ZALTRAP
SANOFI MEDLEY
aflibercepte
Tratamento da DMAI.
Apresentações.
Solução concentrada para diluição para infusão 100 mg/4 mL: Embalagem com 1 frasco-ampola de 4 mL.
Solução concentrada para diluição para infusão 200 mg/8 mL: Embalagem com 1 frasco-ampola de 8 mL.
USO INTRAVENOSO
USO ADULTO
Composição.
Cada mL de ZALTRAP contém 25 mg de aflibercepte.
Excipientes: sacarose, cloreto de sódio, citrato de sódio di-hidratado, ácido cítrico monoidratado, polissorbato 20, fosfato de sódio dibásico heptaidratado, fosfato de sódio monobásico monoidratado, hidróxido de sódio e/ou ácido clorídrico e água para injetáveis.
Informações técnicas.
1. INDICAÇÕES
ZALTRAP, em combinação com 5-fluoruracila, leucovorina, irinotecano (FOLFIRI), é indicado para pacientes com câncer colorretal metastático (CCRM) resistentes a ou que tenham progredido após um esquema contendo oxaliplatina (vide "Resultados de eficácia").
2. RESULTADOS DE EFICÁCIA
A eficácia e segurança de ZALTRAP foram avaliadas em um estudo randomizado, duplo-cego, placebo-controlado em pacientes com câncer colorretal metastático que haviam sido tratados previamente a base de oxaliplatina com ou sem bevacizumabe prévio. Um total de 1226 pacientes foram randomizados (1:1) para receber ZALTRAP (N=612; 4 mg/kg como uma infusão IV de 1 hora no dia 1) ou placebo (N=614) em combinação com 5-fluoruracila mais irinotecano [FOLFIRI: infusão IV de 180 mg/m² de irinotecano por 90 minutos e infusão IV de 400 mg/m² de leucovorina (ácido folínico) (dl racêmico) por 2 horas ao mesmo tempo no dia 1 usando linha Y, seguido por bolus IV de 400 mg/m² de 5-FU, seguido por infusão IV contínua 2400 mg/m² de 5-FU por 46 horas].Os ciclos de tratamento em ambos os braços do estudo foram repetidos a cada 2 semanas. Os pacientes foram tratados até a progressão da doença ou toxicidade inaceitável. O desfecho primário de eficácia foi a sobrevida global. A atribuição do tratamento foi estratificada pelo status do desempenho ECOG (0 versus 1 versus 2) e de acordo com a terapia prévia com bevacizumabe (sim ou não).
Os dados demográficos foram equilibrados entre os dois braços de tratamento (idade, raça, status do desempenho ECOG e utilização de bevacizumabe previamente. Dos 1226 pacientes randomizados no estudo, a idade mediana foi de 61 anos, 58,6% eram do sexo masculino e 97,8% tinham um nível basal de índice ECOG SP (status performance) de 0 ou 1. Entre estes 1226 pacientes randomizados, 89,4% e 90,2% dos pacientes tratados com regimes placebo/FOLFIRI e ZALTRAP/FOLFIRI respectivamente, receberam quimioterapia combinada prévia baseada em oxaliplatina em conjuntos avançados/metastáticos. Aproximadamente 10% dos pacientes (10,4% e 9,8% dos pacientes tratados com regimes placebo/FOLFIRI e ZALTRAP/FOLFIRI respectivamente) receberam quimioterapia adjuvante prévia baseada em oxaliplatina e progredido em ou dentro de 6 meses da conclusão da quimioterapia adjuvante. Regimes baseados em oxaliplatina foram administrados em combinação com bevacizumabe em 373 pacientes (30,4%).
Resultados de eficácia global para regime ZALTRAP/FOLFIRI versus regimes placebo/FOLFIRI estão resumidos na tabela 1.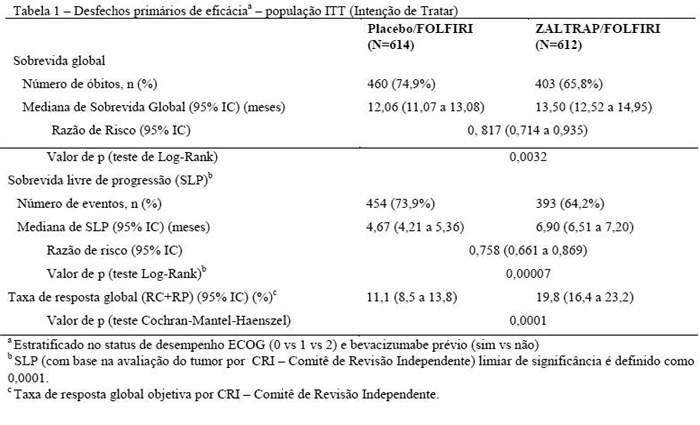
Foram realizadas análises da sobrevida global (SG) e da sobrevida livre de progressão (SLP) pelos fatores de estratificação. Um efeito de tratamento consistente para SG foi observado em favor de pacientes tratados com regime ZALTRAP/FOLFIRI para pacientes tratados com o uso prévio de bevacizumabe bem como em pacientes sem uso prévio de bevacizumabe. Os resultados para exposição prévia ao bevacizumabe estão resumidos na Tabela 2.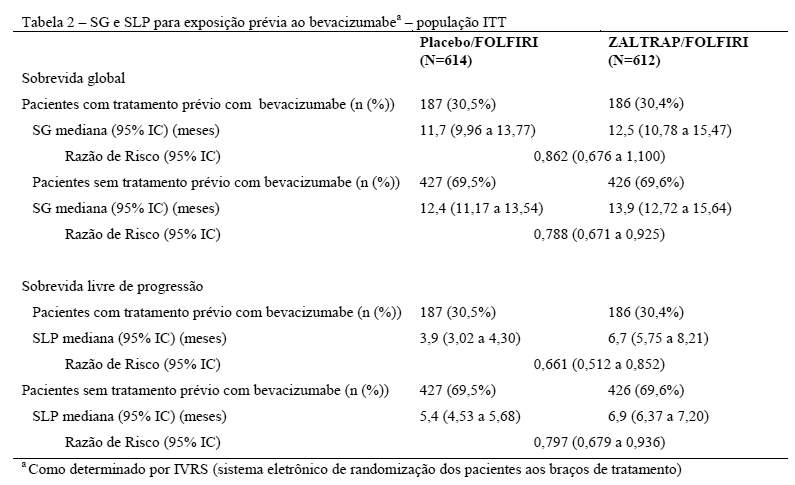
A análise para SG e SLP por ECOG SP também foi realizada. A razão de risco (95% IC) da sobrevida global foi de 0,77 (0,64 a 0,93) para status de performance ECOG 0 e 0,87 (0,71 a 1,06) para status de performance 1. A razão de risco (95% IC) da sobrevida livre de progressão foi de 0,76 (0,63 a 0,91) para status de performance ECOG 0 e 0,75 (0,61 para 0,92) para status de performance ECOG 1.
