AMGEVITA
AMGEN
adalimumabe
Anti-reumático.
Apresentações.
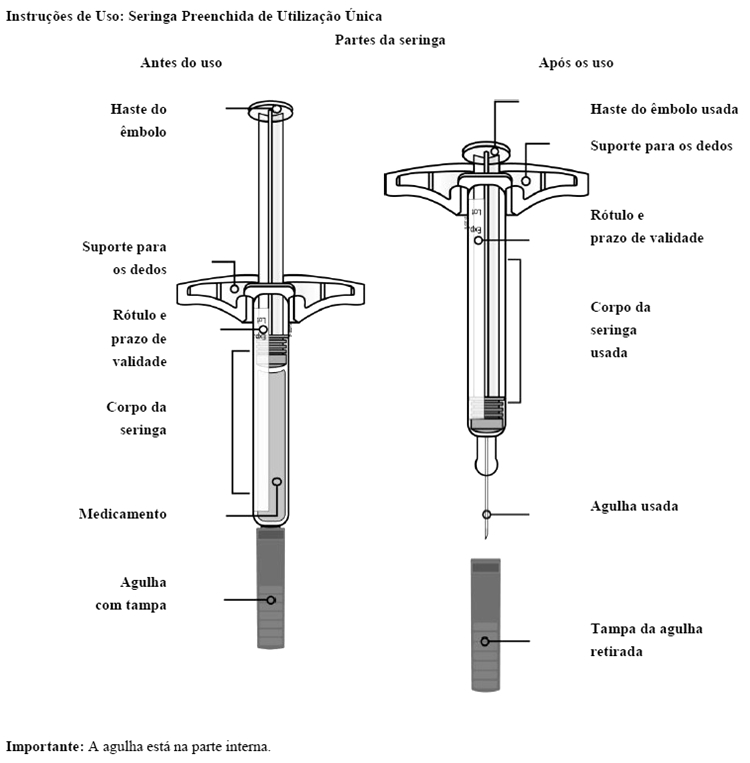
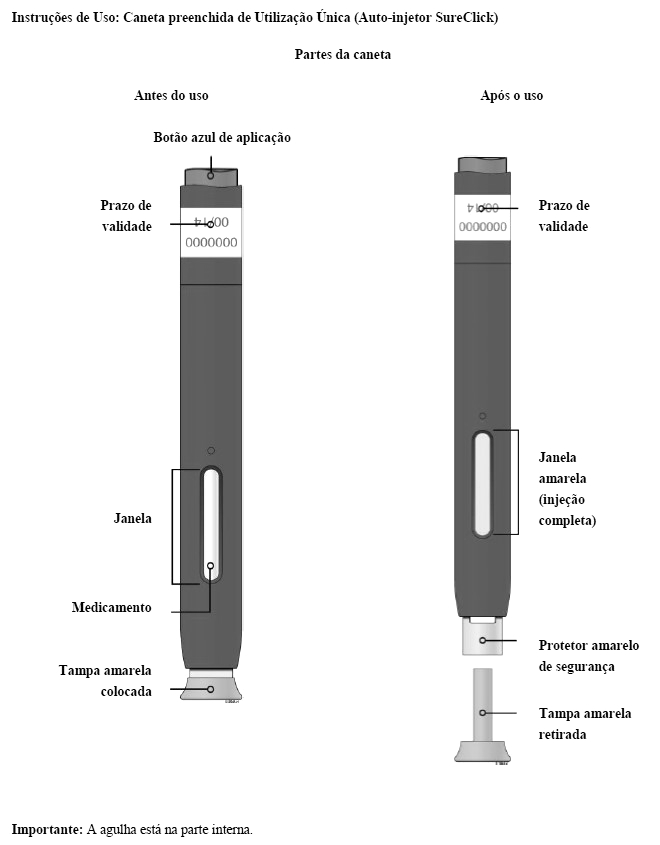
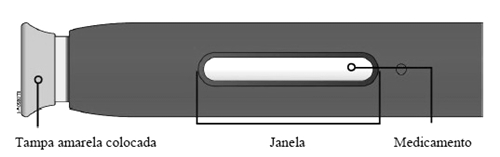
Solução injetável em uma seringa preenchida ou em uma caneta preenchida (SureClick®)*.
AMGEVITA contém 20 mg de adalimumabe em embalagens com 1 seringa preenchida ou 40 mg de adalimumabe em embalagens com 1 ou 2 seringas preenchidas ou canetas preenchidas.
* Caneta - consiste em uma seringa preenchida de dose única descartável (sistema auto-injetor).
USO SUBCUTÂNEO
USO ADULTO E PEDIÁTRICO ACIMA DE 2 ANOS
Composição.

Informações técnicas.
1. INDICAÇÕES
Este medicamento é destinado ao tratamento de:
Artrite reumatoide em pacientes adultos
AMGEVITA é destinado para reduzir os sinais e sintomas, induzir uma resposta clínica e remissão clínica maior, inibir a progressão dos danos estruturais e melhorar a capacidade física em pacientes adultos com artrite reumatoide (AR) ativa de intensidade moderada a grave, que apresentaram resposta inadequada a uma ou mais drogas anti-reumáticas modificadoras do curso da doença (DMARDs).
AMGEVITA é destinado ao tratamento da artrite reumatoide grave, ativa e progressiva em pacientes não tratados com metotrexato previamente.
AMGEVITA pode ser utilizado isoladamente ou em combinação com metotrexato ou outra DMARD.
Artrite psoriásica em pacientes adultos
AMGEVITA é destinado para reduzir os sinais e sintomas da artrite psoriásica (APs). O medicamento demonstrou reduzir a taxa de progressão das lesões articulares periféricas, conforme medido por raio-X em pacientes com subtipos poliarticular simétrico da doença, e melhora da função física.
AMGEVITA pode ser utilizado isoladamente ou em combinação a drogas anti-reumáticas modificadoras do curso da doença DMARDs.
Espondiloartrite axial em pacientes adultos
- Espondilite anquilosante (EA)
AMGEVITA é destinado ao tratamento da espondilite anquilosante ativa em pacientes que responderam inadequadamente à terapia convencional.
- Espondiloartrite axial não radiográfica (espondiloartrite axial sem evidência radiográfica de EA)
AMGEVITA é destinado ao tratamento de pacientes adultos com espondiloartrite axial grave sem evidência radiográfica de EA que possuam sinais objetivos de inflamação (PCR elevada e/ou ressonância magnética) e que responderam inadequadamente ou que sejam intolerantes aos medicamentos anti-inflamatórios não esteroidais
Doença de crohn em pacientes adultos
AMGEVITA é destinado para reduzir sinais e sintomas, induzir e manter a remissão clínica em pacientes adultos com doença de Crohn (DC) ativa de intensidade moderada a grave, que apresentaram resposta inadequada à terapia convencional.
AMGEVITA também é destinado para reduzir sinais e sintomas e induzir remissão clínica em pacientes que perderam resposta ou são intolerantes ao infliximabe.
Colite ulcerativa ou retocolite ulcerativa
AMGEVITA é destinado ao tratamento da colite ulcerativa ou retocolite ulcerativa ativa moderada a grave em pacientes adultos, que apresentaram uma resposta inadequada à terapia convencional incluindo corticosteroides e/ou 6-mercaptopurina (6-MP) ou azatioprina (AZA), ou em pacientes que são intolerantes ou contraindicados para estas terapias. AMGEVITA induz e mantém a cicatrização da mucosa nestes pacientes, reduz a hospitalização relacionada com a doença e suas causas e, melhora a qualidade de vida. O uso de corticosteroide pode ser reduzido ou descontinuado.
Psoríase em placas em pacientes adultos
AMGEVITA é destinado ao tratamento de psoríase em placas crônica moderada a grave em pacientes adultos com indicação de terapia sistêmica.
Hidradenite Supurativa em pacientes adultos
AMGEVITA é destinado para reduzir os sinais e sintomas de hidradenite supurativa ativa moderada a grave em pacientes adultos, nos quais a terapia antibiótica foi inadequada, incluindo o tratamento de lesões inflamatórias e prevenção do agravamento de abscessos e fístulas.
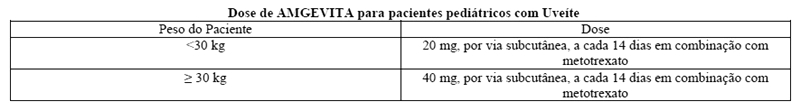
Uveíte em pacientes adultos
AMGEVITA é destinado ao tratamento de uveíte não infecciosa intermediária, posterior ou pan-uveíte, em pacientes adultos que tenham resposta inadequada ao uso de corticosteroides, que necessitem de redução/retirada de corticosteroides (corticosteroid-sparing) ou nos pacientes no qual o uso de corticosteroides é inapropriado.
