ELIGARD®
ADIUM
leuprorrelina
Análogo LH-RH.
Apresentações.
Pó liófilo para suspensão injetável 7,5 mg: cartucho contendo duas seringas (Seringa "B" contém pó liófilo de acetato de leuprorrelina e Seringa "A" contém (diluente) sistema polimérico ATRIGEL®), agulha e sachês dessecantes para controle de umidade do produto.
Pó liófilo para suspensão injetável 22,5 mg: cartucho contendo duas seringas (Seringa "B" contém pó liófilo de acetato de leuprorrelina e Seringa "A" contém (diluente) sistema polimérico ATRIGEL®), agulha e sachês dessecantes para controle de umidade do produto.
USO SUBCUTÂNEO
USO ADULTO
Composição.
Eligard 7,5 mg. Cada seringa "B" contém 10,2 mg* de acetato de leuprorrelina. Cada seringa "A" contém diluente (sistema polimérico) ATRIGEL® com 218 mg* de N-metil-2-pirrolidona e 112 mg* de Poli (DL-lactídeo-co- glicolídeo). *É fornecida uma quantidade superior de cada componente devido à retenção da suspensão injetável na seringa e na agulha, após a administração do produto no paciente. O desenvolvimento da formulação foi realizado de modo que forneça aproximadamente 250 mg de suspensão injetável contendo 7,5 mg de acetato de leuprorrelina,(equivalente a 7 mg de leuprorrelina base) para o paciente. Eligard 22,5 mg. Cada seringa "B" contém 28,0 mg* de acetato de leuprorrelina. Cada seringa "A" contém diluente (sistema polimérico) ATRIGEL® com 242 mg* de N-metil-2-pirrolidona e 198 mg* de Poli(DL-lactídeo-co- glicolídeo). * É fornecida uma quantidade superior de cada componente devido à retenção da suspensão injetável na seringa e na agulha, após a administração do produto no paciente. O desenvolvimento da formulação foi realizado de modo que forneça aproximadamente 375 mg de suspensão injetável contendo 22,5 mg de acetato de leuprorrelina,(equivalente a 21,0 mg de leuprorrelina base) para o paciente.
Indicações.
ELIGARD® é indicado para o tratamento paliativo do câncer de próstata avançado.
Resultados de eficácia.
Estudos com o ELIGARD® 7,5 mg
Em um estudo aberto e multicêntrico (AGL9904), 120 pacientes que apresentavam câncer de próstata avançado foram tratados com seis injeções mensais de ELIGARD®. Oitenta e nove pacientes apresentavam doença estágio C e 31 pacientes apresentavam doença estágio D. Esse estudo avaliou a manutenção da supressão da testosterona sérica durante seis meses de tratamento.
A concentração média de testosterona aumentou de 361,3 ng/dL (valor inicial) para 574,6 ng/dL no Dia 3 após a injeção subcutânea inicial. A concentração sérica média de testosterona posteriormente foi reduzida a níveis abaixo dos basais no Dia 10, sendo de 21,8 ng/dL no Dia 28. Ao término do estudo (6° mês), a concentração média de testosterona foi de 6,1 ng/dL (Figura 1).
A testosterona sérica foi suprimida a níveis abaixo do limiar de castração ( < 50 ng/dL) no Dia 28 (Semana 4) em 112 de 119 (94,1%) pacientes que permaneceram no estudo. Os sete pacientes restantes atingiram níveis de castração até o Dia 42. Depois de ter atingido a supressão da testosterona em concentrações séricas de 50 ng/dL ou abaixo, nenhum paciente (0%) apresentou exacerbação (concentração acima de 0 ng/dL) em nenhum momento do estudo. Todos os 117 pacientes avaliáveis no estudo ao Mês 6 (dois pacientes se retiraram por razões não relacionadas ao ELIGARD®) apresentaram concentrações de testosterona ≤ 50 ng/dL.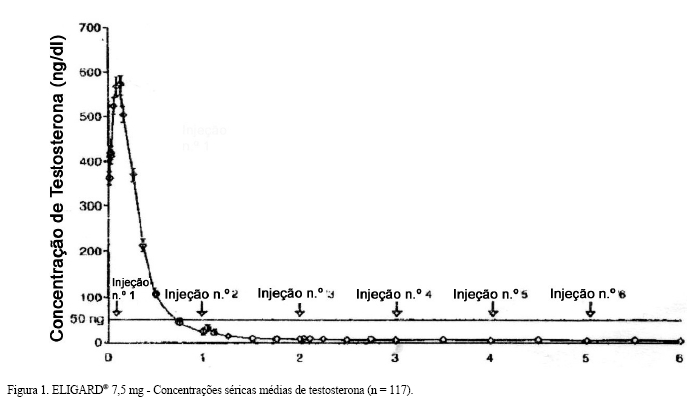
