CELSENTRI ®
GLAXOSMITHKLINE
maraviroque
Anti-retroviral.
Apresentações.
Celsentri 150 mg ou 300 mg em embalagens contendo 30, 60, 90 ou 180 comprimidos revestidos.
USO ADULTO USO ORAL
Composição.
Cada comprimido revestido de Celsentri® 150 mg ou 300 mg contém o equivalente a 150 mg ou 300 mg de maraviroque, respectivamente. Excipientes: celulose microcristalina, fosfato de cálcio dibásico anidro, amidoglicolato de sódio, estearato de magnésio e Opadry® II azul (álcool polivinílico, dióxido de titânio, macrogol, talco, lecitina de soja, corante FD&C n° 2).
Indicações.
Celsentri® (maraviroque), em combinação com outros medicamentos antirretrovirais, é indicado para pacientes adultos, previamente experimentados a tratamento, e infectados somente com o vírus HIV-1 CCR5-trópico detectado (ver Posologia e Modo de Usar). Esta indicação é baseada nos dados de segurança e eficácia de dois estudos duplo-cegos, controlados com placebo, com 48 semanas de duração, em pacientes experimentados a tratamento (ver Características Farmacológicas).
Tropismo
Na maioria dos casos, a falha no tratamento com maraviroque foi associada à detecção de vírus CXCR4-trópico (por ex., CXCR4 ou tropismo duplo/misto) que não foi detectado pelo teste de tropismo antes do tratamento. O vírus CXCR4-trópico foi detectado em aproximadamente 55% dos indivíduos que não obtiveram sucesso no tratamento com maraviroque na semana 48 quando comparado aos 9% dos indivíduos experientes a tratamento com falha no braço placebo. Para se investigar a provável origem deste vírus CXCR4-trópico no tratamento, uma análise clonal detalhada foi realizada em vírus de 20 indivíduos representativos (16 indivíduos no braço com maraviroque e 4 indivíduos do braço placebo) em cujos vírus CXCR4 foi detectado na falha do tratamento. Da análise das diferenças da sequência do aminoácido e dos dados de filogenética foi determinado que o vírus CXCR4-trópico nos pacientes surgiu de um reservatório não detectado no início do tratamento pelo teste de tropismo, e não de uma mutação do vírus CCR5-trópico para CXCR4-trópico previamente existente. A detecção do vírus CXCR4-trópico antes do início da terapia foi associada à redução de resposta virológica ao maraviroque. Além disso, os pacientes que falharam no tratamento com maraviroque 2 vezes ao dia com vírus CXCR4-trópico tiveram um aumento médio menor na contagem de células CD4+ do basal (+41 células/mm3) que aqueles pacientes que falharam com vírus CCR5-trópico (+162 células/mm3). O aumento médio na contagem das células CD4+ em pacientes que falharam no tratamento no braço placebo foi +7 células/mm3.
Resultados de eficácia.
Resultados Clínicos
Estudos em Pacientes CCR5-trópicos Experimentados ao Tratamento
A eficácia clínica de Celsentri® (maraviroque) (em combinação com outros medicamentos antirretrovirais) nos níveis de HIV RNA e contagem de células CD4+ plasmáticas foram investigadas em dois estudos multicêntricos, pivotais, randomizados, duplo-cegos (MOTIVATE-1 e MOTIVATE-2, n=1049) em pacientes infectados com HIV-1 CCR5-trópico (como avaliado pelo teste Trofile). O endpoint primário de eficácia foi a semana 48.
Os pacientes que foram elegíveis para estes estudos tiveram exposição prévia a pelo menos 3 classes de medicamentos antirretrovirais [≥ 1 inibidor da transcriptase reversa análogo de nucleosídeo (ITRN), ≥ 1 inibidor da transcriptase reversa não-análogo de nucleosídeo (ITRNN), ≥ 2 inibidores de protease (IPs) e/ou enfuvirtida] ou resistência documentada a pelo menos um membro de cada classe. Os pacientes foram randomizados na razão de 2:2:1 para maraviroque 300 mg (dose equivalente) a cada 24 horas, a cada 12 horas ou placebo, em combinação com uma Terapia de Base Otimizada (TBO) consistindo de 3 a 6 medicamentos antirretrovirais (excluindo baixa dose de ritonavir). A TBO foi selecionada com base no histórico prévio de tratamento do paciente e mensurações basais do genótipo e do fenótipo de resistência viral.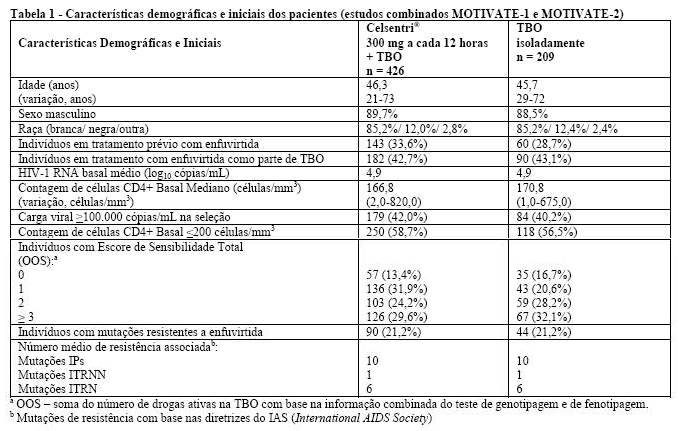
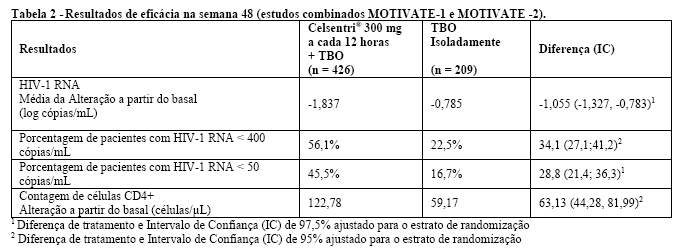
Celsentri® 300 mg a cada 12 horas + TBO foi superior ao TBO isoladamente em todos os subgrupos dos pacientes analisados (ver Tabela 3).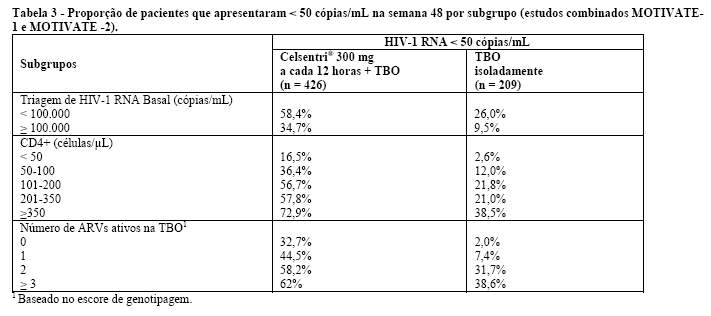
A triagem do tropismo para inscrição dos indivíduos no estudo MOTIVATE foi realizado utilizando um teste de tropismo fenotípico (Trofile). Um ensaio de tropismo fenotípico mais sensível (Trofile ES-) substituiu este, e uma reanálise retrospectiva de eficácia utilizando este ensaio foi realizada em pacientes com vírus R5 trópico. Os resultados desta análise retrospectiva são apresentados na Tabela 4.
