CAMPTOSAR®
PFIZER
irinotecano
Antineoplásico.
Apresentações.
Camptosar® solução injetável 20 mg/mL em embalagens contendo 1 frasco-ampola de plástico âmbar com 2 ou 5 mL de solução.
VIA DE ADMINISTRAÇÃO: INFUSÃO INTRAVENOSA
USO ADULTO
CUIDADO: AGENTE CITOTÓXICO
Composição.
Cada mL da solução injetável de Camptosar® contém 20 mg de cloridrato de irinotecano tri-hidratado equivalente a 17,33 mg de irinotecano. Excipientes: sorbitol, ácido láctico, solução de hidróxido de sódio, solução de ácido clorídrico e água para injetáveis.
Indicações.
Camptosar® (cloridrato de irinotecano tri-hidratado) solução injetável é indicado como agente único ou combinado no tratamento de pacientes com:
Carcinoma metastático do cólon ou reto não tratado previamente;
Carcinoma metastático do cólon ou reto cuja moléstia tenha recorrido ou progredido após terapia anterior com 5-fluoruracila;
Neoplasia pulmonar de células pequenas e não pequenas; Neoplasia de colo de útero;
Neoplasia de ovário;
Neoplasia gástrica recorrente ou inoperável.
Camptosar® está indicado para tratamento como agente único de pacientes com:
Neoplasia de mama inoperável ou recorrente;
Carcinoma de células escamosas da pele;
Linfoma maligno.
Resultados de eficácia.
Câncer colorretal: Foram realizados estudos clínicos com a administração de irinotecano em combinação com 5fluorouracila (5-FU) e leucovorin (LV) e como agente único. Quando utilizado como um componente do esquema combinado, o irinotecano foi utilizado com um esquema semanal de bolo de 5-FU/LV ou em um esquema a cada 2 semanas de infusão de 5-FU/LV. O esquema semanal e o esquema a cada 3 semanas foi utilizado com o irinotecano como agente único. Dois estudos fase III, randomizados, multinacionais, suportam o uso de Camptosar® como tratamento de 1a linha em pacientes com carcinoma metastático do cólon e reto. Em cada um dos estudos, a combinação de irinotecano e 5-FU/LV foi comparada a 5-FU/LV isolado. O estudo 1 comparou a combinação de irinotecano com 5-FU/LV em bolo em esquema semanal, com um regime padrão de 5-FU/LV em bolo, administrado por 5 dias a cada 4 semanas. O estudo 2 avaliou 2 diferentes esquemas de administração de 5-FU/LV infusional, com ou sem irinotecano. Em ambos os estudos, a combinação de irinotecano + 5-FU/LV resultou em significativa melhora das taxas de resposta objetivas, tempo para progressão do tumor e sobrevida, quando comparado ao braço que utilizou 5-FU/LV isoladamente. Foram incluídos 457 pacientes no estudo 1 e 385 no estudo 2. A taxa de resposta no grupo com Camptosar foi de 39 vs 21 no estudo 1 e 35 vs 22 no estudo 2. O tempo para progressão do tumor mediano no grupo com Camptosar foi de 7 meses vs 4,3 meses no estudo 1 e 6,7 meses vs 4,4 meses no estudo 2. A sobrevida global mediana no grupo com Camptosar foi de 14,8 meses vs 12,6 meses no estudo 1 e 17,4 meses vs 14,1 meses no estudo 2. Dados de 3 estudos abertos, com agente único, envolvendo 304 pacientes em 59 centros, suportam o uso de Camptosar® no tratamento de pacientes com câncer metastático de cólon e reto que recorreram ou progrediram após tratamento com 5-FU/LV. Esses estudos foram desenhados para avaliar a taxa de resposta tumoral. Em todos os estudos, Camptosar® foi administrado em ciclos de 6 semanas, consistindo de 1 infusão semanal durante 90 minutos (com doses de 100 mg/m2, 125 mg/m2 e 150 mg/m2 por infusão) por 4 semanas, seguidas de 2 semanas de descanso. Na análise ITT dos dados agrupados dos 3 estudos, 193 dos 304 pacientes iniciaram a terapia com a dose recomendada de 125 mg/m2. Entre esses pacientes, a taxa de resposta global foi de 15% (2 respostas completas e 27 respostas parciais). A maioria das respostas foi observada nos primeiros 2 ciclos de tratamento e a duração mediana da resposta foi de 5,8 meses. A resposta não variou com relação ao sexo, idade (menores e maiores de 65 anos), presença de metástases únicas ou múltiplas, localização do tumor primário (cólon vs. reto) e irradiação prévia. Dois estudos multicêntricos e randomizados suportam o uso de irinotecano no esquema a cada 3 semanas em pacientes com câncer colorretal metastático que recorreu ou progrediu após tratamento com 5-FU/LV. No primeiro estudo, o tratamento de 2a linha com irinotecano + Melhores Cuidados de Suporte (MCS) foi comparado com os MCS isoladamente. No segundo estudo, o tratamento de 2a linha com irinotecano foi comparado com 5-FU/LV em infusão. Em ambos os estudos, os pacientes receberam o irinotecano em uma dose inicial de 350 mg/m2 em infusão, durante 90 minutos, uma vez a cada 3 semanas. Um total de 535 pacientes foram randomizados nos 2 estudos. Os estudos demonstram uma vantagem de sobrevida significativa para irinotecano quando comparado com os MCS (p=0,0001) e com a terapia com 5-FU/LV (p=0,035). No estudo 1, a sobrevida mediana para os pacientes tratados com irinotecano foi de 9,2 meses comparado a 6,5 meses para os pacientes que receberam os MCS. No estudo 2, a sobrevida mediana para os pacientes tratados com irinotecano foi de 10,8 meses comparado com 8,5 meses