XPREZA
NATCOFARMA
azacitidina
Antineoplásico.
Apresentações.
Xpreza pó liofilizado para suspensão injetável, disponível em frasco ampola com 100 mg de azacitidina.
USO SUBCUTÂNEO
USO ADULTO
Composição.
Cada frasco ampola contém 100 mg de azacitidina e 100 mg de manitol como excipiente.
Informações técnicas.
1. INDICAÇÕES
Xpreza é indicado para o tratamento de pacientes com Síndrome Mielodisplásica dos subtipos: anemia refratária com excesso de blastos (AREB), de acordo com a classificação FAB; leucemia mielóide aguda com 20
- 30% de blastos na medula óssea com displasia multilinhagem de acordo com a classificação OMS; e leucemia mielomonocítica crônica (classificação FAB modificada).
2. RESULTADOS DE EFICÁCIA
Estudos Clínicos
Estudo de número 01: foi um ensaio clínico controlado aberto randomizado, realizado em 53 locais nos Estados Unidos, comparou a segurança e eficácia de azacitidina por via subcutânea, mais tratamento de suporte, com somente tratamento de suporte ("Observação") em pacientes com qualquer dos cinco subtipos FAB de síndromes mielodisplásicas (SMD): anemia refratária (AR), AR com sideroblastos em anel (ARSA), AR com excesso de blastos (AREB), AREB em transformação (AREB-T) e leucemia mielomonocítica crônica (LMMoC). Pacientes com AR e ARSA foram incluídos se estes cumprissem um ou mais dos seguintes critérios: transfusões de concentrado de hemácias (packed RBC) requeridas; apresentar contagens de plaquetas ≤50,0 x 109/L; transfusões de plaquetas requeridas; ou serem neutropênicos (ANC < 1,0 x 109/L) com infecções requerendo tratamento com antibióticos. Pacientes com leucemia mielóide aguda (LMA) não deveriam ser incluídos. Característica basal do paciente e da doença é resumida na Tabela 1; os dois grupos foram similares. Azacitidina foi administrado a uma dose subcutânea de 75 mg/m2 diariamente por sete dias a cada quatro semanas. A dose foi aumentada para 100 mg/m2 se nenhum efeito benéfico fosse observado após dois ciclos de tratamento. A dose foi reduzida e/ou retardada com base na resposta hematológica ou evidências de toxicidade renal. Pacientes no braço de observação podiam, pelo protocolo, mudar para azacitidina se estes apresentassem aumentos nos blastos da medula óssea, ou reduções na hemoglobina, aumentos nos requerimentos de transfusão de hemácias, ou reduções em plaquetas, ou se estes requeressem uma transfusão de plaquetas, ou desenvolvessem uma infecção clínica requerendo tratamento com antibióticos. Para fins de avaliação de eficácia, o objetivo primário foi a taxa de resposta (conforme definido na Tabela 2).
Dos 191 pacientes incluídos no estudo, uma revisão independente (diagnóstico adjudicado) descobriu que 19 apresentavam o diagnóstico de LMA no basal. Estes pacientes foram excluídos da análise primária de taxa de resposta embora tenham sido incluídos na análise de intenção de tratamento (ITT) de todos os pacientes randomizados. Aproximadamente 55% dos pacientes randomizados para observação mudaram para receber o tratamento com azacitidina.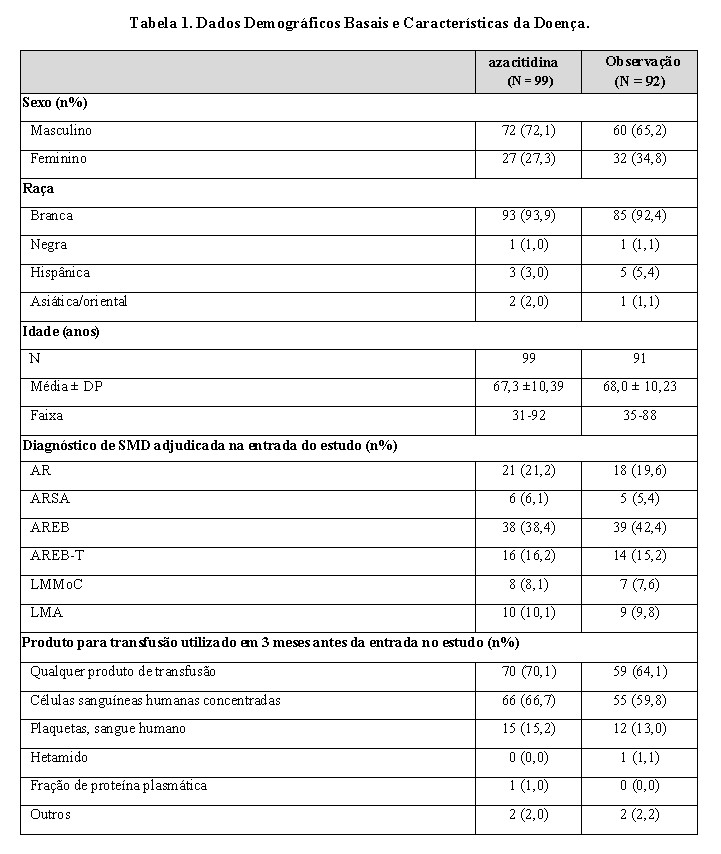
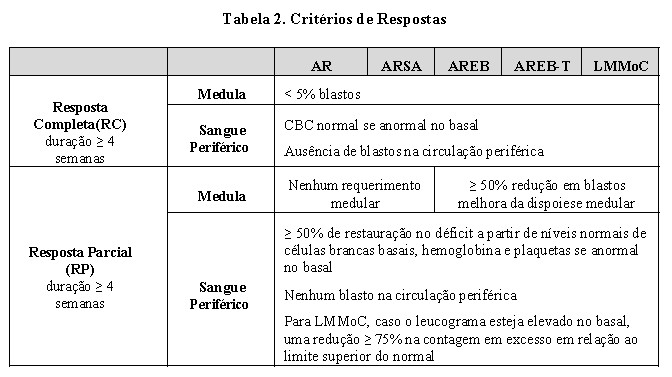
A taxa de resposta geral (RC + RP) de 15,7% em pacientes tratados com azacitidina sem LMA (16,2% para todos os pacientes randomizados para receber azacitidina incluindo LMA) foi maior, de forma estatisticamente significativa, que a taxa de resposta de 0% no grupo de observação (p < 0,0001) (Tabela 3). A maioria dos pacientes que atingiu RC ou RP apresentava 2 ou 3 anormalidades de linhagem celular no basal (79%; 11/14) e apresentava blastos de medula óssea elevados ou eram dependentes de transfusão no basal. Pacientes respondendo a azacitidina apresentaram uma redução na porcentagem de blastos da medula óssea ou um aumento nas plaquetas, hemoglobina ou contagem de leucócitos. Mais de 90% dos responsivos inicialmente demonstraram estas alterações no 5° ciclo de tratamento. Todos os pacientes dependentes de transfusão se tornaram independentes de transfusão durante RP ou RC. A duração média e mediana da resposta clínica de RP, ou melhor, foi estimada como 512 e 330 dias respectivamente, 75% dos pacientes responsivos estavam ainda em RP, ou melhor, na finalização do tratamento. A resposta ocorreu em todos os subtipos SMD, assim como em pacientes com diagnóstico basal adjudicado de LMA.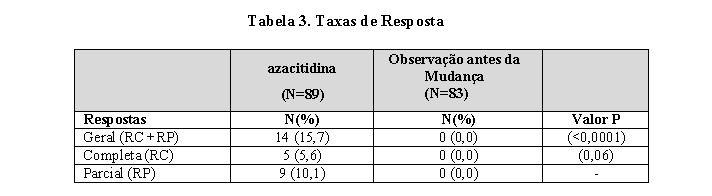
Pacientes no grupo de observação que mudaram para receber o tratamento com azacitidina (47 pacientes) apresentaram uma taxa de resposta de 12,8%.