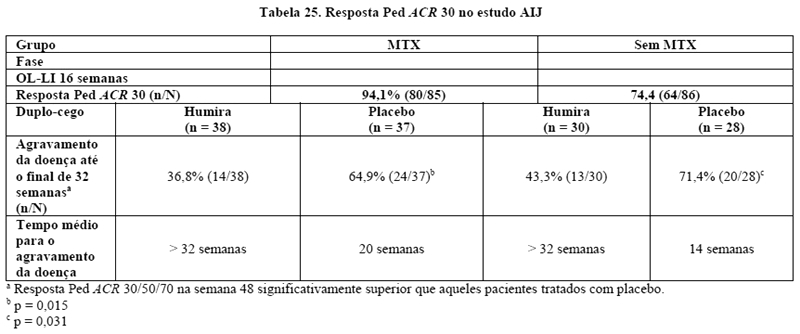
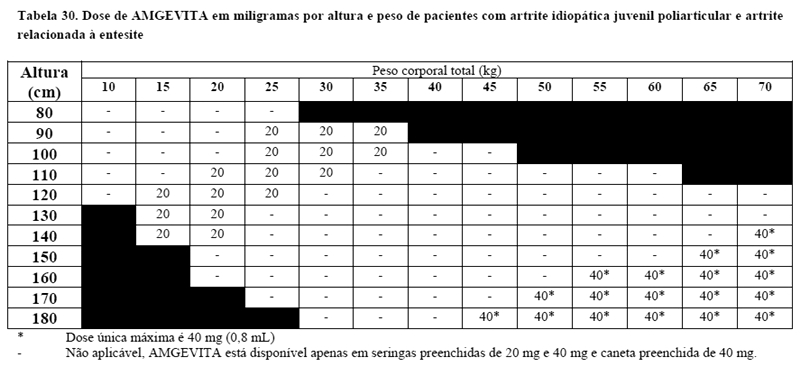
Artrite idiopática juvenil poliarticular em pacientes pediátricos
AMGEVITA, em combinação com metotrexato, é indicado para reduzir os sinais e sintomas da artrite idiopática juvenil poliarticular ativa moderada a grave em pacientes pediátricos acima de 2 anos de idade que apresentaram resposta inadequada a pelo menos um DMARD.
AMGEVITA pode ser utilizado em monoterapia naqueles indivíduos intolerantes ao metotrexato ou quando o uso concomitante com metotrexato é inapropriado.
Artrite relacionada à entesite em pacientes pediátricos
AMGEVITA é destinado ao tratamento de artrite relacionada à entesite em pacientes acima de 6 anos que apresentaram uma resposta inadequada ou que são intolerantes à terapia convencional.
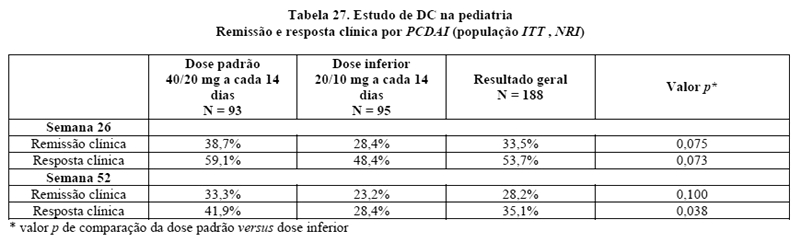
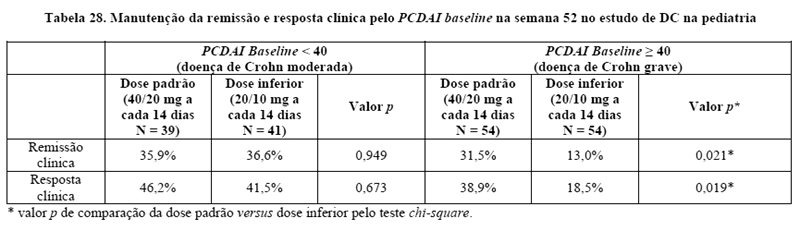
Doença de Crohn em pacientes pediátricos
AMGEVITA é destinado para reduzir sinais e sintomas e induzir e manter a remissão clínica em pacientes pediátricos a partirde 6 anos, com doença de Crohn ativa de intensidade moderada a grave, que apresentaram resposta inadequada à terapia convencional.
Uveíte pediátrica
AMGEVITA é destinado ao tratamento de uveíte não infecciosa, anterior, crônica em pacientes pediátricos com 2 anos de idade ou mais, que apresentaram uma resposta inadequada ou que são intolerantes à terapia convencional, ou quando a terapia convencional é inapropriada.
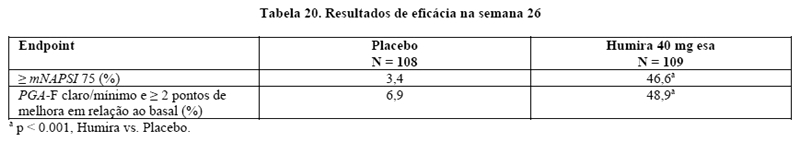
2. RESULTADOS DE EFICÁCIA
AMGEVITA é um medicamento biológico desenvolvido pela via de comparabilidade. O seu programa de desenvolvimento clínico foi projetado para demonstrar comparabilidade entre AMGEVITA e o produto comparador Humira® (adalimumabe)*. AMGEVITA é aprovado como um biossimilar de Humira.
* Medicamento biológico comparador (produto comparador): Humira (adalimumabe), registrado por Abbvie Farmacêutica Ltda.
Artrite reumatoide em pacientes adultos
Dados de eficácia de AMGEVITA
Estudo comparativo de Artrite Reumatoide (AR) entre AMGEVITA e Humira (AR ABP-Estudo 1) - A eficácia e a segurança de AMGEVITA em comparação com Humira foram avaliadas em um estudo duplo-cego, randomizado e controlado por medicamento em pacientes ≥ 18 anos de idade com artrite reumatoide (AR) ativa diagnosticada de acordo com os critérios do American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) de 2010. Os pacientes tinham AR há pelo menos 3 meses e pelo menos 6 articulações dolorosas e inchadas e VHS ou PCR elevada ao ingressar no estudo. Os pacientes tinham fator reumatoide ou positividade para peptídeos citrulinados anticíclicos. O estudo avaliou 526 pacientes que apresentaram uma resposta inadequada ao metotrexato (MTX) nas doses de 7,5 a 25 mg. Os pacientes receberam 40 mg de AMGEVITA ou Humira por via subcutânea a cada duas semanas por até 22 semanas.
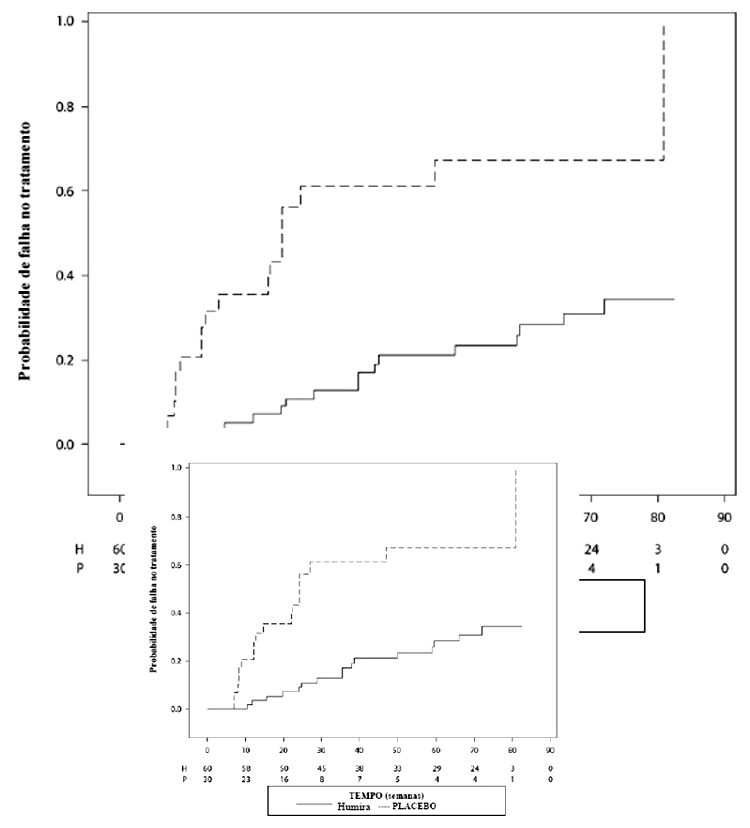
Resposta Clínica - A Tabela 1 apresenta a porcentagem de indivíduos tratados com AMGEVITA que alcançaram ACR 20 na semana 24 no AR ABP-Estudo 1. A razão de risco (RR) do parâmetro final primário ACR 20 ficou dentro da margem de equivalência pré-especificada e demonstrou equivalência clínica entre AMGEVITA e Humira.
