XENPOZYME
SANOFI MEDLEY
alfaolipudase
Tratamento da ASMD.
Apresentações.
XENPOZYME 20 mg pó liofilizado para solução injetável - 1 frasco-ampola contendo 21,2 mg de alfaolipudase, com uma dose extraível de 20 mg após reconstituição.
USO INTRAVENOSO
USO ADULTO E PEDIÁTRICO
Composição.
Cada frasco-ampola de XENPOZYME 20 mg contém 21,2 mg de alfaolipudase, com uma dose extraível de 20 mg após reconstituição.
Excipientes: sacarose, levometionina, fosfato de sódio dibásico heptaidratado e fosfato de sódio monobásico monoidratado.
Informações técnicas.
1. INDICAÇÕES
XENPOZYME é indicado como terapêutica de reposição enzimática para o tratamento de manifestações não relacionadas ao sistema nervoso central (SNC) de deficiência de esfingomielinase ácida (Acid Sphingomyelinase Deficiency, ASMD) em pacientes pediátricos e adultos com tipo A/B ou tipo B.
2. RESULTADOS DE EFICÁCIA
A eficácia de XENPOZYME foi avaliada em 3 estudos clínicos (estudo DFI12712/ASCEND em pacientes adultos, estudo DFI13803/ASCEND-Peds em pacientes pediátricos e estudo de extensão LTS13632 em pacientes adultos e pediátricos) envolvendo um total de 61 pacientes com ASMD.
Estudo clínico em pacientes adultos:
O estudo ASCEND é um estudo multicêntrico, randomizado, duplo-cego, controlado por placebo, de dose repetida fase II/III em pacientes adultos com ASMD (diagnóstico clínico consistente com ASMD tipo B e A/B). Um total de 36 pacientes foram randomizados em uma proporção 1:1 para receber XENPOZYME ou placebo. O tratamento foi administrado em ambos os grupos como uma infusão intravenosa uma vez a cada 2 semanas. Os pacientes que receberam XENPOZYME tiveram a dose escalonada de 0,1 mg/kg para uma dose alvo de 3 mg/kg. O estudo foi dividido em 2 períodos consecutivos: um período de análise primária (PAP) controlado por placebo randomizado e duplo-cego que durou até à semana 52, seguido de uma extensão do período de tratamento (ETP) por até 4 anos.
Os pacientes do braço placebo passaram a serem tratados com escalonamento de dose até 3 mg/kg de XENPOZYME no ETP, enquanto os pacientes no braço original de XENPOZYME continuaram o tratamento. A duração do estudo mais longo por paciente foi de até 5 anos e 3 meses.
Pacientes em condição clínica grave, incluindo malignidade, doença cardíaca significativa, uso de suporte ventilatório invasivo, hepatite B ou C ativa ou infecção pelo vírus da imunodeficiência humana (HIV), pacientes com contagem de plaquetas < 60x103/mL, alanina aminotransferase (ALT) ou aspartato aminotransferase (AST) > 250 UI/L, razão internacional normalizada (INR) > 1,5 foram excluídos.
Os pacientes incluídos no estudo tinham uma capacidade de difusão pulmonar para o monóxido de carbono (DLco) ≤70% do valor normal previsto, um volume do baço ≥6 múltiplos do normal (MN) medido por imagem de ressonãncia magnètica (MRI) e pontuações ≥5 na pontuação relacionada â esplenomegalia (SRS). Em geral, as características demográficas e da doença no momento basal foram semelhantes entre os dois grupos de tratamento. A idade média do paciente foi de 30 anos (variação: 18-66). Dois pacientes (11,1%) com insuficiência renal leve (60 mL/min. depuração de creatinina < 90 mL/min) foram incluídos em cada grupo. Não houve pacientes com insuficiência renal moderada ou grave.
Este estudo incluiu 2 desfechos primários de eficácia separados: a alteração percentual em DLco (em % prevista de normal) e o volume do baço (MN), conforme medido por MRI, do momento basal para a semana
52.
Os desfechos secundários de eficácia incluíram a alteração percentual no volume hepático (em MN) e a contagem de plaquetas do momento basal para a semana 52.
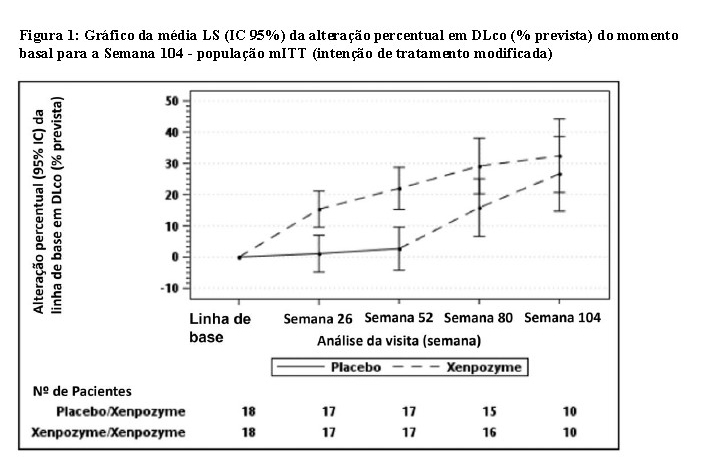
Melhorias na alteração percentual média em % prevista de DLco (p=0,0004) e volume do baço (p < 0,0001), bem como no volume médio do fígado (p < 0,0001) e contagem de plaquetas (p=0,0185) foram observados no grupo XENPOZYME em comparação com o grupo placebo durante o período de análise primária de 52 semanas (ver Tabela 1, Figura 1 e Figura 2). Na semana 26 de tratamento, a primeira avaliação do desfecho pós-dose, observou-se uma melhoria significativa na alteração percentual média da % prevista de DLco, volume do baço, volume hepático e contagem de plaquetas.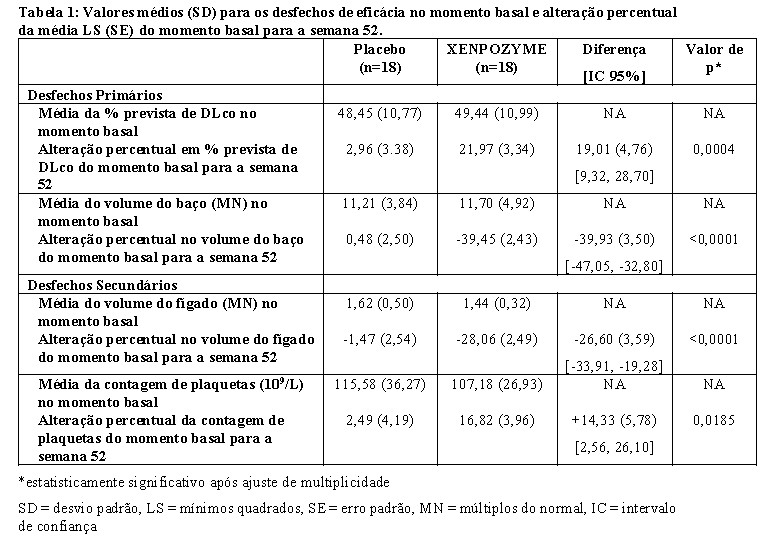
As barras verticais representam o IC 95% para a média LS.
