16.
Tumores neuroendócrinos avançados de origem gastrintestinal, pulmonar ou pancreática
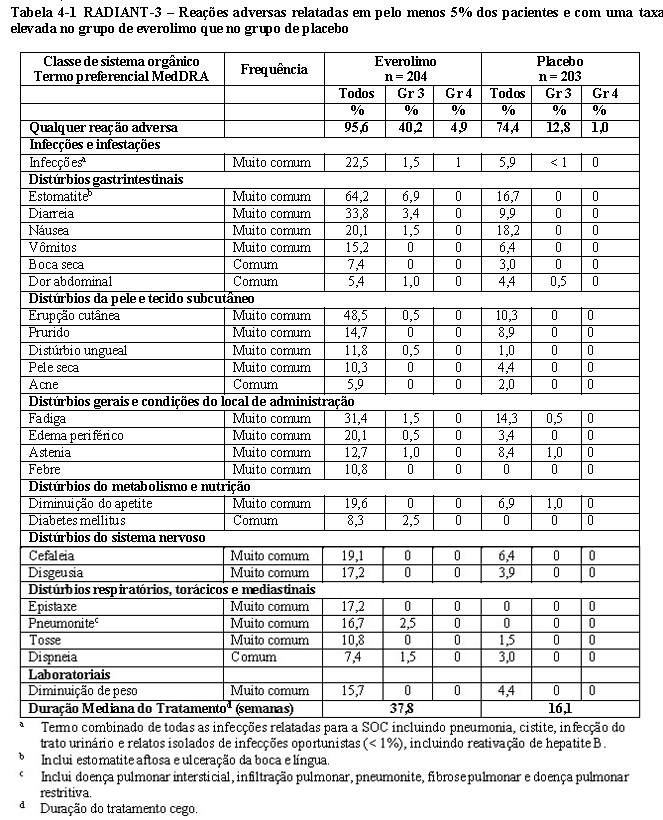
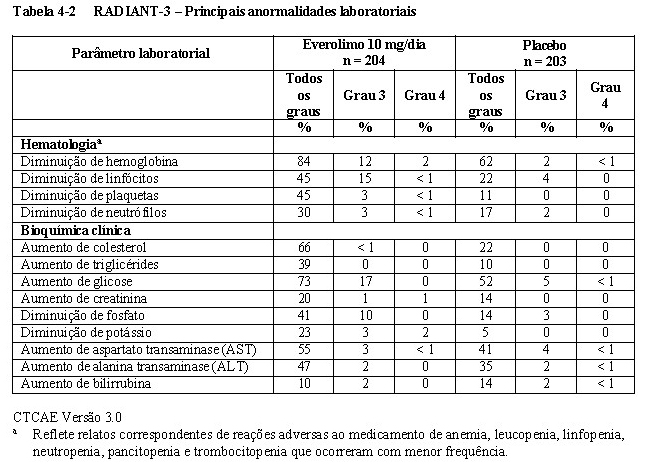
O RADIANT-3 (Estudo CRAD001C2324), um estudo de fase III, randomizado, duplo-cego e multicêntrico de everolimo mais o "melhor tratamento de suporte" (BSC) versus placebo mais o BSC em pacientes com tumores neuroendócrinos pancreáticos (pNETs) avançados, demonstrou um benefício clínico estatisticamente significativo do everolimo em relação ao placebo por um prolongamento de 2,4 vezes na sobrevida livre de progressão (SLP) mediana (11,04 meses versus 4,6 meses), resultando em uma redução de risco de 65% na SLP (RR de 0,35; IC de 95%: 0,27, 0,45; p < 0,0001) (veja a Tabela 1-3 e a Figura 1-5)1.
O RADIANT-3 incluiu pacientes com pNET avançado cuja doença havia progredido dentro dos 12 meses anteriores. Os pacientes foram estratificados por quimioterapia citotóxica anterior (sim/não) e por capacidade funcional de acordo com a OMS (0 vs. 1 e 2). O tratamento com análogos da somatostatina foi permitido como parte do BSC.
O desfecho primário para o estudo foi a SLP avaliada pelos RECIST (Critérios de Avaliação de Resposta em Tumores Sólidos, versão1.0), conforme revisão radiológica do investigador. Após a progressão radiológica documentada, os pacientes puderam ter a quebra do código cego pelo investigador: pacientes randomizados para o placebo puderam, então, receber o everolimo aberto.
Os desfechos secundários incluem a segurança, a taxa de resposta objetiva (TRO) (resposta completa (RC) ou resposta parcial (RP), a duração da resposta e a sobrevida geral (SG).
No total, 410 pacientes foram randomizados, a 1:1, para receber 10 mg/dia de everolimo (n = 207) ou placebo (n = 203). Os dados demográficos foram bem equilibrados (idade mediana de 58 anos, 55% homens, 78,5% caucasianos). A duração mediana de tratamento do estudo cego, foi de 37,8 semanas para pacientes recebendo everolimo e de 16,1 semanas para pacientes recebendo placebo.1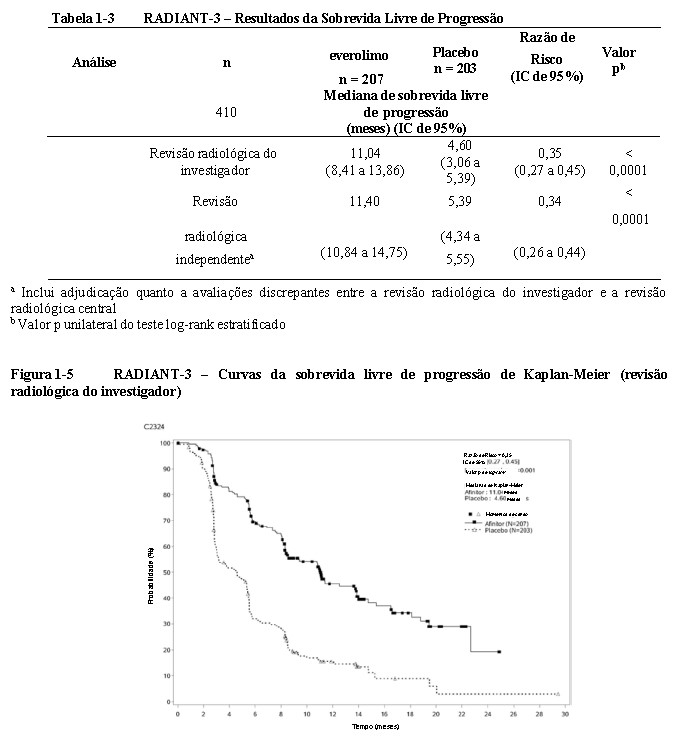
As taxas de SLP de dezoito meses foram de 34,2% para a terapia com everolimo em comparação a 8,9% para o placebo.
A taxa de resposta objetiva de acordo com a avaliação do investigador foi de 4,8% para o braço de everolimo em comparação com 2,0% para o braço de placebo. A redução tumoral também foi evidente segundo o gráfico em cascata correspondente. Os resultados indicam que 64,4% dos pacientes no braço de everolimo apresentaram regressão tumoral em comparação com 20,6% no braço de placebo (Figura 1-6)1.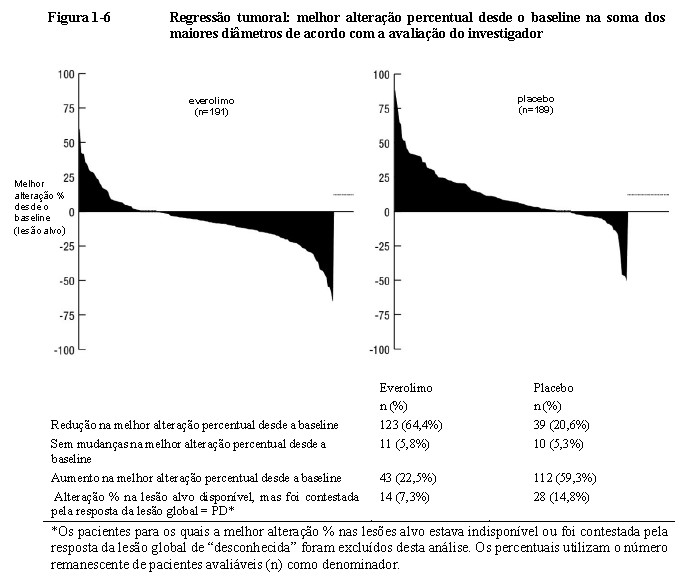
No momento da análise final da sobrevida global (SG), a duração mediana de sobrevida global foi 44 meses para o braço de everolimo versus 37,7 meses para o braço de placebo, respectivamente [Razão de Risco = 0,94 (IC de 95% 0,73 a 1,20)]; p=0,300 (Figura 1-7). Seguindo a progressão da doença, o cruzamento de everolimo aberto ocorreu em 172 dos
203 pacientes (84,7%) randomizados para placebo e provavelmente confundiu a detecção de qualquer diferença relacionada ao tratamento na SG12,22.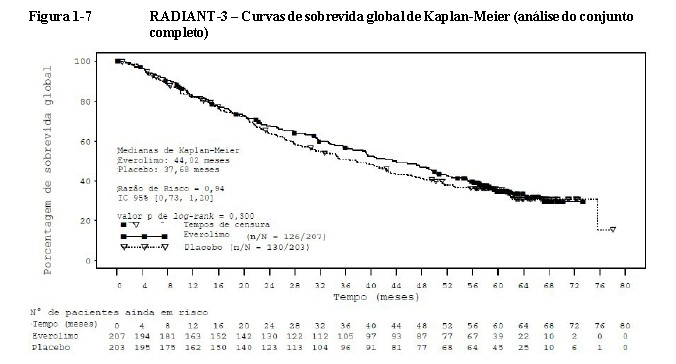
RADIANT-4 (Estudo CRAD001T2302), um estudo de fase III, randomizado, duplo-cego e multicêntrico de everolimo mais o "melhor tratamento de suporte" (BSC) versus placebo mais o BSC foi conduzido em pacientes com tumores avançados neuroendócrinos não funcionais (NET) de origem gastrointestinal ou pulmonar sem histórico ou sintomas ativos relacionados à síndrome carcinóide. A randomização foi estratificada pelo uso prévio de análogo da somatostatina (SSA), origem do tumor e classificação de desempenho da OMS.