Todos os pacientes com níveis de PSA acima do normal, tiveram seus níveis reduzidos. Os valores médios foram reduzidos em 94% a partir do nível basal até o 6° mês. No 6° mês, os níveis de PSA haviam sido reduzidos aos limites normais em 94% dos pacientes que apresentaram níveis elevados ao nível basal. A análise da eficácia inclui os objetivos secundários utilizando a escala de estadiamento de performance de OMS, objetivando dor óssea, dor ao urinar e sinais e sintomas urinários. No nível basal, 88% dos pacientes foram classificados como "totalmente ativos" pela escala de desempenho da OMS (Status=0) e 11% como "restritos em atividade extenuante, porém capazes de realizar trabalho de natureza leve ou sedentária" (Status=1). Essas porcentagens não foram alteradas ao Mês 6. Durante a visita basal, os pacientes apresentaram pouca dor óssea, com pontuação média de 1,22 (variação de 1-9) em uma escala de 1 (sem dor) a 10 (pior dor possível). Ao Mês 6, a pontuação média de dor óssea se apresentou essencialmente inalterada em 1,26 (variação de 1-7). A dor ao urinar, pontuada na mesma escala, foi igualmente baixa, com média de 1,12 à visita basal (variação de 1-5) e 1,07 ao Mês 6 (variação de 1-8). Os sinais e sintomas urinários foram igualmente baixos à visita basal, e foram discretamente reduzidos ao Mês 6. Além disso, houve uma redução nos pacientes que apresentaram anormalidades prostáticas detectadas durante o exame físico, de 102 (85%) à Triagem para 77 (64%) ao 6° mês.
Estudos com o ELIGARD® 22,5 mg
Em um estudo aberto e multicêntrico (AGL9909), 117 pacientes que apresentavam câncer de próstata avançado foram tratados com no mínimo uma injeção de ELIGARD®. Desses, 113 pacientes receberam no total duas injeções de ELIGARD® 22,5 mg, administradas uma a cada três meses. Dois pacientes apresentavam doença estágio A, 19 pacientes apresentavam doença estágio B, 60 pacientes apresentavam doença estágio C e 36 pacientes apresentavam doença estágio D. Esse estudo avaliou a manutenção da supressão da testosterona sérica em níveis de castração durante seis meses de terapia. No total, 111 pacientes concluíram o estudo. A concentração média de testosterona aumentou de 367,1 ng/dL (valor inicial) basal para 588,0 ng/dL após 2 dias da injeção subcutânea. A concentração sérica média de testosterona posteriormente foi reduzida a níveis abaixo dos basais no Dia 14, sendo de 27,7 ng/dL no Dia 21. Ao término do estudo (Mês 6), a concentração média de testosterona foi de 10,1 ng/dL (Figura 2). Dos 117 pacientes, um recebeu menos de uma dose completa de ELIGARD® 22,5 mg durante a visita basal, e nunca apresentou supressão, e foi retirado no Dia 73, tendo recebido um tratamento alternativo. Nos 116 pacientes restantes tratados com a dose completa durante a visita basal, a testosterona sérica foi suprimida aos níveis abaixo do limiar de castração ( < 50 ng/dL) no Dia 28 (Semana 4) em 115 dos 116 (99%). Até o Dia 35, todos os 116 pacientes (100%) tratados com uma dose completa durante a visita basal atingiram o limiar de castração. Depois de ter atingido a supressão da testosterona em concentrações séricas de 50 ng/dL ou abaixo, apenas um paciente ( < 1%) apresentou aumento (concentração acima de 50 ng/dL) após a injeção inicial; este paciente permaneceu abaixo do limiar de castração após a segunda injeção. Todos os 111 pacientes avaliáveis no estudo apresentaram concentrações de testosterona < 50 ng/dL ao 6° mês. Todos os pacientes não avaliáveis que atingiram níveis de castração no dia 28 mantiveram os mesmos resultados em todas as visitas de avaliação, incluindo o momento da retirada.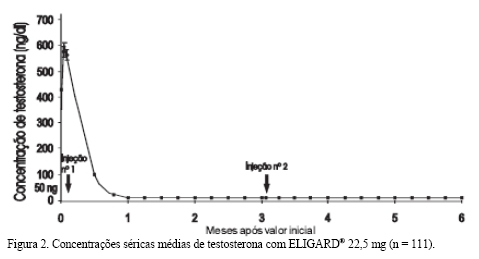
Todos os pacientes com níveis de PSA acima do normal tiveram seus níveis reduzidos. Os valores médios foram reduzidos em 98% a partir do nível basal até o Mês 6. No Mês 6, os níveis de PSA haviam sido reduzidos aos limites normais em 91% dos pacientes que apresentaram níveis elevados à visita basal. A análise da eficácia inclui o objetivo secundário utilizando a escala de estadiamento de performance da OMS, objetivando dor óssea, dor ao urinar e sinais e sintomas urinários. Nos níveis basais, 94% dos pacientes foram classificados como "totalmente ativos" pela escala de status de desempenho