Estudos com Pacientes com HIV-1 não CCR5-trópicos Experimentados a Tratamento
O estudo A4001029 foi um estudo multicêntrico, duplo-cego, randomizado e exploratório para determinar a segurança e eficácia de maraviroque em indivíduos infectados com vírus HIV-1 de tropismo duplo/misto ou CXCR4 trópico. Os critérios de inclusão/exclusão foram similares aos utilizados nos estudos MOTIVATE-1 e MOTIVATE-2 mencionados acima e os indivíduos foram randomizados a uma razão de 1:1:1 para maraviroque uma vez ao dia, duas vezes ao dia, ou placebo. Não se observou aumento no risco de infecção ou progressão da doença pelo HIV nos indivíduos que receberam maraviroque. O uso de maraviroque não foi associado à diminuição significativa de HIV-1 RNA quando comparado ao placebo nestes indivíduos e não se notou efeitos adversos na contagem de CD4.
Caract. farmacológicas.
Propriedades Farmacodinâmicas
Mecanismo de Ação
Celsentri® é uma molécula pequena, antagonista reversível da interação entre o CCR5 humano e a gp120 do HIV-1. O bloqueio desta interação previne a entrada do vírus HIV-1 CCR5-trópico nas células. O maraviroque é membro da classe terapêutica conhecida como antagonistas do correceptor CCR5. Portanto, o maraviroque liga-se seletivamente ao receptor de quimiocina humana CCR5, prevenindo a entrada do vírus HIV-1 CCR5-trópico nas células. A entrada do HIV-1 CXCR4-trópico e duplo trópico não é inibida pelo maraviroque.
Atividade antiviral em cultura celular
O ajuste do valor da EC90 sérica em 43 isolados clínicos primários de HIV-1 CCR5-trópico foi de 0,57 (0,06 - 10,7) ng/mL (fração nãoligada) sem alterações significativas entre os diferentes subtipos testados. Em cultura de células, maraviroque não apresenta atividade contra os vírus que podem utilizar o CXCR4 como correceptor de entrada (os vírus duo-trópicos e/ou CXCR4-trópicos). A atividade do maraviroque contra HIV-2 não foi avaliada. Em cultura de células, a combinação do maraviroque com ITRNs, ITRNNs, IPs ou inibidor da fusão do HIV (enfuvirtida) não se mostrou antagônica.
Resistência
Durante o uso do maraviroque, o escape viral pode ocorrer por 2 vias: seleção de vírus que utilizam o CXCR4 como correceptor de entrada (vírus CXCR4-trópicos) ou seleção de vírus que continuam a usar exclusivamente o CCR5 (vírus CCR5-trópicos).
Resistência em cultura de células
As variantes de HIV-1 com suscetibilidade reduzida ao maraviroque foram selecionadas em cultura de células, após a passagem em série de dois vírus CCR5-trópicos isolados clinicamente. Os vírus resistentes ao maraviroque permaneceram como CCR5-trópico e não houve conversão de vírus CCR5-trópico a vírus que usam o CXCR4.
Resistência fenotípica
A curva concentração resposta dos vírus resistentes ao maraviroque foi caracterizada pelas curvas que não atingiram 100% de inibição em ensaios utilizando diluições em série de maraviroque. A tradicional mudança no fold-change da EC50 não foi um parâmetro útil para medir a resistência fenotípica ao maraviroque, considerando que este valor permaneceu inalterado apesar da redução significativa da suscetibilidade.
Resistência genotípica
Foi observado que mutações se acumularam na glicoproteína do envelope (gp120 -proteína viral que se liga ao correceptor CCR5). Entretanto, as posições destas mutações não foram consistentes entre os diferentes isolados. Consequentemente, não se sabe a relevância destas mutações na suscetibilidade ao maraviroque em outros vírus.
Resistência cruzada
Os vírus HIV-1 isolados clínicos resistentes aos inibidores da transcriptase reversa análogos de nucleosídeos (ITRNs), inibidores da transcriptase reversa não-análogos de nucleosídeos (ITRNNs), inibidores de protease (IPs) e enfuvirtida, foram todos suscetíveis ao maraviroque em cultura de células. Os vírus resistentes ao maraviroque que surgiram em cultura de células permaneceram sensíveis ao inibidor de fusão enfuvirtida e ao inibidor de protease saquinavir.