O desfecho primário do estudo era a sobrevida livre de progressão (SLP) avaliada pelos Critérios de Avaliação de Resposta em Tumores Sólidos (RECIST modificado versão 1.0), com base na avaliação radiológica independente. A análise da SLP de apoio foi baseada na avaliação do investigador local.
Os desfechos secundários incluíram sobrevida global (SG), taxa de resposta global (TRG), taxa de controle da doença (TCD = proporção de pacientes com uma melhor resposta global da resposta completa, resposta parcial ou doença estável), segurança, mudança na qualidade de vida (QoL) através da FACT-G e tempo da deterioração da PS OMS.
Um total de 302 pacientes foi randomizado em uma proporção de 2:1 para receber everolimo (10 mg diáriamente) (n = 205) ou placebo (n = 97). Os dois grupos de tratamento foram geralmente balanceados em relação à linha base demográfica, às características da doença e ao histórico prévio de utilização de análogos de somatostatina (SSA). A idade mediana dos pacientes foi de 63 anos (variação de 22 a 86) e 76% eram caucasianos. A duração média do tratamento cego foi de 40,4 semanas para os pacientes que receberam everolimo e 19,6 semanas para aqueles que receberam o placebo. Os pacientes no braço do placebo não cruzaram para o everolimo no momento da progressão26, 27. Os resultados de eficácia foram obtidos a partir da análise final do SLP após 178 eventos de SLP terem sido observados por análise radiológica independente.
O estudo demonstrou um benefício clínico estatisticamente significativo de everolimo sobre o placebo por um prolongamento de 2,8 vezes do SLP médio (11,01 meses versus 3,91 meses), resultando em uma redução do risco de 52% de progressão ou morte (razão de risco 0,48; IC de 95%: 0,35,0,67; valor p unilateral do teste log-rank estratificado < 0,0001) por avaliação independente (ver Tabela 1-4 e Figura 1-8)26,27.
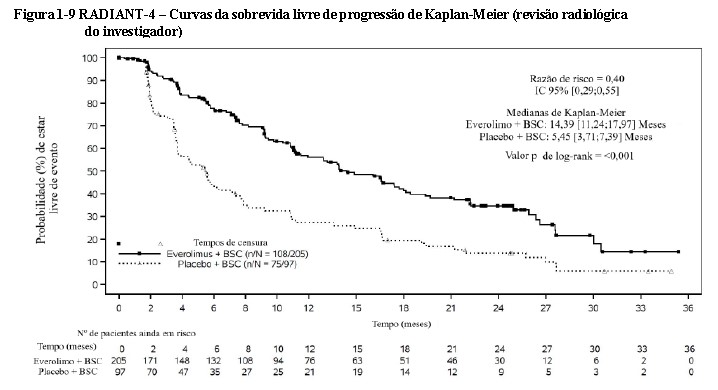
A análise de SLP com base na avaliação do investigador local apoiou e demonstrou um prolongamento de 2,5 vezes na sobrevida livre de progressão média (13,96 meses versus 5,45 meses), resultando em uma redução de risco de 61% de progressão ou morte (razão de risco 0,39; IC de 95%: 0,28, 0,54; valor p unilateral do teste log-rank estratificado < 0,0001) (ver Tabela 1-4 e Figura 1-9)29.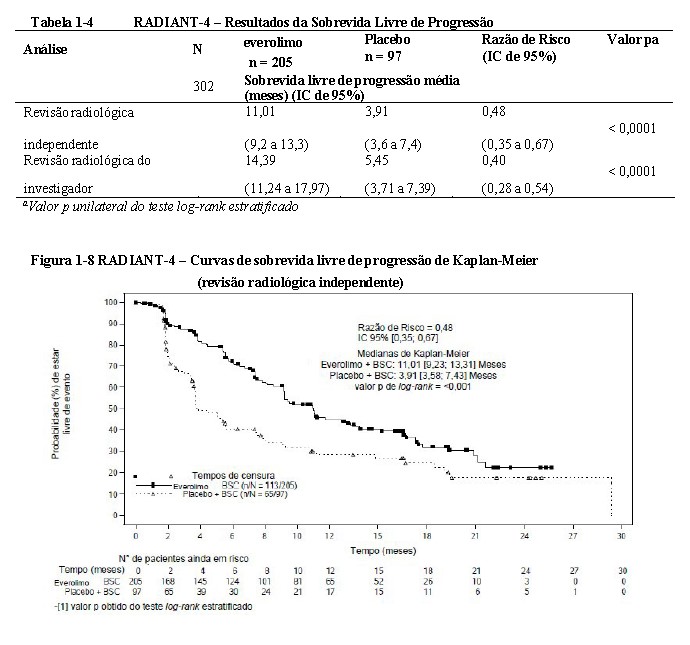
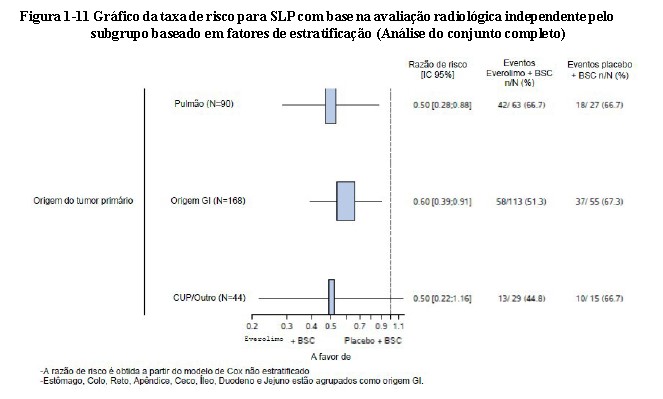
O benefício global na SLP favoreceu o everolimo nos subgrupos predefinidos demograficamente e na estratificação prognóstica (Figura 1-10). Uma análise do subgrupo post-hoc de SLP em locais cuja origem tumoral seja gastrointestinal, pulmonar e carcinoma de origem primária/outra desconhecida mostrou um benefício positivo na SLP (Veja a Figura 1-11)26,27.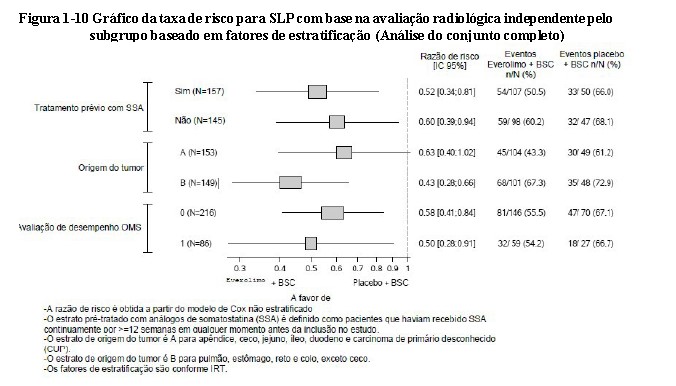
A taxa de resposta global por avaliação independente foi de 2% no braço do everolimo vs. 1% no braço do placebo. A Taxa de Controle da Doença (RC ou RP ou SD) para everolimo foi de 82,4% vs. 64,9% no braço do placebo. A redução tumoral também foi evidente segundo o gráfico em cascata correspondente. Os resultados indicam que 63,6% dos pacientes no braço de everolimo experimentaram redução do tumor contra 25,9% para o placebo (ver Figura 1-12)26,27.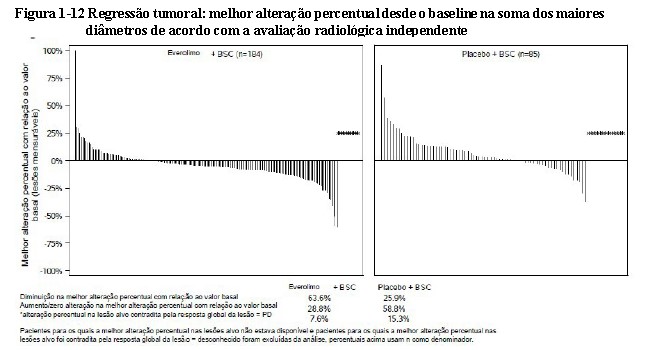