Estudo número 2: foi realizado o estudo de número 2 um estudo multicêntrico aberto, de braço único, incluindo 72 pacientes com AREB, AREB-T, LMMoC ou LMA. O tratamento com azacitidina subcutâneo resultou em uma taxa de resposta (RC + RP) de 13,9%, utilizando critérios similares aos descritos acima. A duração média e mediana da resposta clínica de RP ou melhora, foram estimadas como sendo 810 e 430 dias respectivamente, 80% dos pacientes responsivos ainda estavam em RP ou melhora no momento da finalização do envolvimento no estudo.
Estudo de número 3: outro estudo aberto de um braço com 48 pacientes com AREB, AREB-T ou LMA o tratamento com azacitidina intravenoso resultou em uma taxa de resposta de 18,8%, novamente utilizando critérios similares aos descritos acima. A duração média e mediana da resposta clínica de RP ou melhora foram estimadas como 389 e 281 dias, respectivamente, 67% dos pacientes responsivos ainda estavam em RP ou melhora no momento da finalização do tratamento. A resposta ocorreu em todos os subtipos de SMD assim como em pacientes com diagnóstico basal adjudicado de LMA em ambos os estudos. Os regimes de dosagem de azacitidina nestes dois estudos foram similares ao regime utilizado no estudo controlado.
Benefício foi observado em pacientes que não cumpriram os critérios para RP ou melhora, mas foram considerados como "com melhora". Cerca de 24% dos pacientes tratados com azacitidina foram considerados como "com melhora" e cerca de 2/3 destes perderam a dependência à transfusão. No grupo de observação somente 5/83 pacientes cumpriram os critérios para melhora, nenhum perdeu a dependência à transfusão. Em todos os três estudos, cerca de 19% dos pacientes cumpriram os critérios para melhora com uma duração mediana de 195 dias. Estimativas de taxa de resposta foram similares independentemente da idade e sexo.
Estudo de número 4: foi um estudo internacional multicêntrico, aberto e aleatório, em pacientes com SMD que tinham AREB, AREB-T ou LMMoC, modificada de acordo com a classificação FAB e risco intermediário2 e alto risco, de acordo com a classificação pelo sistema internacional de prognóstico (International Prognosis Score System - IPSS). Dos 358 pacientes inscritos no estudo, 179 foram randomizados para receber azacitidina além de melhor cuidado de apoio (MCA) e 179 foram randomizados para receber regimes de cuidados convencionais (RCC) mais MCA (105 randomizados para MCA apenas, 49 randomizados para receber dose baixa de citarabina e 25 randomizados para receber quimioterapia com citarabina e antraciclina). O critério de avaliação primário de eficácia foi a sobrevida geral.
Os grupos recebendo azacitidina e RCC foram comparáveis quanto aos parâmetros basais. A idade mediana dos pacientes foi de 69 anos (variando entre 38-88 anos) 98% eram da raça branca e 70% eram pacientes masculinos. A nível basal 95% dos pacientes se encontravam em um risco mais elevado pela classificação FAB: AREB (58%), AREB-T (34%) e LMMoC (3%). De acordo com a classificação IPSS, 87% se encontravam em um risco mais elevado: Intermediário-2 (41%), Alto (47%). A nível basal, 32% dos pacientes atendiam aos critérios da OMS para LMA.
A azacitidina foi administrada por via subcutânea a uma dose de 75 mg/m2 diariamente durante 7 dias consecutivos a cada 28 dias (que constituía um ciclo de terapia). Os pacientes continuaram a receber tratamento até a ocorrência de progressão da doença recidiva depois da resposta ou toxicidade inaceitável. Os pacientes que receberam azacitidina foram tratados com uma mediana de 09 ciclos (variando de 1 a 39) os pacientes recebendo apenas MCA com uma mediana de 7 ciclos (variando de 1 a 26); os pacientes recebendo baixa dose de citarabinacom uma mediana de 4,5 ciclos (variando de 1 a 15) e os pacientes recebendo quimioterapia com citarabina e antraciclina com uma mediana de 1 ciclo (variando de 1 a 3, isto é, indução mais um ou dois ciclos de consolidação).