A média LS e IC 95% são baseados em um modelo misto para abordagem de medidas repetidas, usando dados até a Semana 104.
Os pacientes do grupo placebo/ XENPOZYME receberam placebo até a semana 52 e mudaram para XENPOZYME depois disso.
As barras verticais representam IC 95% para a média LS.
A média LS e IC 95% são baseados em um modelo misto para abordagem de medidas repetidas, usando dados até a Semana104.
Os pacientes do grupo placebo/XENPOZYME receberam placebo até a semana 52 e mudaram para XENPOZYME depois disso.
Dezessete dos 18 pacientes que receberam previamente placebo e 18 dos 18 pacientes previamente tratados com XENPOZYME durante 52 semanas (PAP) iniciaram ou continuaram o tratamento com XENPOZYME, respectivamente, por até 4 anos. Na semana 104, os pacientes inicialmente randomizados para placebo receberam XENPOZYME por 52 semanas e demonstraram as seguintes mudanças nos parâmetros clínicos a partir do momento basal (antes da primeira administração de XENPOZYME): o aumento percentual médio (SE) do DLco previsto foi de 28,04% (6,16) (ver Figura 1); a redução percentual média (SE) no volume do baço (MN) foi de 35,93% (2,99) (ver Figura 2); a redução percentual média (SE) no volume hepático (MN) foi de 30,66 (2,45), o aumento percentual médio (SE) na contagem de plaquetas foi de 21,73 (6,43). Os pacientes do grupo anterior XENPOZYME demonstraram uma melhoria sustentada do momento basal para a semana 104 nos seguintes parâmetros: aumento percentual médio (SE) na % prevista DLco foi de 28,49 (6,16) (ver Figura 1); redução percentual média (SE) no volume do baço (MN) foi de 46,95 (2,65) (ver Figura 2); redução percentual média (SE) no volume do fígado (MN) foi de 33,42 (2,16); aumento percentual médio (SE) na contagem de plaquetas foi de 24,94 (6,91).
Estudo clínico em pacientes pediátricos
O estudo ASCEND-Peds (Estudo clínico fase 1/2) é um estudo multicêntrico, aberto, de dose repetida para avaliar a segurança e tolerabilidade do XENPOZYME administrado durante 64 semanas em pacientes pediátricos com menos de 18 anos de idade com ASMD (diagnóstico clínico consistente com ASMD tipo B e A/B). Além disso, os desfechos de eficácia exploratórios relacionados à visceromegalia, funções pulmonares e hepáticas, e crescimento linear foram avaliados na semana 52.
Pacientes com anormalidades neurológicas agudas ou rapidamente progressivas, em condição clínica grave, incluindo malignidade, doença cardíaca significativa, uso de suporte ventilatório invasivo, hepatite B ou C ativa, pacientes com contagem média de plaquetas < 60 x 103-mL, alanina aminotransferase (ALT) ou aspartato aminotransferase (AST) > 250 UI/L ou bilirrubina total > 1,5 mg/dL, ou uma razão internacional normalizada (INR) > 1,5 e pacientes que são homozigotos para as mutações do gene SMPD1 p.Arg498Leu, p.Leu304Pro ou p.Phe333SerfsTer52 ou qualquer combinação dessas 3 mutações foram excluídos.
Não há dados de eficácia e segurança da utilização de Xenpozyme em pacientes pediátricos com ASMD tipo A.
Os pacientes foram distribuídos em todas as idades de 1,5 a 17,5 anos em geral, com ambos os sexos representados em cada coorte de idade e igualmente representados no grupo geral (total de 10 homens e 10 mulheres). Os pacientes foram diagnosticados por volta dos 2 anos de idade (mediana), com os sintomas aparecendo por volta de 1 ano de idade (mediana). No início da doença, 18 pacientes (90,0%) apresentavam hepatomegalia e/ou esplenomegalia e 7 pacientes (35,0%) apresentavam doença respiratória. Doze pacientes (60,0%) tiveram esplenomegalia grave ( > 15 MN) no momento basal, e 1 paciente (11,0%) teve um valor percentual de DLco previsto severamente reduzido ( < 40,0%) entre aqueles que puderam realizar
o teste.
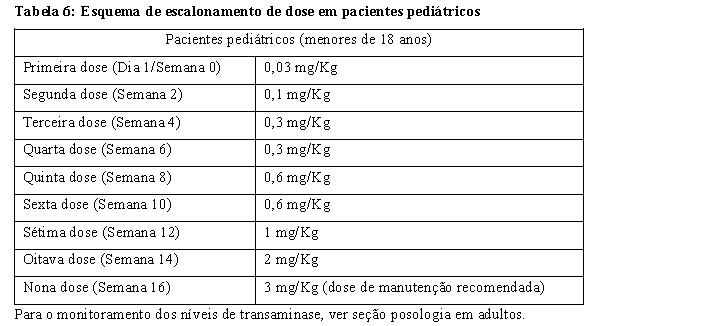
Um total de 20 pacientes (4 adolescentes de 12 a 18 anos, 9 crianças de 6 a 12 anos e 7 bebês/crianças < 6 anos) foram titulados com XENPOZYME através de um esquema de escalonamento de dose de 0,03 mg/kg para uma dose alvo de 3 mg/kg (ver Tabela 6 para informação sobre escalonamento de dose). O tratamento foi administrado como uma infusão intravenosa uma vez a cada 2 semanas por até 64 semanas.
Os pacientes incluídos no estudo tinham um volume de baço ≥5 MN medido por ressonãncia magnètica. Os pacientes foram distribuídos por todos os grupos etários de 1,5 a 17,5 anos de idade, com ambos os sexos igualmente representados.