A análise de sobrevida global (SG) ainda não está madura. Na primeira análise interina, 42 (20,5%) dos óbitos foram observados no braço do everolimo vs. 28 (28,9%) dos óbitos no braço do placebo. No entanto, os resultados desta análise não atingiram o ponto de análise pré-especificado para que possa ser considerado estatisticamente significativo [razão de risco = 0,64 (IC de 95%: 0,40 para 1,05; p = 0,037)] (ver Figura 1-13)29.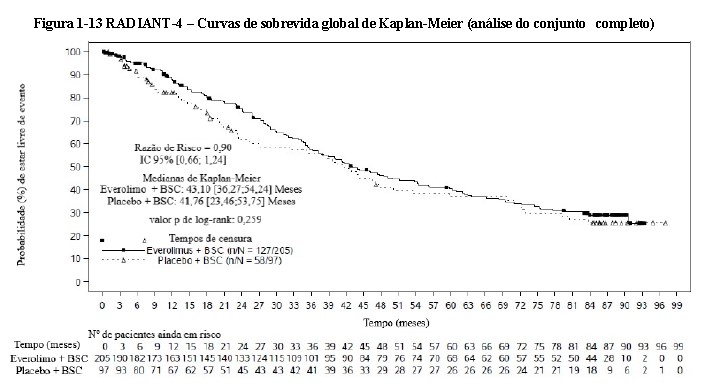
Diferenças clinicamente ou estatisticamente significativas não foram observadas entre os dois braços de tratamento em relação ao tempo para deterioração da OMS PS (Razão de risco: 1,02; IC de 95%: 0,65, 1,61) e ao tempo para deterioração da classificação total da FACT-G (Razão de risco: 0,74; IC de 95%: 0,50, 1,10)29.
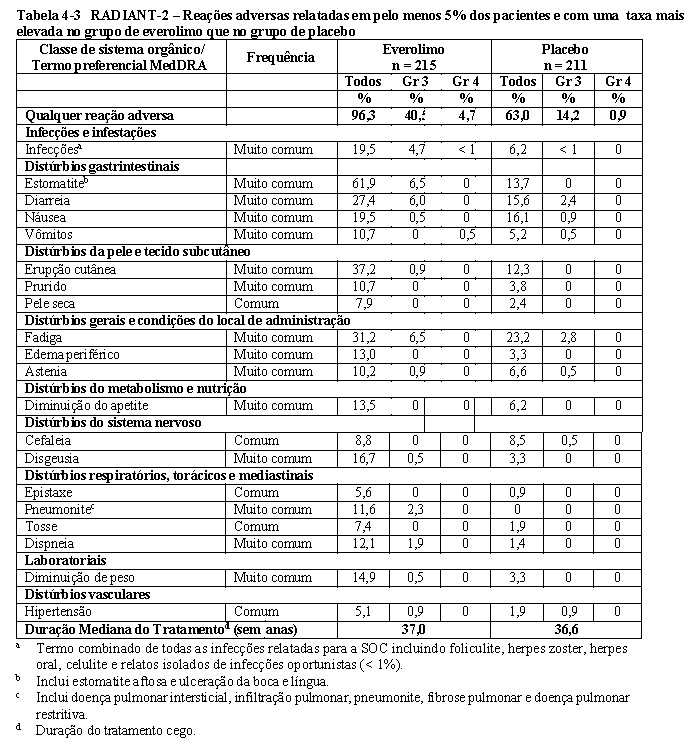
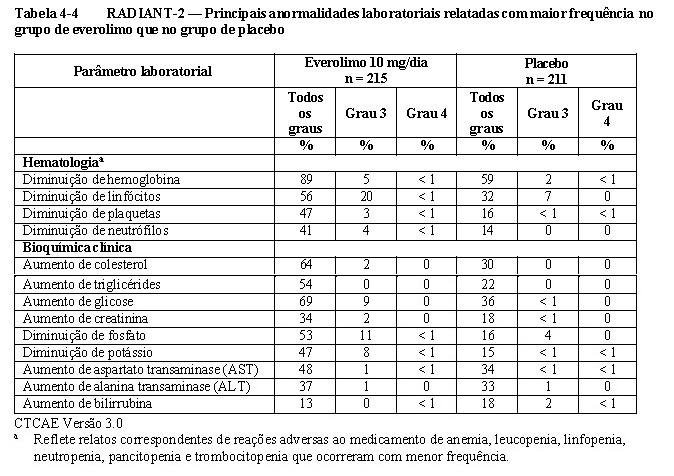
O RADIANT-2 (Estudo CRAD001C2325), um estudo de fase III, randomizado, duplo-cego e multicêntrico de everolimo mais octreotida de depósito (Sandostatin LAR®) versus placebo mais octreotida de depósito em pacientes com tumores neuroendócrinos (tumor carcinoide) avançados primariamente de origem gastrintestinal ou pulmonar, mostrou evidências de benefício clínico do everolimo em relação ao placebo por um prolongamento de 5,1 meses na SLP mediana (16,43 meses versus 11,33 meses; RR de 0,77; IC de 95%: 0,59 a 1,00; p = 0,026 unilateral), resultando em uma redução de risco de 23% na SLP primária (vide a Tabela 1-5 e a Figura 1-14). Embora a significância estatística não tenha sido alcançada para a análise primária (o limite para significância estatística foi de p = 0,0246), as análises que realizaram um ajuste em relação à censura informativa e aos desequilíbrios nos dois braços de tratamento mostraram um efeito do tratamento a favor do everolimo.
O RADIANT-2 incluiu pacientes com tumores neuroendócrinos (tumor carcinoide) avançados, primariamente de origem gastrintestinal ou pulmonar, cuja doença havia progredido dentro dos 12 meses anteriores e que apresentavam histórico de sintomas secretores. 80,1% dos pacientes no grupo de everolimo receberam uma terapia com análogo da somatostatina antes da entrada no estudo em comparação a 77,9% no grupo placebo2.
O desfecho primário é a SLP avaliada pelo RECIST conforme revisão radiológica independente. Após a progressão radiológica documentada, os pacientes puderam ter a quebra do código cego pelo investigador: aqueles randomizados para o placebo puderam, então, receber o everolimo aberto.
Os desfechos secundários incluem a segurança, resposta objetiva, a duração da resposta e a sobrevida geral. No total, 429 pacientes foram randomizados, a 1:1, para receber 10 mg/dia de everolimo (n = 216) ou placebo (n = 213), além de 30 mg de octreotida de depósito (Sandostatin LAR®, administrado intramuscularmente) a cada 28 dias. A duração mediana de tratamento no estudo cego, foi de 37,0 semanas para os pacientes recebendo everolimo e de 36,6 semanas para os pacientes recebendo placebo. Desequilíbrios notáveis foram evidentes para diversos fatores prognósticos importantes de baseline, principalmente a favor do grupo de placebo.