Estão resumidas na Tabela 3 as análises post-hoc excluindo pacientes que progrediram durante ou dentro dos 6 meses de terapia adjuvante por pacientes com ou sem tratamento prévio com bevacizumabe.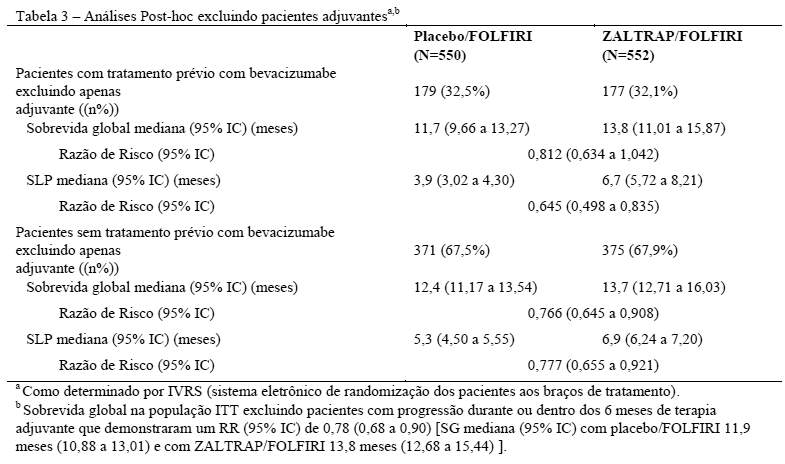
Análises de subgrupos para sobrevida global e sobrevida de progressão livre de acordo com a idade ( < 65 anos;≥ 65 anos), gênero, uso prévio de bevacizumabe, índice ECOG SP 0 e 1, presença somente de metástase hepática, histórico de hipertensão prévia e número de órgãos envolvidos mostraram um efeito de tratamento que favorece o regime ZALTRAP/FOLFIRI sobre o regime placebo/FOLFIRI.
Nas análises de subgrupo de sobrevida global, foi observado um benefício consistente com a população global em pacientes < 65 anos e ≥65 anos que receberam o regime ZALTRAP/FOLFIRI.
Em uma análise retrospectiva do estudo VELOUR baseada na mutação do gene RAS, em 482 dos 1226 pacientes (aproximadamente 39%; n = 240 aflibercepte; 242 placebo), não houve evidência de heterogeneidade no efeito do tratamento (teste de interação não-significante).
O RR (95% IC) da sobrevida global (SG) foi de 0,696 (0,501 - 0,967) com a mediana de SG de 16 meses (95% CI: 12,7 - 22,8) para pacientes com tumor RAS tipo selvagem tratados com aflibercepte e 11,7 meses (10,1 - 15,9) para os pacientes tratados com placebo.
O RR (95%) da SG foi 0,926 (0,698 - 1,23) com mediana de SG de 12,6 meses (95% IC: 10,7 - 14,5) para pacientes com tumor RAS mutante tratados com aflibercepte e 11,2 meses (9,9 - 13,8) para pacientes tratados com placebo.
Referências bibliográficas
Rougier P, Riess H, Manges R, et al. Randomised, placebo-controlled, double-blind, parallel-group phase III study evaluating aflibercept in patients receiving first-line treatment with gemcitabine for metastatic pancreatic cancer. European Journal of Cancer (2013) 49, 2633- 2642.
Cutsem EV, Tabernero J, Lakomy R, et al. Addition of Aflibercept to Fluorouracil, Leucovorin, and Irinotecan Improves Survival in a Phase III Randomized Trial in Patients With Metastatic Colorectal Cancer Previously Treated With an Oxaliplatin-Based Regimen. J Clin Oncol. 2012 Oct 1;30(28):3499-506.
Ramlau R, Gorbunova V, Ciuleanu TE, et al. Aflibercept and Docetaxel Versus Docetaxel Alone After Platinum Failure in Patients With Advanced or Metastatic Non-Small-Cell Lung Cancer: A Randomized, Controlled Phase III Trial. J Clin Oncol. 2012 Oct 10;30(29):3640-7.
Allegra CJ, Rumble RB, Schilsky RL. Extended RAS Gene Mutation Testing in Metastatic Colorectal Carcinoma to Predict Response to Anti-Epidermal Growth Factor Receptor Monoclonal Antibody Therapy: American Society of Clinical Oncology Provisional Clinical Opinion Update 2015 Summary. J Oncol Pract. 2016;12(2):180-1.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Grupo farmacoterapêutico: Agentes antineoplásicos, outros agentes antineoplásicos. Código ATC: L01XX44.
