VYZULTA
BAUSCH & LOMB
latanoprosta
Antiglaucomatoso.
Apresentações.
Solução oftálmica tópica com 0,024% de latanoprosteno bunode: embalagem contendo 1 frasco de 2,5 mL ou 1 frasco de 5 mL.
USO OFTÁLMICO TÓPICO
USO ADULTO
Composição.
Cada mL de solução oftálmica tópica contém 0,24 mg de latanoprosteno bunode. Excipientes: cloridrato de benzalcônio, polissorbato 80, edetato dissódico di-hidratado, citrato de sódio di-hidratado, ácido cítrico, glicerol, água.
Uma gota da solução contém aproximadamente 7,2mg de latanoprosteno bunode.
Cada 1 mL da solução oftálmica de VYZULTA® corresponde a aproximadamente 33 gotas.
Informações técnicas.
1. INDICAÇÕES
O VYZULTA® (solução oftálmica de 0,024% latanoprosteno bunode) é indicado para a redução da pressão intraocular (PIO) em pacientes com glaucoma de ângulo aberto ou hipertensão ocular.
2. RESULTADOS DE EFICÁCIA
Os estudos de Fase 3 de LBN (latanotoprosteno bunode) 0,024% para o tratamento de glaucoma de ângulo aberto e hipertensão ocular incluíram os estudos APOLLO e LUNAR de desenho semelhante, onde ambos tiveram uma fase de estudo de eficácia duplo-mascarado de três meses seguida de uma fase de extensão de estudo de segurança aberto de três meses (LUNAR) ou nove meses (APOLLO).
O estudo APOLLO foi um estudo clínico de grupo paralelo, fase 3, randomizado, multicêntrico, duplo mascarado, realizado em 45 locais de investigação nos Estados Unidos e na Europa.
O estudo foi composto por 2 fases: uma fase de eficácia controlada ativamente por 3 meses, seguida por uma fase de extensão de 9 meses de segurança em estudo aberto. O principal objetivo da fase de eficácia foi avaliar a não-inferioridade do LBN 0,024% uma vez por dia à noite em comparação com o timolol 0,5% duas vezes ao dia em relação à redução média da PIO em cada ponto de tempo ao longo dos 3 meses de tratamento. Se o LBN 0,024% uma vez por dia à noite era determinado como não-inferior ao timolol 0,5% duas vezes ao dia, o objetivo secundário era avaliar a superioridade do LBN 0,024% uma vez por dia à noite ao timolol 0,5% duas vezes ao dia.
Indivíduos
O estudo envolveu homens e mulheres com idade > 18 anos com diagnóstico de glaucoma de ângulo-aberto (incluindo GAA pigmentar ou pseudoexfoliativa) ou HO (hipertensão ocular) em um ou ambos os olhos. A pressão intraocular foi avaliada uma vez na triagem e às 8h, 12h e 16h no início do estudo para estabelecer a elegibilidade e os valores basais.
Tratamento e Avaliações do Estudo
Dados básicos, incluindo dados demográficos, histórico médico e ocular relevantes e medicamentos concomitantes, foram registrados durante a visita de triagem. Os indivíduos elegíveis que receberam tratamento hipotensivo ocular tópico na triagem foram obrigados a interromper o tratamento e passar por um período de washout antes da visita de base (dia 0), variando em duração dependendo da medicação de redução de PIO utilizada.
Os indivíduos foram retirados do estudo se sua PIO era > 36 mmHg em qualquer dos olhos em qualquer ponto durante o período de washout.
Após medidas de PIO de base, os indivíduos elegíveis foram aleatorizados 2:1 para receber LBN 0,024% uma vez por dia à noite e veículo todas as manhãs ou timolol 0,5% duas vezes ao dia durante 3 meses. Para fins de mascaramento, cada tratamento foi rotulado com rótulos investigacionais idênticos e embalados em caixas de kits idênticos. O medicamento em estudo foi distribuído através de um sistema de Tecnologia de Resposta Interativa. Os cronogramas de randomização foram criados por um estatístico desmascarado designado usando o SAS Versão 9.2 (SAS Institute, Inc., Cary, NC). Cada indivíduo recebeu kits de estudo contendo 4 frascos de colírios e foi instruído a instilar 1 gota do medicamento em estudo do frasco dosador "noturno" no(s) olho(s) afetado(s) por volta das 8 da noite a cada dia e 1 gota do medicamento em estudo do frasco dosador "diurno" por volta das 8 da manhã a cada dia (com exceção da manhã das visitas programadas à clínica, quando o sujeito instilou o medicamento em estudo após as avaliações clínicas).