da OMS (Status=0) e 6% como "restritos em atividade extenuante, porém capazes de realizar trabalho de natureza leve ou sedentária" (Status=1). Ao Mês 6, essa porcentagem foi alterada para 96% (Status=0) e 4% (Status=1). Durante a visita basal, os pacientes apresentaram pouca dor óssea, com pontuação média de 1,20 (variação de 1-9) em uma escala de 1 (sem dor) a 10 (pior dor possível). Ao Mês 6, a pontuação média de dor óssea se apresentou essencialmente inalterada em 1,22 (variação de 1-5). A dor ao urinar, pontuada na mesma escala, foi igualmente baixa, com média de 1,02 à visita basal (variação de 1-2) e 1,10 ao Mês 6 (variação de 1-8). Os sinais e sintomas urinários apresentaram uma pontuação média de 1,09 à visita basal (variação de 1-4), sendo elevada para 1,18 ao Mês 6 (variação de 1-7). Além disso, houve uma redução nos pacientes com anormalidades prostáticas detectadas durante o exame físico, de 96 (82%) para 76 (65%) ao 6° mês.
Caract. farmacológicas.
Descrição
ELIGARD® 7,5 mg é uma formulação estéril de matriz polimérica de acetato de leuprorrelina para injeção subcutânea, elaborado para administrar 7,5 mg de acetato de leuprorrelina em velocidade controlada durante um período terapêutico de 1 mês.
ELIGARD® 22,5 mg também é uma formulação estéril de matriz polimérica de acetato de leuprorrelina para injeção subcutânea, elaborado para administrar 22,5 mg de acetato de leuprorrelina em velocidade controlada durante um período terapêutico de 3 meses.
O acetato de leuprorrelina é um análogo nonapeptídeo sintético do hormônio natural de liberação de gonadotrofina (GnRH ou LH-RH) que, quando administrado continuamente, inibe a secreção hipofisária de gonadotrofina e suprime a esteroidogênese testicular e ovariana. O análogo apresenta maior potência que o hormônio natural. A denominação química é acetato de 5-oxo-L-prolil-Lhistidil-L-triptofil-L-seril-L-tirosil-D-leucil-L-leucil-L-arginil-N-etil-L-prolinamida.
Farmacologia Clínica
O acetato de leuprorrelina, um agonista de LH-RH, atua como um potente inibidor da secreção de gonadotrofina, quando administrado continuamente em doses terapêuticas. Estudos em animais e em humanos indicam que após um estímulo inicial, a administração crônica de acetato de leuprorrelina resulta em uma supressão da esteroidogênese ovariana e testicular. Esse efeito é reversível após a descontinuação do tratamento.
Em humanos, a administração de acetato de leuprorrelina resulta em um aumento inicial dos níveis circulantes do hormônio luteinizante (LH) e hormônio folículo estimulante (FSH), causando um aumento temporário nos níveis do esteroides gonadais (testosterona e diidrotestosterona em homens, e estrona e estradiol em mulheres em pré-menopausa). Entretanto, a administração contínua de acetato de leuprorrelina resulta em redução dos níveis de LH e FSH. Em homens, a testosterona é reduzida ao limiar abaixo dos níveis de castração ( < 50 ng/dL). Essa redução ocorre no período de duas a quatro semanas após o início do tratamento. Estudos a longo prazo demonstraram que a continuação da terapia à base de acetato de leuprorrelina mantém os níveis de testosterona abaixo dos níveis de castração durante até sete anos.
Propriedades Farmacodinâmicas
Após a primeira dose de ELIGARD®, as concentrações séricas médias de testosterona foram elevadas temporariamente, e posteriormente foram reduzidas abaixo do limiar de castração ( < 50 ng/dL) no período de três semanas (Figuras 3 e 4). O tratamento contínuo manteve a supressão de testosterona similar à da castração durante todo o estudo. Não houve exacerbação das concentrações de testosterona acima do limiar de castração ( > 50 ng/dL) em nenhum momento do estudo após ter atingido a supressão semelhante à castração.
O acetato de leuprorrelina não é ativo quando administrado por via oral.