Na análise de intenção de tratamento, os pacientes tratados com azacitidina demonstraram uma diferença estatisticamente significativa na sobrevida geral quando comparados aos pacientes tratados com RCC (sobrevida mediana de 24,5 meses em contraposição há 15,0 meses, teste de log rank estratificado de p=0,0001). O índice derisco descrevendo o efeito deste tratamento foi de 0,58 (95%, IC: 0,43 - 0,77).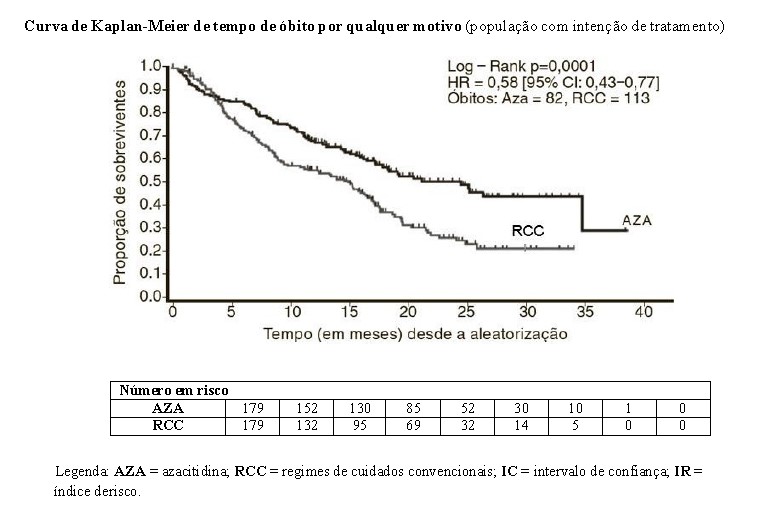
O tratamento com azacitidina levou à redução da necessidade de transfusões de hemácias (veja a Tabela 4). Nos pacientes tratados com azacitidina que dependiam de transfusão de hemácias a nível basal e se tornaram independentes de transfusão, a duração mediana da independência de transfusão de hemácias foi de 13,0 meses.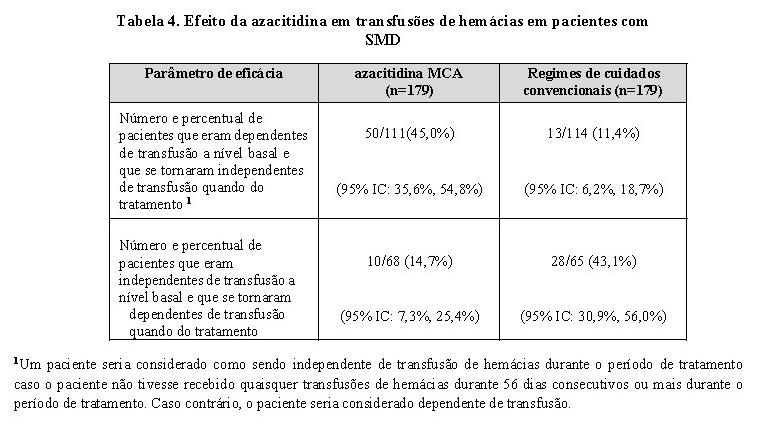
Referências bibliográficas:
1. Estudo clínico CALGB 9221: A Randomized Phase III Controlled Trial of Subcutaneous 5-Azacitidine (NSC #102816) vs. Observation in Myelodysplastic Syndromes, 2003.
2. Estudo clínico CALGB 8921: A Phase II Study of Subcutaneous 5-Azacitidine in Myelodisplastic Syndromes, 2003.
3. Estudo clínico CALGB 8421: 5-Azacitidine to Induce Differentiation in Myelodisplastic Syndromes: A Phase II Pilot Study, 2003.
4. Fenaux, P. and Garcia-Manero,G. Hypomethylating Agents and Other Novel Strategies in Myelodysplasic Syndromes. J Clin Oncol 29:516-523, 2011
5. Sullivan, M. et all. Azacitidine: A novel agent for myelodysplastic syndromes. Am J Health-Syst Pharm
62: 1567-1573, 2005.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Mecanismo de ação
Acredita-se que azacitidina exerça seus efeitos antineoplásicos por causar hipometilação do DNA e citotoxicidade direta em células hematopoiéticas anormais na medula óssea. A concentração de azacitidina requerida para inibição máxima da metilação do DNA in-vitro não causa supressão de porte na síntese de DNA. A hipometilação pode restaurar a função normal a genes que sejam críticos para diferenciação e proliferação. Os efeitos citotóxicos da azacitidina causam a morte de células que se dividem rapidamente, incluindo células cancerosas que não respondam aos mecanismos de controle de crescimento normal. As células não proliferativas são relativamente insensíveis ao azacitidina.
Farmacocinética
A farmacocinética da azacitidina foi estudada em seis pacientes com SMD (Síndrome Mielodisplásica) após uma dose subcutânea (SC) única de 75 mg/m2 e uma dose intravenosa (IV) única de 75 mg/m2. A azacitidina é rapidamente absorvida após administração SC; a concentração plasmática de pico da azacitidina de 750 ± 403 mg/mL ocorreu em 0,5 hora. A biodisponibilidade de azacitidina SC em relação á azacitidina IV é de aproximadamente 89% baseada na área sob a curva. O volume médio de distribuição após administração IV é de 76 ± 26 L. A depuração aparente média após administração SC é de 167 ± 49 L/hora e a meia-vida média após administração SC de 41± 8 minutos.
Estudos publicados indicam que a excreção urinária é a via primária de eliminação da azacitidina e seus metabólitos. Após administração IV de azacitidina radioativa a pacientes com câncer, a excreção urinária cumulativa foi 85% da dose radioativa. A excreção fecal foi responsável por < 1% da radioatividade administradadurante três dias. A excreção média de radioatividade na urina após administração SC de 14C-azacitidina foi de 50%. As meias-vidas de eliminação médias da radioatividade total (azacitidina e seus metabólitos) foram similares após administrações IV e SC, cerca de 4 horas.
4. CONTRAINDICAÇÕES
Xpreza é contraindicado em pacientes com hipersensibilidade conhecida a azacitidina ou manitol. Xpreza é também contraindicado em pacientes com tumores hepáticos malignos avançados (Ver seção 5. ADVERTÊNCIAS E PRECAUÇÕES).
5. ADVERTÊNCIAS E PRECAUÇÕES
Xpreza pode causar danos fetais quando administrado a uma mulher grávida. Estudos de embriotoxicidade iniciais em camundongos revelaram uma frequência de 44% de morte embrionária intrauterina (reabsorção aumentada) após injeção IP (intraperitoneal) única de 6 mg/m2 (aproximadamente 8% da dose diária humana recomendada com base em mg/m2) de azacitidina no dia 10 da gestação. Anormalidades no desenvolvimento no cérebro foram detectadas em camundongos recebendo azacitidina no dia 15, ou antes, do dia 15 da gestação desta em doses de ~3 - 12 mg/m2 (aproximadamente 4% -16% da dose diária humana recomendada com base em mg/m2).
Em ratos a azacitidina foi claramente embriotóxica quando administrada IP nos dias 4 - 8 da gestação (pósimplantação) a uma dose de 6 mg/m2 (aproximadamente 8% da dose diária humana recomendada com base em mg/m2), embora o tratamento no período pré-implantação (nos dias 1-3 de gestação) não tenha apresentado nenhum efeito adverso nos embriões. A azacitidina causou múltiplas anormalidades fetais em ratos após uma dose IP única de 3 a 12 mg/m2 (aproximadamente 8% da dose diária humana recomendada com base em mg/m2) administrado nos dias de gestação 9, 10, 11 ou 12. Neste estudo, a azacitidina causou morte fetal quando administrada a 3-12 mg/m2 nos dias 9 e 10 da gestação; a média de animais vivos por ninhada foi reduzida para 9%, do controle na dose mais alta no dia 9 da gestação. As anormalidades fetais incluíram anomalias no SNC (exencefalia / encefalocele), anormalidades nos membros (micromelia, pé torto, sindactilia, oligodactilia) e outros (micrognatia, gastrosquise, edema e anormalidades nas costelas).