As análises adicionais para a revisão radiológica independente, que realizaram um ajuste em relação à censura informativa e aos desequilíbrios nos dois braços de tratamento, mostraram um efeito do tratamento a favor do everolimo. Os resultados de uma análise multivariada ajustada adicional, que realizou uma correção em relação aos desequilíbrios entre os braços de tratamento, produziram uma Razão de Risco de 0,73 (IC de 95% de 0,56 a 0,97). Um modelo de Cox com Probabilidade Inversa dos Pesos de Censura (IPCW) foi usado para abordar e realizar uma correção em relação à censura informativa e aos desequilíbrios nas características de baseline entre os dois braços do estudo. A Razão de Risco estimada (IC de 95%) a partir da análise de IPCW foi de 0,60 (0,44 a 0,84), a favor do everolimo.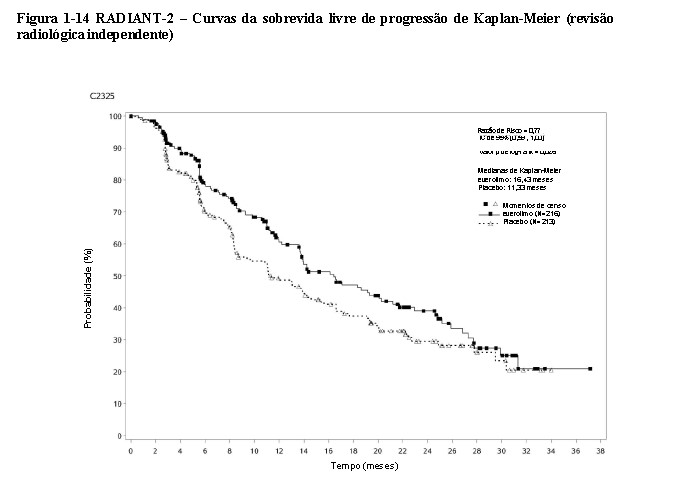
As taxas de SLP aos dezoito meses foram de 47,2% para a terapia com everolimo mais octreotida de depósito (Sandostatin LAR®) em comparação a 37,4% para placebo mais octreotida de depósito (Sandostatin LAR®).
A taxa de resposta objetiva de acordo com a revisão radiológica independente foi de 2,3% para o braço de everolimo mais octreotida de depósito (Sandostatin LAR®) em comparação com 1,9% para o braço de placebo mais octreotida de depósito (Sandostatin LAR®). A redução tumoral também foi evidente segundo o gráfico em cascata correspondente. Os resultados indicam que 75,0% dos pacientes no braço de everolimo mais octreotida de depósito (Sandostatin LAR®) apresentaram regressão tumoral em comparação com 44,8% no braço de placebo mais octreotida de depósito (Sandostatin LAR®) (Figura 1-15)12.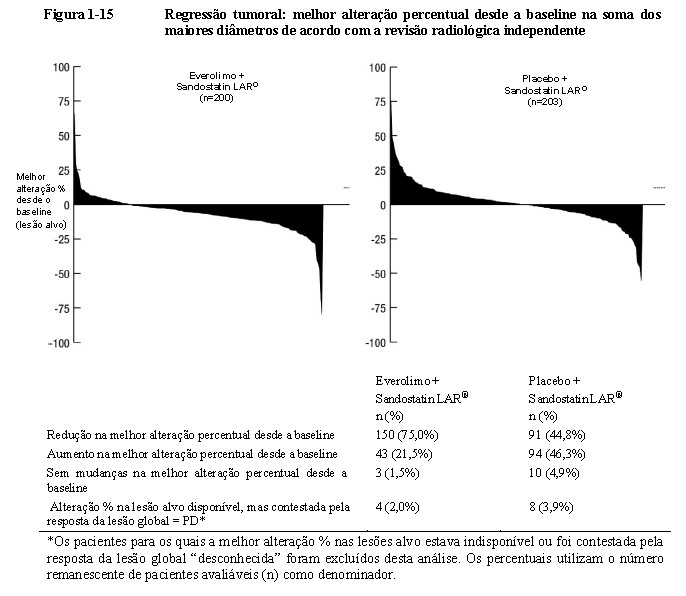
A análise final de sobrevida global não mostra nenhuma diferença estatisticamente significativa na SG (Razão de Risco
= 1,16 (IC 95%: 0,91 a 1,49))13,14. Houve 133 mortes (61,6%), no braço everolimo + octreotida de depósito, e 120 (56,3%), no braço placebo + octreotida de depósito. O cruzamento de 58% dos pacientes do placebo para o everolimo em regime aberto, seguindo a progressão da doença, o desequilíbrio entre os braços de tratamento em uso subsequente de octreotida e desequilíbrio nos fatores prognósticos chave na baseline, provavelmente confundiram a detecção de qualquer diferença relacionada ao tratamento na SG. Quando ajustados para fatores prognósticos importantes, a razão de risco da SG inclinou em direção à unidade (Razão de risco 1,06; IC 95%: 0,82; 1,36).
Carcinoma avançado de células renais
O RECORD-1 (Estudo CRAD001C2240), um estudo duplo-cego, randomizado, multicêntrico, internacional de fase III, foi conduzido comparando everolimo 10 mg/dia e placebo, ambos em conjunto com o melhor tratamento de suporte em pacientes com carcinoma metastático de células renais cuja doença evoluiu apesar do tratamento prévio de terapia (sunitinibe, sorafenibe ou ambos sunitinibe e sorafenibe) com VEGFR-TKI (inibidor da quinase de tirosina do receptor do fator de crescimento endotelial vascular). A terapia prévia com bevacizumabe e alfainterferona também foi permitida. Os pacientes foram estratificados de acordo com critérios prognósticos do Memorial Sloan-Kettering Cancer Center (MSKCC) (grupos de risco favorável vs intermediário vs desfavorável) e terapia prévia anticâncer (1 vs 2 VEGFR-TKIs anteriores) 3,4.
A sobrevida livre de progressão, documentada usando RECIST (Critérios de Avaliação da Resposta em Tumores Sólidos) e avaliada por meio de uma análise central cega e independente, foi o objetivo primário. Os objetivos secundários incluíam segurança, taxa de resposta objetiva tumoral, sobrevida global, sintomas relacionados à doença e qualidade de vida. Após a progressão radiológica documentada, o investigador podia revelar o caráter cego aos pacientes: aqueles randomizados para placebo puderam receber 10 mg/dia de everolimo em regime aberto. O Comitê Independente de Monitoramento de Dados recomendou o término deste estudo no momento da segunda análise interina assim que o objetivo primário foi alcançado.3
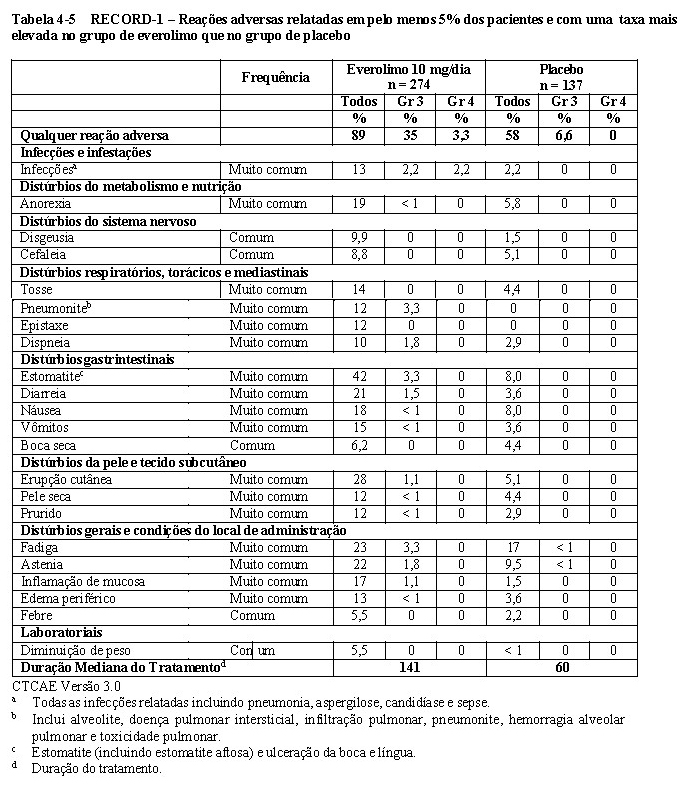
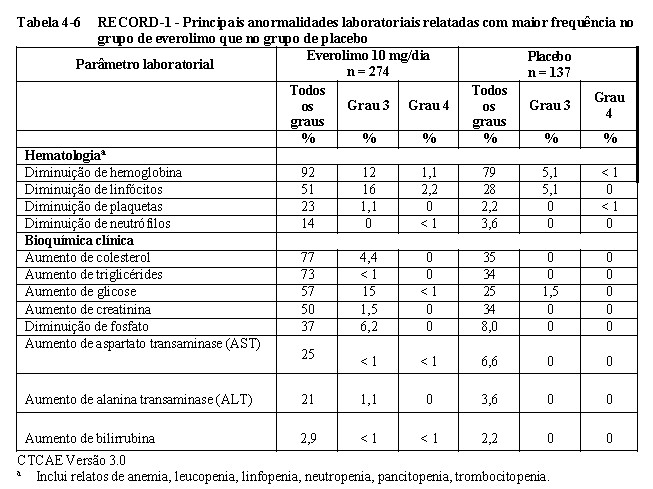
No total, 416 pacientes foram randomizados na proporção 2:1 para receber everolimo (n = 277) ou placebo (n= 139). Os dados demográficos foram bem equilibrados (idade mediana agrupada de 61 anos [variação entre 27 a 85], 77% eram 3,5,6 homens, 88% caucasianos e 74 % receberam pelo menos uma terapia prévia com VEGFR- TKI. A duração mediana do tratamento no estudo cego foi de 141 dias para pacientes recebendo everolimo e 60 dias para aqueles recebendo placebo.