para os pacientes que receberam 5-FU/LV infusional. Além da sobrevida, a utilização de irinotecano foi positiva em outros aspectos como no tempo para aparecimento de dor, tempo para deterioração do PS, tempo para perda de peso > 5% e em alguns itens da avaliação de qualidade de vida. Câncer de Pulmão de Células Não Pequenas (CPCNP): O irinotecano, particularmente em regimes de combinação (por exemplo, cisplatina, cisplatina/vindesida, etoposídeo), mostrou eficácia antitumoral no câncer de pulmão de células não pequenas. Taxas de resposta de até 54% foram observadas em pacientes tratados com o regime irinotecano/cisplatina. Dados em monoterapia: a utilização semanal de irinotecano (100 mg/m2) produziu taxas de resposta de aproximadamente 30% (apenas Respostas Parciais-RP) em pacientes previamente não tratados com câncer de pulmão de células não pequenas (CPCNP), com uma duração mediana de resposta de 15 semanas. Dados em combinação: uma taxa de resposta de 52% (1 RC; 32 RP) foi obtida com a combinação de irinotecano e cisplatina no CPCNP avançado. Nesse estudo de fase II, 70 pacientes foram incluídos e a posologia utilizada do irinotecano foi de 60 mg/m2 no d1, d8 e d15, a cada 4 semanas. A dose de cisplatina utilizada foi de 80 mg/m2 no d1, a cada 21 dias. A duração mediana de resposta foi de 19 semanas e a sobrevida mediana foi de 44 semanas. O tempo para se alcançar a remissão foi, em média, de 28 dias. Câncer de Pulmão de Células Pequenas (CPCP): Dados em monoterapia: em estudos pequenos, o tratamento com irinotecano como agente único (100 mg/m2 por semana) produziu uma alta taxa de respostas objetivas (33% a 47%) em pacientes com CPCP previamente tratados, recidivados ou refratários. Uma taxa de resposta de 50% foi observada nos pacientes previamente não tratados. Dados em combinação: em um estudo fase III, o esquema de irinotecano+cisplatina foi comparado ao esquema etoposídeo+cisplatina no tratamento de pacientes com CPCP extensivo (n=154). O esquema contendo irinotecano resultou em uma maior sobrevida significativa (12,8 meses vs. 9,4 meses, p=0,002) e uma maior taxa de resposta tumoral global (84,4% vs. 67,5%, p=0,02). A sobrevida em 1 e 2 anos também foi significativamente maior no regime contendo o irinotecano (sobrevida 1 ano: 58,4% vs. 37,7%; sobrevida em 2 anos: 19,5% vs. 5,2%). O tamanho da amostragem proposto inicialmente nesse estudo era de 230 pacientes, mas o estudo foi interrompido precocemente, pois na análise interina já se demonstrou uma diferença significativa na sobrevida global. Câncer de colo de útero: Um estudo de fase II avaliou o uso de Camptosar® + cisplatina no tratamento de 1a linha do câncer de colo de útero avançado. Nesse estudo, foram avaliadas 29 mulheres. A dose de irinotecano utilizada foi de 60 mg/m2 no d1, d8 e d15, a cada 4 semanas, enquanto a dose de cisplatina foi de 60 mg/m2 no d1, a cada 4 semanas. A resposta global nesse estudo foi de 59% (7% de RC e 52% de RP), com sobrevida mediana de 27,7 meses. Câncer de ovário: O Camptosar® foi avaliado no tratamento de 2a linha do câncer recorrente de ovário em associação à cisplatina. Em um estudo fase II, 25 pacientes foram tratados com a associação de Camptosar®: 50 ou 60 mg/m2 no d1, d8 e d15, a cada 4 semanas e cisplatina: 50 ou 60 mg/m2 no d1, a cada 4 semanas. A resposta global de tratamento foi de 40%, com 2 respostas completas e 8 respostas parciais. A sobrevida mediana alcançada nesse estudo foi de 12 meses. Câncer de estômago: Dados em monoterapia: em pacientes com câncer gástrico avançado previamente tratados ou virgens de tratamento, o uso de irinotecano como agente único na dose de 100 mg/m2/semana ou 150 mg/m2 a cada 2 semanas, promoveu 23% de resposta parcial (33% em pacientes virgens de tratamento). Dados em combinação: a utilização combinada de irinotecano e cisplatina produziu taxa de resposta global de 48% (1 RC; 20RP) em pacientes com câncer gástrico metastático (n=44). A dose de irinotecano utilizada foi de 70 mg/m2, administrada no d1 e d15 a cada 4 semanas; a dose de cisplatina utilizada foi de 80 mg/m2, administrada no d1 a cada 4 semanas. O tempo mediano para resposta foi de 40 dias e a duração mediana de resposta foi de 176 dias. A sobrevida mediana dos pacientes foi de 272 dias.
Referências
1. Saltz LB, Cox JV, Blanke C, Rosen LS, Fehrenbacher L, Moore MJ, Maroun JA, Ackland SP, Locker PK, Pirotta N, Elfring GL, Miller LL. Irinotecan plus fluorouracil and leucovorin for metastatic colorectal cancer. N Engl J Med 2000 Sep 28;343(13):905-14.
2. Douillard JY, Cunningham D, Roth AD, Navarro M, James RD, Karasek P, Jandik P, Iveson T, Carmichael J, Alakl M, Gruia G, Awad L, Rougier P. Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: a multicentre randomised trial. Lancet 2000 Mar 25;355(9209):1041-7.
3. Jagasia MH, Langer CJ, Johnson DH, Yunus F, Rodgers JS, Schlabach LL, Cohen AG, Shyr Y, Carbone DP, Devore RF. Weekly irinotecan and cisplatin in advanced non-small cell lung cancer: a multicenter phase II study. Clin Cancer Res 2001 Jan;7(1):68-73.