Na semana 24, 74,6% (194/260) dos indivíduos no grupo de AMGEVITA e 72,4% (189/261) dos indivíduos no grupo de Humira atingiram os critérios de resposta ACR 20. A RR de ACR 20 para AMGEVITA versus Humira foi 1,039, com o intervalo de confiança (IC) bilateral de 90% de (0,954, 1,133). O IC de 90% ficou dentro da margem de equivalência pré-definida. A diferença de risco (DR) de ACR 20 para AMGEVITA versus Humira foi 2,604%, com o intervalo de confiança (IC) bilateral de 90% de (-3,728%, 8,936%). A equivalência clínica entre AMGEVITA e Humira foi demonstrada.
A Tabela 2 apresenta os resultados desses componentes dos critérios de resposta ACR para o estudo AR ABP-Estudo 1. As taxas de resposta ACR e melhora em todos os componentes da resposta ACR demonstraram uma ausência de diferenças clinicamente significativas entre os dois grupos na semana 24.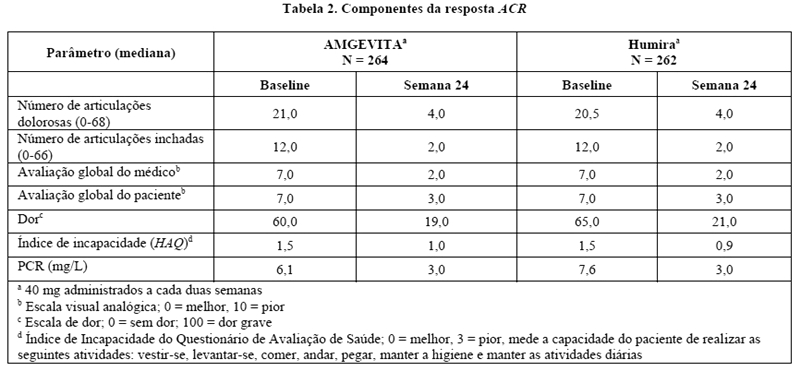
A Figura 1 mostra o curso de tempo de resposta ACR 20.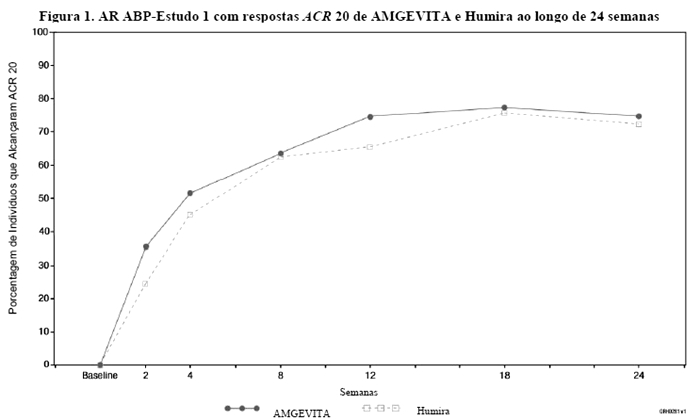
Dados de Eficácia de Humira
Artrite reumatóide
Humira foi avaliado em mais de 3.000 pacientes com artrite reumatoide (AR) em estudos clínicos. Alguns pacientes foram tratados por até 120 meses. A eficácia e a segurança de Humira foram avaliados em cinco estudos clínicos controlados, duplo-cegos e randomizados.1-5
O estudo I (ARMADA)1 avaliou 271 pacientes com artrite reumatoide moderada a grave, com mais de 18 anos de idade, que falharam ao tratamento com pelo menos uma droga modificadora da doença (DMARDs), com resposta insuficiente ao metotrexato em doses constantes de 12,5 a 25 mg/semana (ou 10 mg caso o paciente fosse intolerante ao metotrexato). Os pacientes apresentavam articulações edemaciadas ≥ 6 e articulações doloridas ≥ 9 e com AR diagnosticada de acordo com o critério ACR. Os pacientes receberam placebo ou 20, 40 ou 80 mg de Humira a cada 2 semanas, por 24 semanas, por via subcutânea (SC).
O estudo II (DE011)2 avaliou 544 pacientes com artrite reumatoide moderada a grave, com mais de 18 anos de idade, que falharam ao tratamento com pelo menos um DMARD (metotrexato, sulfassalazina, hidroxicloroquina, ouro oral ou injetável, d-penicilamina, azatioprina). Os pacientes apresentaram articulações edemaciadas ≥ 10 e articulações doloridas ≥ 12 e também diagnosticados de acordo com o critério ACR. Os pacientes foram divididos em 5 grupos: placebo semanal, Humira 20 mg + placebo semanal, Humira 40 mg + placebo semanal, Humira 20 mg + placebo a cada 2 semanas, Humira 40 mg + placebo a cada 2 semanas. Todos os pacientes receberam os tratamentos por via subcutânea (SC). A duração do estudo foi de 26 semanas.
O estudo III (DE019)3 avaliou 619 pacientes com artrite reumatoide moderada a grave, com mais de 18 anos de idade, com resposta insuficiente ao metotrexato em doses constantes semanais de 12,5 a 25 mg/semana (ou 10 mg caso o paciente fosse intolerante ao metotrexato). Diferente do estudo I, os pacientes com AR do estudo III não apresentavam falhas ao tratamento com pelo menos um DMARD. Os pacientes foram divididos em grupos: injeções de placebo semanalmente, injeções de Humira 20 mg semanalmente e injeções de Humira 40 mg a cada duas semanas + placebo nas semanas alternadas. Todos os pacientes receberam os tratamentos por via SC. A duração do estudo foi de 52 semanas. Após este período, os pacientes puderam entrar em um período de extensão aberto no qual se avaliou o uso de Humira 40 mg/metotrexato a cada 2 semanas, por via SC, por até 10 anos.6
O estudo IV (STAR)4 avaliou 636 pacientes com artrite reumatoide moderada a grave, com mais de 18 anos de idade. A população do estudo incluiu pacientes que nunca haviam usado DMARDs ou que estavam em tratamento com DMARDs estável por no mínimo 28 dias. Estes tratamentos incluíram leflunomida, hidroxicloroquina, sulfassalazina e/ou sais de ouro. Os pacientes foram randomizados para receberem Humira 40 mg ou placebo, por via SC, a cada 2 semanas, por 24 semanas.
O estudo V (PREMIER)5 avaliou 799 pacientes com artrite reumatoide de início recente (duração média dos sintomas de menos de 9 meses), moderada a grave, que nunca haviam usado metotrexato. O estudo avaliou a eficácia, a segurança e a progressão radiológica da destruição articular de Humira 40 mg + metotrexato a cada 2 semanas, Humira 40 mg a cada 2 semanas e monoterapia com metotrexato, por 104 semanas. Todos os tratamentos foram por via SC.
As medidas de desfechos primárias dos estudos I, II e III e a medida de desfecho secundária do estudo IV foram a porcentagem de pacientes que atingiu respostas ACR 20 nas semanas 24 ou 26 (diminuição de 20% dos critérios do American College of Rheumatology). A medida de desfecho primária do estudo V foi a porcentagem de pacientes que atingiu respostas ACR 50 (diminuição de 50% nos critérios do American College of Rheumatology) na semana 52. Os estudos III e V também tiveram a inibição da progressão da doença (medida por exames de raios-X) como medida de desfecho co-primária na semana 52. O estudo III também avaliou mudanças em escores de qualidade de vida como medida de desfecho co-primária.
Os principais resultados de eficácia destes estudos são apresentados a seguir.