Os resultados da análise interina mostraram que o everolimo foi superior ao placebo para o objetivo primário de sobrevida livre de progressão, com uma redução estatisticamente significativa de 67% no risco de progressão ou morte (vide Tabela 1-6 e Figura 1-16). 3,5,6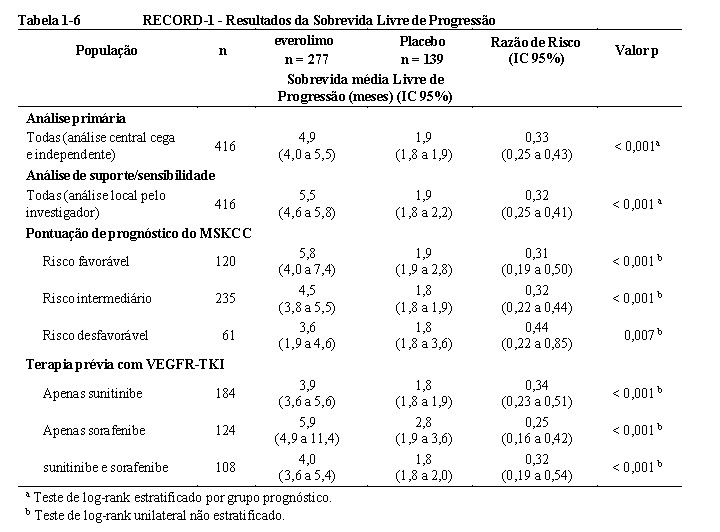
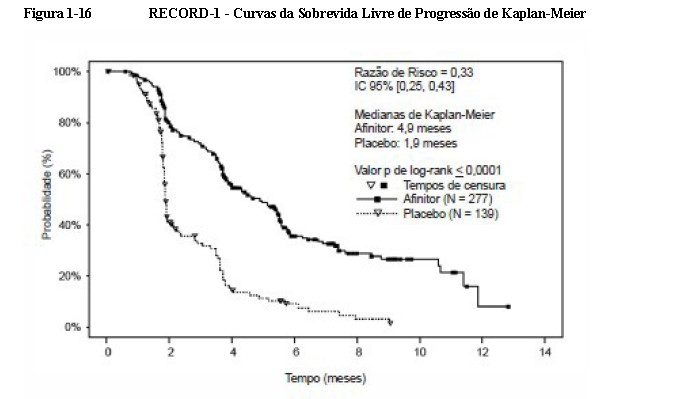
A taxa de sobrevida livre de progressão aos seis meses foi de 36 % para a terapia com everolimo comparado com 9 % para placebo5,6.
Respostas tumorais objetivas confirmadas foram observadas em 5 pacientes (2%) que receberam everolimo enquanto nenhuma foi observada em pacientes que receberam placebo. Portanto, a vantagem da sobrevida livre de progressão reflete principalmente a população com estabilização da doença (correspondente a 67% do grupo de tratamento com everolimo)5.
Os resultados na sobrevida global final, geraram uma tendência a favor do everolimo; a diferença entre os braços de tratamento não foram estatisticamente significativas (Razão de risco 0,90; lC95%: 0,71 a 1,14; p = 0,183). O cruzamento com everolimo em regime aberto após a progressão da doença ocorreu em 111 dos 139 pacientes (79,9%) alocados para placebo e podem ter confundido a detecção de diferenças relacionadas ao tratamento na sobrevida global5,6.
Uma melhora na qualidade de vida, medida através dos sintomas relacionados à doença, foi demonstrada nos pacientes que receberam everolimo (Razão de risco 0,75; 95% IC: 0,53 a 1,06; p = 0,053)5,6.
Complexo de esclerose tuberosa (TSC) com angiomiolipoma renal (em pacientes acima de 18 anos)
EXIST-2 (Estudo CRAD001M2302), estudo de fase III, randomizado, duplo-cego, multicêntrico de everolimo versus placebo foi conduzido em pacientes com TSC que apresentavam angiomiolipoma (n = 113) ou LAM esporádica e que apresentavam angiomiolipoma (n = 5). Os pacientes foram randomizados em uma proporção de 2:1 para receber everolimo ou placebo correspondente. A presença de pelo menos um angiomiolipoma ≥ 3 cm no maior diâmetro por TC/RMI (com base em avaliação radiológica local) foi exigida para inclusão19.
O desfecho de eficácia primário foi a taxa de resposta para angiomiolipoma com base na revisão radiológica central independente. A análise foi estratificada pelo uso de medicamentos antiepilépticos indutores de enzima (EIAEDs) na randomização (sim/não)19.
Os desfechos secundários principais incluíram tempo para progressão de angiomiolipoma e taxa de resposta para lesão cutânea19.
Um total de 118 pacientes foi randomizado, 79 para everolimo 10 mg, diariamente, e 39 para placebo. Os dois braços de tratamento foram, em geral, bem equilibrados no que se refere às características demográficas de baseline da doença e história de terapias anti-angiomiolipomas anteriores. A idade média foi de 31 anos de idade (faixa: 18 a 61; 46,6% tinham < 30 anos na inclusão), 33,9% eram homens e 89,0% eram caucasianos.
Dos pacientes incluídos, 83,1% apresentavam angiomiolipomas ≥ 4 cm (sendo 28,8% com angiomiolipomas ≥ 8 cm), 78,0% apresentavam angiomiolipomas bilaterais, e 39,0% haviam se submetido a nefrectomia/embolização renal anterior; 96,6% apresentavam lesões cutâneas e 44,1% apresentavam SEGAs (pelo menos um SEGA ≥ 1 cm no maior diâmetro)18,19. A duração média do tratamento do estudo cego foi de 48,1 semanas (intervalo de 2 a 115) para pacientes que receberam everolimo e 45,0 semanas (intervalo de 9 a 115) para aqueles que receberam placebo.
Os resultados mostraram que everolimo foi superior ao placebo em relação ao desfecho primário de melhor resposta geral do angiomiolipoma (p < 0,0001); a diferença observada foi tanto clinicamente relevante quanto estatisticamente significante. A melhor taxa de resposta global foi de 41,8% (IC de 95%: 30,8, 53,4) para o braço de everolimo em comparação a 0% (IC de 95%: 0,0, 9,0) para o braço de placebo (Tabela 1-7)18,19,20.
Pacientes inicialmente tratados com placebos foram permitidos ao cruzamento para everolimo no momento da progressão da angiomiolipoma e após o reconhecimento que o tratamento com everolimo foi superior ao tratamento com o placebo. No momento da análise final (4 anos após a última randomização de pacientes), a duração média de exposição ao everolimo foi 204,1 semanas (intervalo de 2 a 278). A melhor taxa de resposta global de angiomiolipoma havia aumentado para 58,0% (IC 95%: 48,3, 67,3), com taxa de doença estável de 30,4%21,25.