Propriedades Farmacocinéticas
Absorção: a farmacocinética/farmacodinâmica observada durante três injeções mensais (ELIGARD® 7,5 mg) ou duas injeções a cada 3 meses - a primeira administrada no tempo zero e a outra após 3 meses - (ELIGARD® 22.5 mg), em 20 e 22 pacientes, respectivamente, que apresentavam carcinoma prostático avançado é ilustrada nas Figuras 3 e 4. A concentração sérica média de leuprorrelina após a injeção inicial elevou-se até (Cmax) 25,3 ng/mL (dose 7,5 mg) e 127 ng/mL (dose 22,5 mg), aproximadamente 5 horas após a primeira injeção e de 107 ng/mL após a segunda dose de ELIGARD® 22,5 mg. Após o aumento inicial (após cada injeção), a concentração sérica permaneceu relativamente constante (0,28 - 2,00 ng/mL) para ambas as doses. Não houve evidências de acúmulo significativo durante a dose repetida. Foram observadas concentrações plasmáticas não detectáveis de leuprorrelina durante a administração crônica de ELIGARD® 7,5 mg ou 22,5 mg, porém os níveis de testosterona foram mantidos nos níveis de castração. 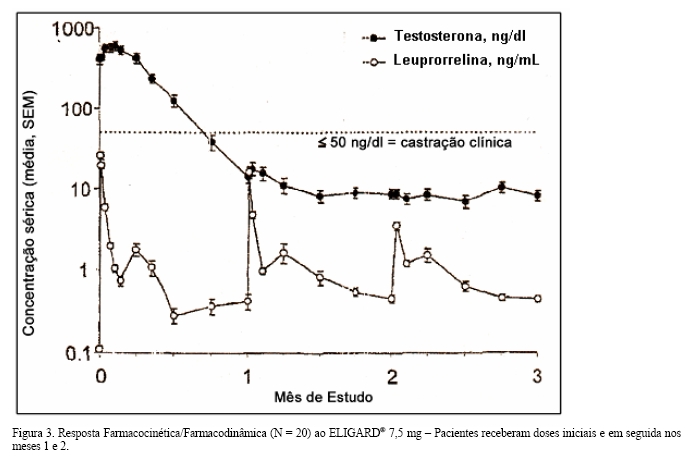
Um número reduzido de visita de avaliação de amostragem resultou na redução aparente dos valores de Cmax na segunda e terceira doses de ELIGARD® 7,5 mg (Figura 3).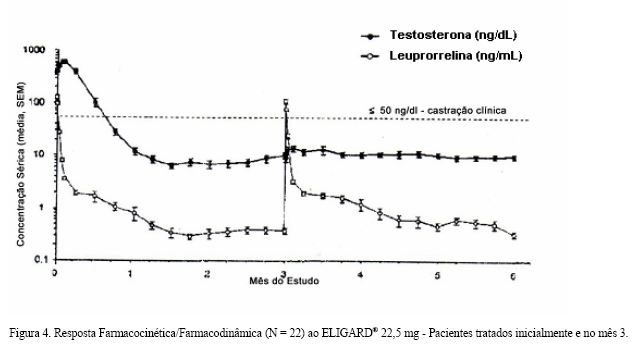
Distribuição: o volume de distribuição médio em estado de equilíbrio da leuprorrelina após a administração intravenosa em bolus em voluntários saudáveis do sexo masculino foi de 27 L. A ligação in vitro às proteínas plasmáticas variou de 43% a 49%.
Metabolismo: em voluntários saudáveis do sexo masculino, 1 mg de leuprorrelina (em bolus) administrado por via intravenosa resultou na depuração sistêmica média de 8,34 L/h, com meia-vida de eliminação terminal de aproximadamente 3 horas, com base em um modelo de dois compartimentos. O estudo de metabolismo do fármaco não foi realizado com ELIGARD®. Após a administração com diferentes formulações à base de acetato de leuprorrelina, o principal metabólito do acetato de leuprorrelina é um metabólito pentapeptídeo (M-1).
Excreção: não foi realizado estudo de excreção do medicamento ELIGARD®.
Populações Especiais
Geriátrica: aproximadamente 70% dos pacientes estudados tinha 70 anos ou mais.
Pediátrica: a segurança e a eficácia de ELIGARD® em pacientes pediátricos não foram estabelecidas.
Raça: nos pacientes estudados (26 brancos, 2 hispânicos - ELIGARD® 7,5 mg - e 19 brancos, 4 negros, 2 hispânicos - ELIGARD® 22,5 mg), a concentração sérica média de leuprorrelina foi similar.
Insuficiência hepática e renal: a farmacocinética de ELIGARD® em pacientes que apresentavam comprometimento hepático e renal não foi determinada.
Contraindicações.