A avaliação exploratória de eficácia de XENPOZYME indica que houve melhorias na alteração percentual média em % prevista dos volumes de DLco, baço e fígado, contagem de plaquetas e progressão linear do crescimento (medida pelas pontuações Z de Altura) na semana 52 em comparação com o momento basal (ver Tabela 2).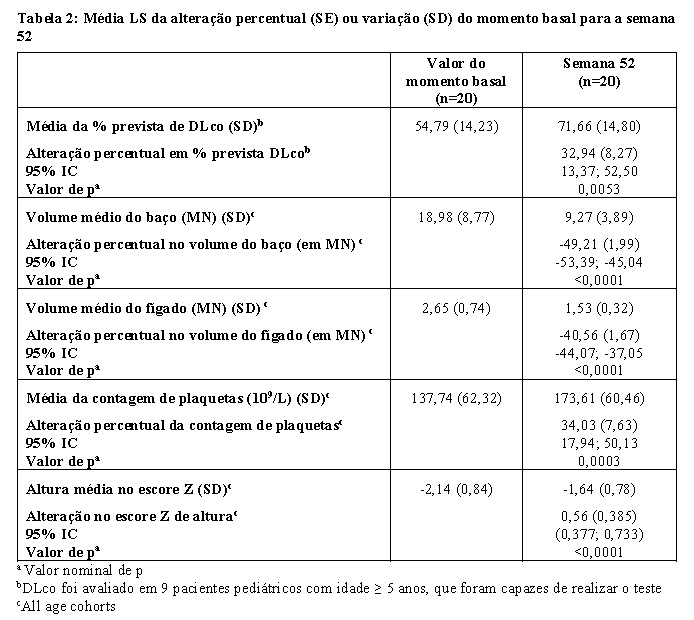
Os efeitos de XENPOZYME nos volumes do baço e do fígado, e o escore Z de altura foram semelhantes em todas as coortes de idade pediátrica incluídas no estudo.
Embora diferentes desenhos de estudos tenham sido empregados, os resultados da eficácia em populações adultas e pediátricas foram consistentes.
Olipudase alfa foi geralmente bem tolerada em todas as faixas etárias. A maioria dos TEAEs foram leves ou moderados. Cinco pacientes tiveram um total de 12 TEAEs graves, incluindo 3 pacientes que tiveram 5 TEAEs graves potencialmente relacionados (2 eventos de aumento de ALT em 1 paciente; urticária difusa e erupção cutânea difusa em 1 paciente e reação anafilática em 1 paciente). Nenhum paciente descontinuou permanentemente o tratamento devido a TEAEs ou se retirou do estudo, e nenhum paciente morreu. Todos os pacientes foram escalados para a dose alvo de 3,0 mg/kg.
Entre os 11 pacientes (55,0%) que apresentaram RIs especificadas pelo protocolo (6 pacientes na coorte infantil e 5 pacientes na coorte infantil/infantil), 2 pacientes (na coorte infantil/infantil) apresentaram RIs graves.
Um total de 7 pacientes (35,0%) apresentou aumento transitório de transaminases durante a fase de escalonamento de dose e isso levou à repetição ou diminuição de uma dose. Além disso, 16 dos 16 pacientes com ALT basal elevada apresentaram ALT dentro da faixa normal e 16 dos 17 pacientes com AST basal elevada apresentaram AST dentro da faixa normal ao longo de 52 semanas de tratamento com alfaolipudase.
Todos os casos de reduções de dose relacionadas ao medicamento ocorreram durante a fase de escalonamento de dose, exceto 1 caso em um paciente que teve uma reação de hipersensibilidade seguida por consecutivas doses perdidas não relacionadas.
Outras medidas laboratoriais, sinais vitais e ECGs não levantaram nenhuma preocupação de segurança.
Estudo de extensão em pacientes adultos e pediátricos
Os pacientes que participaram do estudo DFI13412 (estudo de dose ascendente aberto em pacientes adultos ASMD) ou estudos DFI13803/ASCEND-Peds continuaram o tratamento em um estudo de extensão aberto (LTS13632). Cinco pacientes adultos do estudo DFI13412 e 20 pacientes pediátricos do estudo DFI13803/ASCEND-Peds receberam XENPOZYME a 3 mg/kg uma vez a cada 2 semanas por infusão intravenosa por até 7 anos e 5 anos em pacientes adultos e pediátricos, respectivamente.
Melhorias sustentadas na % prevista de DLco, volume do baço e fígado e contagem de plaquetas, em comparação ao momento basal, foram observadas em pacientes adultos e pediátricos durante o curso do estudo. Além disso, pacientes pediátricos (todas as coortes de idade) apresentaram uma melhora contínua no escore Z de Altura e uma melhora na idade óssea (por raio-X manual) no mês 48, indicando que a idade óssea estava se aproximando da idade cronológica.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Mecanismo de ação
A deficiência de esfingomielinase ácida (ASMD) é um distúrbio de armazenamento lisossomal raro e potencialmente fatal. Pacientes com ASMD têm prejuízo variável no metabolismo da esfingomielina devido a variantes patogênicas no SMPD1, o gene que codifica a esfingomielinase ácida (ASM) que resulta na expressão de ASM defeituosa com atividade reduzida. Como a ASM catalisa a hidrólise da esfingomielina em ceramida e fosfocolina, a redução da atividade da ASM resulta no acúmulo progressivo de esfingomielina lisossomal principalmente dentro das células da linhagem de monócitos/macrófagos que residem em tecidos reticuloendoteliais, ou seja, no baço, fígado, pulmão, medula óssea, e linfonodos. Com a doença grave, os neurônios também podem ser afetados. O espectro fenotípico varia desde a forma neurovisceral infantil grave (ASMD tipo A) para a forma visceral crônica (ASMD tipo B), com uma apresentação fenotípica neurovisceral intermediária ou crônica também sendo descrita (ASMD tipo A/B). A alfaolipudase é uma esfingomielinase ácida humana recombinante expressa em células de ovário de hamster chinês, desenvolvida como uma terapia de reposição enzimática para o tratamento de manifestações não relacionadas ao SNC. Não se espera que XENPOZYME atravesse a barreira hematoencefálica ou que apresente atividade em relação às manifestações da doença no SNC.
Propriedades farmacodinâmicas
A ceramida é um catabólito direto do metabolismo da esfingomielina mediado pela alfaolipudase e a lisoesfingomielina é a forma desacetilada da esfingomielina (SM); ambos os parâmetros são usados para avaliar os efeitos farmacodinâmicos do XENPOZYME.
Após a administração repetida de XENPOZYME em pacientes adultos e pediátricos, os níveis plasmáticos de ceramida mostraram um aumento transitório após cada dose (pós-infusão), com uma diminuição gradual nos níveis plasmáticos durante o período de tratamento. No estudo DFI12712/ASCEND, a alteração percentual da média dos mínimos quadrados (LS) desde o início até a semana 52 (erro padrão, SE) no nível de ceramida plasmática pré-infusão foi de -36,4% (5,3) no grupo em tratamento com XENPOZYME em comparação com -0,2% (5,6) no grupo placebo. Em pacientes pediátricos, a média LS do nível de ceramida plasmática pré-infusão foi reduzido em 57% (SE: 5,1) em comparação com o momento basal após 52 semanas de tratamento.