Propriedades farmacodinâmicas
-Mecanismo de ação
Os fatores de crescimento endotelial vascular tipo A e B (VEGF-A, VEGF-B) e o fator de crescimento placentário (PIGF) são membros da família VEGF de fatores angiogênicos que podem atuar como potentes fatores mitogênicos, quimiotáxicos e como fatores de permeabilidade vascular para células endoteliais. VEGF-A atua através de dois receptores tirosina quinase, VEGFR-1 e VEGFR-2, presentes na superfície das células endoteliais. PIGF e VEGF-B ligam-se apenas ao VEGFR-1, que estão presentes também na superfície dos leucócitos. A ativação excessiva destes receptores pelo VEGF-A pode resultar na neovascularização patológica e permeabilidade vascular excessiva. PIGF também está ligado à neovascularização patológica e recrutamento de células inflamatórias em tumores.
O aflibercepte, também conhecido como VEGF TRAP na literatura científica, é uma proteína recombinante fundida que consiste de porções para a ligação VEGF de domínios extracelulares dos receptores VEGF 1 e 2 humanos fundidos à porção Fc de IgG1 humana. O aflibercepte atua como um receptor atrativo solúvel que se liga ao VEGF-A, com afinidade superior ao receptor natural, bem como os ligantes PIGF e VEGF-B. Atuando como uma armadilha ligante, o aflibercepte evita que os ligantes endógenos se liguem aos respectivos receptores e assim bloqueia a mediação do sinal no receptor.
O aflibercepte bloqueia a ativação dos receptores VEGF e a proliferação de células endoteliais, inibindo desse modo o crescimento de novos vasos que suprem os tumores com oxigênio e nutrientes.
O aflibercepte se liga ao VEGF-A humano (constante de equilíbrio de dissociação KD de 0,5 pM para VEGF A165 e 0,36 pM para VEGF A121), ao PIGF humano (KD de 39pM para PIGF-2) e ao VEGF-B humano (KD de 1,92 pM) para formar um complexo estável e inerte que tem atividade biológica não detectável.
-Características farmacodinâmicas
A administração de aflibercepte a camundongos com tumores xenotransplantados ou alotransplantados inibiu o crescimento de vários tipos de cânceres.
Propriedades farmacocinéticas
Ambas farmacocinéticas não-clínica e clínica foram avaliadas para aflibercepte.
A análise farmacocinética populacional foi realizada com dados de 1507 pacientes com vários tipos de neoplasias malignas avançadas, os quais receberam aflibercepte em monoterapia ou em combinações nas faixas de doses de 2 a 9 mg/kg administrados a cada 2 a 3 semanas como infusão intravenosa de 1 hora. As concentrações plasmáticas de aflibercepte ligado ou livre foram medidas usando um método de ensaio imuno-enzimático (ELISA) específico.
-Absorção
Em modelos tumorais pré-clínicos, doses biologicamente ativas de aflibercepte correlacionaram-se com aquelas necessárias para produzir concentrações circulantes de aflibercepte livre em excesso de aflibercepte ligado a VEGF. Concentrações circulantes de aflibercepte ligado a VEGF aumentam com a dose de aflibercepte até que a maioria disponível de VEGF se ligue. Elevações adicionais da dose de aflibercepte levam ao aumento relacionado à dose nas concentrações de aflibercepte livre circulante, mas levam apenas a pequenas elevações adicionais na concentração de aflibercepte ligado a VEGF.
Em pacientes, ZALTRAP é administrado em doses IV de 4 mg/kg a cada 2 semanas para que haja um excesso de aflibercepte circulante livre comparado ao aflibercepte ligado ao VEGF. Consistente com objetivo mediado de disponibilidade do medicamento, aflibercepte livre exibe clearance não linear na dose abaixo de 2mg/kg, provavelmente devido à alta afinidade de ligação de aflibercepte ao VEGF endógeno. O clearance linear observado na faixa de dose de 2 a 9 mg/kg é provavelmente devido ao mecanismo de eliminação biológico não saturável, tal como o catabolismo proteico.
No regime de dose recomendado de 4 mg/kg a cada duas semanas, a concentração de aflibercepte livre está próxima dos níveis de estado de equilíbrio no segundo ciclo, essencialmente sem acúmulo (taxa de acúmulo de 1,2 no estado de equilíbrio comparado com a primeira administração).
-Distribuição
O volume de distribuição de aflibercepte livre no estado de equilíbrio é de 8 L.
-Metabolismo
Não foram conduzidos estudos de metabolismo com aflibercepte uma vez que se trata de uma proteína. Espera-se que aflibercepte seja degradado em pequenos peptídeos e aminoácidos individuais.
-Eliminação
O aflibercepte livre é primariamente eliminado ligado ao VEGF endógeno para formar um complexo inerte e estável. Como nas demais proteínas grandes, ambas as formas de aflibercepte ligado ou livre devem ser eliminados mais lentamente por outros mecanismos biológicos tais como o catabolismo proteolítico. O aflibercepte ligado a VEGF é eliminado sem qualquer grau apreciável de dissociação reversível ou de formação de imunocomplexos de maior ordem.
Em doses superiores de 2mg/kg, o clearance de aflibercepte livre foi de 1,0 L/dia com meia-vida de 6 dias.
Proteínas de alto peso molecular não são depuradas por via renal, portanto a eliminação renal de aflibercepte deverá ser mínima.
POPULAÇÕES ESPECIAIS
-Crianças
Após a administração intravenosa de ZALTRAP 2,0 mg/kg, 2,5 mg/kg, ou de 3,0 mg/kg a cada duas semanas a 8 pacientes pediátricos com tumores sólidos (com idade de 5 a 17 anos), a meia-vida de eliminação média de aflibercepte livre, determinada após a primeira dose, foi de aproximadamente 4 dias (intervalo de 3-6 dias).
-Idosos
Não há efeito da idade na farmacocinética de aflibercepte.