Respostas ACR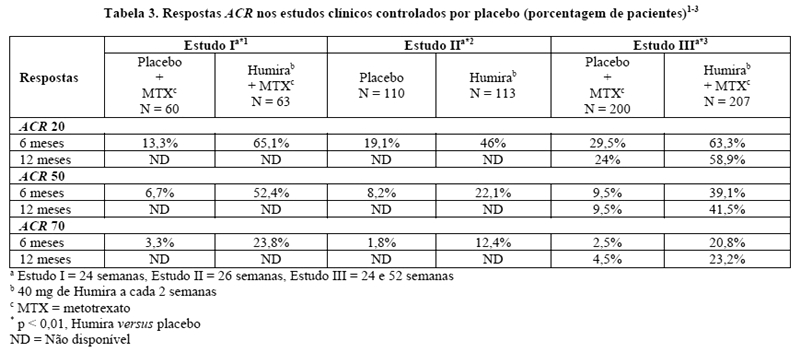
No estudo IV, a resposta ACR 20 dos pacientes tratados com Humira foi significativamente melhor do que os pacientes tratados com placebo (p < 0,001).4
Nos estudos I-IV todos os componentes individuais dos critérios de resposta ACR (número de articulações dolorosas, número de articulações edemaciadas, avaliações da atividade da doença e da dor pelo médico, avaliações da atividade da doença e da dor pelo paciente, escores do índice de incapacidade [HAQ - Health Assessment Questionnaire] e valores de PCR [proteína C-reativa] em mg/dL) melhoraram em 241,3,4 ou 26 semanas2, quando comparados ao placebo. No estudo III, estas melhoras foram mantidas ao longo de 52 semanas.3
Além disto, as taxas de respostas ACR foram mantidas na maioria dos pacientes seguidos na fase de extensão aberta do estudo III.114/207 pacientes continuaram com Humira 40 mg SC a cada 2 semanas por 60 meses. Destes, 65%, 58% e 35% apresentaram respostas ACR 20/50/70, respectivamente, no mês 60.6
Nos estudos I-V, os pacientes tratados com Humira atingiram melhores respostas ACR 20 e 50, quando comparados ao placebo, de forma estatisticamente significante, após 1 ou 2 semanas após o início do tratamento.1-5
No estudo V, o tratamento combinado de Humira com metotrexato, em pacientes com artrite reumatoide inicial, levou a respostas ACR maiores e mais rápidas do que as monoterapias com Humira ou metotrexato, na semana 52, mantidas na semana 104 (Tabela 4).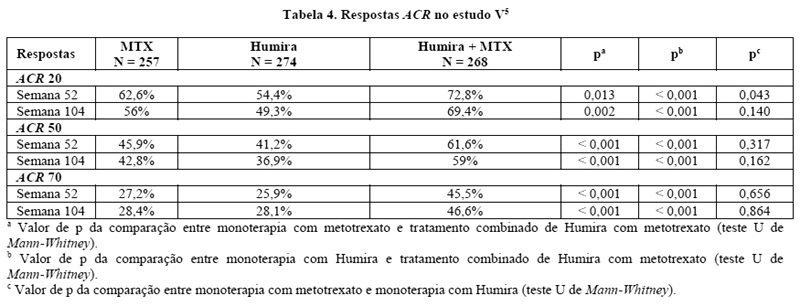
Na semana 52, 42,9% dos pacientes que receberam o tratamento combinado de Humira com metotrexato atingiram remissão clínica (DAS 28 < 2,6), comparados a 20,6% dos pacientes que receberam monoterapia com metotrexato e 23,4% dos que receberam monoterapia com Humira. O tratamento combinado de Humira com metotrexato foi superior às monoterapias com metotrexato e Humira (ambos p < 0,001) em atingir baixa atividade da doença em pacientes com artrite reumatoide recentemente diagnosticada de moderada a grave intensidade. A resposta entre ambas monoterapias foi semelhante (p = 0,447).5
Progressão radiográfica - No estudo III, no qual os pacientes tratados com Humira apresentaram uma duração média da artrite reumatoide de aproximadamente 11 anos, o dano articular estrutural foi avaliado radiograficamente e expresso por meio da mudança no Escore Total de Sharp modificado e seus componentes (escores de erosão e de estreitamento dos espaços articulares). Os pacientes tratados com Humira e metotrexato apresentaram significativamente menos progressão radiográfica do que os pacientes tratados apenas com metotrexato, após 6 e 12 meses (Tabela 5).3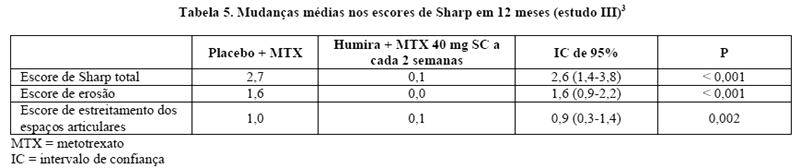
Dados da fase de extensão indicaram que a redução na taxa de progressão do dano estrutural é mantido por 60 meses em um subgrupo de pacientes. 113/207 pacientes originalmente tratados com Humira 40 mg SC a cada 2 semanas foram avaliados após 5 anos. Destes, 66 pacientes não mostraram nenhuma progressão do dano estrutural, definida por mudança no escore total de Sharp de zero ou menos.6
No estudo V, o dano estrutural foi avaliado radiograficamente e também expresso por meio das mudanças no Escore Total de Sharp modificado e seus componentes, de acordo com a Tabela 6.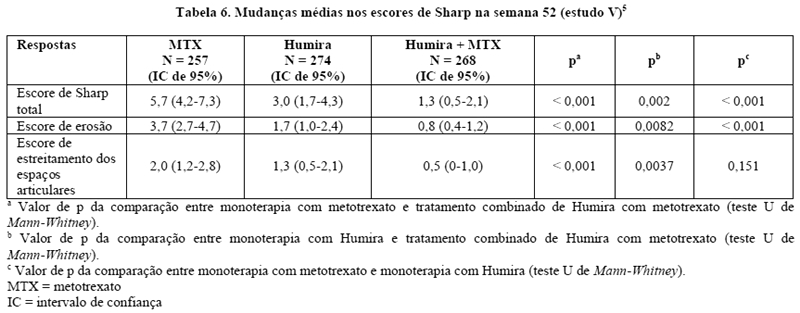
Após 52 e 104 semanas de tratamento, a porcentagem de pacientes sem progressão (mudança no escore total de Sharp modificado < 0,5) foi significativamente maior no grupo de tratamento combinado de Humira mais metotrexato (63,8% e 61,2%, respectivamente), quando comparado ao grupo que recebeu monoterapia com Humira (50,7%, p < 0,002, e 44,5%, p < 0,001, respectivamente) e monoterapia com metotrexato (37,4% e 33,5%, respectivamente, ambos p < 0,001).5
Qualidade de vida e função física - Qualidade de vida e função física foram avaliados pelo HAQ (Health Assessment Questionnaire), em todos os estudos de Humira, com placebo como comparador, sendo uma medida de desfecho co-primária no estudo III. Todos os grupos tratados com Humira apresentaram melhora significativamente maior que o placebo no índice de incapacidade do HAQ, após 6 meses, o mesmo acontecendo no estudo III após 52 semanas. Nestes estudos, uma melhora do componente físico do Short Form 36 (SF-36) também suporta estes achados. No estudo V, a melhora do índice de incapacidade do HAQ e do componente físico do SF-36 foi significativamente maior para o grupo tratado com Humira e metotrexato, quando comparada aos grupos tratados com monoterapia com Humira e metotrexato (p < 0,001).1-4