A liso-esfingomielina é substancialmente elevada no plasma de pacientes adultos e pediátricos com ASMD. Após a administração repetida de XENPOZYME, os níveis de liso-esfingomielina plasmática diminuíram significativamente, refletindo a redução do conteúdo de esfingomielina no tecido. No estudo DFI12712/ASCEND, a alteração percentual média LS do momento basal para a Semana 52 (SE) do nível
da liso-esfingomielina plasmática pré-infusão foi de -77,7% (3,9) no grupo em tratamento com XENPOZYME em comparação com -5% (4,2) no grupo placebo. Em pacientes pediátricos, a média LS do nível de liso-esfingomielina plasmática pré-infusão foi reduzido em 87,2% (SE: 1,3) em comparação com o momento basal após 52 semanas de tratamento.
Em pacientes adultos, o conteúdo de esfingomielina do fígado, conforme avaliado pela histopatologia, diminuiu 92% (SE: 8,1) do momento basal para a semana 52 no grupo em tratamento com XENPOZYME (comparado com +10,3% (SE: 7,8) no grupo placebo).
Propriedades farmacocinéticas
A farmacocinética (PK) de alfaolipudase foi avaliada em 49 pacientes adultos com ASMD de todos os estudos clínicos, recebendo uma ou várias administrações.
A alfaolipudase exibiu farmacocinética linear na faixa de doses de 0,03 a 3 mg/kg. Seguindo um esquema de escalonamento de dose de 0,1 mg/kg para a dose de manutenção de 3 mg/kg administrada uma vez a cada 2 semanas, houve um acúmulo mínimo nos níveis plasmáticos de alfaolipudase. Na dose de 3 mg/kg administrada uma vez a cada 2 semanas, a média (coeficiente de variação percentual, CV%) de concentração máxima (Cmax) e área sob a curva de tempo-concentração ao longo de um intervalo de dosagem (AUC0-t) no estado estacionário foram de 30,2 mg/mL (17%) e 607 mg.h/mL (20%), respectivamente.
Absorção
Não há absorção uma vez que XENPOZYME é administrado por via intravenosa.
Distribuição
A média estimada (CV%) do volume de distribuição de alfaolipudase é de 13,1 L (18%).
Metabolismo
A alfaolipudase é uma enzima recombinante humana e espera-se que seja eliminada através da degradação proteolítica em pequenos peptídeos e aminoácidos.
Eliminação
A média (CV%) de depuração de alfaolipudase é de 0,331 L/h (22%). A meia-vida terminal média (t1/2) variou de 31,9 a 37,6 horas.
Populações especiais
Gênero
Não houve diferenças clinicamente relevantes na farmacocinética de alfaolipudase com base no sexo.
Raça
Existe informação limitada da farmacocinética de alfaolipudase em grupos étnicos não caucasianos.
Pacientes idosos (≥ 65 anos de idade)
Existe informação limitada sobre farmacocinética de alfaolipudase em pacientes idosos (apenas 2 pacientes entre 65 e 75 anos de idade incluídos em estudos clínicos com XENPOZYME).
Pacientes pediátricos
A PK de alfaolipudase foi avaliada em 20 pacientes pediátricos, incluindo 4 pacientes adolescentes, 9 pacientes crianças e 7 pacientes crianças/recém-nascidos (Tabela 3). As exposições de alfaolipudase foram menores em pacientes pediátricos do que em pacientes adultos. Estas diferenças não foram consideradas como clinicamente relevantes.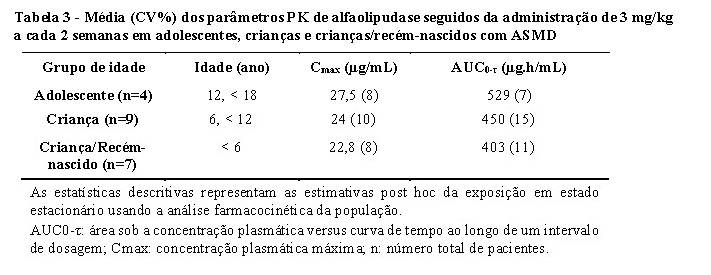
Deficiência Hepática
A alfaolipudase é uma proteína recombinante e espera-se que seja eliminada por degradação proteolítica. Portanto, não se espera que o comprometimento da função hepática afete a farmacocinética de alfaolipudase.
Deficiência Renal
Não houve diferenças clinicamente relevantes na farmacocinética de alfaolipudase em pacientes com comprometimento renal leve. O impacto da insuficiência renal moderada a grave na farmacocinética de alfaolipudase não é conhecido. Não se espera que alfaolipudase seja eliminada por excreção renal. Portanto, não se espera que a insuficiência renal afete a farmacocinética de alfaolipudase.
4. CONTRAINDICAÇÕES
XENPOZYME é contraindicado a pacientes que já demonstraram hipersensibilidade grave (reação anafilática) à alfaolipudase ou a qualquer um dos componentes do medicamento quando a dessensibilização sob medida não foi bem-sucedida (ver seção 5. Advertências e Precauções).