4. J H Schiller, D Harrington, A Sandler, C Belani, C Langer, J Krook, D H Johnson, Eastern Cooperative Oncology Group, Boston, MA. A Randomized Phase III Trial of Four Chemotherapy Regimens in Advanced Non-Small Cell Lung Cancer (NSCLC). Proc. Am. Soc. Clin. Oncol.,19: 1a, 2000.
5. Lynch TJ Jr. Lung cancer highlights. Oncologist 2000;5(4):274-9.
6. K. Noda, Y. Nishiwaki, M. Kawahara, S. Negoro, T. Sugiura, A. Yokoyama, M. Fukuoka, K. Mori, K. Watanabe, T. Tamura, N. Saijo, K. Yoshimura. Randomized Phase III Study of Irinotecan (CPT-11) and Cisplatin Versus Etoposide and Cisplatin in Extensive-Disease Small-Cell Lung Cancer: Japan Clinical Oncology Group Study (JCOG9511). Proc. Am. Soc. Clin. Oncol.,19: 483a, 2000.
7. K. Noda, Y. Nishiwaki, M. Kawahara, S. Negoro, T. Sugiura, A. Yokoyama, M. Fukuoka, K. Mori, K. Watanabe, T. Tamura, N. Saijo, K. Yoshimura. Randomized Phase III Study of Irinotecan (CPT-11) and Cisplatin Versus Etoposide and Cisplatin in Extensive-Disease Small-Cell Lung Cancer: Japan Clinical Oncology Group Study (JCOG9511). Cancer Abstracts and Summaries - 9th World Conference on Lung Cancer, 2000.
8. Ajani JA, Fairweather J, Pisters PW. et al. Phase II Study of CPT-11 Plus Cisplatin in Patients With Advanced Gastric and GE Junction Carcinomas. Proc. Am. Soc. Clin. Oncol.,18: 241a, 1999.
9. Boku N, Ohtsu A, Shimada Y, Shirao K, Seki S, Saito H, Sakata Y, Hyodo I. Phase II study of a combination of irinotecan and cisplatin against metastatic gastric cancer. J Clin Oncol 1999 Jan;17(1):319-23.
10. Sugiyama T, Yakushiji M, Noda K, Ikeda M, Kudoh R, Yajima A, Tomoda Y, Terashima Y, Takeuchi S, Hiura M, Saji F, Takahashi T, Umesaki N, Sato S, Hatae M, Ohashi Y.Phase II study of irinotecan and cisplatin as first-line chemotherapy in advanced or recurrent cervical cancer. Oncology 2000;58(1):31-7.
11. Sugiyama T, Yakushiji M, Nishida T, Ushijima K, Okura N, Kigawa J, Terakawa N. Irinotecan (CPT-11) combined with cisplatin in patients with refractory or recurrent ovarian cancer. Cancer Lett 1998 Jun 19;128(2):211-8.
12. Informações para Prescrição do Camptosar (Irinotecano) - PDR (US).
13. Base de Dados Micromedex (Acessada em 01/03/2004).
Caract. farmacológicas.
Propriedades Farmacodinâmicas
O cloridrato de irinotecano tri-hidratado é um agente antineoplásico da classe dos agentes inibidores da topoisomerase I, que contém na formulação o irinotecano, um derivado semissintético da camptotecina, um alcaloide extraído de vegetais como, por exemplo, a Camptotheca acuminata ou sintetizada quimicamente.
O irinotecano e seu metabólito ativo SN-38 se liga ao complexo DNA-topoisomerase I e impede a religação das fitas únicas. Pesquisas atuais sugerem que a citotoxicidade do irinotecano é devido ao dano na fita dupla de DNA produzido durante a síntese de DNA, quando as enzimas de replicação interagem com o complexo terciário formado pela topoisomerase I, DNA e pelo irinotecano ou SN-38.
O irinotecano é um precursor hidrossolúvel do metabólito lipofílico SN-38. O SN-38 é formado a partir do irinotecano, por clivagem da ligação carbamato entre a fração camptotecina e a cadeia lateral dipiperidina mediada pela carboxilesterase. Em linhagens de células tumorais de humanos e roedores o SN-38 inibe a topoisomerase I com potência aproximadamente 1.000 vezes maior do que o irinotecano. Testes de citotoxicidade in vitro mostraram que a potência relativa do SN-38 varia de 2-a 2000-vezes a do irinotecano. Entretanto, os valores da área sob a curva de concentração plasmática versus tempo (AUC) para SN-38 são de 2% a 8% do irinotecano. Noventa e cinco por cento do SN-38 se liga às proteínas plasmáticas comparado a aproximadamente 50% do irinotecano. A contribuição precisa do SN-38 para a atividade do irinotecano é desconhecida.
Ambos, irinotecano e o SN-38, ocorrem sob forma ativa de lactona e sob forma inativa como ânion hidroxiácido. Entre as duas formas há um equilíbrio pH-dependente, de tal maneira que um pH ácido promove a formação da lactona, enquanto que um pH mais básico resulta na forma aniônica do hidroxiácido.
Propriedades Farmacocinéticas
Absorção e Distribuição
Após a infusão intravenosa do produto em humanos, as concentrações plasmáticas do irinotecano decaem de forma multiexponencial, com uma meia-vida média de eliminação de cerca de 6 horas; sendo que a meia-vida média de eliminação do SN-38 é de cerca de 10 horas. A meia-vida da lactona, forma ativa do irinotecano e a do SN-38, é similar àquela observada no irinotecano total e no SN-38, conforme a lactona e a forma hidroxiácido estão em equilíbrio.
Sobre a variação da dose recomendada de 50 a 350 mg/m2, a AUC de irinotecano aumenta linearmente com a dose. Proporcionalmente, a AUC do SN-38 aumenta menos do que a do irinotecano com a dose. As concentrações máximas do metabólito ativo SN-38 são atingidas, geralmente, dentro de 1 hora após o término de uma infusão de 90 minutos do irinotecano.