Uma diminuição significativa da fadiga, medida pelo escore FACIT (Functional Assessment of Chronic Illness Therapy) foi observada nos estudos I, III e IV, onde tal instrumento foi usado.1,3,5 No estudo III, a melhora da função física foi mantida por até 60 meses da fase de extensão aberta. A qualidade de vida foi medida até a semana 156 (36 meses) e a melhora foi mantida por este período.3
Artrite psoriásica em pacientes adultos
Humira 40 mg SC a cada duas semanas, foi avaliado em pacientes com artrite psoriásica moderada a grave em 2 estudos controlados por placebo. No estudo I, foram observados 313 pacientes adultos com resposta inadequada a anti-inflamatórios não esteroidais (AINEs), por 24 semanas.7 No estudo II, 100 pacientes com resposta inadequada a DMARDs foram observados por 12 semanas.8 Os pacientes de ambos os estudos puderam entrar em uma fase aberta, onde todos receberam Humira 40 mg SC a cada 2 semanas, por até 144 semanas.9,10
As respostas ACR no estudo I foram semelhantes com e sem tratamento concomitante com metotrexato (aproximadamente 50% dos pacientes foram tratados concomitantemente com metotrexato) (Tabela 7).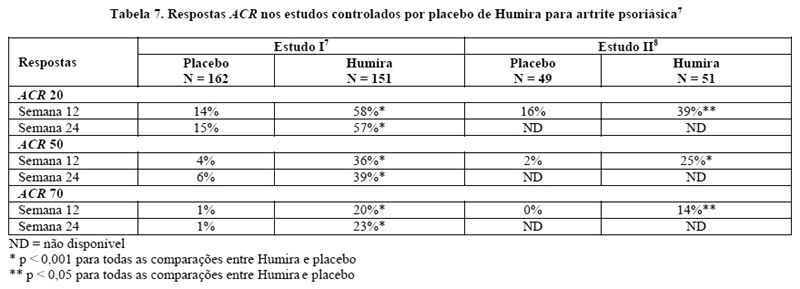
As respostas ACR foram mantidas na fase de extensão aberta por até 136 semanas.9
As mudanças radiográficas também foram avaliadas nos estudos de artrite psoriásica. Radiografias de mãos, punhos e pés foram obtidas no início do estudo e nas semanas 24 (fase duplo-cega do estudo I)7 e semana 48 (fase aberta).10 Um escore de Sharp modificado (mTSS), que incluiu as articulações interfalangianas distais, foi usado para medir a progressão radiográfica. Humira reduziu a taxa de progressão do dano articular periférico, quando comparado com o placebo (mudança média do mTSS = 0,8 ± 2,42 no grupo placebo na semana 24 comparado a 0,1 ± 1,95 no grupo tratado com Humira na semana 48, p < 0,001).9,10 Nos pacientes tratados com Humira sem progressão radiográfica do início do estudo até a semana 48 (n = 102), 84% continuaram a demonstrar ausência de progressão por até 144 semanas de tratamento.9,10
Os pacientes tratados com Humira demonstraram melhora significativa na função física, avaliada pelo HAQ e pelo SF-36 comparados aos pacientes que receberam placebo, na semana 24.7 A melhora da função física continuou durante a fase de extensão aberta até a semana 136.9
Espondilite anquilosante em pacientes adultos
Humira 40 mg SC a cada duas semanas foi avaliado em dois estudos duplo-cegos, placebo-controlados, de 24 semanas, em pacientes com espondilite anquilosante ativa, sem resposta adequada ao tratamento convencional.11-13 O período cego foi seguido de uma fase de extensão aberta, na qual os pacientes receberam apenas Humira 40 mg SC a cada 2 semanas.14
No estudo I, com 315 pacientes, os resultados apresentaram melhora significativa dos sinais e sintomas da espondilite anquilosante nos pacientes tratados com Humira, quando comparados aos tratados com placebo. Uma resposta significativa foi observada na semana 2, e mantida ao longo de 24 semanas (Tabela 8).11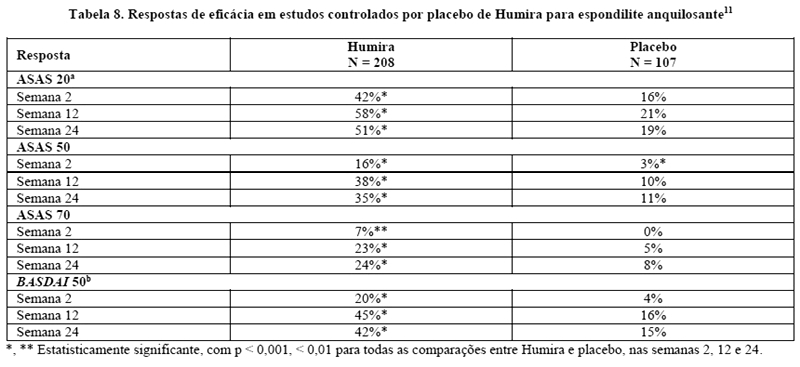
A melhora nas respostas ASAS e nos escores BASDAI foi mantida por até 2 anos.14
Pacientes tratados com Humira apresentaram melhora significativamente maior nos escores de dor, fadiga e rigidez,15 e nos escores de qualidade de vida (SF-36 e ASQoL - Questionário de Qualidade de Vida para Espondilite Anquilosante), quando comparados aos que receberam placebo, na semana 24.16
Tendências semelhantes (nem todas estatisticamente significantes) foram observadas no estudo II, realizado com 82 pacientes adultos com espondilite anquilosante ativa.12,13
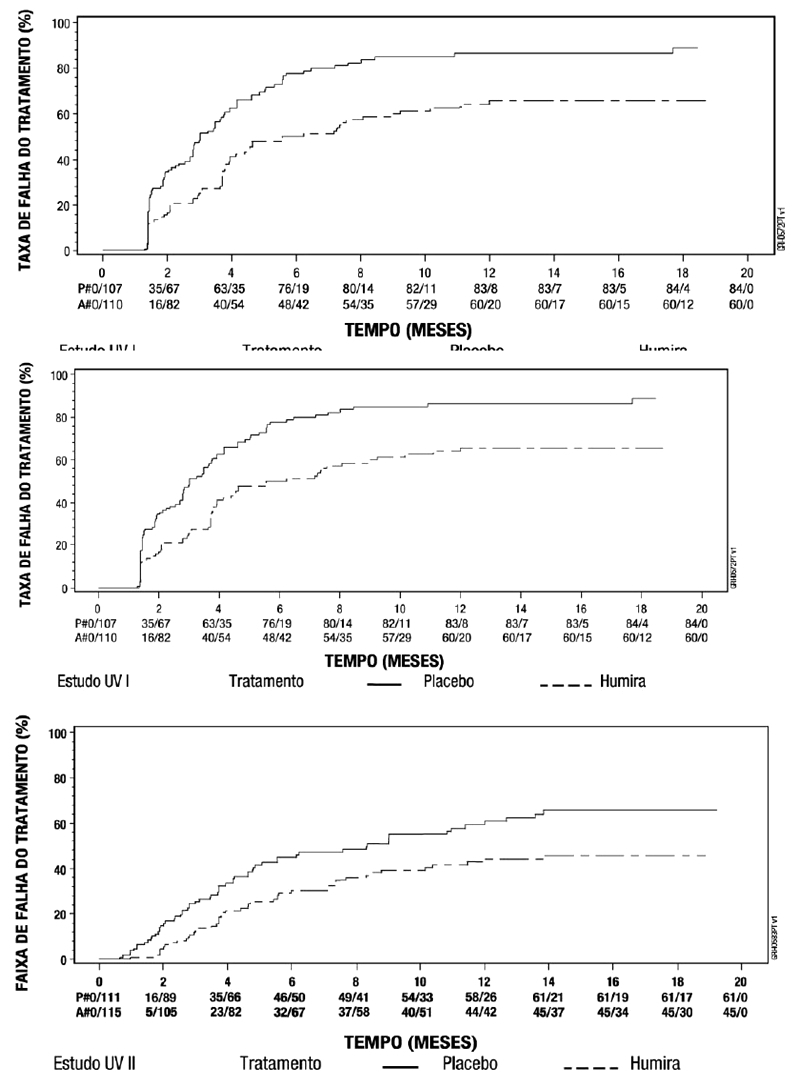
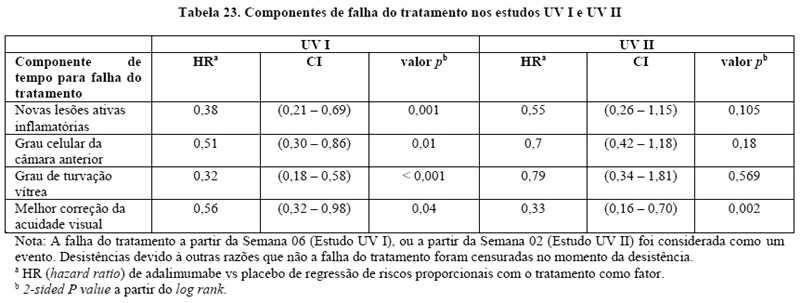
Espondilite axial não radiográfica (espondiloartrite axial sem evidência radiográfica de EA) em pacientes adultos
A segurança e eficácia de Humira foram avaliadas em dois estudos randomizados, duplo cego controlados por placebo, em pacientes com espondiloartrite axial não-radiográfica (nr-axSpA). O estudo nr-axSpA I avaliou pacientes com nr-axSpA ativa. O estudo nr-axSpA II foi um estudo de descontinuação de tratamento em pacientes com nr-axSpA ativa que atingiram a remissão durante o tratamento aberto com Humira.