Entre os pacientes tratados com everolimo durante o estudo, nenhum caso de angiomiolipoma relacionado à nefrectomia e apenas um caso de embolização renal foram reportados21,25.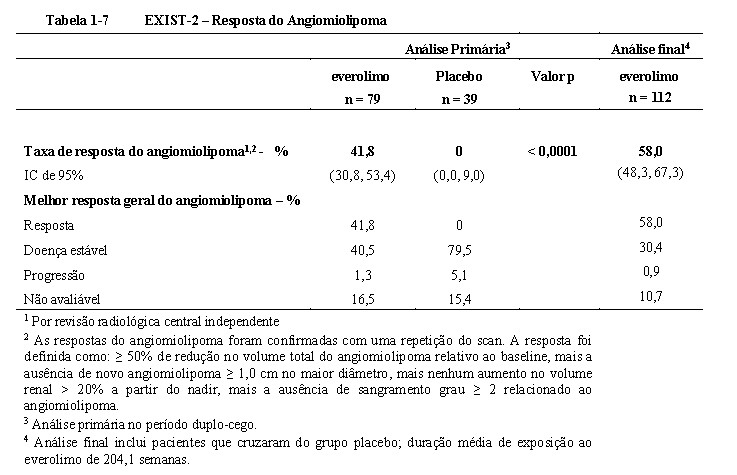
Efeitos consistentes do tratamento foram observados em todos os subgrupos avaliados (i.e., uso de EIAED
versus não uso de EIAED, sexo, idade e etnia) (Tabela 1-8, Figura 1-17) 18,19,20. 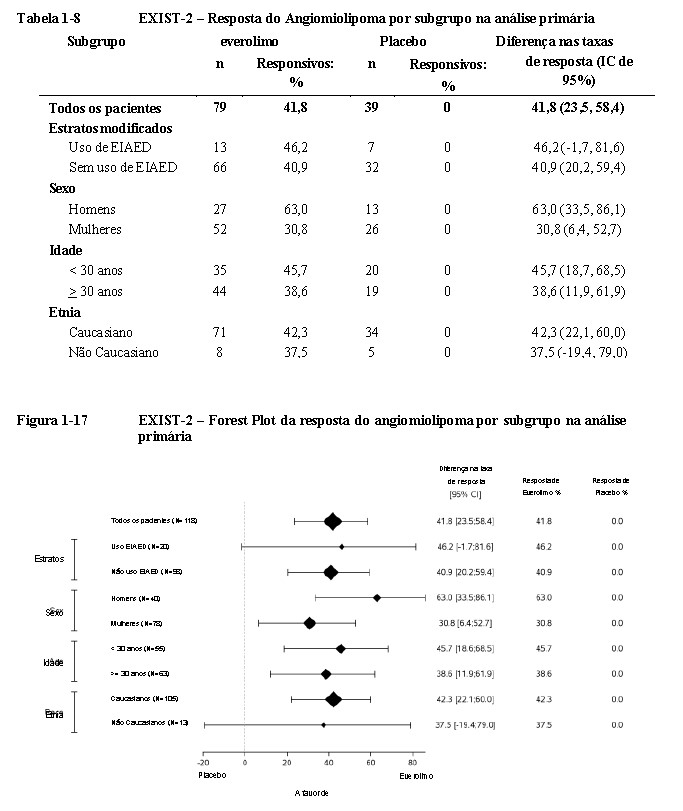
O Gráfico Cascata (waterfall plot) fornece uma representação gráfica da redução no volume do angiomiolipoma (Figura 1-18) na análise primária; 95,5% dos pacientes no braço do everolimo apresentaram redução do angiomiolipoma versus 59,4% no braço de placebo18,19,20.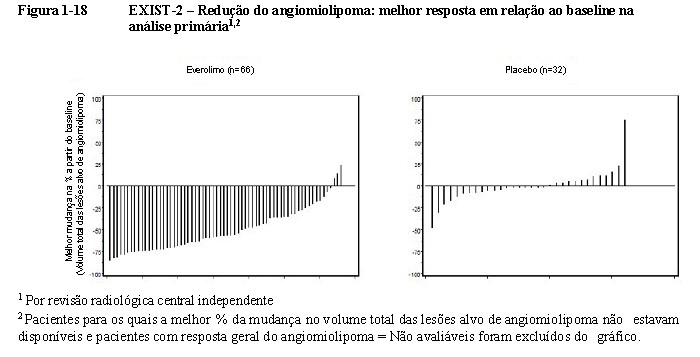
Na análise final, redução em do volume de angiomiolipoma aumentou com tratamento a longo prazo com everolimo. Nas semanas 12, 96 e 192, ≥ 30% de reduções no volume foram observadas em 75,0% (78/104), 80,6% (79/98) e 85,2% (52/61) dos pacientes tratados, respectivamente. Similarmente, nos mesmos períodos de tempo, ≥50% de reduções no volume foram observadas em 44,2% (46/104), 63,3% (62/98) e 68,9% (42/61) dos pacientes tratados, respectivamente21,25.
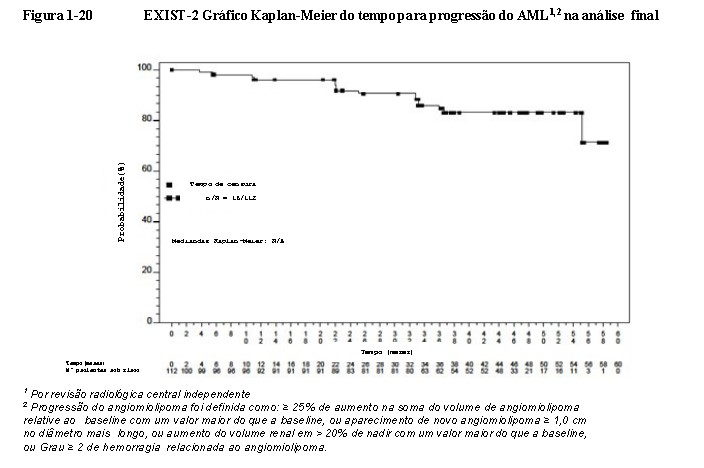
Everolimo foi associado a um prolongamento clinicamente relevante e estatisticamente significativo no tempo de progressão do angiomiolipoma (HR 0,08; IC de 95%: 0,02, 0,37; p < 0,0001) (Figura 1-11) na análise primária. O tempo médio de progressão do angiomiolipoma foi de 11,4 meses no braço de placebo e não foi alcançado no braço de everolimo. Progressões foram observadas em 3,8% (3/79) dos pacientes no braço de everolimo em comparação com 20,5% (8/39) no braço de placebo. As taxas estimadas livres de progressão em 6 meses foram de 98,4% para o braço de everolimo e de 83,4% para o braço de placebo18,19,20. No final da análise, o tempo médio de progressão do angiomiolipoma não foi atingido. Progressões do angiomiolipoma não foram observadas em 14,3% dos pacientes (16/112). As taxas estimadas de angiomiolipoma livre de progressão aos 24 meses e 48 meses foram 91,6% (IC de 95%: 84,0%, 95,7%) e 83,1% (IC de 95%: 73,4%,
89,5%) respectivamente (vide Figura 1-20)21,25.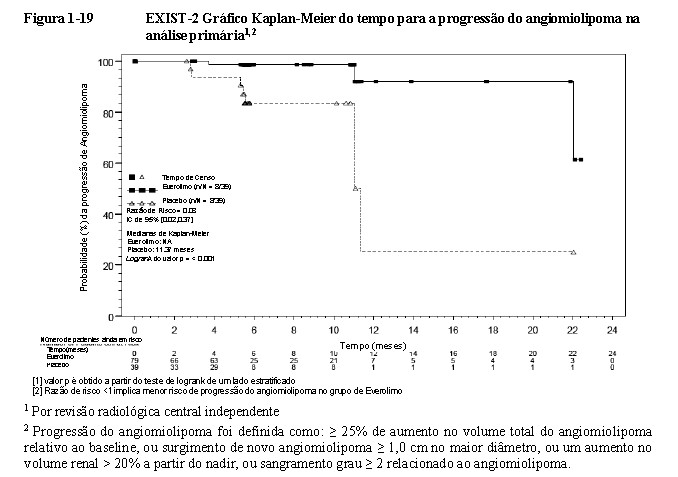
Na análise primária, everolimo também demonstrou melhoras clinicamente relevantes e estatisticamente significativas na resposta de lesões cutâneas (p = 0,0002), com taxas de resposta de 26,0% (20/77) (IC de 95%: 16,6, 37,2) para o braço de everolimo e de 0% (0/37) (IC de 95%: 0,0, 9,5) para o braço de placebo (Tabela 1- 9)18,19,20. No final da análise, a taxa de resposta de lesão cutânea aumentou para 68,2% (73/107) (IC de 95%: 58,5%, 76,9%) (vide Tabela 1-9), com um paciente reportando uma resposta clínica de lesão cutânea completa confirmada e nenhum paciente experimentando doença progressiva como sua melhor resposta21,25.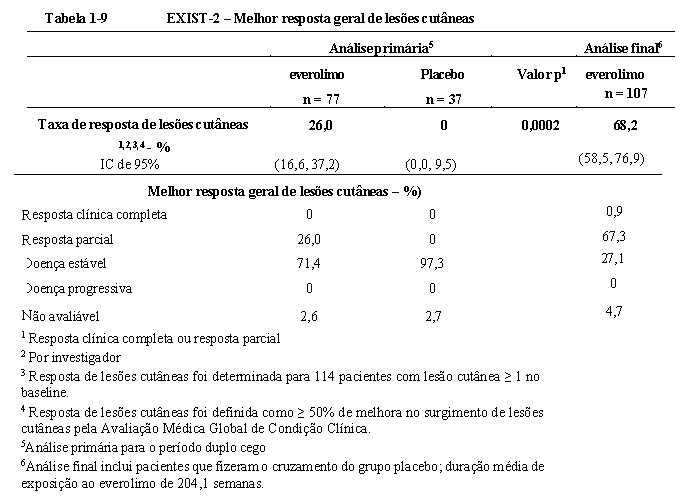
Em uma análise exploratória de pacientes com TSC com angiomiolipoma que também tiveram SEGA, a taxa de resposta do SEGA (proporção de pacientes com redução ≥ 50% da baseline em volumes de lesão alvo na ausência de progressão) foi 10,3% (4/39) no braço de everolimo na análise primária (versus nenhum resposta reportada nos 13 pacientes randomizados para o placebo com lesão SEGA na baseline) e aumentou para 48,0% (24/50) no final da análise21,25.