O irinotecano apresenta ligação moderada às proteínas plasmáticas (de 30 a 68%). O SN-38 é altamente ligado às proteínas plasmáticas em humanos (aproximadamente 95%). A principal proteína plasmática de ligação de ambos é a albumina.
Metabolismo e Excreção
O irinotecano (CPT-11) está sujeito à conversão metabólica extensa por vários sistemas enzimáticos, incluindo esterases, para formar o metabólito ativo SN38, e a UGT1A1 faz a mediação da glucuronidação do SN-38 para formar o metabólito inativo glucuronida SN-38G. O irinotecano (CPT-11) pode sofrer também metabolismo oxidativo mediado por CYP3A4 a diversos produtos de oxidação farmacologicamente inativos, um dos quais pode ser hidrolisado por carboxilesterase para liberar o SN-38.A atividade UGT1A1 é reduzida em indivíduos com polimorfismo genético que leva à redução da atividade enzimática tal como o polimorfismo UGT1A1*28 (vide item 5. Advertências e Precauções). O SN-38 glicuronídeo teve 1/50 a 1/100 a atividade do SN-38 em estudos de citotoxicidade utilizando duas linhas de células in vitro.
A eliminação do irinotecano ainda não foi completamente elucidada em humanos. A excreção urinária do irinotecano é 11% a 20%; SN-38 < 1% e SN-38-glicuronídeo, 3%. A excreção urinária e biliar acumulada de irinotecano e de seus metabólitos (SN-38 e SN-38-glicuronídeo), por um período de 48 horas após a administração de irinotecano, em dois pacientes, variou de aproximadamente 25% (100 mg/m2) a 50% (300 mg/m2).
Populações Especiais
Pacientes Idosos
A farmacocinética do irinotecano administrado em esquema posológico semanal foi avaliada em um estudo prospectivo com 183 pacientes para avaliar o efeito da idade em relação à toxicidade do irinotecano. Os resultados dos estudos indicaram que não há diferença na farmacocinética do irinotecano, SN-38 e SN-38 glicuronídeo em pacientes < 65 anos quando comparados com pacientes ≥ 65 anos. Em um estudo não prospectivo com 162 pacientes para avaliar o efeito da idade, foram observadas diferenças menores (menos de 18%), mas estatisticamente significativas, nos parâmetros farmacocinéticos dose-normalizada do irinotecano em pacientes < 65 anos quando comparado com pacientes ≥ 65 anos. Embora a AUC0-24 dose-normalizada para o SN-38 em pacientes ≥ 65 anos foi 11% maior do que em pacientes < 65 anos, essa diferença não foi estatisticamente significativa.
Pacientes Pediátricos (vide item 5. Advertências e Precauções)
A farmacocinética de irinotecano e seus principais metabólitos na população pediátrica foi investigada em estudos clínicos conduzidos nos EUA e na Europa. Geralmente, resultados e conclusões gerais considerando a farmacocinética do irinotecano foram comparáveis nos estudos americanos e europeus. Qualquer diferença nos resultados entre esses estudos são, provavelmente atribuíveis às diferenças nas doses investigadas (20 a 200 mg/m2 e 200 a 720 mg/m2 nos estudos americanos e europeus, respectivamente) e na variabilidade dos valores interpacientes determinada para os parâmetros farmacocinéticos do irinotecano e do SN-38.
Estudos americanos
Parâmetros farmacocinéticos do irinotecano e do SN-38 foram determinados em 2 estudos pediátricos em tumores sólidos com doses de 50 mg/m2 (infusão de 60 minutos, n=48) e 125 mg/m2 (infusão de 90 minutos, n=6). O clearance do irinotecano foi 17,3 ± 6,7 L/h/m2 (média ± desvio padrão) para a dose de 50 mg/m2 e 16,2 ± 4,6 L/h/m2 para a dose de 125 mg/m2, que é um pouco maior que em adultos. Em crianças, que receberam o irinotecano 1 vez/dia por 5 dias a cada 3 semanas ou 1 vez/dia por 5 dias por 2 semanas a cada 3 semanas, observou-se acumulação mínima de irinotecano e SN-38 (vide item "Advertências - Populações Especiais"). O resultado em que os valores de AUC de SN-38 dose-normalizada foi comparável entre adultos e crianças foi inconsistente com o aumento do clearance de irinotecano observado na população pediátrica e provavelmente foi reflexo da variabilidade interpacientes (% dos valores de CV para AUC de SN-38 foi de 84 a 120%). De fato, a exposição de SN-38 em pacientes pediátricos foi aproximadamente 30% menor que em adultos quando uma comparação foi feita sem considerar a variabilidade dos dados.
Estudos europeus
A farmacocinética do irinotecano e seus principais metabólitos foi investigada em pacientes pediátricos com tumores sólidos em estudo fase I nas doses de 200 a 720 mg/m2 (infusão de 2 horas, n=77). A exposição sistêmica do irinotecano, SN-38, APC (7-etil-10-[4-N-(5-ácido aminopentoico)-1-piperidino]-carboniloxicamptotecina) e NPC [7etil-10-(4-amino-1-piperidino)-carboniloxicamptotecina] foi dose-proporcional. Parâmetros farmacocinéticos do irinotecano e seus metabólitos demonstraram variabilidade interpacientes com valores (média ± desvio padrão) para clearance plasmático do irinotecano de 18 ± 8 L/h/m² e volume de distribuição no estado de equilíbrio de 104 ± 84 L/m². O clearance de irinotecano foi 26% menor em adolescentes que em crianças e exposições de SN-38 dosenormalizada e SN-38G foram 52% e 105% maiores em adolescentes que em crianças, respectivamente. O clearance de irinotecano foi maior e valores doses-normalizadas para exposições de SN-38, SN-38G e APC foram menores na população pediátrica que na de adultos.