ELIGARD® é contraindicado em pacientes com hipersensibilidade ao LHRH, análogos agonistas de LHRH ou qualquer um dos componentes de ELIGARD®. Foram relatadas reações anafiláticas ao LHRH sintético ou aos análogos agonistas do LHRH.
ELIGARD® é contra-indicado em mulheres e em pacientes pediátricos, não sendo estudado nessas populações.
Categoria de risco na gravidez: X
Este medicamento não deve ser utilizado por mulheres grávidas ou que possam ficar grávidas durante o tratamento.
Este medicamento causa malformação ao bebê durante a gravidez.
Este medicamento é contraindicado para uso por mulheres.
Este medicamento é contraindicado para uso por pacientes pediátricos.
Advertências e precauções.
ELIGARD®, como outros agonistas do LH-RH, ocasiona na primeira semana de tratamento um aumento temporário nas concentrações séricas de testosterona. Os pacientes poderão apresentar piora dos sintomas da doença de base ou aparecimento de novos sinais e sintomas durante as primeiras semanas de tratamento, incluindo dor óssea, neuropatia, hematúria, ou obstrução do trato urinário.
Foram observados casos isolados de obstrução ureteral e/ou compressão da medula espinhal, que podem contribuir para paralisia (com ou sem complicações fatais), durante o tratamento paliativo do câncer de próstata avançado com agonistas de LH-RH. Caso haja desenvolvimento de compressão da medula espinhal ou comprometimento renal, o tratamento padrão dessas complicações deverá ser instituído.
Os pacientes que apresentam lesões vertebrais metastáticas e/ou obstrução do trato urinário deverão ser observados com cautela durante as primeiras semanas de terapia.
Alterações nos exames laboratoriais: A terapia com acetato de leuprorrelina resulta na supressão do sistema hipofisário-gonadal. Os resultados de testes diagnósticos das funções hipofisária gonadotrófica e gonadal realizados durante e após a terapia com leuprorrelina podem ser afetados.
Testes laboratoriais: a resposta ao ELIGARD® deverá ser monitorizada por meio da avaliação periódica das concentrações séricas de testosterona e antígeno prostático específico (PSA). Na maioria dos pacientes, os níveis de testosterona foram elevados acima dos níveis basais durante a primeira semana, posteriormente sendo reduzidos aos níveis basais ou abaixo deles ao final da segunda semana. Os níveis de castração foram geralmente atingidos no período de duas a quatro semanas e, depois de atingidos, foram mantidos durante todo o tratamento. Não houve aumento acima dos níveis de castração em nenhum dos pacientes. Os resultados das determinações de testosterona dependem da metodologia do ensaio. É aconselhável estar alerta ao tipo e precisão da metodologia do ensaio, para tomar decisões clínicas e terapêuticas adequadas.
Gravidez e lactação
Categoria de risco na gravidez: X
Este medicamento não deve ser utilizado por mulheres grávidas ou que possam ficar grávidas durante o tratamento.
Este medicamento causa malformação ao bebê durante a gravidez.
O acetato de leuprorrelina poderá causar lesão fetal quando administrado a gestantes. Foram observadas lesões fetais importantes após a administração de acetato de leuprorrelina em coelhos, o que não ocorreu em ratos. Houve aumento da mortalidade fetal e redução do peso fetal em ratos e coelhos. Os efeitos sobre a mortalidade fetal são consequências esperadas das alterações nos níveis hormonais causados por este medicamento. Há possibilidade de ocorrência de aborto espontâneo.
Carcinogênese, mutagênese e comprometimento da fertilidade
Foram realizados estudos de carcinogenicidade (dois anos de duração) com o acetato de leuprorrelina, em ratos e camundongos. Em ratos, foi observado um aumento relacionado à dose, da hiperplasia benigna hipofisária e adenomas hipofisários benignos aos 24 meses, quando o fármaco foi administrado por via subcutânea em elevadas doses diárias (0,6 a 4 mg/Kg). Houve um aumento significativo, porém não relacionado à dose nos adenomas de células das ilhotas pancreáticas em fêmeas e de adenomas de células intersticiais testiculares em machos (maior incidência no grupo de dose baixa). Em camundongos, não foram observados tumores induzidos pelo acetato de leuprorrelina ou anormalidades hipofisárias nas doses até 60 mg/Kg durante os dois anos de estudo. Pacientes foram tratados com acetato de leuprorrelina com doses de até 10 mg/dia, por até três anos, e com doses de até 20 mg/dia durante dois anos, sem a observação de anormalidades hipofisárias. Não foram realizados estudos de carcinogenicidade com ELIGARD®.
Foram realizados estudos de mutagenicidade com o acetato de leuprorrelina, utilizando sistemas bacterianos e mamíferos e com ELIGARD® em sistemas bacterianos. Esses estudos não forneceram evidências de potencial mutagênico.