In vivo
Ambas as rotas para resistência foram observadas em estudos clínicos de pacientes virgens e previamente experimentados a tratamento. A presença do vírus que utiliza CXCR4 na falha virológica parece originar de uma população viral pré-existente. Teste pré-tratamento para a presença desta forma de vírus pode reduzir a incidência de falha através deste mecanismo.
O maraviroque ainda pode ser considerado ativo se o valor da porcentagem de inibição máxima (PIM) for ≥95% (Phenosense entry assay), somente em pacientes com o vírus R5 que falharam ao tratamento. A atividade residual in vivo para vírus com valor de PIM < 95% não foi determinada. A resistência do vírus R5 através do aumento de EC50 não parece ser um mecanismo importante de falha.
Resistência genotípica: mutação chave (V3-loop) atualmente pode não ser sugerida devido à alta variabilidade de sequência V3 e ao pequeno número de amostras analisadas.
Pacientes previamente experimentados a tratamento: nos estudos pivotais (MOTIVATE 1 e MOTIVATE 2), 7,6% dos pacientes apresentaram mudança do resultado do tropismo de CCR5-trópico para CXCR4-trópico ou tropismo duplo/misto entre a seleção e o início do tratamento (período de 4-6 semanas).
Falha terapêutica com aparecimento de vírus CXCR4
Em vigência de falha terapêutica, vírus que utilizam o CXCR4 foram detectados em aproximadamente 55% dos indivíduos que não obtiveram sucesso no tratamento com maraviroque e em 6% dos indivíduos que não obtiveram sucesso no tratamento com TBO (Terapia de Base Otimizada) isoladamente (placebo).
Para se investigar a provável origem deste vírus que utiliza o CXCR4 que surgiu durante o tratamento, uma análise clonal detalhada foi realizada em vírus de 20 indivíduos, representativos daqueles onde foram detectados vírus CXCR4-trópicos (16 indivíduos no braço do maraviroque e 4 indivíduos no braço da TBO isoladamente). Esta análise indicou que o vírus que utiliza o CXCR4 surgiu a partir de reservatório não detectado no início do tratamento, e não a partir de mutação do vírus CCR5-trópico presente no início.
Análise do tropismo, após falha da terapia com maraviroque com vírus CXCR4 em pacientes com vírus CCR5 no início, demonstrou que a população de vírus reverte-se a CCR5-trópico em 33 de 36 pacientes com mais de 35 dias de acompanhamento. De acordo com dados disponíveis, no momento da falha terapêutica com vírus que utiliza o CXCR4, o padrão de resistência aos outros antirretrovirais foi semelhante ao previamente observado na população de vírus CCR5-trópico.
Com isso, para a seleção do esquema de tratamento, pode-se assumir que a população de vírus não-detectável que utiliza o CXCR4 (isto é, população viral minoritária) apresenta o mesmo padrão de resistência que a população CCR5-trópica detectada.
Falha Terapêutica com permanência de vírus CCR5-trópico
Resistência fenotípica: em pacientes com vírus CCR5-trópico, no momento da falha terapêutica com maraviroque, 22 de 58 pacientes tinham vírus com sensibilidade reduzida ao mesmo. Adicionalmente, o vírus CCR5-trópico de 2 indivíduos que falharam ao tratamento tinham ≥3 vezes os valores de EC50 para maraviroque na falha, mas o significado disso não é claro. Nos outros pacientes não houve evidência de vírus com sensibilidade reduzida ao maraviroque conforme identificação por análise virológica exploratória no grupo representante. Este último grupo apresentou evidências de baixa exposição, em alguns casos associados com baixa adesão.
Propriedades Farmacocinéticas
Absorção
A absorção do maraviroque é variável com múltiplos picos. O pico médio da concentração plasmática de maraviroque é atingido em 2 horas (intervalo de 0,5 a 4 horas) após doses orais únicas de 300 mg do comprimido comercial administrado em voluntários sadios. A farmacocinética do maraviroque oral não é proporcional à variação da dose de 1-1200 mg. A biodisponibilidade absoluta de uma dose de 100 mg é 23% e presume-se que seja 33% com uma dose de 300 mg. O maraviroque é um substrato da glicoproteína P (bomba de efluxo). A coadministração de um comprimido de 300 mg com café da manhã rico em gordura reduziu a Cmáx e a AUC do maraviroque em 33% em voluntários sadios. Não houve restrições com relação à alimentação nos estudos que demonstraram a eficácia e a segurança do maraviroque (ver Propriedades Farmacodinâmicas). Portanto, Celsentri® pode ser administrado com ou sem alimentos nas doses recomendadas (ver Posologia e Modo de Usar).
Distribuição
O maraviroque se liga às proteínas plasmáticas humanas (aproximadamente 76%), e demonstra afinidade moderada pela albumina e pela alfa-1 glicoproteína ácida. O volume de distribuição do maraviroque é aproximadamente 194 L.
Metabolismo
Estudos em humanos e estudos in vitro utilizando enzimas microssomais hepáticas e enzimas de expressão humanas demonstraram que maraviroque é metabolizado principalmente pelo sistema do citocromo P450 a metabólitos que são essencialmente inativos contra o HIV-1. Estudos in vitro indicam que CYP3A é a principal enzima responsável pelo metabolismo do maraviroque. Estudos in vitro também indicam que as enzimas polimórficas CYP2C9, CYP2D6 e CYP2C19 não contribuem significativamente para o metabolismo do maraviroque. O maraviroque é o principal componente circulante (aproximadamente 42% de radioatividade) após dose oral única de 300 mg. O metabólito circulante mais significativo em humanos é uma amina secundária (aproximadamente 22% de radioatividade) formado pela N-desalquilação. Este metabólito polar não apresenta atividade farmacológica significativa. Outros metabólitos são produtos de mono-oxidações e são componentes secundários da radioatividade plasmática.