5. ADVERTÊNCIAS E PRECAUÇÕES
Reações associadas à infusão (RIs)
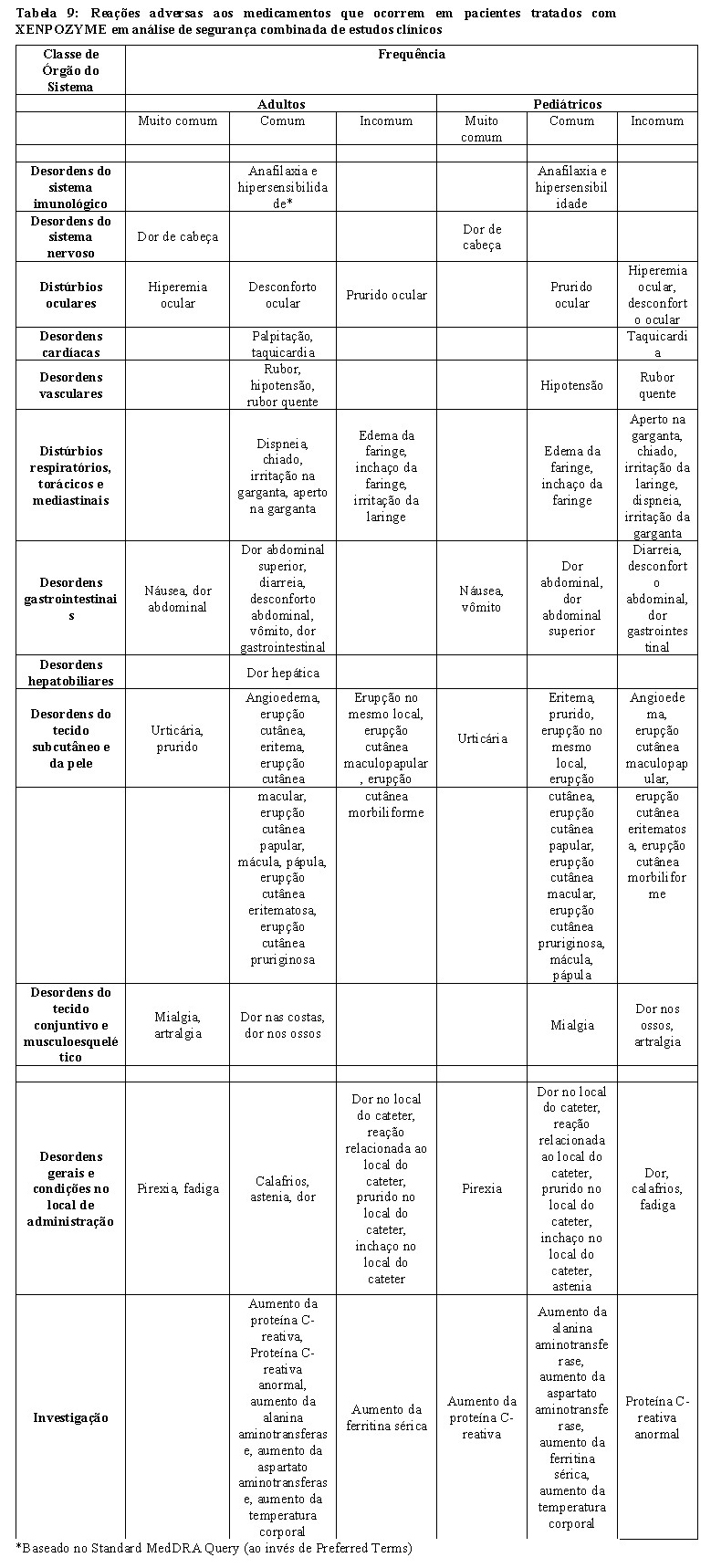
Em estudos clínicos, as RIs ocorreram em aproximadamente 58% dos pacientes tratados com XENPOZYME. Estas RIs incluem reações de hipersensibilidade e reações de fase aguda (ver seção 9. Reações Adversas). As RIs mais frequentes foram dor de cabeça, urticária, pirexia, náusea e vômito (ver seção 9. Reações Adversas). As RIs normalmente ocorreram entre o momento da infusão e até 24 horas após a conclusão da infusão.
• Reações de hipersensibilidade/anafilaxia
Reações de hipersensibilidade, incluindo anafilaxia, foram relatadas em pacientes tratados com XENPOZYME (ver seção 9. Reações Adversas). Em estudos clínicos, as reações de hipersensibilidade ocorreram em 7 (17,5%) pacientes adultos e 9 (45%) pacientes pediátricos, incluindo um paciente pediátrico com anafilaxia. Independente do programa de estudo clínico, um paciente de 16 meses de idade com ASMD tipo A tratado com XENPOZYME teve 2 reações anafiláticas. Em ambos os pacientes com anafilaxia, foram detectados anticorpos anti-alfaolipudase IgE (ver seção 9. Reações Adversas).
Reações de hipersensibilidade leve a moderada relatadas em mais de um paciente adulto, incluíram urticária, eritema e prurido (ver seção 9. Reações Adversas). Em pacientes pediátricos, as reações de hipersensibilidade leve a moderada relatadas em mais de um paciente incluíram urticária, eritema e erupção cutânea (ver seção 9. Reações Adversas).
• Manejo
Observe atentamente os pacientes durante e por um período de tempo apropriado após a infusão, com base na avaliação clínica. Informar os pacientes sobre os potenciais sintomas de hipersensibilidade/anafilaxia e instruí-los a procurar cuidados médicos imediatos caso os sintomas ocorram. O manejo das RIs deve ser
baseado na gravidade dos sinais e sintomas e pode incluir a interrupção temporária da infusão, a redução da taxa de infusão e/ou tratamento médico apropriado.
Se ocorrer hipersensibilidade grave ou anafilaxia, XENPOZYME deve ser descontinuado imediatamente e deve ser iniciado o tratamento médico apropriado. O paciente que apresentou anafilaxia no estudo clínico foi submetido a um regime de dessensibilização individualizado que permitiu ao paciente retomar o tratamento com XENPOZYME. Os riscos e benefícios da readministração de XENPOZYME após anafilaxia ou reação de hipersensibilidade grave devem ser considerados. Ao se considerar a readministração de XENPOZYME após anafilaxia, o médico deve contatar o Serviço de atendimento ao consumidor através do 0800-703-0014 para aconselhamento. Nestes pacientes, deve-se ter extremo cuidado, com medidas apropriadas de reanimação disponíveis no momento da readministração.
Os médicos podem considerar testar anticorpos anti-alfaolipudase IgE em pacientes que tenham sofrido reações de hipersensibilidade graves (ver seção 9. Reações Adversas). Contatar o Serviço de atendimento ao consumidor através do 0800-703-0014 para informação sobre o teste.
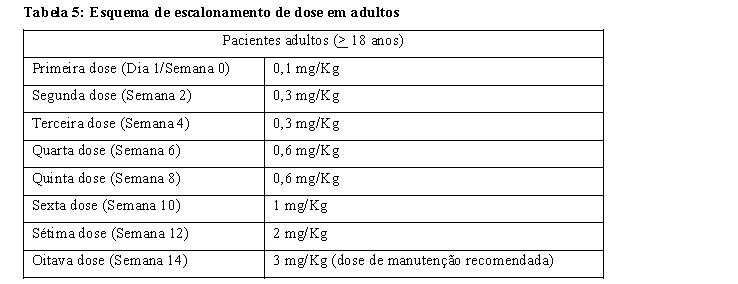
Se ocorrerem RIs leves ou moderadas, a taxa de infusão pode ser diminuída ou parada temporariamente, a duração de cada etapa de uma infusão individual pode ser aumentada, e/ou a dose de XENPOZYME diminuída. Se um paciente necessitar de uma redução de dose, o reescalonamento deve seguir o escalonamento de dose descrito na Tabela 5 e Tabela 6 para pacientes adultos e pediátricos, respectivamente (ver seção 8. Posologia e Modo de usar).