Não houve nenhum estudo adequado e bem controlado em mulheres grávidas utilizando azacitidina.Se esta droga for utilizada durante a gravidez, ou se a paciente ficar grávida enquanto recebe esta droga a paciente deve ser avisada sobre o perigo potencial ao feto. Mulheres com potencial de terem crianças devem ser aconselhadas a evitar a gravidez enquanto receberem o tratamento com azacitidina.
Geral
O tratamento com Xpreza está associado a neutropenia e trombocitopenia. Contagens sanguíneas completas devem ser realizadas, conforme necessidade a fim de monitorar a resposta e toxicidade, mas no mínimo, antes de cada ciclo de dosagem. Após administração da dosagem recomendada para o primeiro ciclo a dosagem para ciclossubsequentes deve ser reduzida ou retardada com base nas contagens de nadir e resposta hematológica conforme descrito em POSOLOGIA.
A segurança e eficácia de azacitidina em pacientes com SMD e comprometimento hepático ou renal não foram estudadas, pois estes pacientes foram excluídos dos estudos clínicos.
Como a azacitidina é potencialmente hepatotóxica em pacientes com comprometimento hepático pré- existente severo, cautela é necessária em pacientes com doença hepática. Pacientes com carga de tumor extensivo devido à doença metastática foram raramente relatados como apresentando coma hepático progressivo e morte durante tratamento com azacitidina, especialmente naqueles pacientes com albumina basal < 30 g/L. A azacitidina é contraindicada em pacientes com tumores hepáticos malignos avançados.
Anormalidades renais, variando de creatinina sérica elevada a insuficiência renal e morte, foram raramente relatadas em pacientes tratados com azacitidina intravenosa em combinação com outros agentes quimioterápicos para condição não SMD. Em adição, ocorreu desenvolvimento de acidose tubular renal definida como uma queda em bicarbonato sérico para < 20 mEq/L em associação com urina alcalina e hipocalemia (potássio sérico < 3 mEq/L), em 5 pacientes com LMC tratados com azacitidina e etoposide. Se ocorrerem reduções não explicadas no bicarbonato sérico < 20 mEq/L ou elevações de BUN ou creatinina sérica, a dosagem deve ser reduzida ou mantida conforme descrito em seção 8. POSOLOGIA E MODO DE USAR.
Pacientes com comprometimento renal devem ser muito bem monitorados quanto à toxicidade, já que azacitidina e seus metabólitos são primariamente excretados pelos rins.
Casos de síndrome de diferenciação (também conhecida como síndrome do ácido retinóico) foram relatados em pacientes recebendo azacitidina injetável. A síndrome de diferenciação pode ser fatal e os sintomas e achados clínicos incluem dificuldade respiratória, infiltrados pulmonares, febre, erupção cutânea, edema pulmonar, edema periférico, rápido ganho de peso, derrames pleurais, derrames pericárdicos, hipotensão e disfunção renal (ver seção 9. REAÇÕES ADVERSAS). O tratamento com altas doses de corticosteroides IV e o monitoramento hemodinâmico devem ser considerados no início dos sintomas ou sinais sugestivos de síndrome de diferenciação. A descontinuação temporária da azacitidina injetável deve ser considerada até a resolução dos sintomas e, se retomada, recomenda-se cautela.
Testes laboratoriais
Contagens sanguíneas completas devem ser realizadas conforme necessidade para monitorar a resposta e a toxicidade, mas no mínimo antes de cada ciclo. Química do fígado e creatinina sérica devem ser obtidos antes do início da terapia.
Carcinogênese, Mutagênese, Comprometimento da Fertilidade
A carcinogenicidade potencial da azacitidina foi avaliada em camundongos e ratos. A azacitidina induziu tumores do sistema hematopoiético em camundongos fêmea a 2,2 mg/Kg (6,6 mg/m2 aproximadamente 8% dados e diária humana recomendada com base em mg/m2) administrada IP três vezes por semana por 52 semanas. Uma maior incidência de tumores no sistema linforreticular, pulmão, glândula mamária e pele foi observada em camundongos tratados com azacitidina IP a 2,0 mg/Kg (6,0 mg/m2, aproximadamente 8% da dose diária humana recomendada com base em mg/m2) uma vez por semana durante 50 semanas. Um estudo de tumorigenicidade em ratos administrados duas vezes por semana a 15 ou 60 mg/m2 (aproximadamente 2080% da dose diária humana recomendada com base em mg/m2) revelou uma maior incidência de tumores testiculares em comparação aos controles.
O potencial mutagênico e clastogênico da azacitidina foi avaliado em sistemas bacterianos in vitro,Salmonella typhimurium cepas TA100 e várias cepas de trpE8, cepas de Escherichia coli WP14 Pro, WP3103P, WP3104P e CC103; ensaio de mutação genética forward in vitro em células de linfoma de camundongo e células linfoblásticas humanas, e em um ensaio de micronúcleo in vivo em células de linfoma de camundongo L5178Y e células embrionárias de hamster Sírio.
A azacitidina foi mutagênica em sistemas bacterianos e células de mamíferos. O efeito clastogênico da azacitidina foi mostrado pela indução do micronúcleo em células de camundongo L5178Y e células embrionárias de hamster Sírio. A administração de azacitidina a camundongos machos a 9,9 mg/m2 (aproximadamente 9% da dose diária humanarecomendada com base em mg/m2) diariamente, por 3 dias antes do acasalamento com camundongos fêmeas não tratadas, resultou em uma fertilidade reduzida e perda da prole durante subsequente desenvolvimento embrionário e pós-natal. O tratamento de ratos machos três vezes por semana por 11 ou 16 semanas em doses de 15 a 30 mg/m2(aproximadamente 20 - 40% da dose diária humana recomendada com base em mg/m2) resultou em peso reduzido dos testículos e epidídimos e contagem de esperma reduzida acompanhadas por menores taxas de gravidez e maior perda de embriões nas fêmeas acasaladas. Em um estudo relacionado, os ratos machos tratados por 16 semanas a 24 mg/m2 resultaram em um aumento em embriões anormais em fêmeas acasaladas quando examinados no dia 2 da gestação.
Uso em idosos, crianças e outros grupos de risco
Uso Geriátrico
Do número total de pacientes dos três estudos clínicos descritos na seção 2. RESULTADOS DE EFICÁCIA, 62% possuíam 65 anos ou mais e 21% possuíam 75 anos ou mais. Nenhuma diferença na eficácia foi observada entre estes pacientes e pacientes mais jovens. Em adição, não houve nenhuma diferença relevante na frequência de eventos adversos observada em pacientes com 65 anos ou mais em comparação com pacientes mais jovens.