Estudo nr-axSpA I
No estudo nr-axSpA I, Humira 40 mg a cada duas semanas foi avaliado em 185 pacientes por 12 semanas, em um estudo randomizado, duplo-cego, placebo-controlado, em pacientes com nr-axSpA ativa (média basal da atividade da doença [BASDAI - Bath Ankylosing Spondylitis Disease Activity Index] de 6,4 para pacientes tratados com Humira e 6,5 para pacientes recebendo placebo) que responderam inadequadamente ou que são intolerantes a ≥ 1 AINEs, ou que apresentam contraindicação a AINEs. Os pacientes incluídos foram classificados de acordo com o critério ASAS de EpA axial, excluindo pacientes que satisfizeram os critérios de New York modificados para espondilite anquilosante e aqueles com psoríase ou artrite psoriásica. O objetivo primário foi a porcentagem de pacientes que alcançaram o critério de resposta ASAS 40 na semana 12.17
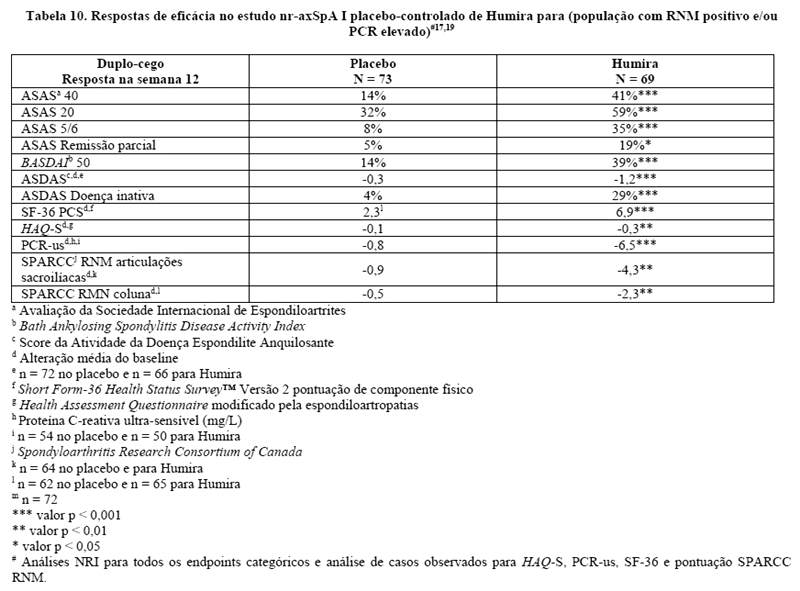
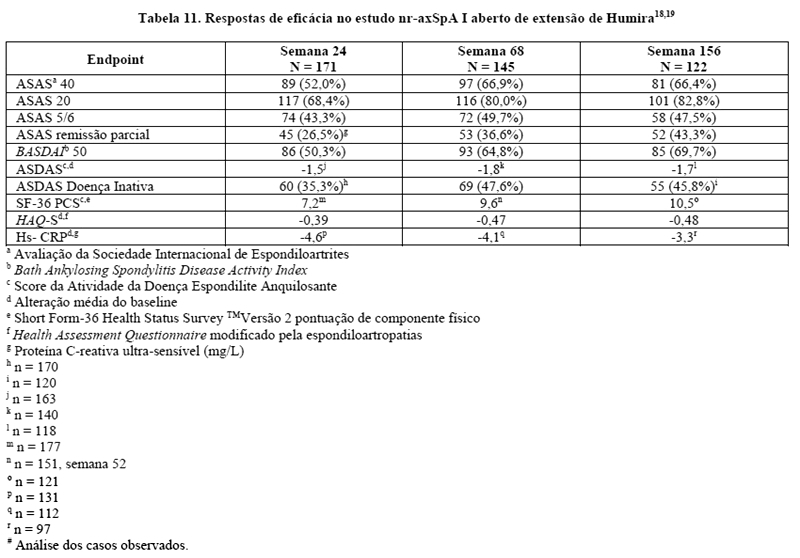
Trinta e três (18%) dos pacientes foram tratados concomitantemente com drogas modificadoras do curso da doença (DMARDs), e 146 (79%) dos pacientes, com AINEs no baseline. O período duplo-cego foi seguido de uma fase de extensão aberta, no qual, os pacientes receberam Humira 40 mg, por via subcutânea, a cada duas semanas por 144 semanas adicionais. A semana 12 mostrou uma melhora estatisticamente significante dos sinais e sintomas da nr-axSpA ativa em pacientes tratados com Humira comparado com placebo tanto na população geral quanto em pacientes com ressonância nuclear magnética (RNM) positivo ou PCR elevada (Tabelas 9 e 10). Variáveis que demonstram uma redução dos sinais e sintomas da nr-axSpA foram sustentadas ou continuaram a melhorar na semana 24 e na semana 68 e foram mantidas até a semana 156 (Tabelas 9 e 10).17-19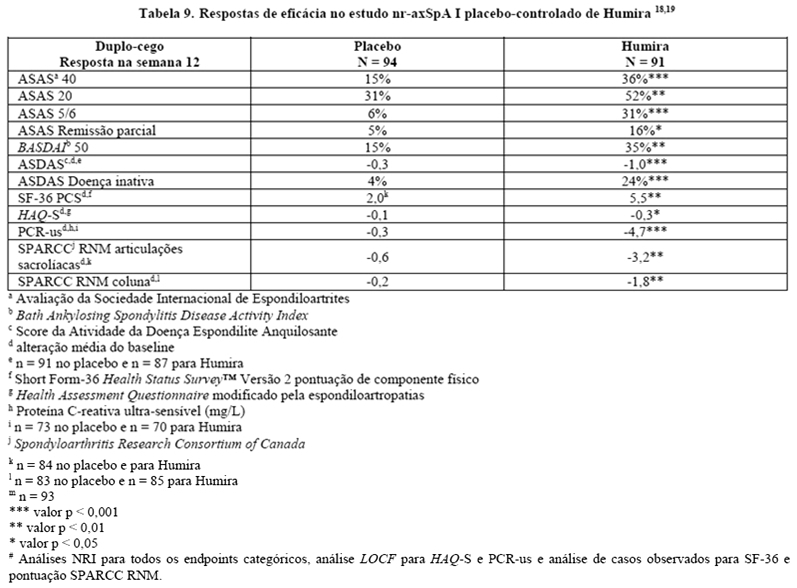
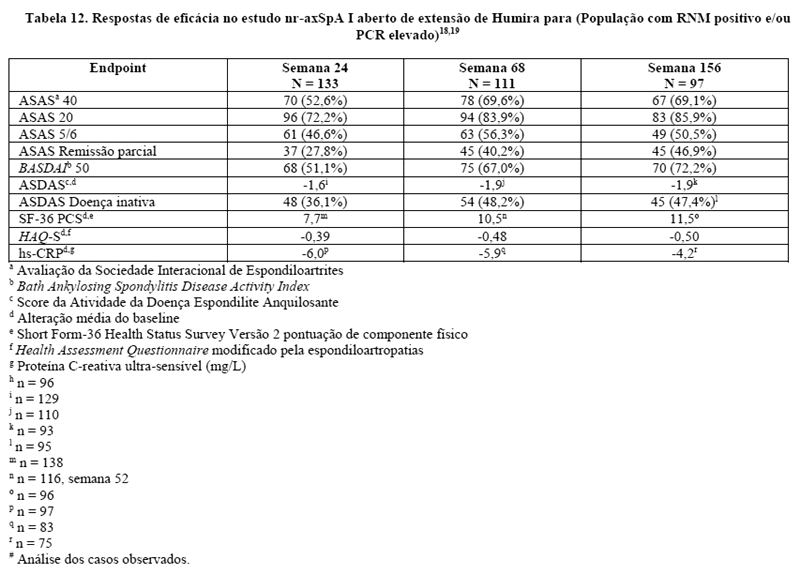
Inibição da inflamação
Foram mantidas melhoras significantes dos sinais da inflamação como medidos pelo hs-CRP e MRI para as articulações sacroilíacas e a coluna em pacientes tratados com Humira durante a semana 156 e semana 104, respectivamente. SPARCC MRI para articulações sacroilíacas estavam disponíveis para 131 pacientes e SPARCCC MRI para coluna estavam disponíveis para 130 pacientes com uma alteração média do baseline de -3,8 e -1,4, respectivamente, na semana 104.
Qualidade de vida e capacidade física
A qualidade de vida relacionada com a saúde e a capacidade física foram avaliadas através dos questionários HAQ-S e SF-36. Humira mostrou uma melhora estatisticamente significativa na nota total do HAQ-S e na pontuação do componente físico do SF-36 (PCS) do início até a semana 12 comparados com o placebo. Os resultados para SF-36 (PCS) e HAQ-S foram sustentados durante as semanas 52, 68 e 156, respectivamente.17-19
Estudo nr-axSpA II
673 pacientes com nr-axSpA ativa (atividade média base da doença [BASDAI] foi de 7,0) os quais tiveram uma resposta inadequada a ≥2 AINEs ou uma intolerância ou contraindicação para os AINEs incluídos no período de estudo aberto nr-axSpA II durante o qual receberam Humira 40 mg a cada duas semanas durante 28 semanas. Esses pacientes também apresentaram evidência objetiva de inflamação nas articulações sacroilíacas ou coluna vertebral na ressonância magnética ou elevação da PCR (proteina C reativa). Os pacientes que alcançaram remissão sustentada durante pelo menos 12 semanas (N = 305) (ASDAS < 1,3 nas semanas 16, 20, 24 e 28) durante o período aberto foram então aleatorizados para receberem tratamento continuado com Humira 40 mg a cada duas semanas (N = 152) ou placebo (N = 153) por um periodo adicional de 40 semanas em um periodo duplo-cego, controlado por placebo (duração total do estudo de 68 semanas). Pacientes que apresentaram flare da doença durante o período duplo-cego foram autorizados a terapia de resgate Humira 40 mg por pelo menos 12 semanas.