-Sexo
Apesar de diferenças no clearance de aflibercepte livre e no volume de distribuição entre homens e mulheres, não foi observada nenhuma diferença de exposição do medicamento relacionado ao sexo na dose de 4 mg/kg no estudo pivotal.
-Peso
O peso teve um efeito no clearance de aflibercepte livre e no volume de distribuição, levando ao aumento de 29% na exposição do medicamento em pacientes com peso ≥100 kg.
-Raça
Não há efeito de grupos étnicos e da raça na farmacocinética de aflibercepte.
-Insuficiência hepática
Não houve estudo clínico formal de ZALTRAP em pacientes com insuficiência hepática.
Em uma análise de farmacocinética populacional com dados de 1507 pacientes, com vários tipos de neoplasias malignas avançadas recebendo ZALTRAP com ou sem quimioterapia, 63 pacientes com insuficiência hepática leve (bilirrubina total > 1,0 x - 1,5 x ULN e qualquer AST) e 5 pacientes com insuficiência hepática moderada (bilirrubina total > 1,5 x - 3x ULN e qualquer AST) foram tratados com ZALTRAP. Não houve efeito no clearance de aflibercepte nos pacientes com insuficiência hepática leve e moderada. Não há dados disponíveis para pacientes com insuficiência hepática severa (bilirrubina total > 3x ULN e qualquer AST).
-Insuficiência renal
Não houve estudo clínico formal de ZALTRAP em pacientes com insuficiência renal.
Foi conduzida uma análise de farmacocinética populacional com dados de 1507 pacientes, com vários tipos de neoplasias malignas avançadas, recebendo ZALTRAP com ou sem quimioterapia. Esta população incluía 549 pacientes com insuficiência renal leve (CLCR entre 50-80 mL/min), 96 pacientes com insuficiência renal moderada (CLCR entre 30-50 mL/min) e 5 pacientes com insuficiência renal severa (CLCR < 30 mL/min). Esta análise de farmacocinética populacional não revelou diferenças na exposição sistêmica (AUC) de aflibercepte livre entre pacientes de vários graus de insuficiência renal na dose de 4 mg/mL de ZALTRAP.
Dados de segurança pré-clínicos
-Farmacologia em animais
A administração de aflibercepte levou a um retardo na cicatrização de feridas em coelhos. Em modelos de feridas cutâneas excisionais e incisionais a administração de aflibercepte reduziu a resposta fibrótica, neovascularização, hiperplasia epidermal /re-epitelialização e resistência a tensão.
O aflibercepte não exacerbou a formação de trombos venosos e arteriais em coelhos.
O aflibercepte aumentou a pressão arterial em roedores normotensos.
-Toxicidade aguda
Uma única injeção intravenosa de aflibercepte a 50, 150 ou 500 mg/kg em ratos resultou em lesões mínimas (vermelhidão, inchaço e/ou crostas) no local da injeção e redução moderada no ganho de peso corpóreo e consumo de alimento.
-Toxicidade crônica
A administração IV de aflibercepte semanal ou a cada 2 semanas em macacos cynomolgus (sexualmente maduros) por até 6 meses resultou em alterações ósseas (efeitos sobre a placa de crescimento e no esqueleto axial e apendicular), cavidade nasal, rins, ovários e glândula adrenal.
Os principais achados relacionados ao aflibercepte foram observados a partir da menor dose testada correspondente a exposições plasmáticas próximas àquelas dos pacientes na dose terapêutica.
Em outro estudo em macacos cynomolgus sexualmente imaturos (tratados por IV por 3 meses), foram observados efeitos similares em exposições inferiores à dos pacientes na dose terapêutica.
Em ambos os macacos maduros e imaturos sexualmente, a maior parte dos efeitos induzidos por aflibercepte foram reversíveis após o período de 5 meses livre do medicamento, com exceção dos achados no esqueleto e na cavidade nasal. A maioria dos achados foi relacionada à atividade farmacológica do aflibercepte.
Os efeitos nos ossos incluem espessamento da placa de crescimento e exostose osteocartilaginosa no esqueleto axial e apendicular.
Os efeitos na cavidade nasal incluíram degeneração/regeneração do epitélio respiratório e olfativo, atrofia/perda do septo nasal e/ou cornetas nasais frequentemente associados com hemorragia e exsudação supurativa.
Os efeitos nos rins incluíram aumento da matriz mesangial glomerular, diminuição das proteínas séricas totais e dos níveis de albumina, e aumento sérico BUN e dos níveis de proteína na urina e/ou microalbumina.
Os efeitos no ovário incluíram diminuição do número de folículos em maturação, células granulosas, e/ou células theca. Em macacos machos, foram observados diminuição da motilidade do esperma e aumento da incidência de anormalidades morfológicas dos espermatozoides.
Outros efeitos incluem proliferação/degeneração vascular focal (sistema digestivo, bexiga urinária, coração e cérebro) e aumento nos níveis das enzimas hepáticas com inflamação e necrose hepática em exposições próximas àquelas dos pacientes nas doses terapêuticas recomendadas.
-Carcinogenicidade, mutagenicidade e genotoxicidade
Não foram conduzidos estudos para avaliar a carcinogenicidade, mutagenicidade e genotoxicidade de aflibercepte.
-Teratogenicidade
O aflibercepte mostrou-se ser embriotóxico e teratogênico quando administrado intravenosamente a coelhas grávidas a cada 3 dias durante o período de organogênese (dias de gestação 6 a 18) em doses aproximadamente 1 a 15 vezes a dose humana de 4 mg/kg a cada 2 semanas. Os efeitos observados incluem decréscimo no peso corpóreo materno, aumento no número de reabsorções fetais e aumento da incidência externa (incluindo anasarca, hérnia umbilical, hérnia diafragmática e gastroquise, fenda palatina, ectrodactilia e atresia), viscerais (no coração, grandes vasos e artérias) e esqueléticas (incluindo vértebras fundidas, externo e costelas, arcos e costelas supranumerárias, e ossificação incompleta) de malformações fetais.