A azacitidina e seus metabólitos são substancialmente excretados pelos rins e o risco de reações tóxicas a esta droga pode ser maior em pacientes com função renal comprometida. Como pacientes idosos possuem maior probabilidade de terem função renal comprometida pode ser útil monitorar a função renal (ver seção 8. POSOLOGIA).
Uso no Sexo Masculino
Não existem dados sobre o efeito da azacitidina na fertilidade. Em animais, os efeitos adversos da azacitidina na fertilidade masculina têm sido documentados. Os homens devem ser aconselhados a não serem pais de uma criança durante o tratamento e por pelo menos 3 meses após a última dose (ver seção 5. ADVERTÊNCIAS E PRECAUÇÕES).
Carcinogênese, Mutagênese e Comprometimento da Fertilidade para obter uma discussão sobre os efeitos pré-acasalamento da exposição a azacitidina na fertilidade de machos e viabilidade embrionária). Homens devem ser avisados a não fecundarem mulheres enquanto receberem tratamento com azacitidina.
Gravidez - Efeitos teratogênicos: Gravidez Categoria D
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
A azacitidina pode causar danos fetais quando administrada a uma mulher grávida. A azacitidina foi teratogênica em animais (ver seção 5. ADVERTÊNCIAS E PRECAUÇÕES). Mulheres com potencial para engravidar devem ser aconselhadas para evitar a gravidez durante o tratamento com Xpreza. Se esta droga for usada durante a gravidez ou se a pacienteengravidar durante tomando este medicamento, a paciente deve ser informada do perigo potencial para o feto. Aconselhe as mulheres sobre o potencial reprodutivo para usar um método eficaz de contracepção durante o tratamento com Xpreza e durante pelo menos 6 meses após a última dose. Mulheres parceiras de pacientes do sexo masculino recebendo Xpreza não devem engravidar (ver seção
5. ADVERTÊNCIAS E PRECAUÇÕES).
Mães em Aleitamento
Não é conhecido se a azacitidina ou seus metabólitos são excretados no leite humano. Devido ao potencial para
tumorigenicidade demonstrado para azacitidina em estudos animais e o potencial de reações adversas graves, mulheres tratadas com azacitidina não devem amamentar.
Uso Pediátrico
A segurança e eficácia em pacientes pediátricos não foram estabelecidas.
Populações Especiais
Os efeitos do comprometimento renal ou hepático, sexo, idade ou raça na farmacocinética de azacitidina não foram estabelecidas.
Síndrome de lise tumoral
Pacientes em risco de síndrome de lise tumoral são aqueles com elevada carga tumoral antes do tratamento. Estes pacientes devem ser cuidadosamente monitorados e tomadas precauções apropriadas.
6. INTERAÇÕES MEDICAMENTOSAS
Nenhum estudo clínico formal de interações medicamentosas com azacitidina foi conduzido.
Um estudo in vitro da incubação de azacitidina em frações de fígado humano indicou que azacitidina pode ser metabolizada pelo fígado. Se o metabolismo de azacitidina pode ser afetado por inibidores de enzimas microssomais conhecidas ou indutores não foi estudado.
Estudos in vitro com culturas de hepatócitos humanos indicam que a azacitidina a concentrações de 1,0 mM a 100 mM (ou seja, até 30 vezes maior que concentrações clinicamente viáveis) não induz o citocromo P450 (CYP) isoenzimas CYP 1A2, 2C19 ou 3A4/5.
Azacitidina não mostrou inibição notável de isoenzimas P450 (CYP 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 e 3A4)na faixa de concentração de 0,1 a 100 mM. Portanto, efeitos inibitórios ou indutivos clinicamente relevantes sobre o metabolismo de substratos do citocromo P450 são improváveis.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original. Antes de usar, observe o aspecto do medicamento.
Este medicamento possui prazo de validade de 24 meses a partir da data de fabricação.
Xpreza é um pó ou pedaço liofilizado branco a esbranquiçado.
Armazene os frascos não reconstituídos em temperatura ambiente (temperatura entre 15 e 30°C).
CUIDADOS DE CONSERVAÇÃO DEPOIS DE ABERTO
Estabilidade da Suspensão
Xpreza reconstituído com água para injetáveis para administração subcutânea pode ser armazenado por até 22 horas entre 2°C e 8°C, se a água para injetáveis for refrigerada, ou por até 8 horas entre 2°C e 8°C, se água utilizada não for refrigerada. Descartar a porção não utilizada.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
Preparação de Xpreza
Xpreza é uma droga citotóxica e, assim como outros compostos potencialmente tóxicos, cautela deve ser tomada durante a manipulação e preparação de suspensões de Xpreza. (Ver a seção Manipulação e Descarte). Se Xpreza reconstituído entrar em contato com a pele, lave muito bem com água e sabão imediatamente. Se entrar em contato com membranas mucosas, enxágue muito bem com água. O frasco ampola de Xpreza é para uso único e não contém conservantes. Porções não utilizadas de cada frasco devem ser adequadamente descartadas (ver a seção Manipulação e Descarte). Não armazene qualquer porção não usada para administração posterior.
Preparação para Administração Subcutânea
Xpreza deve ser reconstituído assepticamente com 4 mL de água estéril para injeção. O diluente deve ser injetado lentamente no frasco. Agite vigorosamente o frasco ou rotacionando até obter uma suspensão uniforme. A suspensão será turva. A suspensão resultante irá conter 25 mg/mL de azacitidina. Não filtre a suspensão após a reconstituição. Fazer isso pode remover a substância ativa.
Preparação para Administração Subcutânea Imediata:
Quando mais de um frasco-ampola for necessário, os passos de preparação anteriores devem ser repetidos. Para doses que requeiram mais de um frasco-ampola, a dose deve ser dividida igualmente (ex.: dose de 150mg = 6mL, 2 seringas com 3 mL em cada seringa) e injetadas em dois locais separados. Devido à retenção no frasco e agulha, pode não ser viável remover toda a suspensão do frasco-ampola. O produto pode ser mantido à temperatura ambiente por até uma hora, mas deve ser administrado dentro de 1 hora após a reconstituição.
Preparação para Administração Subcutânea Retardada:
O produto reconstituído pode ser mantido no frasco ou ser retirado para uma seringa. Doses que requeiram mais de um frasco-ampola devem ser divididas igualmente em duas seringas. O produto deve ser refrigerado imediatamente e pode ser mantido sob condições refrigeradas (2°C - 8°C) por até (8 horas água para injetáveis não refrigerada) ou 22 horas (água para injetáveis refrigerada). Após remoção das condições refrigeradas, a suspensão pode ser deixada para equilibrar a temperatura ambiente (20°C a 25°C) por até 30 minutos antes da administração. Se o tempo decorrido for superior a 30 minutos, a suspensão deve ser descartada adequadamente e preparada uma nova dose.