Os pacientes não foram rotineiramente pré-medicados antes da infusão de XENPOZYME nos estudos clínicos. Medicamentos (por exemplo, anti-histamínicos, antipiréticos, glicocorticoides) foram usados antes ou depois das infusões em alguns pacientes. No entanto, a eficácia destes tratamentos em melhorar as reações de hipersensibilidade recorrente leve-moderada não foi estabelecida.
Elevação transitória de transaminases
Em estudos clínicos, elevações transitórias de transaminase (ALT ou AST) dentro de 24 a 48 horas após infusões foram relatadas em 4 pacientes adultos e 7 pediátricos durante a fase de escalonamento de dose com XENPOZYME (ver seção 9. Reações Adversas). No momento da próxima infusão programada, esses níveis elevados de transaminase geralmente retornavam aos níveis observados antes da infusão de XENPOZYME.
Após 52 semanas de tratamento, a maioria dos pacientes com níveis elevados de transaminase no momento basal tinham valores dentro da faixa normal (ver seção 9. Reações Adversas).
Os níveis de transaminases (ALT e AST) devem ser obtidos dentro de 1 mês antes do início do tratamento (ver seção 8. Posologia e Modo de usar). Durante o escalonamento da dose ou ao retomar o tratamento após as doses perdidas, os níveis de transaminases devem ser obtidos dentro de 72 horas antes da próxima infusão programada (ver seção 8. Posologia e Modo de usar). Se o momento basal ou o nível de transaminase préinfusão for > 2 vezes o limite superior da normalidade (ULN) durante o aumento da dose, então níveis adicionais de transaminase devem ser obtidos dentro de 72 horas após o final da infusão. Se os níveis de transaminase forem elevados acima do momento basal e > 2 vezes da ULN, a dose XENPOZYME pode ser ajustada (dose anterior repetida ou reduzida) ou o tratamento pode ser temporariamente suspenso, com base na avaliação clínica.
Ao atingir a dose de manutenção recomendada, os testes de transaminase podem ser realizados como parte do gerenciamento clínico de rotina da ASMD.
Gravidez
Mulheres com potencial para engravidar:
Recomenda-se a realização de um teste de gravidez antes do início do tratamento com XENPOZYME.
Mulheres com potencial para engravidar são aconselhadas a usar métodos contraceptivos eficazes durante o tratamento e por 14 dias após a última dose se XENPOZYME for descontinuado.
Não há dados disponíveis sobre uso de XENPOZYME em mulheres grávidas. Estudos em animais tem mostrado toxicidade no desenvolvimento (ver seção Toxicidade reprodutiva e de desenvolvimento). XENPOZYME não é recomendado durante a gravidez e em mulheres com potencial para engravidar que não utilizam métodos contraceptivos eficazes, a menos que os benefícios potenciais para a mãe superem os riscos potenciais, incluindo aqueles para o feto.
Categoria de risco de gravidez: C
ESTE MEDICAMENTO NÃO DEVE SER UTILIZADO POR MULHERES GRÁVIDAS SEM ORIENTAÇÃO MÉDICA OU DO CIRURGIÃO-DENTISTA.
Lactação
Não existem dados disponíveis sobre a presença de XENPOZYME no leite humano, efeitos na produção de leite ou no lactante amamentado. A alfaolipudase foi detectada no leite de camundongos lactantes (ver seção Toxicidade reprodutiva e de desenvolvimento). Quando o fármaco está presente no leite animal, pode estar presente no leite humano. Não é possível tirar conclusões se XENPOZYME é ou não seguro para utilização durante o aleitamento materno. XENPOZYME não deve ser utilizado durante a amamentação a menos que os potenciais benefícios para a mãe superem os riscos potenciais, incluindo aqueles para a criança amamentada.
Fertilidade
Não existem dados disponíveis para humanos para determinar os potenciais efeitos de XENPOZYME na fertilidade de homens e mulheres. Os dados dos animais não mostraram qualquer efeito na fertilidade masculina ou feminina em camundongos (ver seção Toxicidade reprodutiva e de desenvolvimento).
Efeitos sobre a capacidade de dirigir veículos ou operar máquinas
Não foram realizados estudos sobre a capacidade de dirigir veículos ou operar máquinas. Como foi relatado hipotensão em estudos clínicos, XENPOZYME pode ter uma pequena influência na capacidade de conduzir e operar máquina (ver seção 9. Reações Adversas).
Abuso e dependência
XENPOZYME não atravessa a barreira hematoencefálica em modelos animais; portanto, não é previsto efeito de abuso ou dependência.
Atenção diabéticos: contém açúcar.
Dados de segurança não clínica
Toxicidade de dose única
Em camundongos, ratos, cães e macacos, uma única administração IV de alfaolipudase foi bem tolerada até 12, 30, 30, e 30 mg/kg (doses mais altas testadas em cada estudo), respectivamente. Em camundongos com inativação da esfingomielinase ácida ("acid sphingomyelinase knockout" -ASMKO), um modelo para a doença ASMD, doses önicas de alfaolipudase resultaram em mortalidade nas doses ≥10 mg/kg administrado como injeção IV em bolus.
Algumas manifestações (letargia, frieza ao toque e relutância em se mover), combinadas com hemorragia adrenal, sugerem que choque hipotensivo pode ter sido a causa de morte. Estes achados foram acompanhados por elevações de ceramida, esfingosina e esfingosina 1-fosfato no soro, catabólitos de esfingomielina acumulada, bem como por elevações nas concentrações séricas de mediadores inflamatórios, tais como citocinas e proteínas de fase aguda.
Foram observados achados microscópicos relacionados à dosagem que consistem em áreas focais de necrose e apoptose no fígado e glândulas supra-renais, e hemorragia nas glândulas supra-renais.
A falta de achados adversos em camundongos BALB/c, C57BL/6, ratos, cães e macacos em doses comparáveis de alfaolipudase sugere que a toxicidade relacionada com a dose observada em camundongos ASMKO pode ser devida à taxa e quantidade de degradação do substrato.
Toxicidade por dose repetida
Estudos de toxicologia por dose repetida foram realizados em camundongos ASMKO, ratos Sprague-Dawley e macacos Cynomolgus.