-Alterações da fertilidade
Não foram conduzidos estudos específicos com aflibercepte em animais para avaliar o efeito sobre a fertilidade.
Entretanto, os resultados do estudo de toxicidade de repetidas doses sugerem que haja um potencial para aflibercepte prejudicar a função reprodutiva e a fertilidade.
Em macacas sexualmente maduras tratadas com IV por 6 meses, a inibição da função ovariana e do desenvolvimento folicular foi evidenciada pelo decréscimo do peso ovariano, decréscimo da quantidade de tecido lúteo, decréscimo do número de folículos em maturação, atrofia do endométrio uterino e miométrio, atrofia vaginal, anulação dos picos de progesterona e sangramento menstrual com administração de 3 mg/kg.
Em macacos cynomolgus sexualmente maduros tratados com IV por 6 meses, foram notados o decréscimo da motilidade do esperma e o aumento da incidência de anormalidades morfológicas dos espermatozoides com administração de 3 mg/kg.
Os efeitos induzidos por aflibercepte IV sobre a função reprodutiva e fertilidade de macacos ocorreram em exposições próximas a dos pacientes na dose terapêutica recomendada. Estes efeitos foram completamente reversíveis dentro de 8-18 semanas após a última injeção.
4. CONTRAINDICAÇÕES
ZALTRAP é contraindicado a pacientes com hipersensibilidade severa conhecida ao aflibercepte ou a qualquer um dos excipientes (vide "Advertências e precauções").
Para contraindicações relacionadas ao irinotecano, ao 5-Fu e à leucovorina (ácido folínico), consulte as informações atualizadas dos produtos nas respectivas bulas.
5. ADVERTÊNCIAS E PRECAUÇÕES
-Hemorragia
Pacientes tratados com ZALTRAP tem um risco aumentado de hemorragia, incluindo eventos hemorrágicos severos e algumas vezes fatais (vide "Reações adversas").
Os pacientes devem ser monitorados quanto a sinais e sintomas de hemorragia gastrintestinal e outras hemorragias severas. Não administrar ZALTRAP a pacientes com hemorragia severa (vide "Posologia e modo de usar").
-Insuficiência cardíaca e fração de ejeção diminuída
Insuficiência cardíaca e fração de ejeção diminuída foram relatadas em pacientes tratados com ZALTRAP. Os pacientes devem ser monitorados para sinais e sintomas de insuficiência cardíaca e fração de ejeção diminuída.
Descontinuar ZALTRAP em pacientes que apresentam insuficiência cardíaca e fração de ejeção diminuída.
-Perfuração gastrintestinal
Foi relatada perfuração gastrintestinal (GI), incluindo perfuração gastrintestinal fatal em pacientes tratados com ZALTRAP (vide "Reações adversas").
Os pacientes devem ser monitorados quanto a sinais e sintomas de perfuração gastrintestinal. Descontinuar a terapia com ZALTRAP em pacientes que sofrem perfuração gastrintestinal (vide "Posologia e modo de usar").
-Formação de fístula
Ocorreu a formação de fístula em regiões gastrintestinais e não-gastrintestinais em pacientes tratados com ZALTRAP (vide "Reações adversas").
Descontinuar a terapia com ZALTRAP em pacientes que desenvolverem fístula (vide "Posologia e modo de usar").
-Hipertensão
Foi observado um risco aumentado de hipertensão grau 3-4 (incluindo hipertensão e um caso de hipertensão essencial) em pacientes que receberam regime ZALTRAP/FOLFIRI (vide "Reações adversas").
Durante o tratamento com ZALTRAP recomenda-se monitorar a pressão arterial a cada duas semanas ou como indicado clinicamente. Em caso de hipertensão, tratar com terapia anti-hipertensiva adequada e monitorar a pressão arterial regularmente. Suspender a terapia com ZALTRAP em pacientes com hipertensão não controlada. Na recorrência de hipertensão severa, suspender até o controle e reduzir a dose de ZALTRAP a 2 mg/kg nos ciclos subsequentes. ZALTRAP deve ser descontinuado permanentemente se ocorrer crise hipertensiva ou encefalopatia hipertensiva (vide "Posologia e modo de usar").
A administração de ZALTRAP deve ser cautelosa em pacientes com história clínica de doença cardiovascular significativa, tais como doença arterial coronariana ou insuficiência cardíaca congestiva. Não há experiência nos estudos clínicos da administração de ZALTRAP a pacientes com insuficiência cardíaca classe III ou IV (NYHA).
-Eventos tromboembólicos arteriais
Foram observados eventos tromboembólicos arteriais (ETA) (incluindo ataque isquêmico transitório, acidente vascular cerebral, angina de peito, trombo intracardíaco, infarto do miocárdio, embolia arterial e colite isquêmica) em pacientes que receberam ZALTRAP (vide "Reações adversas").
Descontinuar ZALTRAP em pacientes que vivenciaram um ETA (vide "Posologia e modo de usar").
-Eventos tromboembólicos venosos (ETV)
Eventos tromboembólicos venosos (TEV), incluindo trombose venosa profunda (TVP) e embolismo pulmonar (raramente fatal) têm sido relatados em pacientes tratados com aflibercepte. O tratamento com ZALTRAP deve ser interrompido em pacientes com eventos tromboembólicos fatais (grau 4), incluindo embolia pulmonar. Pacientes com trombose venosa profunda Grau 3 devem ser tratados com anticoagulantes como clinicamente indicado, e a terapia com ZALTRAP deve ser continuada. Em caso de recorrência ocorre apesar da terapia com anticoagulantes apropriados, suspender a administração de ZALTRAP. Pacientes com eventos tromboembólicos Grau 3 ou inferior devem ser cuidadosamente monitorizados.