Eliminação
Um estudo de balanço de massa/excreção foi conduzido utilizando dose única de 300 mg de maraviroque marcado com 14C.
Aproximadamente 20% do fármaco radiomarcado foi recuperado na urina e 76% foi recuperado nas fezes por 168 horas. O maraviroque foi o principal componente presente na urina (média de 8% da dose) e nas fezes (média de 25 % da dose). O restante foi excretado na forma de metabólitos. Após administração intravenosa (30 mg) a meia-vida de maraviroque foi 13,2 h, 22% da dose foi excretada inalterada na urina e os valores do clearance total e clearance renal foram 44,0 L/h e 10,2 L/h respectivamente.
Crianças
A farmacocinética do maraviroque em pacientes pediátricos não foi estabelecida (ver Posologia e Modo de Usar).
Idosos
Foi conduzida uma análise de população nos estudos fase I/IIa e III (16-65 anos de idade) e nenhum efeito da idade foi observado. A farmacocinética de maraviroque em pacientes com mais de 65 anos de idade não foi estabelecida (ver Posologia e Modo de Usar).
Insuficiência Renal
Um estudo comparou a farmacocinética de maraviroque 300 mg dose única em pacientes com disfunção renal grave (clearance de creatinina < 30 mL/min, n=6) e com doença renal em estágio final (ESRD) contra voluntários saudáveis (n=6). As médias geométricas para AUCinf (CV%) de maraviroque foi a seguinte: voluntários saudáveis (função renal normal) 1348,4 ng. h/mL (61%); função renal grave 4367,7 ng.h/mL (52%); ESRD (dose pós-diálise) 2677,4 ng.h/mL (40%) e ESRD (dose pré-diálise) 2805,5 ng.h/mL (45%). A Cmáx (CV%) foi 335,6 ng/mL (87%) em voluntários saudáveis (função renal normal); 801,2 ng/mL (56%) em função renal grave; 576,7 ng/mL (51%) em ESRD (dose após diálise) e 478,5 ng/mL (38%) em ESRD (dose antes da diálise). A diálise teve efeito mínimo na exposição em pacientes com ESRD. Exposições observadas em pacientes com disfunção renal grave e ESRD estavam dentro da faixa observada nos estudos com maraviroque 300 mg dose única em voluntários saudáveis com função renal normal. Portanto, nenhum ajuste de dose é necessário em pacientes com insuficiência renal recebendo maraviroque sem um potente inibidor de CYP3A (ver Posologia e Modo de Usar, Advertências e Precauções e Interações Medicamentosas).
Adicionalmente, o estudo comparou a farmacocinética de doses múltiplas de maraviroque em combinação com saquinavir/ritonavir 1000/100 mg a cada 12 horas (uma combinação de inibidor potente de CYP3A) por 7 dias em pacientes com insuficiência renal leve (CLcr > 50 e ≤ 80 mL/min, n=6) e insuficiência renal moderada (clearance de creatinina ≥ 30 e ≤ 50 mL/min, n=6) contra voluntários sadios (n=6). Pacientes recebendo maraviroque 150 mg em diferentes frequências de dose (voluntários sadios - cada 12 horas; insuficiência renal leve - cada 24 horas; insuficiência renal moderada - cada 48 horas). A concentração média Cméd do maraviroque por mais de 24 horas foi de 445,1 ng/mL, 338,3 ng/mL e 223,7 ng/mL para pacientes com função renal normal, disfunção renal leve e disfunção renal moderada, respectivamente. O Cméd de maraviroque de 24-48 horas para pacientes com insuficiência renal moderada foi baixa (Cméd 32,8 ng/mL). Portanto, em pacientes com insuficiência renal moderada (e pela extrapolação em insuficiência renal grave) frequências de dose superiores a 24 horas podem resultar em exposição inadequada entre 24-48 horas. Em pacientes com insuficiência renal recebendo maraviroque com potente inibidor de CYP3A, é recomendada uma dose de 150 mg a cada 24 horas (ver Posologia e Modo de Usar, Advertências e Precauções e Interações Medicamentosas).
Insuficiência Hepática
O maraviroque é metabolizado e eliminado primariamente pelo fígado. Um estudo comparou a farmacocinética de uma dose única de 300 mg de maraviroque em pacientes com disfunção hepática leve (classe A de Child-Pugh, n=8), e moderada (classe B de Child-Pugh, n=8) comparado a indivíduos sadios (n=8). As taxas médias geométricas para a Cmáx e a AUClast foram respectivamente 11% e 25% superior para indivíduos com disfunção hepática leve, e respectivamente 32% e 46% superior para indivíduos com disfunção hepática moderada comparado a indivíduos com função hepática normal. Os efeitos da insuficiência hepática moderada podem estar subestimados devido aos dados limitados em pacientes com capacidade metabólica reduzida e clearance renal maior nesses indivíduos. Os resultados devem, portanto, ser interpretados com cuidado. A farmacocinética do maraviroque não foi estudada em indivíduos com disfunção hepática grave (ver Posologia e Modo de Usar e Advertências e Precauções).
Outras características dos pacientes
Raça
A análise da farmacocinética populacional dos dados combinados dos estudos fase I/IIa indica que a exposição é 26,5% maior em asiáticos (n=95) quando comparado a não-asiáticos (n=318). No entanto, estudo realizado para avaliar as diferenças entre a farmacocinética entre caucasianos (n=12) e asiáticos (n=12) não mostrou diferença entre estas duas populações. Apenas 14 indivíduos negros foram incluídos na análise farmacocinética populacional. Não é necessário ajuste da dose com relação à raça.