O olho em estudo foi o olho que se qualificou por critério de inclusão no dia 0; se ambos os olhos se qualificaram, então o olho em estudo foi o olho com o maior valor médio de PIO diurna média no dia 0 ou o olho direito se ambos os olhos tinham o mesmo valor médio de PIO diurna média. Se ambos os olhos de um indivíduo tinham um diagnóstico de GAA ou HO, então ambos os olhos foram tratados durante a duração do estudo, mesmo se apenas 1 olho qualificado no dia 0.
Após a randomização, os indivíduos completaram 3 visitas de estudo: semana 2 (+-2 dias), semana 6 (+-3 dias), e mês 3 (+-10 dias). A cada visita, a PIO foi medida em ambos os olhos às 8h, 12h e 16h, usando um tonômetro de aplanação de Goldmann. Sempre que possível, o mesmo operador mediu a PIO, e o mesmo tonômetro foi usado em cada visita para um determinado indivíduo.
O investigador classificou a hiperemia conjuntival em uma escala de 1 a 4 usando padrões fotográficos (1 ¼ nenhum, 4 ¼ severo).
Desfechos
O desfecho primário de eficácia foi ao PIO no olho de estudo do indivíduo medido às 8h, 12h e 16h em cada visita pós-base (semana 2, semana 6, e mês 3). Os desfechos secundários de eficácia foram a proporção de indivíduos com PIO < 18 mmHg consistentemente em todos os 9 pontos de tempo nos primeiros 3 meses e a proporção de indivíduos com redução de PIO > 25% consistentemente em todos os 9 pontos de tempo nos primeiros 3 meses. Os desfechos secundários de eficácia adicionais incluíram a alteração da linha de base (ALB) em PIO medida às 8h, 12h e 16h em cada visita pós-base e o ALB em PIO diurno (definido como a média da PIO às 8h, 12h e 16h) em cada visita pós-randomização. Os desfechos de segurança durante a fase de eficácia incluíram MAVC (Melhor Acuidade Visual Corrigida), avaliação de hiperemia conjuntival e incidência de eventos adversos oculares e sistêmicos.
Análise estatística
A estimativa do tamanho da amostra foi baseada em um teste de não-inferioridade da diferença entre LBN 0,024% versus timolol 0,5% com relação à PIO na população por protocolo (PP), assumindo um desvio padrão (DP) de 3,75 mmHg, uma potência de 90%, uma margem de 2 lados a=0,05, e uma margem de não-inferioridade de 1,5 mmHg.
O DP foi baseado em dados de um estudo fase 2b de LBN 0,024% e o braço timolol de um estudo fase 3. Para responder por possíveis desistências e violações de protocolo, 393 indivíduos deveriam ser randomizados para os grupos de tratamento LBN 0,024% e timolol 0,5% em uma proporção de 2:1.
A análise de eficácia primária foi realizada utilizando a análise de covariância (ANCOVA) na população de intenção de tratar (IDT), que incluiu todos os indivíduos randomizados que instilaram pelo menos 1 dose do medicamento em estudo e tiveram pelo menos 1 avaliação da PIO após a linha de base.
No modelo ANCOVA, o tratamento era uma variável e a PIO média de linha de base correspondia ao tempo, o que significa que a PIO era uma covariância. Os dados faltantes foram inseridos usando o método da última observação levada adiante (UOLA). Os 2 grupos de tratamento foram comparados para cada ponto de tempo por visita, e a média dos mínimos quadrados de cada grupo de tratamento, a diferença na média dos mínimos quadrados (LBN 0,024%-timolol 0,5%), e o intervalo de confiança de 95% (IC) dos 2 lados para a diferença foram obtidos. A não-inferioridade foi determinada se o limite superior dos ICs para a diferença não excederam 1,5 mmHg em todos os 9 pontos de tempo e não excederam 1,00 mmHg para > 5 dos 9 pontos de tempo.
Se a não-inferioridade foi determinada, a superioridade em cada ponto de tempo foi demonstrada se o limite superior do IC 95% não excedeu 0 mmHg em todos os 9 pontos de tempo. Para complementar as análises primárias, a ANCOVA foi repetida para a população PP (ou seja, todos os indivíduos da população IDT que permaneceram no estudo até o mês 3 sem falta de avaliações do PIO pós-base e sem violações importantes do protocolo). Além disso, a análise primária foi repetida na população do IDT usando o método da pior observação levada adiante (POLA).
Se a não-inferioridade da LBN 0,024% para o timolol 0,5% foi mostrada, os desfechos secundários analisados foram baseados na população do IDT com UOLA, com análises de apoio realizadas usando a população PP. A proporção de indivíduos com PIO < 18 mmHg em todos os pontos de tempo nos primeiros 3 meses e a proporção com redução da PIO > 25% em todos os pontos de tempo nos primeiros 3 meses foram resumidas categoricamente. A redução percentual a partir da linha de base foi calculada como 100 x (linha de base média de PIO - PIO pós-base média) / PIO média de linha de base média. Para cada desfecho secundário chave, o IC de 2 lados 95% em torno da diferença de proporções (LBN 0,024%-timolol 0,5%) foi calculado, e o valor de P foi determinado usando o teste qui-quadrado de Pearson. Uma ANCOVA de ALB em PIO foi realizada com termos de efeito fixo para tratamento e linha de base para os pontos de tempo pós-base especificados na semana 2, semana 6 e mês 3.