No EXIST-2, 12 de 16 pacientes que foram avaliados quanto ao volume de angiomiolipoma por até 1 ano após a interrupção do everolimo, apresentaram um aumento no volume tumoral em comparação com a sua mais recente avaliação de volume tumoral realizada antes da descontinuação do tratamento; embora o volume de angiomiolipoma não tenha excedido o valor medido na linha de base. Dois dos 16 pacientes que foram avaliados desenvolveram progressão definida de angiomiolipoma em virtude de sangramento relacionado ao angiomiolipoma (n = 1) e aumento do volume renal (n = 1). Esses achados sugerem que a persistência da redução do volume do angiomiolipoma clinicamente significativo requer tratamento contínuo na maioria dos pacientes.28
Esclerose Tuberosa com SEGA
Estudo de fase III em pacientes com TSC que possuem SEGA
O EXIST-1 (Estudo CRAD001M2301), um estudo de fase III, randomizado, duplo-cego, multicêntrico de everolimo versus placebo, foi conduzido em pacientes com TSC e SEGA, independentemente da idade. Os pacientes foram randomizados a uma razão de 2:1 para receber everolimo ou placebo correspondente. Era exigida a presença de ao menos uma lesão de SEGA ≥ 1,0 cm, no maior diâmetro, definido por ressonância magnética (com base na avaliação radiológica local) para a inclusão. Além disso, era necessária evidência radiológica em série de crescimento do SEGA, presença de uma nova lesão de SEGA ≥ 1 cm, no maior diâmetro, ou hidrocefalia nova ou agravada para a inclusão9,10,11,15.
O desfecho primário de eficácia foi uma taxa de resposta do SEGA baseada em uma análise radiológica central independente. A análise foi estratificada pelo uso de medicamentos antiepiléticos indutores de enzimas (EIAEDs) na randomização (sim / não)9,10,11,15.
Os principais desfechos secundários, em ordem hierárquica de teste incluíram alteração absoluta na frequência do número total de eventos de crise epiléptica por meio de um EEG de 24 horas, desde a baseline até a Semana 24, tempo até a progressão do SEGA e taxa de resposta de lesões cutâneas. A taxa de resposta de angiomiolipoma foi avaliada com uma análise exploratória9,10,11,15.
No total, 117 pacientes foram randomizados, sendo 78 para everolimo e 39 para placebo. Os dois braços de tratamento estavam, bem balanceados em relação as características demográficas e da doença na baseline e o histórico de terapias anteriores anti- SEGA. A idade mediana foi de 9,5 anos de idade (variação: 0,8 a 26,6; 69,2% estavam entre 3 e < 18 anos de idade no momento da inclusão; 17,1% estava com < 3 anos de idade no momento da inclusão), 57,3% eram do sexo masculino e 93,2% eram caucasianos. Dos pacientes incluídos, 79,5% apresentavam SEGAs bilaterais, 42,7% apresentavam ≥ 2 lesões de SEGA, 25,6% apresentavam crescimento inferior, 9,4% apresentavam evidência de invasão profunda no parênquima, 6,8% apresentavam evidência radiológica de hidrocefalia e 6,8% haviam sido submetidos a cirurgia anterior relacionada à SEGA; 94,0% apresentavam lesões cutâneas na baseline e 37,6% apresentavam angiomiolipomas renais (ao menos um angiomiolipoma ≥ 1 cm no maior diâmetro). A duração mediana do tratamento do estudo cego foi de 52,2 semanas (variação 24 a 89) para os pacientes que receberam everolimo e 46,6 semanas (variação 14 a 88) para aqueles que receberam placebo9,10,11,15.
Os resultados demonstram que everolimo foi superior ao placebo com relação ao desfecho primário de melhor resposta geral do SEGA (p < 0,0001). As taxas de resposta foram 34,6% (IC 95%: 24,2; 46,2) para o braço de everolimo comparado a 0% (IC 95%: 0,0, 9,0) para o braço de placebo (Tabela 1-10). Além disso, todos os 8 pacientes do braço de everolimo que apresentaram evidência radiológica de hidrocefalia na baseline apresentaram redução no volume ventricular e nenhuma paciente necessitou de intervenção cirúrgica durante o decorrer deste estudo9,10,11,15.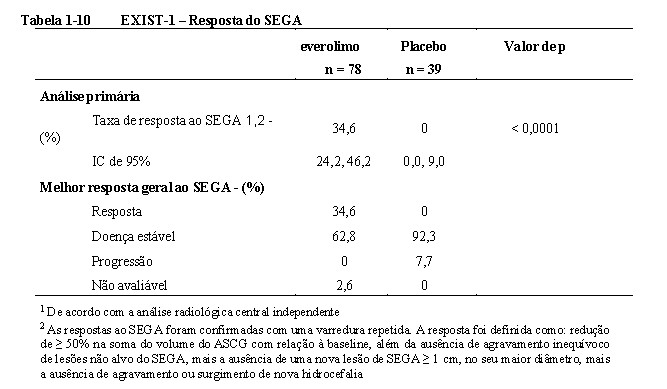
Foram observados efeitos consistentes com o tratamento em todos os subgrupos avaliados (ou seja, uso de EIAED versus sem o uso de EIAED, sexo e idade) (Tabela 1-11, Figura 1-21) 9,10,11,15.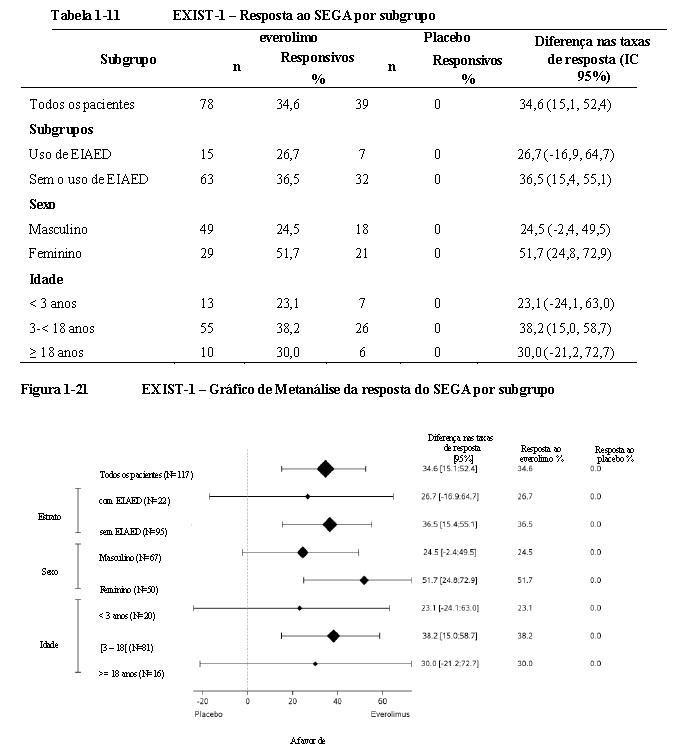
Nas primeiras 12 semanas de tratamento com everolimo, a redução do SEGA foi evidente: 73,0% dos pacientes apresentaram reduções de ≥ 30% e 29,7% apresentaram reduções de ≥ 50% na ocasião da primeira avaliação radiológica (Semana 12). Nos períodos subsequentes, reduções consistentes foram evidentes; na Semana 24, 78,4% dos pacientes apresentaram reduções ≥ 30% e 41,9% apresentaram reduções ≥ 50%9,10,11.
A análise do primeiro desfecho secundário principal, alteração da frequência de crises epilépticas, foi inconclusiva.