Sexo
A análise da farmacocinética populacional dos dados combinados dos estudos fase I/IIa indica que o sexo (mulher n=96, 23,2% da população total) não afeta as concentrações de maraviroque. Não é necessário ajuste da dose com relação ao sexo.
Dados de Segurança Pré-Clínicos
Com base em estudos convencionais de segurança farmacológica, toxicidade de dose repetida, genotoxicidade, potencial carcinogênico e toxicidade na reprodução, dados não-clínicos não revelaram risco especial para humanos.
Carcinogenicidade/mutagenicidade: o maraviroque foi avaliado quanto ao potencial carcinogênico em estudo de 6 meses em camundongos transgênicos e estudo de 24 meses em ratos. Em camundongos, maraviroque não causou aumento estatisticamente significativo na incidência de qualquer tipo de tumor em exposições sistêmicas que variaram de 7 a 39 vezes em relação à exposição humana (com base na mensuração da AUC0-24h do fármaco livre) da dose máxima recomendada de 300 mg a cada 12 horas. Em ratos, a administração de maraviroque resultou em adenomas da tireoide, associados com alterações adaptativas do fígado durante exposição sistêmica 21 vezes maior que a exposição humana de 300 mg a cada 12 horas. Não há indícios de potencial carcinogênico em humanos.
Toxicologia reprodutiva: o maraviroque não foi mutagênico ou genotóxico em vários ensaios in vitro e in vivo, incluindo mutação bacteriana reversa, aberrações cromossômicas em linfócitos humanos e micronúcleos na medula óssea de ratos.
O maraviroque não prejudicou o acasalamento ou a fertilidade de ratos machos e fêmeas e não afetou o esperma de ratos machos tratados com até 1000 mg/kg. Exposição a este nível de dose corresponde a 39 vezes a AUC estimada para uma dose de 300 mg a cada 12 horas.
Estudos de desenvolvimento em embriões e fetos foram conduzidos em ratos e coelhos com doses de até 39 e 34 vezes a AUC para a dose de 300 mg a cada 12 horas. Estudos em animais não revelaram evidência de risco ao feto com maraviroque. Estudos de desenvolvimento pré e pós-natal foram realizados em ratos com doses de até 27 vezes a AUC estimada para a dose de 300 mg a cada 12 horas. O único efeito na prole foi um leve aumento na atividade motora em ratos machos recebendo altas doses, tanto desmamados quanto adultos, enquanto não foi observado efeito nas fêmeas. Outros parâmetros de desenvolvimento destas proles, incluindo fertilidade e desempenho reprodutivo, não foram afetados pela administração materna de maraviroque.
Contraindicações.
Celsentri® é contraindicado a pacientes com hipersensibilidade ao maraviroque ou a qualquer componente da fórmula (ver Composição).
Advertências e precauções.
Segurança Hepática
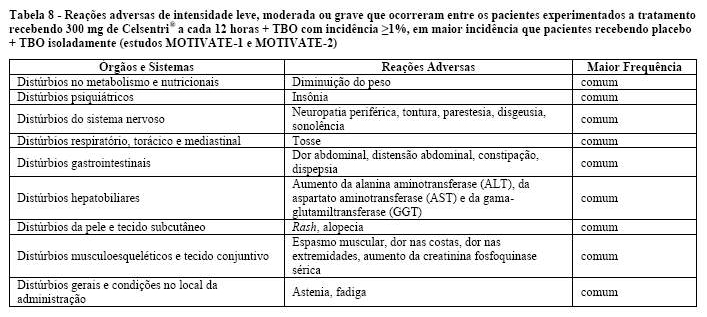
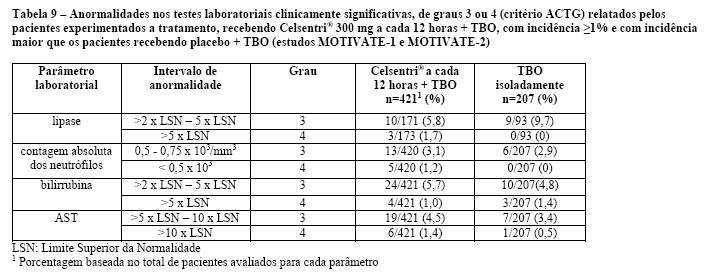
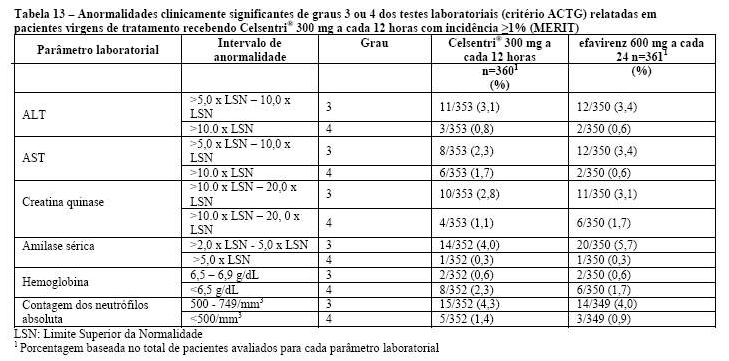
Um aumento dos eventos adversos hepáticos com Celsentri® foi observado durante os estudos em pacientes com infecção por HIV experimentados a tratamento, embora não tenha ocorrido aumento das enzimas hepáticas definidas como anormalidades Grau 3/4 de ACTG (ver Reações Adversas). Foram reportados poucos casos de alterações hepatobiliares em pacientes virgens de tratamento fazendo uso de Celsentri®, quando comparados a efavirenz. No entanto, a incidência geral de eventos adversos hepáticos e anormalidades Grau 3/4 de ACTG nos testes de função hepática foram similares entre Celsentri® e efavirenz.