Uma análise farmacocinética de irinotecano na população foi realizada em 83 crianças e adolescentes com rabdomiossarcoma refratária ou reincidente, tumor neuroectodérmico primitivo (TNEP) incluindo meduloblastoma ou neuroblastoma recebendo 600 mg/m2 de irinotecano em infusões de 1 hora 1 vez a cada 3 semanas como parte de um estudo fase II. Valores médios para clearance e AUC de irinotecano demonstraram uma grande variabilidade inter e intraindividual e foram similares àqueles determinados na mesma dose no estudo pediátrico europeu de fase I.
Sexo
A farmacocinética do irinotecano não parece ser influenciada pelo sexo.
Raça
A influência da raça na farmacocinética do irinotecano não foi avaliada.
Insuficiência hepática
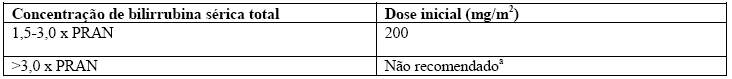
O clearance do irinotecano é diminuído em pacientes com disfunção hepática enquanto a exposição relativa ao metabólito ativo SN-38 é aumentado. A magnitude destes efeitos é proporcional ao grau de comprometimento do fígado, avaliado pelas elevações na concentração sérica de bilirrubina total e transaminases (vide item 8. Posologia e Modo de Usar).
Insuficiência renal
Não foi avaliada a influência da insuficiência renal sobre a farmacocinética do irinotecano (vide item 8. Posologia e Modo de Usar).
Dados de segurança pré-clínicos
Toxicologia
A toxicidade aguda intravenosa de irinotecano em animais é mostrada a seguir. Após doses intravenosas únicas de aproximadamente 111 mg/kg em camundongos e 73 mg/kg em ratos (aproximadamente 2,6 e 3,4 vezes a dose recomendada para humanos de 125 mg/m2, respectivamente) os animais evoluíram para o óbito. As mortes foram precedidas de cianose, tremores, angústia respiratória e convulsões. Estudos de toxicidade subaguda mostraram que irinotecano afeta tecidos com rápida proliferação celular (medula óssea, epitélio intestinal, timo, baço, nodos linfáticos e testículos).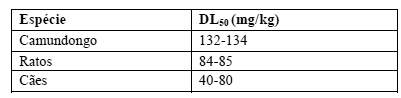
Carcinogenicidade e Mutagenicidade
Não foram conduzidos estudos de carcinogenicidade em longo prazo com irinotecano. Entretanto, foram realizados bioensaios com ratos recebendo por via IV doses de 2 mg/kg ou 25 mg/kg, 1 vez por semana, durante 13 semanas, com um período posterior de observação de 91 semanas (em estudos separados, a dose de 25 mg/kg produziu uma Cmáx e uma AUC para o irinotecano cerca de 7,0 vezes e 1,3 vezes os valores respectivos em pacientes que receberam 125 mg/m2). Nessas condições, houve um aumento linear significativo na incidência de sarcoma e pólipos do estroma uterino.
O irinotecano e o SN-38 não foram mutagênicos na análise de Ames in vitro. No entanto, em testes in vitro em células ovarianas de hamster chinês, o irinotecano produziu um aumento significativo na incidência de aberrações cromossômicas de maneira dose-dependente. Adicionalmente, em testes in vivo em camundongo, uma dose única intraperitoneal de irinotecano variando entre 2,5 a 200 mg/kg, causou um aumento significativo e dose-dependente nos micronúcleos policromáticos eritrocíticos e uma diminuição na taxa de reticulocítico/eritrocítico nas células da medula óssea.
Reprodução
Não foram observados efeitos adversos significativos sobre a fertilidade e desempenho reprodutivo geral após a administração de irinotecano, por via intravenosa, em doses de até 6 mg/kg/dia em ratos. Entretanto, após doses diárias múltiplas de irinotecano observou-se atrofia dos órgãos reprodutores dos machos, tanto em roedores na dose de 20 mg/kg (que, em estudos separados, produziu uma Cmáx e uma área sob a curva para o irinotecano cerca de 5 vezes e 1 vez, respectivamente, os valores correspondentes em pacientes que receberam 125 mg/m2 semanalmente) quanto em cães na dose de 0,4 mg/kg (que, em estudos separados, produziu uma Cmáx e uma área sob a curva para o irinotecano cerca de metade e uma vez e meia, respectivamente, os valores correspondentes em pacientes que receberam 125 mg/m2 semanalmente).
Radioatividade relacionada ao 14C-irinotecano atravessa a placenta de ratas após administração intravenosa de 10 mg/kg (que, em estudos separados produziu uma Cmáx e AUC do irinotecano cerca de 3 e 0,5 vezes, respectivamente, aos valores correspondentes em pacientes recebendo 125 mg/m2). O irinotecano foi teratogênico em ratos com doses maiores que 1,2 mg/kg/dia (que, em estudos separados produziu Cmáx e AUC cerca de 2/3 e 1/40, respectivamente, dos valores correspondentes em pacientes recebendo 125 mg/m2) e em coelhos a 6 mg/kg/dia (cerca de 1,5 da dose humana recomendada semanalmente na base mg/m2). Efeitos teratogênicos incluem uma variedade de anormalidades externas, viscerais e esqueléticas. O irinotecano administrado a ratas durante o período após organogênese até desmame em doses de 6 mg/kg/dia causou diminuição da habilidade de aprendizado e diminuiu o ganho de peso corporal das ratas da ninhada.
Contraindicações.
Camptosar® é contraindicado a pacientes com hipersensibilidade conhecida ao fármaco ou a qualquer componente da fórmula (vide item 5. Advertências e Precauções).
Advertências e precauções.
Administração: Camptosar® deve ser administrado obrigatoriamente sob a supervisão de um médico com experiência no uso de agentes quimioterápicos para neoplasia. O controle apropriado de complicações somente é possível quando estiverem disponíveis os recursos adequados para diagnóstico e tratamento.