Administração Subcutânea
Para proporcionar uma suspensão homogênea o conteúdo da seringa deve ser re-suspenso invertendo-se a seringa 2 -3 vezes e, vigorosamente rolando a seringa entre as palmas da mão por 30 segundos imediatamente antes da administração. A suspensão de Xpreza é administrada subcutaneamente. Doses que requeiram mais de um frasco-ampola
devem ser divididas igualmente em duas seringas e ser injetadas em 2 locais separados. Faça um rodízio dos sítios para cada injeção (coxa, abdômen ou braço superior). Novas injeções devem ser administradas pelo menos uma polegada a partir de um local antigo e nunca devem ser administradas em áreas onde o local seja mole, lesionado, vermelho ou rígido.
Manipulação e descarte
Procedimentos para a manipulação e descarte de drogas anticâncer devem ser aplicados. Várias orientações sobre este assunto foram publicadas. Não existe nenhum acordo geral de que todos os procedimentos recomendados nas orientações sejam necessários ou apropriados.
POSOLOGIA
Primeiro Ciclo de Tratamento
A dose inicial recomendada para o primeiro ciclo de tratamento, para todos os pacientes, independentemente dos valores laboratoriais hematológicos basais, é de 75 mg/m2 por via subcutânea diariamente, durante sete dias. Os pacientes devem ser pré-medicados para náusea e vômitos.
Ciclos Subsequentes de Tratamento
Os ciclos podem ser repetidos a cada quatro semanas. A dose pode ser aumentada para 100 mg/m2 se nenhum efeito benéfico for observado após dois ciclos de tratamento e se nenhuma toxicidade que não seja náusea e vômito ocorrer. É recomendado que os pacientes sejam tratados por um mínimo de 4 a 6 ciclos. Porém, resposta completa ou parcial pode requerer mais que 4 ciclos de tratamento. O tratamento pode continuar desde que o paciente continue a se beneficiar.
Os pacientes devem ser monitorados quanto a resposta hematológica e toxicidade renal, e retardo ou redução de administração, conforme descrito abaixo, pode ser necessário.
Ajuste de Dose Baseado em Valores Laboratoriais Hematológicos
- Para pacientes com contagens basais de leucócitos (início do tratamento) ≥ 3,0 x 109/L, ANC ≥ 1,5 x 109/L e plaquetas ≥ 75,0 x 109/L, ajuste a dose da seguinte forma, baseado em contagens nadir para qualquer determinado ciclo:
- Para pacientes cujas contagens basais sejam WBC < 3,0 x 109/L, ANC < 1,5 x 109/L e plaquetas < 75,0 x109/L, os ajustes de dose devem ser baseados em contagens nadir e celularidade de biópsia de medula óssea no momento do nadir, conforme observado abaixo, a não ser que não exista nenhuma melhora nítida na diferenciação (porcentagem de granulócitos maduros seja maior e ANC seja maior que no início daquele curso) no momento dopróximo ciclo, onde a dose de tratamento atual deve ser continuada.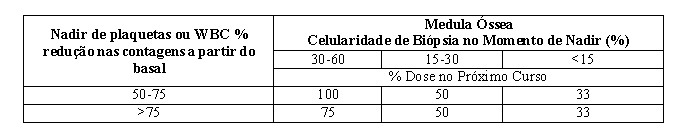
Se tiver ocorrido um nadir, conforme definido na tabela acima o próximo curso de tratamento deve ser administrado 28 dias após o início do curso anterior desde que ambas as contagens de leucócitos e contagens de plaquetas sejam > 25% acima do nadir e em elevação. Se um aumento > 25% acima do nadir não for observado até o dia 28, as contagens devem ser reavaliadas a cada 7 dias. Se um aumento de > 25% não for observado no dia 42, então o paciente deve ser tratado com 50% da dose programada.
Ajuste de Dose Baseado na Função Renal e Eletrólitos Séricos
Se reduções inexplicadas nos níveis de bicarbonato sérico para menos de 20 mEq/L ocorrerem, a dosagem deve ser reduzida em 50% no próximo curso. Similarmente, se elevações inexplicadas de BUN ou creatinina sérica ocorrerem, o próximo ciclo deve ser retardado até que os valores retornem ao normal ou basal e a dose deve ser reduzida em 50% no próximo curso de tratamento (ver a seção 5. ADVERTÊNCIAS E PRECAUÇÕES).
Uso em Pacientes Geriátricos
A Xpreza e seus metabólitos são substancialmente excretados pelos rins e o risco de reações tóxicas a esta droga pode ser maior em pacientes com função renal comprometida. Como pacientes idosos possuem maior probabilidadede apresentarem função renal reduzida, cuidado deve ser tomado na seleção de dose, e pode ser útil monitorar a função renal (ver a seção 5. ADVERTÊNCIAS E PRECAUÇÕES).
9. REAÇÕES ADVERSAS
Reações adversas descritas em outras seções da bula: anemia, neutropenia, trombocitopenia, creatinina sérica elevada, insuficiência renal, acidose tubular renal, hipocalemia, coma hepático.
Reações adversas que ocorrem mais comumente (Via SC): náusea, anemia, trombocitopenia, vômitos, pirexia,leucopenia, diarreia, fadiga, eritema no local de injeção, constipação, neutropenia, equimose.
Reações adversas que mais freqentemente ( > 2%) resultam em intervenção clínica (via SC):
Descontinuação: leucopenia, trombocitopenia, neutropenia.
Suspensão da Administração: leucopenia, neutropenia, trombocitopenia, pirexia, pneumonia, neutropenia febril.
Redução de Dose: leucopenia, neutropenia, trombocitopenia.
Uma vez que os ensaios clínicos são realizados sob condições muito variáveis, os índices de reações adversas observados nos ensaios clínicos de um fármaco não podem ser comparados diretamente aos índices obtidos em ensaios clínicos de outros fármacos e podem não refletir os índices observados na prática.
Os dados descritos a seguir refletem a exposição ao azacitidina por 443 pacientes com SMD a partir de 4 estudos clínicos. O estudo de número 1 foi um ensaio controlado com cuidados de apoio (com administração subcutânea), os estudos de número 2 e 3 foram estudos de braço único (um com administração subcutânea e um com administração intravenosa) e o estudo de número 4 foi um ensaio internacional aleatório (com administração subcutânea).
Nos estudos de número 1, 2 e 3, um total de 268 pacientes foram expostos ao azacitidina incluindo 116 expostos a 6 ciclos (durante aproximadamente 6 meses) ou mais e 60 foram expostos a mais de 12 ciclos (durante aproximadamente um ano). Azacitidina foi estudado primariamente em ensaios quer controlado com cuidados de apoio ou ensaios não controlados (n=150 e n=118 respectivamente). A população nos estudos com administração subcutânea (n=220), tinha entre 23 a 92 anos de idade (média de 66,4 anos), 68% eram pacientes masculinos e 94% eram da raça branca, e tinham SMD ou leucemia mielóide aguda (LMA). A população no estudo com administração intravenosa (n=48) tinha entre 35 a 81 anos de idade (média de 63,1 anos), 65% eram pacientes masculinos e 100% eram da raça branca. A maioria dos pacientes recebeu doses médias diárias entre 50 a 100 mg/m2.