O tempo mediano até a progressão do SEGA com base na análise radiológica central não foi alcançado em nenhum braço de tratamento. Foram somente observadas progressões no braço de placebo (15,4%; p = 0,0002)
(Figura 1-22). As taxas estimadas livres de progressão no sexto mês foram de 100% para o braço de everolimo e 85,7% para o braço de placebo9,10,11,15.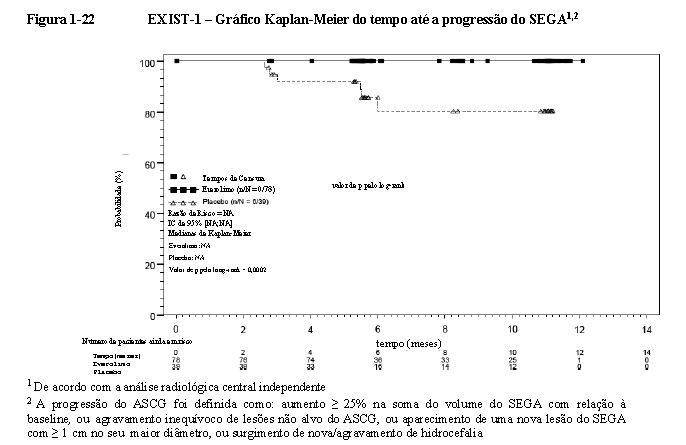
Everolimo demonstrou melhora clinicamente significativa na resposta de lesões cutâneas (p = 0,0004), com taxas de resposta de 41,7% (IC de 95%: 30,2, 53,9) para o braço de everolimo e 10,5% (IC de 95%: 2,9, 24,8) para o braço de placebo (Tabela 1-12)9,10,11,15.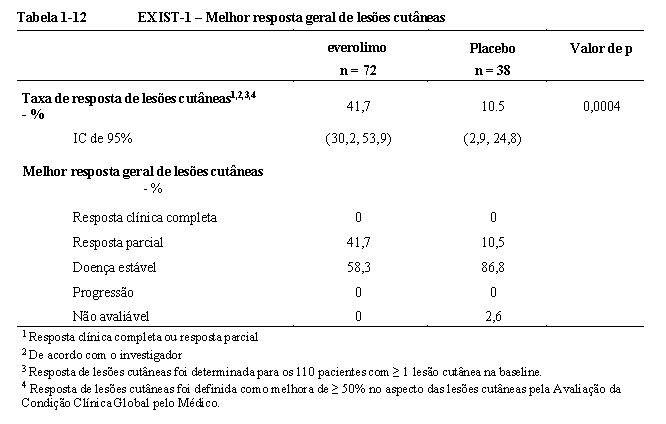
Estudos fase II em pacientes com TSC e SEGA
Um estudo prospectivo (CRAD001C2485), aberto, de fase II foi conduzido para avaliar a segurança e a eficácia de everolimo em pacientes com SEGA. Evidências radiológicas seriadas do crescimento do SEGA foram exigidas para inclusão7, 8.
A alteração no volume do SEGA durante a fase de tratamento central de 6 meses, avaliada por uma revisão radiológica central independente, constituiu o desfecho primário de eficácia. Após a fase de tratamento central, os pacientes poderiam entrar na fase de tratamento de extensão, onde o volume do SEGA foi avaliado a cada 6 meses7,8.
No total, 28 pacientes receberam tratamento com everolimo; a idade mediana correspondeu a 11 anos (variação de 3 a 34), 61% do sexo masculino, 86% caucasianos. Treze pacientes (46%) apresentaram um SEGA secundário menor, incluindo 12 pacientes com SEGA no ventrículo contralateral8. A duração mediana de exposição foi de 45,7 meses (variação 4,7 a 58,5).
O everolimo foi associado a uma redução clinicamente relevante e estatisticamente significativa no volume do SEGA primário após 6 meses em relação à avaliação basal (p < 0,001). A redução do tumor foi mais rápida durante os 3 meses de tratamento iniciais, com evidência de uma resposta mantida em pontos de tempo subsequentes (ver Tabela 1-13). Nenhum paciente desenvolveu novas lesões, agravamento de hidrocefalia, aumento da pressão intracraniana e nenhum necessitou de ressecção cirúrgica ou outra terapia para SEGA.7, 8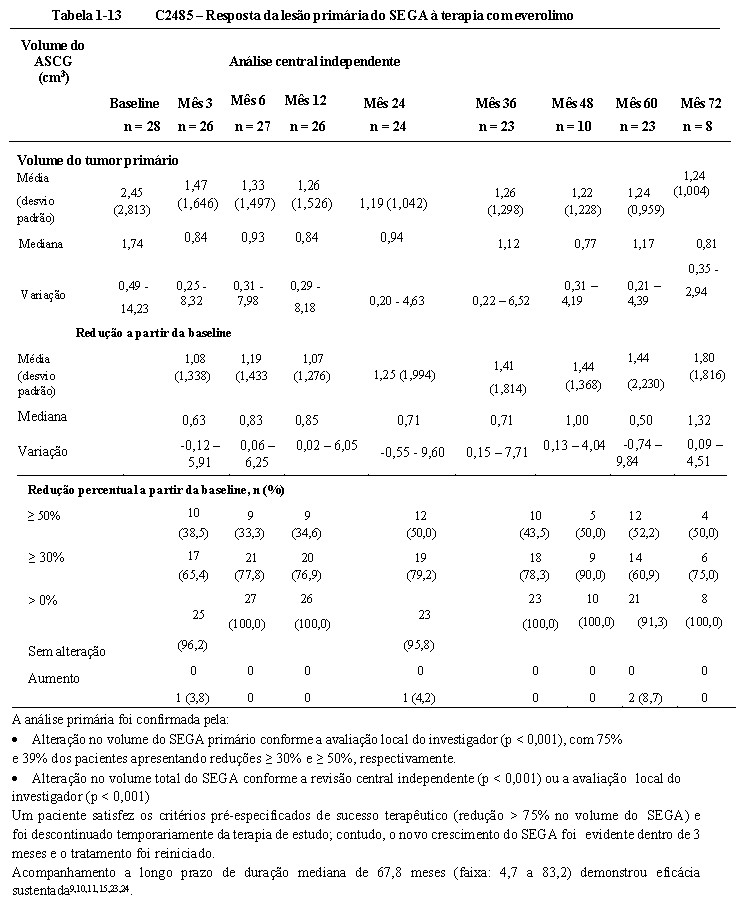
Referências
1. [Study C2324] A randomized double-blind phase III study of RAD001 10 mg/d plus best supportive care versus placebo plus best supportive care in the treatment of patients with advanced pancreatic neuroendocrine tumor (NET). Novartis Pharmaceuticals Corporation. East Hanover, USA. 29-Aug-2010.
2. [Study C2325] A randomized, double-blind, placebo-controlled, multicenter phase III study in patients with advanced carcinoid tumor receiving Sandostatin LAR® Depot and RAD001 10 mg/d or Sandostatin LAR® Depot and placebo. Novartis Pharmaceuticals Corporation. East Hanover, USA. 01-Oct-2010.
3. [Study C2240] A randomized, double-blind, placebo-controlled, multicenter phase III study to compare the safety and efficacy of RAD001 plus Best Supportive Care (BSC) versus BSC plus Placebo in patients with metastatic carcinoma of the kidney which has progressed on VEGF receptor tyrosine kinase inhibitor therapy. Full Clinical Study Report RAD001 C2240. Novartis Pharmaceuticals Corporation. East Hanover, USA. 29 May 08.
4. [Summary of Clinical Efficacy] RAD001 (everolimus) - 2.7.3. Summary of Clinical Efficacy in advanced renal cell carcinoma. Novartis Pharmaceuticals Corporation. East Hanover, USA. 28 May 08.
5. [Study C2240] A randomized, double-blind, placebo-controlled, multicenter phase III study to compare the safety and efficacy of RAD001 plus Best Supportive Care (BSC) versus BSC plus Placebo in patients with metastatic carcinoma of the kidney which has progressed on VEGF receptor tyrosine kinase inhibitor therapy. Full Clinical Study Report-Addendum Report RAD001 C2240. Novartis Pharmaceuticals Corporation. East Hanover, USA.
6. Expert Statement - Update to Warning and Precautions and Adverse Drug Reactions. 02 Apr 09.
7. [Clinical Overview] in Subependymal giant cell astrocytoma associated with tuberous sclerosis. Novartis Pharmaceuticals Corporation. East Hanover, USA. 31-Mar-10.
8. [Study C2485] Everolimus (RAD001) Therapy of Giant Cell Astrocytoma in Patients with Tuberous Sclerosis Complex. Clinical Study Report RAD001 C2485. Novartis Pharmaceuticals Corporation. East Hanover, USA. 31-Mar-10.
9. [Clinical Overview] Tuberous sclerosis complex with subependymal giant cell astrocytoma. Novartis Pharmaceuticals Corporation. East Hanover, USA. 02-Feb-2012.
10. [Study M2301] A randomized, double-blind, placebo-controlled study of RAD001 in the treatment of patients with subepen