Embora a mortalidade tenha sido observada em camundongos ASMKO após uma dose única ≥10 mg/kg administrada como injeção IV em bolus (ver seção 5. Advertências e Precauções -Toxicidade reprodutiva e de desenvolvimento), estudos de dose repetida em camundongos ASMKO mostram que a administração de alfaolipudase através de um esquema de escalonamento de dose, 3 mg/kg administrada IV a cada dois dias, seguido por uma dose IV única de 20 mg/kg 3 dias depois, não resultou em mortalidade relacionada ao composto e reduziu a gravidade de outros achados de toxicidade. Quando a alfaolipudase foi administrada a cada 2 semanas, o nível sem efeito adverso observável (NOAEL) em camundongos ASMKO foi de 3 mg/kg; entretanto, quando um esquema de escalonamento de dose de alfaolipudase foi empregado antes de cada dose de 2 semanas, o NOAEL para alfaolipudase foi ≥30 mg/kg.
A administração bissemanal IV de alfaolipudase a ratos Sprague Dawley (injeção em bolus) e macacos Cynomolgus (infusão de 30 minutos) por 26 semanas não resultou em efeitos adversos relacionados ao composto em doses de até 30 mg/kg/administração. O NOAEL foi considerado como sendo 30 mg/kg/administração, correspondendo a exposições de 2,3 a 3,9 vezes as dos pacientes em dose terapêutica. A avaliação da tolerabilidade local foi incorporada ao estudo de toxicologia por dose repetida em macacos Cynomolgus através da avaliação macroscópica e microscópica dos locais de infusão IV. Não foram observadas descobertas relacionadas com a administração de alfaolipudase.
Genotoxicidade e Carcinogenicidade
Não foram realizados estudos para avaliar a genotoxicidade e carcinogenicidade da alfaolipudase.
Toxicidade reprodutiva e de desenvolvimento
Um estudo combinado de fertilidade masculina e feminina realizado em camundongos CD-1 nas doses de 0, 3,16, 10 e 30 mg/kg através da administração IV em bolus não mostrou efeitos no acasalamento e fertilidade dos ratos machos e fêmeas ou nos parâmetros de gestação precoce dos camundongos fêmeas. A mortalidade que ocorreu em todos os grupos de alfaolipudase foi considerada devido à hipersensibilidade da administração de alfaolipudase. Os NOAELs reprodutivos de machos e fêmeas foram de 30 mg/kg/dose.
A administração IV de alfaolipudase a camundongos CD-1 grávidas uma vez ao dia, desde os dias de gestação (GD) 6 a 15 em doses de 3, 10 ou 30 mg/kg/dia, resultou em efeitos maternos limitados a um aumento da incidência de diminuição da atividade em ≥3 mg/kg/dia. Exencefalia foi observada em 1 ninhada em cada um dos grupos de dose de 10 e 30 mg/kg (2 e 3 fetos, respectivamente). A incidência foi ligeiramente maior do que os dados históricos de controle. O aumento da incidência de exencefalia foi observado em camundongos prenhes em níveis de exposição inferiores à exposição humana na dose terapêutica de manutenção recomendada e frequência. Com base nesses dados, o NOAEL de desenvolvimento para alfaolipudase é de 3 mg/kg/dia (AUC0-24 83,1 mg.hr/mL) correspondendo a 0,14 vezes a exposição na dose terapêutica. A relevância dessa observação para humanos é desconhecida.
A infusão IV (10 minutos) de alfaolipudase em coelhas NZW grávidas uma vez por dia do GD 6 a 19 em doses de 0, 3, 10 ou 30 mg/kg/dia não resultou em efeito adverso materno relacionado à alfaolipudase em qualquer dose. Não houve efeitos de alfaolipudase relacionados à sobrevivência embrionária ou ao peso do corpo fetal, e não houve anormalidades fetais externas, viscerais ou esqueléticas relacionadas a qualquer dose. Portanto, o NOAEL de desenvolvimento foi de 30 mg/kg/dia (AUC0-24 de 6350.hr*mg /mL) correspondendo a 10,5 vezes a exposição na dose terapêutica.
Em um estudo de toxicidade pré e pós-natal, a administração IV de alfaolipudase a camundongos CD-1 grávidas nas doses de 0, 3,16, 10 ou 30 mg/kg uma vez a cada dois dias desde GD6 até o pós-parto Dia 19 ou 20 não produziu qualquer efeito sobre a função reprodutiva materna (F1). Não houve mortalidade relacionada com a alfaolipudase. Com base na hora do óbito após a administração da dose, e em conjunto com observações clínicas adversas, a mortalidade observada foi considerada devido à hipersensibilidade da administração de alfaolipudase.
Não houve diferenças toxicologicamente significativas em quaisquer parâmetros de desenvolvimento e reprodução avaliados na geração F1 de descendentes machos e fêmeas. O NOAEL materno e o NOAEL para reprodução nas mães e para viabilidade e crescimento da descendência foram de 30 mg/kg/dose.
Em camundongos que receberam 3 mg/kg de alfaolipudase no dia 7 pós-parto, a alfaolipudase foi detectada no leite 2 dias após a administração.
Outros estudos de toxicidade
Farmacologia de Segurança
Em cães Beagle e macacos Cynomolgus, a administração IV única de alfaolipudase em doses de até 30 mg/kg não induziu efeitos adversos nas funções cardiovascular e respiratória.
Em camundongos ASMKO, uma redução dose-dependente da frequência cardíaca acompanhada por uma diminuição da atividade motora e seguida por um lento declínio na pressão arterial foi observada após uma única administração IV a 3, 10, e 20 mg/kg. Após 2 doses de alfaolipudase a 3 e 10 mg/kg para camundongos ASMKO, notou-se uma ligeira queda na frequência cardíaca após a segunda administração.
6. INTERAÇÕES MEDICAMENTOSAS
Nenhum estudo de interações medicamentosas foi realizado com alfaolipudase. Como a alfaolipudase é uma proteína recombinante humana, não são esperadas interações medicamentosas mediadas pelo citocromo P450.
Inibidores funcionais da esfingomielinase ácida: com base em dados publicados in silico e in vitro, os antidepressivos tricíclicos e alguns medicamentos anfifílicos catiônicos, incluindo os anti-histamínicos, podem diminuir a atividade da alfaolipudase. A relevância clínica desta inibição funcional não é conhecida. Esta interação teórica deve ser considerada quando XENPOZYME é prescrito concomitantemente com tratamento sistêmico crônico com inibidores funcionais da esfingomielinase ácida.
Incompatibilidades farmacêuticas
Na ausência de estudos de compatibilidade, XENPOZYME não deve ser misturado com outros medicamentos.
7. CUIDADOS DE ARMAZENAGEM
Conservar XENPOZYME sob refrigeração, em temperatura entre 2°C e 8°C.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Prazo de validade: 60 meses a partir da data de fabricação.
A solução reconstituída e diluídas de XENPOZYME devem ser administradas imediatamente. Se o uso imediato não for possível, a solução reconstituída pode ser armazenada por até 24 horas à temperatura entre 2°C e 8°C. Após diluição, a solução pode ser armazenada por até 24 horas à temperatura entre 2°C e 8°C.