Este medicamento pode causar doping.
Interações medicamentosas.
Não foram realizados estudos de interação medicamentosa farmacocinética com ELIGARD®.
Cuidados de armazenamento.
ELIGARD® deve ser conservado sob refrigeração (2° - 8°C), protegido da luz.
ELIGARD® 7,5 mg tem validade de 18 meses.
ELIGARD® 22,5 mg tem validade de 24 meses.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Após preparo, o produto deverá ser administrado no período de 30 minutos. Depois deste período, a suspensão não utilizada deverá ser descartada.
Características físicas e organolépticas
Após preparo a suspensão apresentará uma coloração amarela clara a amarela.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
Posologia e modo de usar.
ELIGARD® deve ser administrado por via subcutânea, onde forma um depósito sólido de liberação do medicamento. O conteúdo da seringa é de dose única.
Assim como os demais medicamentos administrados por injeção subcutânea, o local de injeção deverá ser alterado periodicamente.
ELIGARD® é preenchido e fornecido em duas seringas estéreis separadas, cujo conteúdo deve ser misturado imediatamente antes da administração. As duas seringas são combinadas e o produto de dose única é misturado até a homogeneização.
Importante: Deixar o produto atingir a temperatura ambiente antes de utilizar. Após a reconstituição, o produto deverá ser administrado no período de 30 minutos. Depois deste período, a suspensão injetável não utilizada deverá ser descartada.
ELIGARD® possui 2 blísteres: um identificado como diluente contendo a seringa A estéril preenchida com o diluente (sistema polimérico) ATRIGEL®, um êmbolo branco longo de reposição e sachê dessecante; o outro identificado como ELIGARD® contendo a seringa B estéril preenchida com pó liófilo de acetato de leuprorrelina, agulha estéril descartável para aplicação e sachê dessecante (figura 5).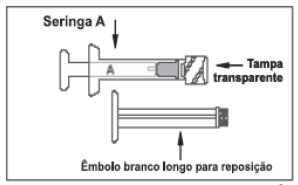
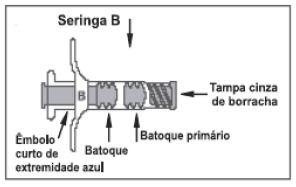
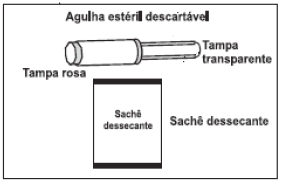
Figura 5: imagens das seringas de ELIGARD®
Seguir as instruções fornecidas para garantir a preparação adequada de ELIGARD® antes da administração:
1. Em um local limpo, abrir todas as embalagens e retirar seu conteúdo. Descartar o sache dessecante.
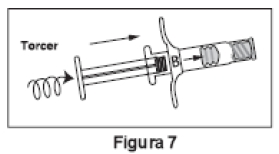
2. Retirar o êmbolo curto de extremidade azul da Seringa B e descartar (Figura 6). Introduzir suavemente o êmbolo branco longo de reposição no batoque primário da Seringa B, girando-o no local (Figura 7).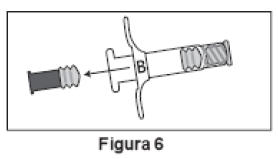
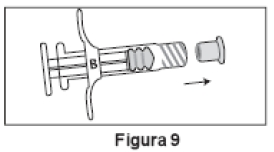
3. Desrosquear a tampa transparente da Seringa A (Figura 8). Remover a tampa cinza de borracha da Seringa B (Figura 9).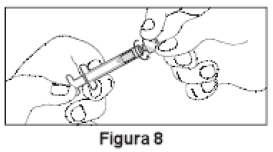
4. Conectar as duas seringas, girando-as até que estejam firmemente conectadas (Figura 10).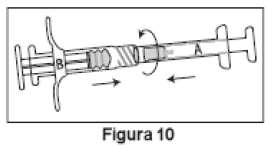
5. Misturar totalmente o produto, empurrando o conteúdo das seringas para frente e para trás entre as seringas (durante aproximadamente 45 segundos) para obter uma suspensão uniforme (Figura 11). Depois de misturada de modo uniforme, a suspensão apresentará uma coloração de amarela clara a amarela.