O uso de Camptosar® nas situações a seguir deve ser avaliado através da análise dos benefícios e riscos esperados, e indicado quando os benefícios superarem os possíveis riscos:
em pacientes que apresentam um fator de risco (particularmente os com performance status = 2 OMS).
em raros casos, onde os pacientes apresentam recomendações relacionadas ao controle de eventos adversos (necessidade de tratamento antidiarreico imediato e prolongado combinado a alto consumo de fluidos no início da diarreia tardia). Recomenda-se estrita supervisão hospitalar a tais pacientes.
Sintomas colinérgicos: os pacientes podem apresentar sintomas colinérgicos como rinite, salivação aumentada, miose, lacrimejamento, diaforese, rubor (vasodilatação), bradicardia e aumento do peristaltismo intestinal, que pode causar cólicas abdominais e diarreia em fase inicial da administração (por ex.: diarreia ocorrendo geralmente durante ou até 8 horas da administração de Camptosar®). Esses sintomas podem ser observados durante, ou logo após, a infusão de Camptosar®. Possivelmente eles se relacionam à atividade anticolinesterásica do fármaco inalterado e são mais frequentes em administração de doses mais altas. Em pacientes com sintomas colinérgicos a administração terapêutica, ou profilática, de atropina 0,25 a 1 mg por via intravenosa ou subcutânea deve ser considerada (a não ser que contraindicada clinicamente).
Extravasamento: embora Camptosar® não seja, sabidamente, vesicante, deve-se tomar cuidado para evitar extravasamento e observar o local da infusão quanto a sinais inflamatórios. Caso ocorra extravasamento, recomendase infusão para "lavar" o local de acesso (flushing) e aplicação de gelo.
Hepático: em estudos clínicos foram observadas, em menos de 10% dos pacientes, anormalidades das enzimas hepáticas de Graus 3 ou 4 de acordo com os Critérios Comuns de Toxicidade do National Cancer Institute (NCI). Esses eventos ocorrem tipicamente em pacientes com metástases hepáticas conhecidas e não estão claramente relacionados ao Camptosar®.
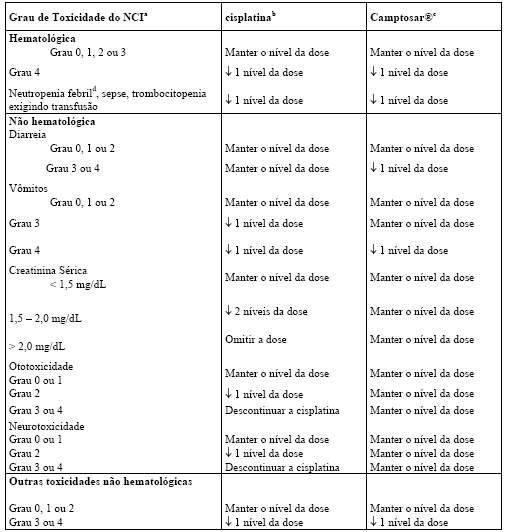
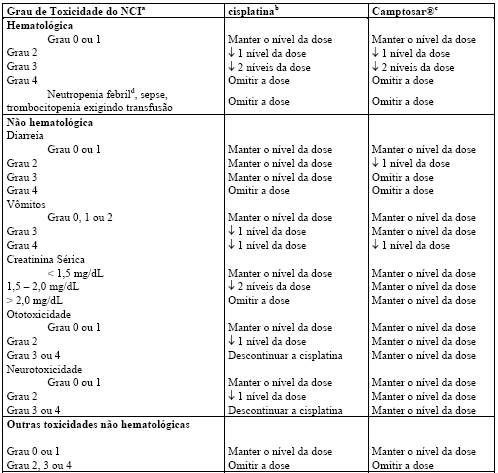
Hematológico: o Camptosar® frequentemente causa neutropenia, leucopenia e anemia, inclusive graves, devendo ser evitado em pacientes com insuficiência aguda grave da medula óssea. A trombocitopenia grave é incomum. Nos estudos clínicos, a frequência de neutropenia Graus 3 e 4 NCI foi significativamente maior em pacientes que haviam recebido previamente irradiação pélvica/abdominal do que naqueles que não haviam recebido tal irradiação. Pacientes com níveis séricos basais de bilirrubina total de 1,0 mg/dL ou mais, também tiveram uma probabilidade significativamente maior de ter neutropenia Grau 3 ou 4 na primeira dose do que aqueles cujos níveis de bilirrubina eram menores do que 1,0 mg/dL. Não houve diferenças significativas na frequência de neutropenia Grau 3 ou 4 por idade ou sexo (vide item 8. Posologia e Modo de Usar).