O desfecho primário de eficácia foi a proporção de pacientes sem agravamento na Semana 68 do estudo. O agravamento foi definido como ASDAS ≥2,1 em duas visitas consecutivas com quatro semanas de intervalo. Uma proporcao maior de pacientes em uso de Humira não apresentou agravamento da doença durante o período duplo-cego, quando comparados com os que receberam placebo (70,4% vs. 47,1%, p < 0,001) (Figura 2).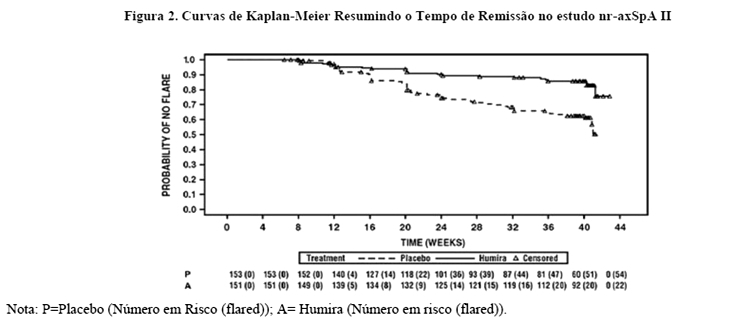
Entre os 68 pacientes que apresentaram flare da doença no grupo alocado para descontinuação do tratamento, 65 completaram 12 semanas de terapia de resgate com Humira, dos quais 37 (56,9%) haviam recuperado a remissão (ASDAS < 1,3) após 12 semanas de reinício do tratamento aberto.
Na Semana 68, os pacientes que receberam tratamento contínuo com Humira apresentaram melhora estatisticamente significativa maior dos sinais e sintomas de nr-axSpA ativa em comparação com os pacientes alocados para descontinuação do tratamento durante o período duplo-cego do estudo (Tabela 13).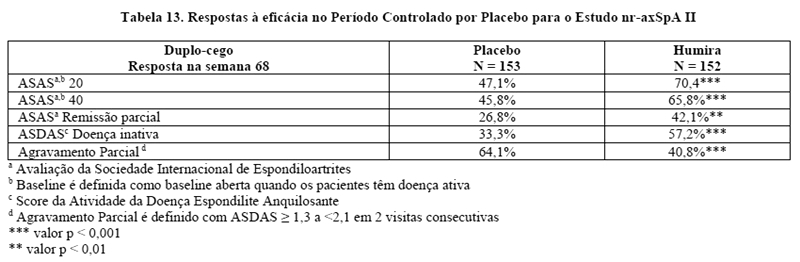
Doença de crohn em pacientes adultos
A segurança e a eficácia de Humira foram avaliadas em mais de 1.400 pacientes com doença de Crohn (DC) ativa, moderada a grave (Crohn's Disease Activity Index [CDAI] ≥ 220 e ≤ 450) em estudos duplo-cegos, randomizados, controlados por placebo. Nestes estudos foi permitido o uso concomitante de doses estáveis de aminossalicilatos, corticosteroides e/ou agentes imunomoduladores.
A indução de remissão clínica (definida como CDAI < 150) foi avaliada em dois estudos, estudo I20 de DC (M02-403) e estudo II21 de DC (M04-691). No estudo I20 de DC, 299 pacientes virgens de antagonistas de TNF foram randomizados para um de quatro grupos de tratamento: placebo nas semanas 0 e 2, 160 mg de Humira na semana 0 e 80 mg na semana 2, 80 mg na semana 0 e 40 mg na semana 2, e 40 mg na semana 0 e 20 mg na semana 2. No estudo II21 de DC, 325 pacientes que tinham perdido resposta ou eram intolerantes ao infliximabe foram randomizados para receber ou 160 mg Humira na semana 0 e 80 mg na semana 2 ou placebo nas semanas 0 e 2. Em ambos os estudos os resultados clínicos foram avaliados na semana 4.
Uma maior porcentagem de pacientes tratados com 160/80 mg de Humira alcançou a indução de remissão clínica, em comparação com o placebo na semana 4, independentemente dos pacientes serem virgens de tratamento com bloqueadores de TNF (estudo I20 de DC), ou terem perdido resposta ou terem sido intolerantes ao infliximabe (estudo II21 de DC) (Tabela 14).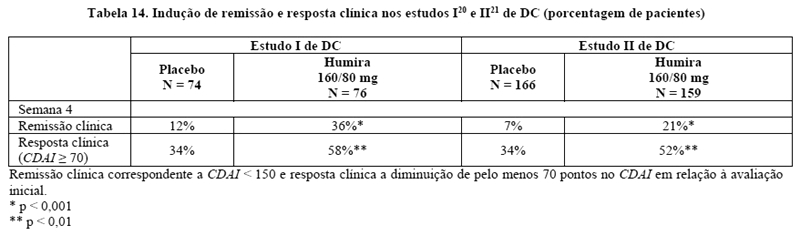
A manutenção da remissão clinica foi avaliada no estudo III22 de DC (M02-404). No estudo III22 de DC, 854 pacientes receberam de forma aberta 80 mg de Humira na semana 0 e 40 mg na semana 2. Na semana 4 os pacientes foram randomizados para receber 40 mg em semanas alternadas, 40 mg todas as semanas, ou placebo com uma duração total do estudo de 56 semanas. Pacientes com resposta clínica (CR-70 = diminuição do CDAI ≥ 70) na semana 4 foram estratificados e analisados separadamente daqueles sem resposta clínica na semana 4.
No estudo III22 de DC (CHARM), na semana 4, 58% (499/854) dos pacientes apresentavam resposta clínica e foram avaliados na análise primária. Os índices de manutenção da remissão e de resposta clínica estão representados na Tabela 15. Os índices de remissão clinica permaneceram relativamente constantes independentemente de uma exposição prévia a um antagonista de TNF.
Nos estudos I20 e II21 de DC, foi observada melhora estatisticamente significante na pontuação total do questionário específico para a doença inflamatória intestinal (IBDQ) alcançada na semana 4 nos pacientes randomizados para Humira 80/40 mg e 160/80 mg comparada a placebo. A melhora também foi vista nas semanas 26 e 56 no estudo III dwe DC entre os grupos de tratamento com Humira comparados com o grupo placebo. No estudo III, houve também uma diminuição estatisticamente significante de hospitalização e cirurgias relacionadas à doença quando comparada com o placebo na semana 56.23
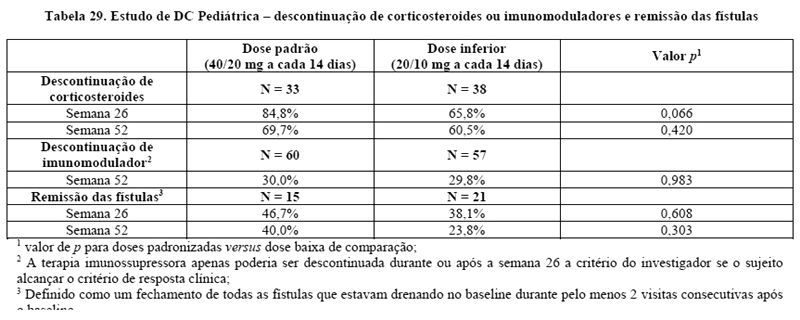
No Estudo I20, 117/276 pacientes com DC e 272/777 pacientes do estudo II21 e III22 foram acompanhados por pelo menos 3 anos em terapia aberta com Humira. Respectivamente 88 (75,2%) e 189 (69,5%) pacientes, continuaram com remissão clínica. A resposta clínica foi mantida em 107 (91,5%) e 248 (91,2%) pacientes, respectivamente. Os 117/854 pacientes (a partir de estudo DC III) apresentaram fístulas drenadas tanto na seleção como no baseline. Para a avaliação da cicatrização das fístulas, os dados de ambas as doses de Humira utilizados no estudo foram agrupados. A proporção de pacientes com cicatrização das fístulas na semana 26 foi estatística e significativamente maior em pacientes tratados com Humira (21/70 [30,0%]) em comparação com placebo (6/47 [12,8%]). A cicatrização completa das fístulas foi mantida até a semana 56 em 23/70 (32,9%) pacientes no grupo com Humira e 6/47 (12,8%) no grupo placebo. Um estudo endoscópico (M05-769) que envolveu 135 pacientes, indicou um efeito de Humira na cicatrização da mucosa. 27,4% dos pacientes tratados com Humira tinham cicatrização da mucosa na semana 12 comparados com 13,1% dos pacientes-placebo (p = 0,056), e 24,2% dos pacientes tratados com Humira na semana 52 contra 0% dos pacientes-placebo (p < 0,001).24
Colite ulcerativa ou retocolite ulcerativa em pacientes adultos
A segurança e eficácia de múltiplas doses de Humira foi testada em pacientes adultos com colite ulcerativa ou retocolite ulcerativa ativa moderada a grave (escore Mayo de 6 a 12 e com subtotal de endoscopia de 2 a 3 pontos) em dois estudos randomizados, duplo-cego, placebo-controlados. Os pacientes tinham de ter um diagnóstico de colite ulcerativa por mais de 90 dias, confirmado por endoscopia. Eles tinham de ter a doença ativa apesar do tratamento com pelo menos um dos seguintes corticosteroides orais ou imunossupressores: prednisona, azatioprina ou 6-mercaptopurina. Os pacientes foram excluídos da participação nos estudos se eles tinham uma história de colectomia subtotal com ileostomia ou proctocolectomia com reservatório ileal e anastomose ileoanal, bolsa de Koch ou ileostomia para retocolite ulcerativa ou se estava planejando uma cirurgia intestinal, ou se eles tinham um diagnóstico vigente de colite fulminante e/ou megacólon tóxico, colite indeterminada, ou doença de Crohn, se sua doença estava limitada ao reto (proctite ulcerativa), ou se eles estavam recebendo nutrição parenteral. Pacientes com Clostridium difficile positivo no exame de fezes, infecções que requerem tratamento intravenoso, que tinha um histórico de malignidade tratada com sucesso diferente de carcinoma cutâneo de células escamosas não metastáticas ou basocelular e/ou carcinoma localizado no colo do útero, ou uma história de listeria, histoplasmose, infecção crônica ou ativa da hepatite B, vírus da imunodeficiência humana, síndrome da imunodeficiência, doenças desmielinizantes do sistema nervoso central, ou tuberculose não tratada (TB) também foram excluídos, bem como os pacientes cuja endoscopia mostrou evidencias de displasia ou malignidade. No estudo UC-I25, 390 pacientes que nunca foram tratados com antagonistas de TNF foram randomizados para receber: placebo nas semanas 0 e 2 ou 160 mg de Humira na semana 0 seguido por 80 mg na semana 2, ou 80 mg de Humira na semana 0 seguindo por 40 mg na semana 2. Depois da semana 2, pacientes que receberam Humira nas semanas anteriores, receberam 40 mg de Humira a cada 14 dias. A remissão clínica (definida como escore Mayo ≤ 2 sem subtotal > 1) foi avaliada na semana 8.