Observação: O produto deverá ser misturado conforme descrito; a agitação não fornecerá a mistura adequada do produto.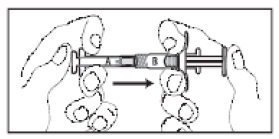
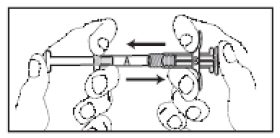
6. Segure as seringas em posição vertical, com a Seringa B para baixo. As seringas deverão permanecer acopladas firmemente. Retirar todo o conteúdo do produto misturado para a Seringa B (seringa curta e larga) pressionando o êmbolo da Seringa A e soltando levemente o êmbolo da Seringa B. Continuar pressionando para baixo o êmbolo da Seringa A no momento que a mesma for desconectada (Figura 12). Observação: é aceitável que pequenas bolhas de ar permaneçam na formulação.
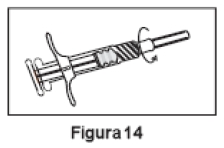
7. Manter a Seringa B em posição vertical. Remover a tampa rosa que protege o cartucho da agulha estéril, através de movimentos giratórios (Figura 13). Conectar a agulha à extremidade da Seringa B (Figura 14) empurrando e girando o cartucho da agulha até que esteja firmemente acoplado. Não encaixar a agulha na seringa antes de remover a tampa rosa. Retirar a tampa transparente da agulha antes da administração (Figura 15). Após a administração, descartar todos os componentes de modo seguro em um recipiente adequado para materiais biológicos.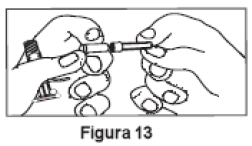
Posologia:
A dose recomendada de ELIGARD® 7,5 mg é mensal. O medicamento é administrado por via subcutânea,e fornece liberação contínua de leuprorrelina por 1 mês.
A dose recomendada de ELIGARD® 22,5 mg é de uma injeção a cada 3 meses (trimestral). O medicamento é administrado por via subcutânea, e fornece liberação contínua de leuprorrelina por 3 meses.
A administração da suspensão injetável de ELIGARD® 7,5 mg fornece 7,5 mg de acetato de leuprorrelina ao paciente. A suspensão injetável de ELIGARD® 22,5 mg fornece 22,5 mg de acetato de leuprorrelina.
Uso em idosos:
Até o dado momento, não há necessidade de ajuste de dosagem em pacientes idosos.
Reações adversas.
A segurança de ELIGARD® foi avaliada em oito homens castrados cirurgicamente (ELIGARD® 7,5 mg) e em pacientes que apresentavam câncer de próstata avançado (120 pacientes para ELIGARD® 7,5 mg e 117 pacientes para ELIGARD® 22,5 mg). ELIGARD®, assim como outros análogos LH-RH, ocasionou um aumento temporário nas concentrações séricas de testosterona durante a primeira semana de tratamento. Portanto, a exacerbação dos sinais e sintomas da doença de base durante as primeiras semanas de tratamento é motivo de preocupação em pacientes que apresentam metástases vertebrais e/ou obstrução do trato urinário ou hematúria. Caso haja piora dessas condições, poderá ocorrer fraqueza e/ou parestesia dos membros inferiores ou piora dos sintomas urinários.
No Estudo AGL9904, 120 pacientes foram tratados com ELIGARD® 7,5 mg por até seis meses e os locais de injeção foram monitorizados com cautela. No total, foram administradas 716 injeções de ELIGARD® 7,5 mg. Foi relatada queimação e dor transitória na "picada" após 248 (34,6%) injeções. A maioria (84%) desses eventos foram leves. Foi relatada dor após 4,3% das injeções em estudo (18,3% dos pacientes), geralmente leve e de curta duração. Foi relatado eritema após 2,6% das injeções (12,5% dos pacientes). Todos esses eventos foram leves, e geralmente apresentaram resolução no período de alguns dias após a injeção. Foi relatado hematoma leve após 2,5% das injeções (11,7% dos pacientes). Prurido, endurecimento e ulceração foram relatados após 1,4% (11 pacientes), 0,4% (3 pacientes), e 0,1% (1 paciente) das injeções em estudo, respectivamente. Esses eventos adversos locais não se repetiram com o decorrer do tempo. Nenhum paciente descontinuou a terapia devido a um evento adverso no local da injeção.
Os seguintes eventos adversos sistêmicos possivelmente ou provavelmente relacionados ao medicamento ocorreram durante os estudos clínicos de até seis meses de tratamento com ELIGARD® 7,5 mg, e foram relatados em ≥ 2% dos pacientes (Tabelas 1 e 2). Frequentemente, a causalidade é de difícil avaliação em pacientes que apresentam câncer de próstata metastático. As reações não consideradas relacionadas ao medicamento foram excluídas.

Além disso, os seguintes eventos adversos sistêmicos possivelmente ou provavelmente relacionados ao medicamento foram relatados por < 2% dos pacientes tratados com ELIGARD® 7,5 mg nos estudos clínicos:
Gerais: sudorese, insônia, síncope.