No estudo de número 4, um total de 175 pacientes com SMD de risco mais elevado (principalmente dos tipos AREB e AREB-T) foram expostos ao azacitidina. Desses pacientes, 119 foram expostos a 6 ou mais ciclos, e 63 foram expostos a pelo menos 12 ciclos. A idade média desta população era de 68,1 anos (variando desde 42 a 83 anos de idade), 74% eram pacientes masculinos e 99%, eram da raça branca. A maioria dos pacientes recebeu doses médias diárias de azacitidina de 75 mg/m2.
A tabela 5 apresenta as reações adversas ocorrendo em pelo menos 5% dos pacientes tratados com azacitidina (administração subcutânea), nos estudos de número 1 e 2. É importante observar que a duração da exposição foi maior no grupo tratado com azacitidina do que o grupo de observação, os pacientes receberam azacitidina durante uma média de 11,4 meses, enquanto que o tempo médio no braço de observação foi de 6,1 meses.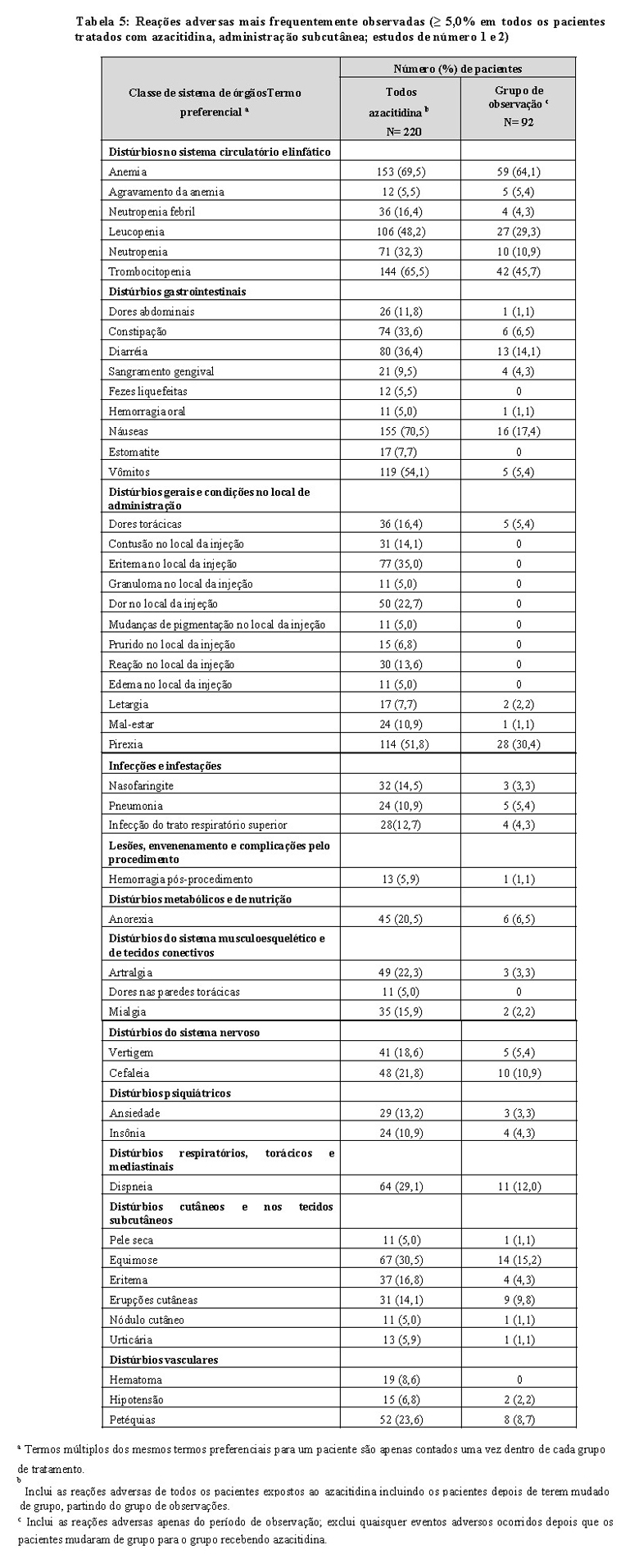
A tabela 6 apresenta as reações adversas ocorrendo em pelo menos 5% dos pacientes tratados com azacitidina no estudo de número 4. Similarmente aos estudos de número 1 e 2 acima, a duração da exposição ao tratamento com azacitidina foi mais demorada (média de 12,2 meses), quando comparada ao melhor cuidado de apoio (média de 7,5 meses).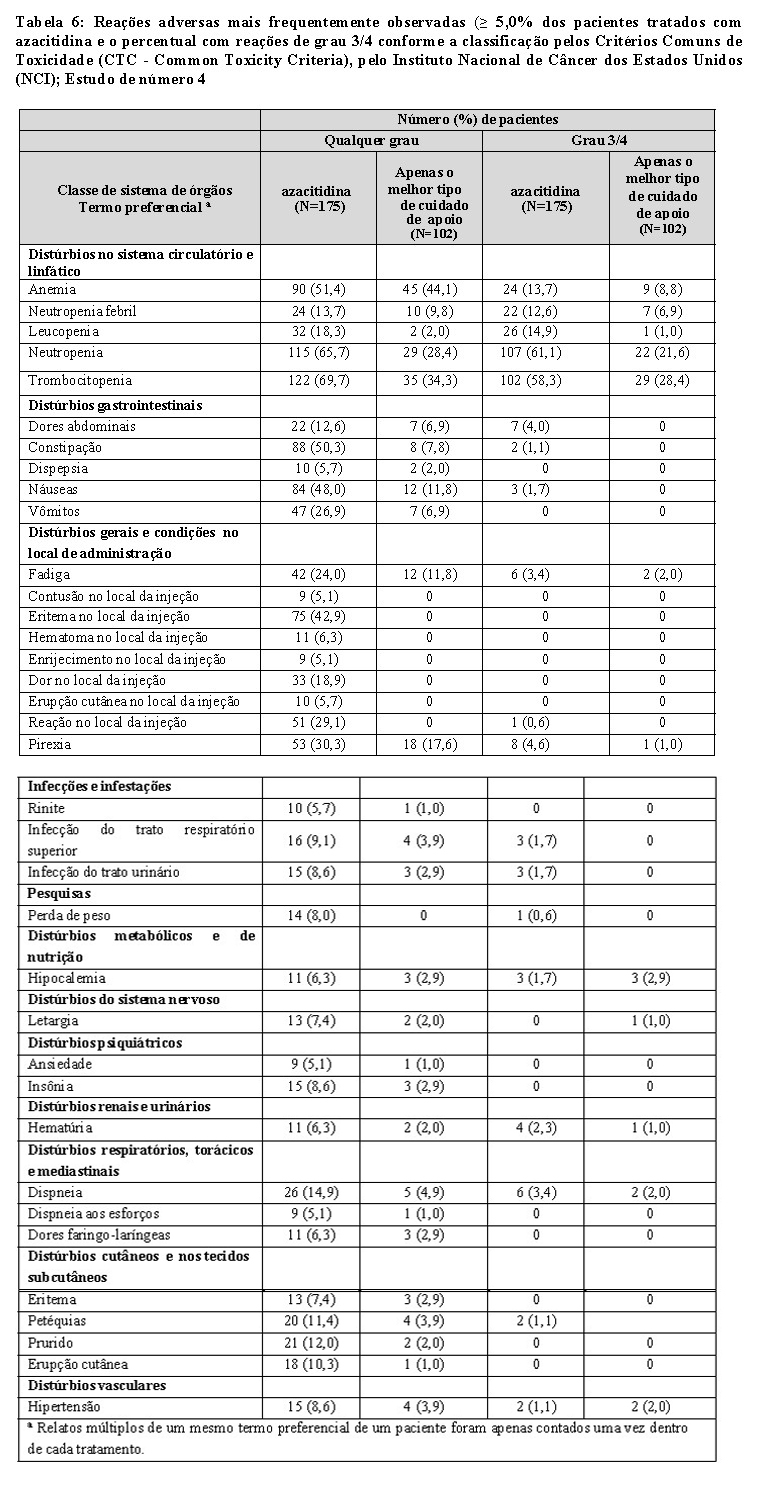
Nos estudos de número 1, 2 e 4 com administração subcutânea de azacitidina as reações adversas de neutropenia, trombocitopenia, anemia, náuseas, vômitos, diarreia, constipação e eritema / reação no local da injeção tenderam a aumentar incidentemente com doses mais elevadas de azacitidina. As reações adversas que tiveram a tendência a serem mais pronunciadas durante os primeiros 1 a 2 ciclos do tratamento subcutâneo, quando comparados a ciclos mais tardios incluíam trombocitopenia, neutropenia, anemia, náuseas, vômitos, eritema / dores / hematoma / reação no local da injeção, constipação, petéquias, vertigem, ansiedade, hipocalemia e insônia. Não pareceu haver quaisquer reações adversas que aumentaram em frequência no curso do tratamento.