-Proteinúria
Foram observadas proteinúria severa, síndrome nefrótica e microangiopatia trombótica (TMA) em pacientes tratados com ZALTRAP (vide "Reações adversas").
Durante a terapia com ZALTRAP, avaliar o desenvolvimento ou agravamento da proteinúria através da análise da urina com tira-teste e/ou a razão de proteína creatinina urinária (RPCU). Pacientes com tira-teste para proteína ≥2+ ou a RPCU > 1 devem ser submetidos a uma coleta de urina de 24 horas.
Suspender a administração de ZALTRAP por proteinúria/24 horas ≥2gramas e retomar quando a proteinúria for < 2 gramas/24 horas. Em caso de recorrência, suspender a administração do medicamento até que a proteinúria seja < 2 gramas/24 horas e então reduzir a dose de ZALTRAP para 2mg/kg. Descontinuar a terapia com ZALTRAP em pacientes que desenvolverem síndrome nefrótica ou TMA (vide "Posologia e modo de usar").
-Neutropenia ou complicações neutropênicas
Uma maior incidência de complicações neutropênicas (neutropenia febril e infecção neutropênica) foi relatada com o regime ZALTRAP/FOLFIRI (vide "Reações adversas").
O monitoramento do hemograma completo com contagem diferencial é recomendado no estado basal e antes do início de cada ciclo com ZALTRAP. A administração de ZALTRAP/FOLFIRI deve ser adiada até que a contagem neutrofílica seja ≥1,5 x109/L (vide "Posologia e modo de usar" para mais detalhes). Em pacientes que podem estar sob risco aumentado para complicações neutropênicas, a utilização terapêutica de G-CSF na primeira ocorrência de neutropenia de grau ≥3 e a profilaxia secundária podem ser consideradas.
-Diarreia e desidratação
Houve maior incidência de diarreia severa com regime ZALTRAP/FOLFIRI (vide "Reações adversas").
A modificação da dose no regime FOLFIRI (vide "Posologia e modo de usar"), o uso de medicamentos antidiarreicos e a reidratação devem ser instituídos conforme necessidade.
-Reações de hipersensibilidade
No estudo pivotal de pacientes CCRM, foram reportadas reações de hipersensibilidade severas em pacientes tratados com regime ZALTRAP/FOLFIRI (vide "Reações adversas").
No caso de eventos de reação de hipersensibilidade severa (incluindo broncoespasmo, dispneia, angioedema e anafilaxia) descontinuar o tratamento e administrar a terapia apropriada (vide "Posologia e modo de usar" e "Contrainidicações").
Em caso de reação de hipersensibilidade leve a moderada (incluindo rubor, erupção cutânea, urticária e prurido) suspender temporariamente o tratamento até que a reação termine. Tratar com corticoesteroides e/ou anti-histamínicos como indicado clinicamente. O pré-tratamento com corticoesteroides e/ou anti-histamínicos deve ser considerado nos ciclos subsequentes (vide "Posologia e modo de usar"). O uso em pacientes com reações de hipersensibilidade prévia deve ser cauteloso uma vez que, apesar do tratamento profilático, foram observadas reações de hipersensibilidade recorrentes em alguns pacientes.
-Complicações na cicatrização de feridas
ZALTRAP prejudica a cicatrização de feridas em modelos animais (vide "Dados de segurança pré-clínicos").
O tratamento com ZALTRAP está associado a um comprometimento potencial da cicatrização de feridas (deiscência da ferida, vazamento anastomótico) (vide "Reações adversas").
Suspender o uso de ZALTRAP por pelo menos 4 semanas antes de cirurgias eletivas.
É recomendável que ZALTRAP não seja iniciado antes de no mínimo 4 semanas após cirurgia de grande porte e não seja iniciado até que a ferida cirúrgica esteja completamente cicatrizada. Para pequenas cirurgias tais como colocação de acessos venosos, biópsia ou extração dentária, ZALTRAP pode ser iniciado/retomado quando a ferida cirúrgica estiver completamente cicatrizada. Descontinuar ZALTRAP em pacientes com cicatrização comprometida de feridas que requeiram intervenção médica (vide "Posologia e modo de usar").
-Síndrome Leucoencefalopática Posterior Reversível (SLPR)
A SLPR (também conhecida como síndrome encefalopática posterior reversível -PRES) não foi relatada no estudo pivotal de Fase III em pacientes com câncer colorretal metastático. A SLPR foi relatada em pacientes tratados com monoterapia de ZALTRAP e em combinação com outras quimioterapias.
A SLPR pode apresentar alteração do estado mental, convulsões, náuseas, vômitos, dor de cabeça ou distúrbios visuais. O diagnóstico de SLPR é confirmado por ressonância nuclear magnética do encéfalo (RNM do encéfalo).
Descontinuar o ZALTRAP em pacientes que desenvolvem SLPR (vide "Posologia e modo de usar").
Gravidez
Os estudos de reprodução em coelhas grávidas mostraram que aflibercepte é embriotóxico e teratogênico (vide "Dados de segurança pré-clínicos - Terarogenicidade").
Não há dados sobre a utilização de aflibercepte em mulheres grávidas. Como a angiogênese é fundamental para o desenvolvimento fetal, sua inibição decorrente da administração de ZALTRAP pode resultar em eventos adversos na gravidez. ZALTRAP não é recomendado durante a gravidez ou para mulheres suscetíveis a engravidar. O aflibercepte só deve ser utilizado durante a gravidez se o benefício potencial justificar o potencial risco para o feto.
Mulheres em idade fértil devem ser aconselhadas a evitar a gravidez durante o uso de ZALTRAP e devem ser informadas do risco potencial para o feto.