Foram relatados casos de hepatotoxicidade e disfunção hepática com quadro alérgico associados à Celsentri® .
A descontinuação de Celsentri® deve ser fortemente considerada em pacientes com sinais ou sintomas de hepatite aguda, em particular se houver suspeita de estar relacionada com hipersensibilidade a medicamentos ou com aumento nas transaminases hepáticas combinado com rash ou outros sintomas sistêmicos de potencial hipersensibilidade (p. ex. rash com prurido, eosinofilia ou IgE elevado).
Uma vez que existem poucos dados em pacientes coinfectados com hepatite B/C, deve-se ter cautela ao prescrever e monitorar o tratamento com Celsentri®. Nos casos de terapia antiviral concomitante para hepatite B e/ou C, verifique também a bula destes medicamentos.
Pacientes com disfunção hepática pré-existente, inclusive hepatite crônica ativa, podem apresentar aumento na frequência de anormalidades da função hepática durante o tratamento antirretroviral combinado e devem ser monitorados de acordo com a prática padrão.
A segurança e eficácia de Celsentri® não foram especificamente estudadas em pacientes com distúrbios hepáticos de base significativos. Portanto, uma vez que há experiência limitada em pacientes com função hepática reduzida, Celsentri® deve ser utilizado com cautela nesta população (ver Posologia e Modo de Usar e Farmacocinética, em Características Farmacológicas).
Reações cutâneas e hipersensibilidade graves
Foram relatadas reações de hipersensibilidade, incluindo eventos graves e potencialmente fatais em pacientes fazendo uso de Celsentri® e, na maioria dos casos, de Celsentri® combinado a outros fármacos associados a esses eventos.
Estas reações foram caracterizadas por sintomas como rash cutâneo, sintomas orgânicos e, em alguns casos, disfunção orgânica e falência hepática. Casos de Síndrome de Stevens-Johnson (SJS), necrólise epidérmica tóxica (TEN) e rash com eosinofilia e sintomas sistêmicos (DRESS) têm sido relatados (ver Reações Adversas).
Celsentri® ou outros fármacos suspeitos devem ser imediatamente descontinuados se os sinais ou sintomas cutâneos graves ou reações de hipersensibilidade se desenvolverem. Um atraso na interrupção do tratamento após o aparecimento de rash cutâneo pode resultar em reações fatais. O quadro clínico do paciente, incluindo o controle das aminotransferases hepáticas, deve ser monitorado e o tratamento adequado, iniciado.
Segurança Cardiovascular
Utilizar com cautela em pacientes com risco aumentado de eventos cardiovasculares. Dez pacientes (1,2%) que receberam Celsentri® apresentaram eventos cardiovasculares que podem estar relacionados a doenças cardíacas coronarianas incluindo isquemia miocárdica e/ou infarto durante os estudos de fase III em pacientes CCR5-trópicos experimentados a tratamento [seis pacientes (1,4%) no grupo de Celsentri® a cada 24 horas e quatro pacientes (0,9%) no grupo de Celsentri® a cada 12 horas)].
Nenhum paciente que recebeu placebo apresentou tais eventos. Estes pacientes geralmente apresentavam doença cardíaca ou fatores de risco cardíaco antes da administração de Celsentri®, e a contribuição relativa de Celsentri® para esses eventos não é conhecida. Na fase IIb/III do estudo em pacientes virgens em tratamento, três pacientes (0,8%) que receberam Celsentri® tiveram eventos adversos relacionados à doença isquêmica do coração e cinco pacientes (1,4%) que receberam efavirenz, tiveram tais eventos (exposição total de 506 e 508 pacientes por ano de maraviroque e efavirenz, respectivamente).
Hipotensão Postural
Em estudos com voluntários sadios, a administração de Celsentri® em doses maiores que as doses recomendadas, foi observada hipotensão postural sintomática em uma frequência maior do que com placebo. Deve-se ter cautela quando Celsentri® for administrado em pacientes com insuficiência renal grave, com histórico e/ou fatores de risco de hipotensão postural ou pacientes fazendo uso concomitantede medicamentos conhecidos por reduzir a pressão sanguínea.
Pacientes com insuficiência renal grave e tratados com inibidores potentes da CYP3A4 ou com booster de inibidores da protease (IP) podem apresentar um risco aumentado de hipotensão postural devido a um aumento da concentração de maraviroque (ver Posologia e Modo de Usar, Interações e Farmacocinética, em Características Farmacológicas).
Pacientes com comorbidades cardiovasculares podem apresentar um risco aumentado de eventos adversos cardiovasculares desencadeados pela hipotensão postural.
Insuficiência Renal
Um estudo avaliou a farmacocinética e a segurança de Celsentri® em pacientes com diferentes graus de disfunção renal e comparou com voluntários sadios. Neste estudo, reduções transitórias na média do clearance de creatinina (CLcr) foram observadas em pacientes com disfunção renal de leve a moderada bem como em voluntários sadios recebendo Celsentri® 150 mg (Frequência de dose dos pacientes: saudáveis - a cada 12 horas; disfunção renal leve-a cada 24 horas; disfunção moderada -a cada 48 horas) e saquinavir/ritonavir 1000/100 mg duas vezes ao dia (BID) que foi resolvida com a dose continuada. Não houve relação entre redução no CLcr médio e a média basal sérica de creatinina. No geral, Celsentri® foi bem tolerado nos estudos, com mais eventos adversos (a maioria leve) reportados em pacientes com disfunção renal leve e moderada, recebendo Celsentri® e saquinavir/ritonavir.