Não agitar as soluções reconstituídas e diluídas.
XENPOZYME é um pó liofilizado estéril branco a quase branco. Após reconstituição, a solução é clara e incolor.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
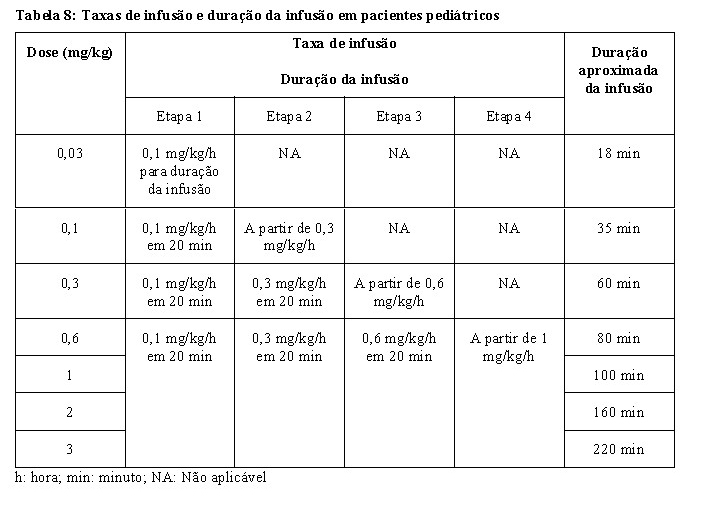
XENPOZYME deve ser reconstituído, diluído e administrado sob a supervisão de um profissional de saúde. As infusões devem ser administradas de forma gradual, de preferência com uma bomba de infusão.
Reconstituição
O pó para solução para infusão deve ser reconstituído com água estéril para injetáveis, diluído com solução de cloreto de sódio 9 mg/mL (0,9%) e, em seguida, administrado por infusão intravenosa.
As etapas de reconstituição e diluição devem ser concluídas em condições assépticas. Os dispositivos filtrantes não devem ser usados em nenhum momento durante a preparação da solução de infusão. Evite a formação de espuma durante as etapas de reconstituição e diluição.
a) Determinar o número de frascos a reconstituir com base no peso individual do paciente e na dose prescrita.
Peso do paciente (kg) x dose (mg/kg) = dose do paciente (em mg). Dose do paciente (em mg) dividida por 20 mg/frasco = número de frascos a reconstituir. Se o número de frascos incluir uma fração, arredonde para
o próximo número inteiro.
b) Retirar o número necessário de frascos da refrigeração e deixá-los atingir a temperatura ambiente por aproximadamente 20 a 30 minutos.
c) Reconstituir cada frasco injetando 5,1 ml de água estéril para injetáveis no frasco de 20 mg, utilizando uma técnica de adição lenta de gota-a-gota na parede interior do frasco.
d) Incline e gire cada frasco suavemente. Cada frasco produzirá uma solução límpida e incolor de 4,0 mg/mL.
e) Inspecionar visualmente a solução reconstituída nos frascos para detecção de material particulado e descoloração. Solução de XENPOZYME deve ser clara e incolor. Não devem ser utilizados frascos com partículas opacas ou descoloração.
Diluição
f) Para volumes reais de infusão com base no peso corporal (ver Tabela 4):
-Preparar uma solução de infusão a 0,1 mg/mL adicionando 0,25 mL (1 mg) da solução reconstituída preparada na etapa c e 9,75 mL de cloreto de sódio a 0,9% para injeção em uma seringa de 10 mL vazia.
-Calcular o volume (mL) necessário para obter a dose (mg) do paciente.
Exemplo: 0,3 mg ÷ 0,1 mg/mL = 3,0 mL
-Transferir o volume necessário de 0,1 mg/mL de solução de infusão para uma seringa esterilizada vazia do tamanho apropriado mais próximo para conter o volume de infusão.
Para volumes fixos de infusão (ver Tabela 4 para o volume total de infusão recomendado com base na idade e/ou peso dos pacientes):
-Retirar o volume de solução reconstituída, correspondente à dose prescrita, do número adequado de frascos e diluir com solução de cloreto de sódio 9 mg/mL (0,9%) em uma seringa ou bolsa de infusão dependendo do volume de infusão (ver Tabela 4 para o volume de infusão total recomendado com base na idade e/ou peso do paciente).
Tabela 4: Volumes de Administração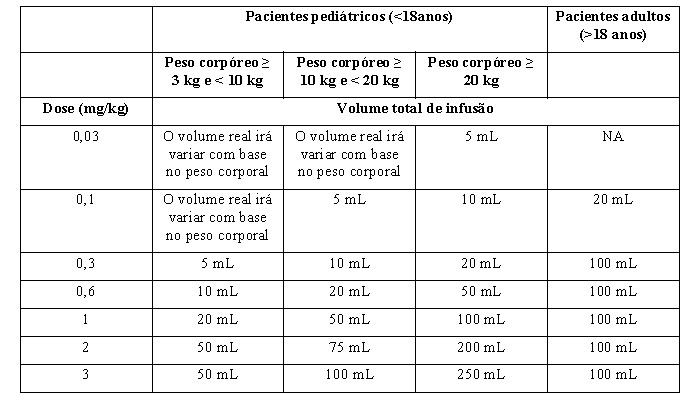
• Instruções de diluição para um volume total entre 5 mL e 20 mL usando uma seringa: -Injete o volume requerido de solução reconstituída lentamente na parede da seringa vazia. -Adicione lentamente a quantidade suficiente de solução de cloreto de sódio 9 mg/mL (0,9%) para obter o
volume total de infusão necessário (evite a formação de espuma dentro da seringa).
• Instruções de diluição para um volume total ≥50 mL usando uma bolsa de infusão:
- Bolsa de infusão vazia:
-Injete lentamente a solução reconstituída a partir da etapa c) na bolsa de infusão estéril de tamanho apropriado.
-Adicione lentamente a quantidade suficiente de solução de cloreto de sódio 9 mg/mL (0,9%) para obter o volume total de infusão necessário (evite a formação de espuma dentro da bolsa de infusão).
- Bolsa de infusão preenchida:
-Retire da bolsa de infusão preenchida com solução de cloreto de sódio 9 mg/mL (0,9%), o volume de solução salina normal para obter um volume final como especificado na Tabela 4.
-Adicione lentamente o volume requerido da solução reconstituída da etapa c) na bolsa de infusão (evite a formação de espuma dentro da bolsa de infusão).
g) Inverta suavemente a seringa ou o saco de infusão para misturar. Não agite. Como se trata de uma solu