De modo geral, as reações adversas foram qualitativamente similares entre os estudos com administração intravenosa e os com administração subcutânea.
Em estudos clínicos com administração subcutânea do azacitidina foi informada a ocorrência das reações adversas graves a seguir a um índice de < 5% (e que não foram descritas nas tabelas 5 ou 6):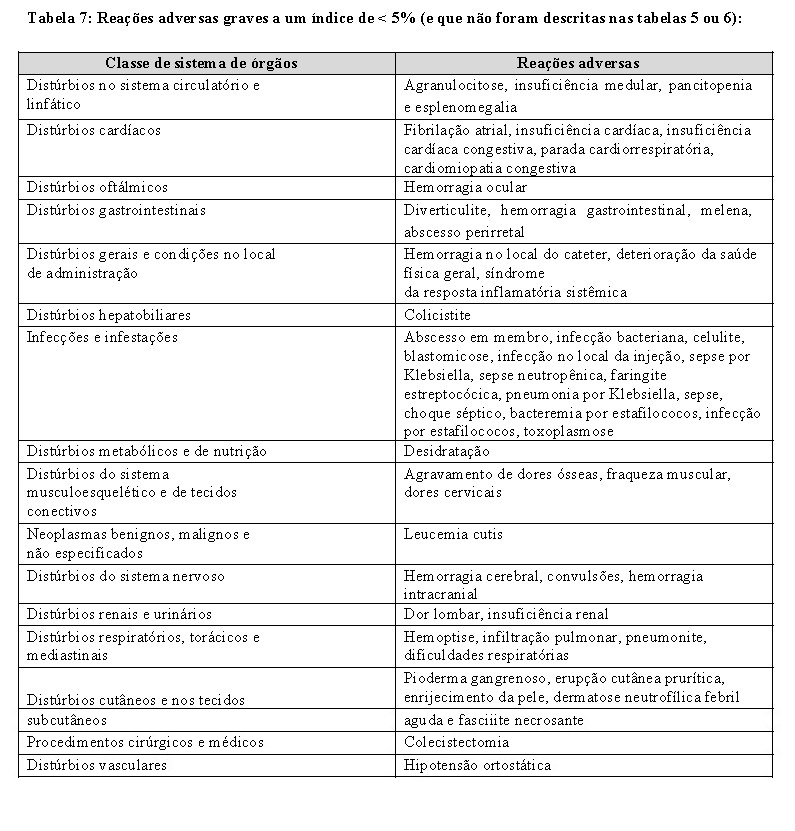
DADOS PÓS-COMERCIALIZAÇÃO
Os seguintes eventos foram relatados no cenário pós-comercialização:
Infecções e infestações: fasciite necrosante
Transtornos do metabolismo e nutrição: síndrome de lise tumoral
Distúrbios respiratórios, torácicos e do mediastino: doença pulmonar intersticial (DPI)
Pele e distúrbios do tecido subcutâneo: dermatose neutrofílica aguda febril e pioderma gangrenosa
Distúrbios gerais e condições no local da administração: necrose no local da injeção
Neoplasias benignas, malignas e não especificadas (incluindo cistos e pólipos): Síndrome de diferenciação
Em casos de eventos adversos, notifique pelo Sistema VigiMed, disponível no Portal da Anvisa.
10. SUPERDOSE
Um caso de superdosagem com azacitidina foi relatado durante estudos clínicos. Um paciente apresentou diarreia, náusea e vômitos após receber uma dose IV única de aproximadamente 290 mg/m2, quase 4 vezes a dose inicial recomendada. Os eventos foram resolvidos sem sequelas e a dose correta continuou no dia seguinte. No evento de superdosagem o paciente deve ser monitorado com contagens sanguíneas apropriadas e deve receber tratamento de suporte, conforme necessário. Não existe nenhum antídoto específico para superdosagem com azacitidina.
Em caso de intoxicação, ligue para 0800 722 6001, se você precisar de mais orientações.
Dizeres legais.
MS - 1.8261.0013
VENDA SOB PRESCRIÇÃO MÉDICA
USO RESTRITO A HOSPITAIS
Esta bula foi atualizada conforme bula padrão aprovada pela ANVISA em: 01/04/2022.