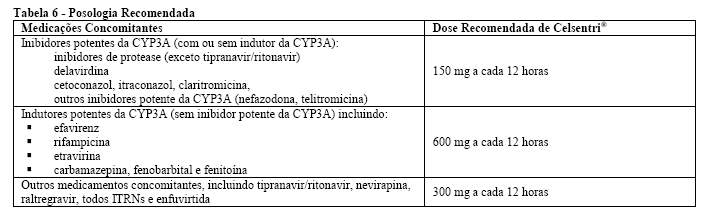
A tabela 6 mostra a diretriz para ajuste de dose e/ou intervalo para pacientes com insuficiência renal com ou sem coadministração de inibidores potentes de CYP3A4 (ver Posologia e Modo de Usar, Interações Medicamentosas e Farmacocinética, em Características Farmacológicas).
Síndrome da Reconstituição Imunológica
Em pacientes infectados com HIV com deficiência imune grave no momento do início da terapia antirretroviral altamente ativa (HAART), uma reação inflamatória a patógenos oportunistas latentes ou assintomáticos pode aparecer e causar condições clínicas graves ou agravamento dos sintomas previamente existentes. Tipicamente, tais reações foram observadas dentro das primeiras semanas ou meses após o início da HAART. Exemplos relevantes são retinite por citomegalovírus, infecções por micobactérias generalizadas e/ou localizadas e pneumonia por Pneumocystis jiroveci (conhecida antigamente como Pneumocystis carinii). Qualquer sintoma inflamatório deve ser avaliado e o tratamento deve ser iniciado quando necessário. Distúrbios autoimunes (tais como Doença de Graves, polimiosite e síndrome de Guillain-Barré) também foram relatados por ocorrerem na reconstituição imunológica. No entanto, o tempo de início é variável e pode ocorrer vários meses após o início do tratamento. Algumas vezes, podem ter uma apresentação atípica.
Tropismo
Celsentri® deve ser administrado como parte do regime antirretroviral combinado. Celsentri® deve ser combinado da forma mais otimizada possível com outros antirretrovirais aos quais o vírus seja sensível (ver Farmacodinâmica, em Características Farmacológicas). Celsentri® deve ser utilizado apenas quando o vírus HIV-1 CCR-5 trópico é detectado (isto é, quando vírus CXCR4-trópicos ou com tropismo duplo/misto não são detectados) conforme determinado por um método de detecção validado e sensível (ver Indicações, Posologia e Modo de Usar e Farmacodinâmica, em Características Farmacológicas). O tropismo viral não pode ser previsto pelo histórico de tratamento ou análise de amostras armazenadas. Somente uma amostra atual do paciente pode ser utilizada para avaliar o tropismo viral. Mudanças no tropismo viral ocorrem ao longo do tempo em pacientes infectados pelo HIV-1. Por esta razão, é necessário iniciar o tratamento logo após a obtenção do resultado do teste de tropismo.
Ajuste de Dose
Os médicos devem assegurar que sejam realizados ajustes apropriados na dose de Celsentri® quando administrado concomitantemente com inibidores e/ou indutores do CYP3A4, uma vez que as concentrações e seus efeitos terapêuticos podem ser afetados (ver Posologia e Modo de Usar e Interações Medicamentosas). Consulte também as bulas dos outros medicamentosutilizados em combinação a Celsentri® .
Informações para os Pacientes
Os pacientes devem ser informados que as terapias antirretrovirais, incluindo Celsentri®, não demonstraram prevenir o risco de transmissão do HIV para os outros através de contato sexual ou contaminação sanguínea. Eles devem continuar a usar as precauções apropriadas. Os pacientes também devem ser informados que Celsentri® não é uma cura para infecção por HIV-1.
Risco Potencial de Infecção
Celsentri® antagoniza o correceptor CCR5 localizado em algumas células do sistema imunológico, e por este motivo pode aumentar potencialmente o risco de desenvolver infecções. A incidência geral e gravidade da infecção, bem como das infecções categoria C definidoras de AIDS, foi comparável nos grupos de tratamento durante os estudos de fase III de Celsentri®. Embora houvesse uma taxa mais alta de certas infecções do trato respiratório superior relatado no braço Celsentri® comparada ao placebo (20,0% versus 11,5%), a taxa de pneumonia foi mais baixa (2,1 % vs 4,8%) em pacientes recebendo Celsentri®. Incidência mais alta de infecções por Herpes vírus (11.4 por 100 pacientes-ano) também foi relatada no braço do Celsentri® quando ajustada para exposição comparada ao placebo (8.2 por 100 pacientes-ano). Os pacientes devem ser monitorados cuidadosamente quanto ao aparecimento de infecções quando estiverem recebendo Celsentri® .
Risco Potencial de Malignidade
Por enquanto nenhum aumento na incidência de malignidades foi observado com Celsentri®. Dado o mecanismo de ação do medicamento, a vigilância imunológica pode ser afetada e provocar um aumento do risco da malignidade. Seguimento de longo prazo é necessário para que este risco seja melhor avaliado.
Uso durante a Gravidez e Lactação
Fertilidade:
Não dá dados disponíveis sobre os efeitos de Celsentri® na fertilidade humana. Em ratos, não foram observados eventos adversos na fertilidade feminina ou masculina (ver Dados de Segurança Pré-Clínicos, em Características Farmacológicas).
Gravidez:
Nenhum dado clínico significativo sobre a exposição durante a gravidez está disponível. Os estudos em animais não indicaram qualquer efeito prejudicial direto ou indireto com relação a gravidez, ao desenvolvimento embrionário/fetal, ao parto ou ao desenvolvimento pós-natal (ver Dados de Segurança Pré-Clínicos, em Características Farmacológicas). Celsentri® deve ser utilizado durante a gravidez somente se o benefício justificar o risco potencial ao feto.