A fertilidade masculina e feminina pode ser comprometida durante o tratamento com ZALTRAP baseado em estudos em macacos (vide "Dados de segurança pré-clínicos -Alterações da fertilidade"). Estes achados foram reversíveis dentro de 8 a 18 semanas após a interrupção do tratamento. Mulheres em idade fértil e homens férteis devem utilizar métodos contraceptivos eficazes durante e por até no mínimo 6 meses após a última dose do tratamento.
Lactação
Não foram realizados estudos para avaliar o impacto de ZALTRAP sobre a produção de leite, sua presença no leite materno ou seus efeitos sobre a criança em amamentação.
Não se sabe se aflibercepte é excretado no leite humano. Como muitos medicamentos são excretados no leite humano e devido ao potencial de reações adversas severas em lactentes decorrente do aflibercepte, deverá ser tomada a decisão de descontinuar a amamentação ou descontinuar a utilização do medicamento, tendo em conta a importância do medicamento para a mãe.
Categoria de risco na gravidez: D. Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
Alterações na capacidade de dirigir veículos e operar máquinas
Não foram estudados os efeitos de aflibercepte sobre a capacidade de conduzir e utilizar máquinas. Os pacientes que tiverem sintomas que afetam sua visão ou concentração, ou sua capacidade de reagir, devem ser aconselhados a não conduzir ou utilizar máquinas.
Atenção diabéticos: ZALTRAP contém açúcar (200 mg/mL de sacarose).
Não se destina à administração intravítrea.
ZALTRAP é uma solução hiperosmótica que não foi formulada para ser compatível com meio intraocular. ZALTRAP não deve ser administrado como uma injeção intravítrea.
6. INTERAÇÕES MEDICAMENTOSAS
Não foram conduzidos estudos formais de interações medicamento-medicamento para aflibercepte.
As concentrações medidas de aflibercepte livre e ligado em estudos combinados são comparáveis àquelas medidas em estudos com monoterapia, sugerindo que estas combinações (incluindo oxaliplatina, cisplatina, 5-FU, irinotecano, docetaxel, pemetrexede, gencitabina e erlotinibe) não têm impacto na farmacocinética do aflibercepte.
Baseado nos estudos combinados Fase I e comparados aos dados históricos e publicados, aflibercepte não tem impacto na farmacocinética do irinotecano, 5-fluoruracila (5-FU), oxaliplatina, cisplatina, docetaxel, pemetrexede, gencitabina e erlotinibe.
Interferências em exames laboratoriais e de diagnóstico
Não foi avaliada a interfererência de aflibercepte em exames laboratoriais e de diagnóstico.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
O frasco-ampola de ZALTRAP deve ser mantido sob refrigeração (2 a 8°C). Proteger da luz.
Não utilizar frascos se houver material particulado ou descoloração.
Prazo de validade: 36 meses a partir da data de fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
As soluções diluídas de ZALTRAP devem ser utilizadas imediatamente. Se não forem usadas imediatamente, as soluções diluídas de ZALTRAP podem ser armazenadas entre 2 a 8°C por até 24 horas, ou a 25°C por até 8 horas uma vez que ZALTRAP não contém conservantes.
Características físicas e organolépticas
Solução límpida incolor a amarelo pálido.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
Dose recomendada e esquema posológico
A dose recomendada de ZALTRAP administrada através de uma infusão intravenosa de 1 hora é de 4 mg/kg de peso corpóreo, seguido pelo regime FOLFIRI (vide "Resultados de Eficácia").
O regime FOLFIRI usado no estudo foi infusão IV de 180 mg/m² de irinotecano por 90 minutos e infusão IV de 400 mg/m² de leucovorina (ácido folínico) (dl racêmico) por 2 horas ao mesmo tempo, no dia 1 e usando linha Y, seguido por bolus IV de 400 mg/m² de 5-fluoruracila (5-FU), seguido por infusão IV contínua de 2400 mg/m² de 5-FU por 46 horas.
Os ciclos de tratamento são repetidos a cada 2 semanas.
O tratamento com ZALTRAP deve continuar até que ocorra progressão da doença ou toxicidade inaceitável.
Modificações de dose/Recomendações para adiar o tratamento
Descontinuar ZALTRAP se ocorrer:
Hemorragia severa (vide "Advertências e precauções");
Perfuração gastrintestinal (vide "Advertências e precauções");
Formação de fístula (vide "Advertências e precauções");
Crise hipertensiva ou encefalopatia hipertensiva (vide "Advertências e precauções");
Eventos tromboembólicos arteriais (vide "Advertências e precauções");
Eventos tromboembólicos venosos grau 4 (incluindo embolia pulmonar);
Síndrome nefrótica ou microangiopatia trombótica (TMA) (vide "Advertências e precauções");
Reações de hipersensibilidade severa (incluindo broncoespasmo, dispneia, angioedema e anafilaxia) (vide "Contraindicações" e"Advertências e precauções");
Cicatrização comprometida de feridas, requerendo intervenção médica (vide "Advertências e precauções");
Síndrome Leucoencefalopática Posterior Reversível (RPLS) (vide "Advertências e precauções").