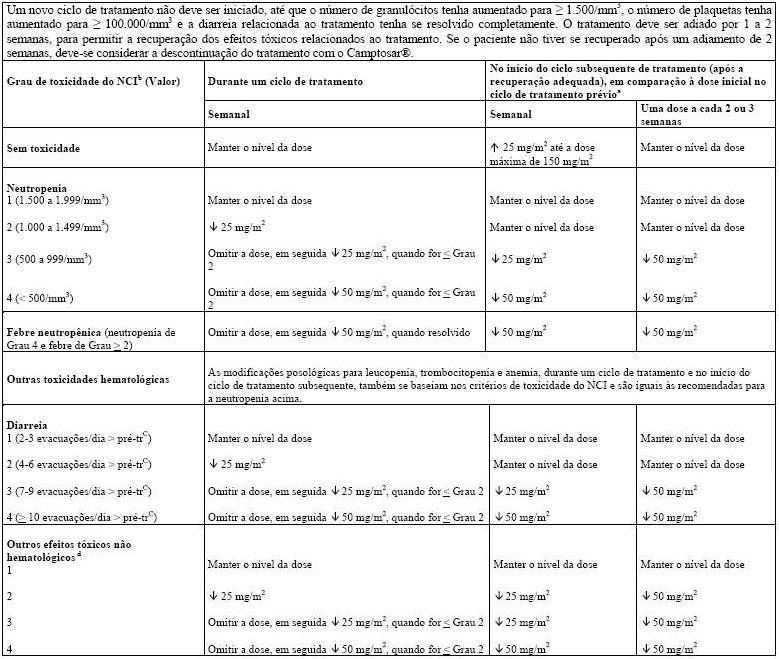
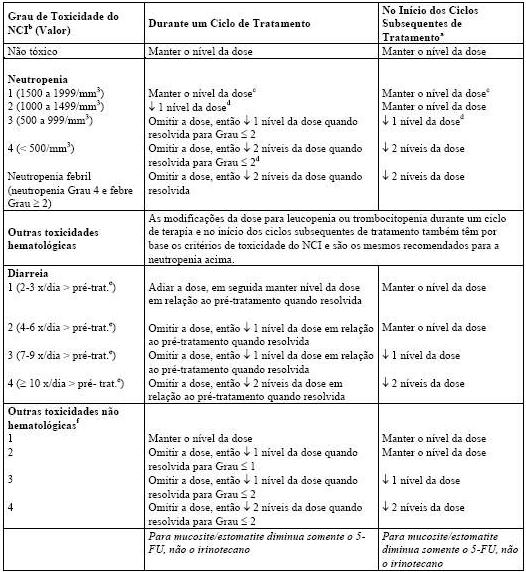
Neutropenia febril (neutropenia Grau 4 NCI e febre Grau ³ 2) ocorreu em menos de 10% dos pacientes nos estudos clínicos. Mortes devido à sepse após neutropenia grave foram relatadas em pacientes tratados com Camptosar®. Complicações neutropênicas devem ser tratadas prontamente com suporte antibiótico. A terapia com Camptosar® deve ser temporariamente descontinuada caso ocorra neutropenia febril ou se a contagem absoluta de neutrófilos cair abaixo de 1000/mm3. A dose do produto deve ser reduzida no caso de ocorrência de neutropenia não febril clinicamente significativa (vide item 8. Posologia e Modo de Usar).
Pacientes com atividade UGT1A1 reduzida.
A conversão metabólica de irinotecano ao metabólito ativo SN-38 é mediada pela enzima carboxilesterase e ocorre primariamente no fígado. Subsequentemente o SN-38 sofre conjugação para formar o metabólito inativo glucuronida SN-38G. Esta reação de glucuronidação é mediada primariamente pela transferase glucuronosil-difosfato uridina 1A1 (UGT1A1), que é codificada pelo gene UGT1A1 (vide item 3. Características Farmacológicas - Propriedades Farmacocinéticas). Este gene é altamente polimórfico, resultando em capacidades metabólicas variáveis entre indivíduos. Uma variação específica do gene UGT1A1 inclui um polimorfismo na região promotora conhecida como alelo variante UGT1A1 28. Esta variação e outras deficiências congênitas na expressão UGT1A1 (tais como Crigler-Najjar e síndrome de Gilbert) estão associadas com a redução da atividade enzimática e exposição sistêmica elevada ao SN-38. Altas concentrações plasmáticas de SN-38 são observadas em indivíduos homozigóticos para o alelo UGT1A1*28 (também referente ao genótipo UGT1A1 7/6) versus pacientes que possuam um ou dois alelos tipo selvagem.
Dados de uma meta-análise de nove estudos envolvendo um total de 821 pacientes indicaram que indivíduos com síndrome Crigler-Najjar (tipos 1 e 2) ou aqueles considerados homozigóticos para o alelo UGT1A1*28 (síndrome de Gilbert) correm um risco elevado de toxicidade hematológica (Graus 3 e 4) seguido de administração de irinotecano de doses moderada à altas ( > 150 mg/m2). A relação entre o genótipo UGT1A1 e a ocorrência do irinotecano induzir diarreia, não foi estabelecida.
Deve ser administrado, em pacientes conhecidos como homozigóticos para UGT1A1*28, a dose inicial normal indicada para irinotecano. Entretanto, estes pacientes devem ser monitorados quanto à toxicidade hematológica. Uma dose inicial reduzida de irinotecano deve ser considerada em pacientes que já tenham sofrido toxicidade hematológica prévia com tratamento anterior. A redução exata da dose inicial nesses pacientes não foi estabelecida e quaisquer modificações de dose subsequente, devem ser baseadas na tolerância individual do paciente ao tratamento.
Reações de hipersensibilidade: foram relatadas reações de hipersensibilidade, inclusive reações anafilática/anafilactoide graves.
Efeitos imunossupressores/ Aumento da suscetibilidade a infecções: a administração de vacinas com microrganismos vivos ou atenuados em pacientes imunocomprometidos por agentes quimioterápicos, incluindo Camptosar®, pode resultar em infecções graves ou fatais. A vacinação com vacinas contendo microrganismos vivos deve ser evitada em pacientes recebendo Camptosar®. As vacinas com microrganismos mortos ou inativados podem ser administradas, no entanto, a resposta a esta vacina pode ser diminuída.