Categoria D de risco na gravidez
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Lactação:
Estudos em ratas lactantes indicaram que maraviroque é extensivamente excretado no leite das ratas. Não se sabe se maraviroque é excretado no leite humano. As mães devem ser instruídas a não amamentar caso estejam recebendo Celsentri® devido ao potencial de transmissão do HIV e qualquer efeito indesejado possível durante a amamentação do lactente.
Efeitos na Habilidade de Dirigir ou Operar Máquinas
Não foram realizados estudos para avaliar os efeitos de Celsentri® na habilidade de realizar tarefas que requerem julgamento e habilidades motoras ou cognitivas. Entretanto, os pacientes devem ser informados sobre a possível ocorrência de sintomas relacionados a hipotensão postural como tontura. Se afetados, eles devem evitar tarefas potencialmente perigosas, tais como dirigir ou operar máquinas.
Uso em idosos, crianças e outros grupos de risco
Ver Posologia e Modo de Usar.
Interações medicamentosas.
O maraviroque é substrato do citocromo P450 CYP3A4. A administração concomitante de Celsentri® (maraviroque) com medicamentos que induzem o CYP3A4 pode reduzir as concentrações de maraviroque e reduzir seus efeitos terapêuticos. A administração concomitante de Celsentri® com medicamentos que inibem o CYP3A4 pode aumentar as concentrações plasmáticas de maraviroque. O ajuste de dose de Celsentri® é recomendado quando este é administrado com inibidores e/ou indutores do CYP3A4 concomitantemente. Os detalhes adicionais da administração concomitante com outros medicamentos são apresentados a seguir (ver Tabela 5, Advertências e Precauções e Tabela 6).
Estudos com sistemas enzimáticos recombinantes e microssomas hepáticos humanos demonstraram que maraviroque não inibe qualquer uma das enzimas P450 importantes em concentrações clinicamente relevantes (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 e CYP3A4). O maraviroque não apresenta efeito clinicamente relevante na farmacocinética do midazolam, dos contraceptivos orais etinilestradiol e levonogestrel, ou na taxa do 6b-hidroxicortisol/cortisol na urina, sugerindo que não há inibição ou indução do CYP3A4 in vivo. Apesar da falta de inibição in vitro de CYP2D6, Celsentri® causou um aumento na variação metabólica da debrisoquina com 600 mg a cada 24 horas, embora não com 300 mg a cada 12 horas. a. Portanto, em alta exposição ao maraviroque, inibição potencial do CYP2D6 não pode ser excluída. Com base nos dados in vitro e nos dados clínicos, o potencial de maraviroque em afetar a farmacocinética de medicamentos coadministrados é baixo.
O clearance renal é estimado em aproximadamente 23% do clearance total do maraviroque quando Celsentri® é administrado sem inibidores do CYP3A. Uma vez que os processos ativos e passivos estão envolvidos, existe o potencial de competição para eliminação com outras substâncias ativas eliminadas por via renal. No entanto, a coadministração de Celsentri® com tenofovir (substrato para eliminação renal) e co-trimoxazol (contém trimetoprima, um inibidor de transporte de cátion renal), não demonstrou efeito na farmacocinética do maraviroque. Além disso, a coadministração de Celsentri® com a lamivudina/zidovudina não demonstrou efeito de maraviroque na farmacocinética da lamivudina (excretada principalmente por via renal) ou da zidovudina (metabolismo não P450 e clearance renal). O maraviroque inibe a glicoproteína-P in vitro (o IC50 é 183 mM). No entanto, o maraviroque não afeta significativamente a farmacocinética da digoxina in vivo, sugerindo que este não inibe ou induz a atividade da glicoproteína -P.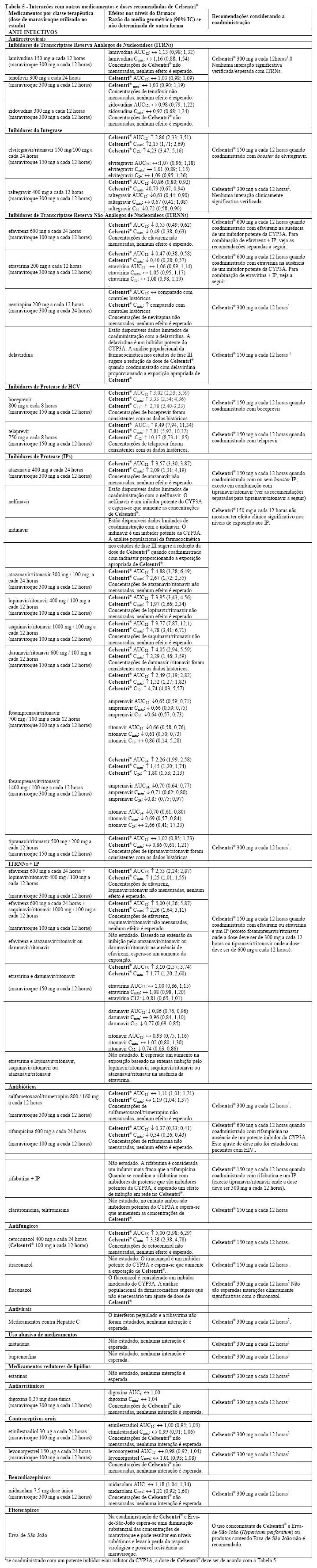
Cuidados de armazenamento.
Celsentri® deve ser conservado em temperatura ambiente (entre 15°C e 30°C), protegido da umidade e pode ser utilizado por 24 meses a partir da data de fabricação.
Número de lote e datas de fabricação e validade: vide embalagem. Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Características físicas e organolépticas
Comprimidos revestidos ovais, biconvexos e azuis.
Antes de usar, observe o aspecto do medicamento. Todo medicamento deve ser mantido fora do alcance das crianças.
Posologia e modo de usar.
O tratamento com Celsentri® deve ser iniciado por um médico com experiência na condução do tratamento da infecção