Suspender temporariamente ZALTRAP por no mínimo 4 semanas antes de cirurgias eletivas (vide "Advertências e precauções").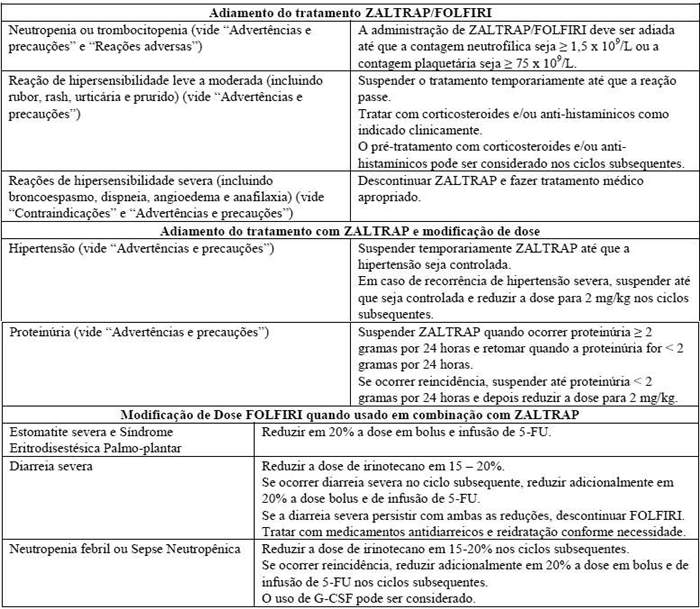
Para toxicidades adicionais relacionadas ao irinotecano, 5-FU e leucovorina (ácido folínico), consultar a respectiva informação de prescrição atualizada.
Administração
ZALTRAP deve ser administrado sob a supervisão de um médico experiente no uso de medicamentos antineoplásicos.
Não administrar a solução concentrada não diluída.
A administração deve ser somente por infusão intravenosa. Não administrar como injeção intravenosa (IV) ou em bólus. Não se destina à injeção intravítrea.
Como todos os produtos de uso parenteral, a solução diluída de ZALTRAP deve ser inspecionada visualmente quanto à presença de material particulado e descoloração antes da administração.
As soluções diluídas de ZALTRAP devem ser administradas utilizando conjuntos para infusão constituídos de um dos seguintes materiais:
- Cloreto de polivinila (PVC) contendo bis(2-etil-hexil) ftalato (DEHP);
- PVC livre de DEHP contendo trioctil trimelitato (TOTM);
- PVC revestido com polietileno, poliuretano ou polipropileno;
- Bolsas para infusão constituídas de PVC contendo DEHP ou poliolefina.
Os conjuntos para infusão devem conter um filtro de polietersulfona de 0,2 micrômetros.
Não use filtros à base de fluoreto de polivinilideno (PVDF) ou nylon.
Na ausência de estudos de compatibilidade, este produto não deve ser misturado a outros produtos ou diluentes exceto aqueles descritos no item "Preparação da solução para infusão".
Não há estudos dos efeitos de ZALTRAP administrado por vias não recomendadas. Portanto, por segurança e para garantir a eficácia deste medicamento, a administração deve ser somente por infusão intravenosa.
Preparação e manipulação -Recomendação para manipulação segura:
ZALTRAP solução concentrada deve ser preparada por um profissional usando técnica asséptica e procedimentos seguros de manipulação.
-Preparação da solução para infusão:
Não utilizar frascos se material particulado ou descoloração estiver presente.
Devem ser utilizadas bolsas para infusão constituídas de PVC contendo DEHP ou poliolefina (livre de PVC e livre de DEHP).
Apenas para infusão IV devido a hiperosmolaridade (1000 mOsmol/kg) do ZALTRAP concentrado.
Não se destina a injeção intravítrea.
ZALTRAP solução concentrada deve ser diluída. Retirar a quantidade necessária de solução e diluir até o volume necessário para administração com cloreto de sódio 0,9% ou solução de dextrose 5% para injeção. A concentração final da solução de ZALTRAP para infusão IV deve ser mantida dentro da faixa de 0,6 - 8 mg/mL de aflibercepte.
As soluções diluídas de aflibercepte devem ser utilizadas imediatamente. Se não forem usadas imediatamente, as soluções diluídas de ZALTRAP podem ser armazenadas entre 2 a 8°C por até 24 horas, ou a 25°C por até 8 horas uma vez que ZALTRAP não contem conservantes.
Descarte:
O frasco-ampola de ZALTRAP é de uso único. Uma vez que o produto não contém conservantes, descartar qualquer porção não utilizada do frasco-ampola. Não perfure novamente o frasco-ampola após a perfuração inicial.
Populações especiais
-Crianças
A segurança e a eficácia em pacientes pediátricos não foram estabelecidas. Em um estudo de dose escalada, de segurança e tolerabilidade, 21 pacientes com idade entre 2 a 21 anos (idade média de 12,9) com tumores sólidos, receberam ZALTRAP em doses que variaram de 2 a 3 mg/kg, IV, a cada duas semanas. A farmacocinética de aflibercepte livre foi avaliada em 8 destes pacientes (com idade entre 5 e 17 anos) (vide "Propriedades farmacocinéticas"). A dose máxima tolerada no estudo foi de 2,5 mg/kg, inferior à dose conhecida ser segura e eficaz em adultos com CCRM.
-Idosos
Não são necessários ajustes de dose de ZALTRAP para pacientes idosos.
-Insuficiência hepática
Não foram conduzidos estudos formais com ZALTRAP em pacientes com insuficiência hepática. Baseado nos dados clínicos, a exposição de aflibercepte em pacientes com insuficiência hepática leve e moderada foi semelhante àquela observada em pacientes com função hepática normal. Os dados clínicos sugerem que nenhuma alteração na dose de aflibercepte é necessária em pacientes com insuficiência hepática leve a moderada. Não há dados sobre a administração de aflibercepte em pacientes com insuficiência hepática severa.
-Insuficiência renal
Não foram conduzidos estudos formais com ZALTRAP em pacientes com insuficiência renal. Baseado nos dados clínicos, a exposição de aflibercepte em pacientes com insuficiência renal leve, moderada ou severa foi semelhante àquela observada em pacientes com função renal normal. Os dados clínicos sugerem que nenhuma alteração na dose inicial de aflibercepte é necessária em pacientes com insuficiência renal leve a moderada. Há dados muito limitados do uso em pacientes com insuficiência renal severa; estes pacientes devem ser tratados co