No estudo UC-II26, 248 pacientes receberam 160 mg de Humira na semana 0, 80 mg na semana 2 e 40 mg a cada 14 dias nas semanas seguintes, e, 246 pacientes receberam placebo. Os resultados clínicos foram avaliados para indução de remissão na semana 8 e para manutenção da remissão na semana 52.
Indivíduos induzidos com 160/80 mg de Humira atingiram a remissão clínica versus o placebo na semana 8 em porcentagens estatística e significativamente maiores no estudo UC-I (18% versus 9%, respectivamente, p = 0,031) e no estudo UC-II (17% versus 9%, respectivamente, p = 0,019). No estudo UC-II, entre os tratados com Humira que estavam em remissão na semana 8, 21/41 (51%) estavam em remissão na semana 52. Os resultados do estudo UC-II são apresentados na Tabela 16 tanto para população total quanto para pacientes que tinham respondido na semana 8 de tratamento por escore total Mayo.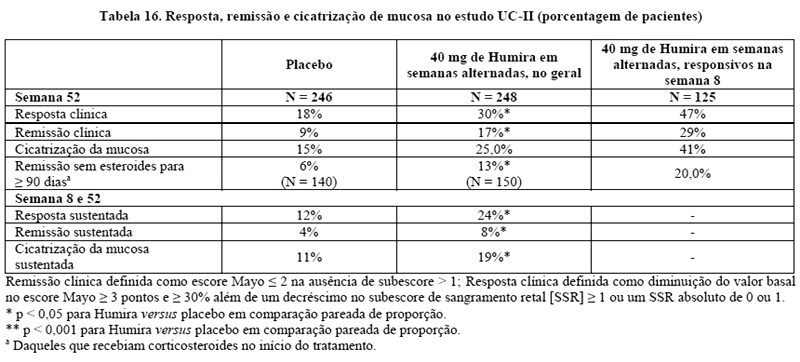
Aproximadamente 40% dos pacientes do estudo UC-II apresentaram falha da terapia primeiro com o tratamento com anti-TNF infliximabe. A eficácia de Humira nestes pacientes foi reduzida, quando comparada com os pacientes que não tiveram um tratamento prévio com anti-TNF. Entre estes pacientes que haviam falhado no tratamento prévio com anti-TNF, a remissão foi alcançada na semana 52 por 3% no grupo placebo e 10% no grupo com Humira.
Os pacientes dos estudos UC-I e UC-II tiveram a opção de continuar um estudo aberto de extensão de longo prazo (UC-III). Após três anos de tratamento com Humira, 74% (268/360) continuaram em remissão clínica por escore parcial Mayo.
Qualidade de vida
No estudo UC-II, uma melhora maior na nota total do questionário de doença específica para doença inflamatória intestinal (IBDQ) foi alcançada na semana 52 em pacientes randomizados para 160/80 mg de Humira comparado com placebo (p = 0,007).
Psoríase em placas em pacientes adultos
Dados de Eficácia de AMGEVITA
Estudo comparativo de Psoríase em Placas (Ps) entre AMGEVITA e Humira (Ps ABP-Estudo 1) - A eficácia e a segurança de AMGEVITA foram avaliadas em um estudo duplo-cego, randomizado e controlado por medicamento em 350 pacientes ≥ 18 anos de idade com psoríase em placas (Ps) de moderada a severa que eram candidatos para terapia sistêmica ou fototerapia. Os pacientes apresentavam Ps em placas estável de moderada a severa há pelo menos 6 meses, uma área de superfície corporal (ASC) ≥ 10%, e Índice da Área e Gravidade da Psoríase (PASI) ≥ 12 ao ingressar no estudo. Os pacientes receberam AMGEVITA ou Humira a uma dose inicial de 80 mg administrados SC na semana 1/dia 1, seguido de 40 mg SC administrados a cada duas semanas, a partir de uma semana após a dose inicial. A melhora percentual no PASI a partir do baseline foi medida e comparada com Humira (Tabela 17) e ficou dentro da margem de equivalência pré-especificada para demonstrar equivalência entre AMGEVITA e Humira.
O endpoint primário foi a melhora percentual no PASI a partir do baseline até a semana 16. Na semana 16, a melhora percentual no PASI a partir do baseline foi 80,9 no grupo de AMGEVITA e 83,1 no grupo de Humira. A diferença média dos mínimos quadrados (LS) da melhora percentual no PASI a partir do baseline até a semana 16 entre AMGEVITA e Humira foi -2,18 com IC de 95% bilateral de (-7,39, 3,02). O IC de 95% ficou dentro da margem de equivalência pré-definida, demonstrando, portanto, equivalência clínica entre AMGEVITA e Humira.
Depois que os indivíduos com resposta PASI 50 (melhora de 50% ou melhor) foram novamente randomizados na semana 16 para continuar no estudo, foram observados resultados semelhantes na semana 50 (fim do estudo), onde a melhora percentual no PASI a partir do baseline foi semelhante entre os grupos de tratamento: AMGEVITA/AMGEVITA foi 87,16, Humira/Humira foi 88,11, e Humira/AMGEVITA foi 85,82.
A Figura 3 mostra a melhora percentual no PASI a partir do baseline ao longo da duração do estudo.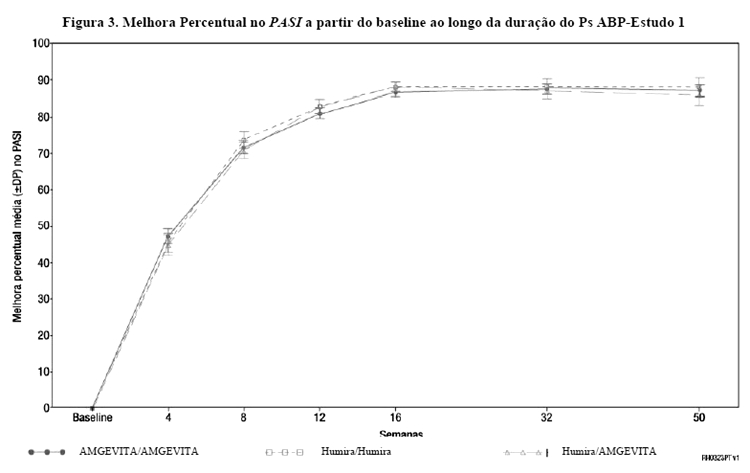
Dados de eficácia de Humira
A segurança e eficácia de Humira foram avaliadas em estudos duplo-cegos, randomizados, realizados em pacientes adultos com psoríase crônica em placas (envolvimento ≥ 10% BSA e Psoriasis Area and Severity Index (PASI) ≥ 12 ou ≥ 10) que eram candidatos a terapia sistêmica ou fototerapia. 73% dos pacientes envolvidos nos estudos de psoríase fase I e II receberam terapia sistêmica prévia ou fototerapia. A segurança e eficácia de Humira também foram avaliadas em estudos duplo-cegos, randomizados, realizado em pacientes adultos com psoríase crônica em placas moderada a grave com acometimento das mãos e/ou pés que eram candidatos a terapia sistêmica.27
O estudo I28 de psoríase (M03-656) avaliou 1.212 pacientes durante três períodos de tratamento. No período A, os pacientes receberam placebo ou Humira na dose inicial de 80 mg seguida por 40 mg em semanas alternadas começando na semana 1, após