Gastrointestinal: flatulência, obstipação.
Hematológico: redução da contagem de eritrócitos, hematócrito e hemoglobina.
Metabólicos: ganho de peso.
Músculo-esqueléticos: tremores, dor nas costas, dor articular.
Neurológicos: distúrbios de olfato e paladar, depressão, vertigem.
Pele: alopécia.
Urogenitais: sensibilidade testicular, impotência*, redução da libido*, ginecomastia, sensibilidade mamária.
* Consequências farmacológicas esperadas da supressão da testosterona. Nas populações de pacientes estudados, um total de 86 eventos adversos de fogachos/sudorese foi relatado em 70 pacientes. Desses, 71 eventos (83%) foram leves; 14 (16%) foram moderados; 1 (1%) foi grave.
No Estudo AGL9909, 117 pacientes foram tratados com ELIGARD® 22,5 mg a cada 3 meses por até seis meses e os locais de injeção foram monitorados com cautela. De modo geral, foram administradas 230 injeções de ELIGARD® 22,5 mg. Foi relatada queimação e dor transitória na "picada" após 50 injeções (21,7%). A maioria (86%) desses eventos foi leve. Foi relatada dor após 3,5% das injeções em estudo (6,0% dos pacientes), geralmente leve e de curta duração. Eritema foi relatado após 2 injeções (0,9% das injeções em estudo, 1,7% dos pacientes). Em um dos relatos o eritema foi caracterizado como leve, com resolução num período de 7 dias. O outro relato foi moderado e apresentou resolução em 15 dias. Nenhum destes pacientes apresentou eritema após várias injeções. Hematoma leve foi relatado após 4 injeções (1,7% das injeções em estudo, 3,4% dos pacientes). Prurido leve foi relatado após 1 injeção (0,4% das injeções em estudo, 0,9% dos pacientes).
Esses eventos adversos localizados não se repetiram com o decorrer do tempo. Nenhum paciente descontinuou a terapia devido a um evento adverso no local da injeção.
Os seguintes eventos adversos sistêmicos (com incidência superior ou igual a 2% dos casos estudados), possivelmente ou provavelmente relacionados ao medicamento ocorreram durante os estudos clínicos de até seis meses de tratamento com ELIGARD® 22,5 mg (Tabela 3). Frequentemente, a causalidade é de difícil avaliação em pacientes que apresentam câncer de próstata metastático e as reações não relacionadas ao ELIGARD® 22,5 mg foram excluídas.
Além disso, os seguintes eventos adversos sistêmicos possivelmente ou provavelmente relacionados foram relatados por < 2% dos pacientes tratados com ELIGARD® 22,5 mg nos estudos clínicos.
Gastrointestinal: dispepsia.
Gerais: tremores, fraqueza, letargia.
Urinária: dificuldades para urinar, dor ao urinar, micção curta, espasmos vesicais, hematúria e retenção urinária.
Gonadais: sensibilidade mamária*,atrofia testicular*,dor testicular, ginecomastia*, impotência*.
Cutâneos: sudorese noturna*, aumento da sudorese*.
Vasculares: hipertensão, hipotensão.
* Consequências farmacológicas esperadas da supressão da testosterona. Nas populações de pacientes estudadas, um total de 84 eventos adversos de calores/sudorese foi relatado em 66 pacientes. Desses, 73 eventos (87%) foram leves; 11 (13%) foram moderados; nenhum como grave.
Alterações na Densidade Óssea: Foi relatada redução da densidade óssea na literatura médica em homens submetidos à orquiectomia ou tratados com análogo agonista do LH-RH. É previsto que longos períodos de castração medicamentosa em homens causarão efeitos sobre a densidade óssea.
Atenção: este produto é um medicamento que possui nova forma farmacêutica no país e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos pelo Sistema de Notificações em Vigilância Sanitária - NOTIVISA, disponível em www.anvisa.gov.br/hotsite/notivisa/index.htm ou para a Vigilância Sanitária Estadual ou Municipal.
Superdose.
Em estudos clínicos que utilizaram acetato de leuprorrelina subcutâneo diário em pacientes que apresentavam câncer de próstata, doses de até 20 mg/dia durante até dois anos não causaram efeitos adversos diferentes dos observados com a dose de 1 mg/dia.
Caso ocorra superdose recomendam-se medidas gerais de monitorização frequente dos sinais vitais e observação estrita do paciente.
Em caso de intoxicação ligue para 0800 722 6001, se você precisar de mais orientações sobre como proceder.
Dizeres legais.
VENDA SOB PRESCRIÇÃO MÉDICA
MS - 1.2214.0074
Fonte: Bulário Eletrônico da Anvisa, 13/08/12.