Diarreia tardia: a diarreia tardia (aquela que ocorre mais de 8 horas após a administração do produto) pode ser prolongada e pode levar à desidratação, desequilíbrio eletrolítico ou sepse, constituindo um risco de morte potencial. Nos estudos clínicos que testaram o esquema posológico a cada 3 semanas, a diarreia tardia foi iniciada, em média, após 5 dias da infusão de Camptosar®; já nos estudos que avaliaram a posologia semanal, este intervalo médio era de 11 dias. Nos pacientes que começaram o tratamento com a dose semanal de 125 mg/m2, o tempo médio de duração de qualquer Grau de diarreia tardia foi de 3 dias. Nos pacientes tratados com a dose semanal de 125 mg/m2 que tiveram diarreia Grau 3 ou 4, o tempo médio de duração de todo o episódio de diarreia foi de 7 dias. Resultados de um estudo prospectivo de um esquema semanal de tratamento não demonstraram diferença na taxa de diarreia tardia em pacientes com 65 anos ou mais em relação a pacientes com menos de 65 anos. Entretanto, pacientes com 65 anos ou mais, devem ser monitorados de perto devido ao risco aumentado de diarreia precoce observada nesta população. Ulceração do cólon, algumas vezes com sangramento, foi observada em associação à diarreia induzida pelo irinotecano.
A diarreia tardia deve ser tratada com loperamida imediatamente após observar-se o primeiro episódio de fezes amolecidas, ou malformadas, ou ainda, na ocorrência de evacuações em frequência maior do que a esperada pelo paciente. O regime de dose recomendado para a loperamida é de 4 mg à primeira ocorrência de diarreia tardia, seguidos de 2 mg a cada 2 horas até que o paciente não apresente diarreia por, pelo menos, 12 horas. Durante a noite, o paciente pode utilizar 4 mg de loperamida a cada 4 horas. O uso de loperamida nestas doses não é recomendado por mais de 48 horas consecutivas (risco de íleo paralítico) e nem por menos de 12 horas. A pré-medicação com loperamida não é recomendada. Pacientes com diarreia devem ser cuidadosamente monitorados e em caso de desidratação, devem ser realizadas reposições hídrica e eletrolítica. Se os pacientes apresentarem íleo paralítico, febre ou neutropenia grave, tratamento de suporte com antibióticos deve ser administrado. Além do tratamento antibiótico, a hospitalização é recomendada para o tratamento de diarreia, nos seguintes casos:
-diarreia com febre; -diarreia grave (requerendo hidratação intravenosa); -pacientes com vômito associado à diarreia tardia; -diarreia persistindo por cerca de 48 horas após o início da terapia com altas doses de loperamida.
Após o primeiro ciclo de tratamento, os ciclos quimioterápicos semanais subsequentes só devem ser iniciados quando a função intestinal do paciente retornar ao padrão pré-tratamento por, pelo menos, 24 horas sem a necessidade de medicação antidiarreica. Se ocorrer diarreia tardia Grau 2, 3 ou 4 (NCI), a administração de Camptosar® deve ser descontinuada e retomada em dose reduzida assim que o paciente se recuperar (vide item 8. Posologia e Modo de Usar).
Doença inflamatória crônica e/ou obstrução intestinal: em caso de obstrução intestinal os pacientes não devem ser tratados com Camptosar®.
Náuseas e vômitos: Camptosar® é emetogênico, como os quadros de náuseas e vômitos podem ser intensos ocorrendo geralmente, durante ou logo após a infusão do irinotecano, recomenda-se que os pacientes recebam antieméticos pelo menos 30 minutos antes da infusão de Camptosar®. O médico também deve considerar a utilização subsequente de esquema de tratamento antiemético se necessário. Pacientes com vômito associado à diarreia tardia devem ser hospitalizados assim que possível para tratamento.
Neurológico: tontura foi observada e pode, algumas vezes, representar evidência sintomática de hipotensão ortostática em pacientes com desidratação.
Renal: elevação dos níveis séricos de creatinina ou ureia foram observados. Ocorreram casos de insuficiência renal aguda. Esses eventos foram atribuídos à complicações infecciosas ou à desidratação, relacionada à náusea, vômitos ou diarreia. Há raros relatos de disfunção renal decorrente de síndrome de lise tumoral.
Respiratório: observou-se dispneia de Grau 3 ou 4 NCI, mas é desconhecido o quanto patologias pré-existentes e/ou envolvimento pulmonar maligno contribuem para o sintoma. Em estudos iniciais no Japão, pequena porcentagem dos pacientes evoluiu com uma síndrome pulmonar, com potencial risco de morte, que se apresenta através de dispneia, febre e de um padrão reticulonodular na radiografia de tórax. Porém, o quanto o Camptosar® contribuiu para estes eventos é desconhecido pois os pacientes também apresentavam tumores pulmonares e, alguns, moléstia pulmonar não maligna pré-existente.
Doença pulmonar intersticial, manifestada através de infiltrado pulmonar, é incomum durante terapia com irinotecano. São fatores de risco para o desenvolvimento desta complicação: doenças pulmonares pr é-existentes, uso de fármacos pneumotóxicos, terapia de radiação e uso de fatores de estimulação de colônias. Na presença de um ou mais destes fatores o paciente deve ser cuidadosamente monitorado quanto a sintomas respiratórios antes e durante a terapia com Camptosar®.
Outros: uma vez que este produto contém sorbitol, não é recomendado o uso em pacientes com intolerância hereditária à frutose.
Atenção: Este medicamento contém Açúcar, portanto, deve ser usado com cautela em portadores de Diabetes.
Uso durante a Gravidez
Camptosar® pode causar danos ao feto quando administrado a mulheres grávidas. Estudos mostram que o irinotecano é teratogênico em ratos e coelhos (vide item 3. Características Farmacológicas - Dados de Segurança Pré-Clínicos). Não foram conduzidos estudos adequados e bem controlados com mulheres grávidas. As mulheres em idade fértil devem ser orientadas a evitar a gravidez enquanto estiverem sendo tratadas com este produto. Caso o fármaco seja utilizado durante a gravidez ou a paciente fique grávida enquanto estiver recebendo esse fármaco, ela deve ser informada dos riscos potenciais ao feto.
Camptosar® é um medicamento classificado na categoria D de risco de gravidez. Portanto, este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. A paciente deve informar imediatamente
o médico em caso de suspeita de gravidez.
Uso durante a Lactação
Cinco minutos após a administ