Além disso, uma ANCOVA de ALB em PIO média diurna foi realizada com termos de efeito fixo para tratamento e PIO diurna de linha de base a cada visita pós-base. A ALB em PIO foi resumida usando estatísticas descritivas, e foram realizados testes t pareados dentro do grupo de tratamento para a PIO média ALB em cada ponto de cada visita.
Eficácia
Resultados de Eficácia Primária
A PIO média no olho do estudo foi significativamente menor no grupo LBN 0,024% (faixa, 17,8-18,7 mmHg) do que no grupo timolol 0,5% (faixa, 19,1-19,8 mmHg) em todos os 9 pontos de tempo de eficácia avaliados (8 AM, 12 PM, e 4 PM na semana 2, semana 6, e mês 3) (Figura 1).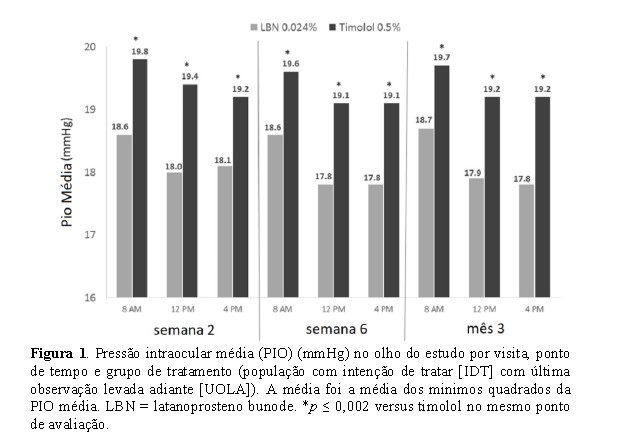
A não-inferioridade do LBN 0,024% ao timolol 0,5% na população do IDT foi demonstrada com base na ANCOVA para a comparação da PIO média entre os grupos de tratamento: O limite superior dos 95% ICs para a diferença entre tratamentos foi < 1,0 mmHg para todos os 9 pontos de tempo de eficácia (Tabela 1).
Da mesma forma, os resultados da ANCOVA demonstraram a superioridade de LBN 0,024% ao timolol 0,5% na população IDT porque o limite superior dos 95% ICs para a diferença entre tratamentos foi < 0 mmHg em todos os 9 pontos de tempo (Tabela 1).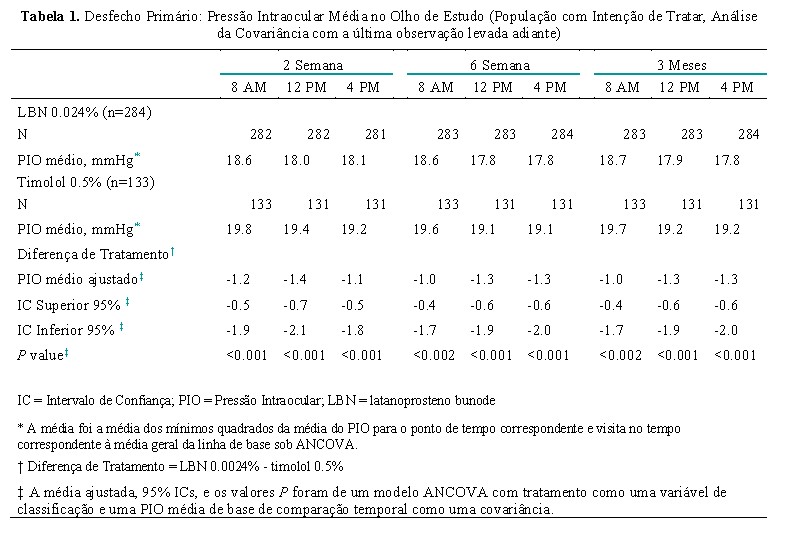
Os principais resultados finais foram suportados pelos resultados de uma análise ANCOVA na população PP e com a POLA na população IDT.
Além disso, os resultados do desfecho primário não foram afetados pelo status do tratamento prévio (indivíduos em tratamento na inscrição versus indivíduos sem tratamento na inscrição), idade do sujeito ( < 65 versus > 65 anos), ou uso simultâneo de betabloqueadores sistêmicos.
Resultados da Eficácia Secundária
Uma porcentagem maior de indivíduos no grupo LBN 0,024% (22,9%) em comparação com o grupo timolol 0,5% (11,3%) teve uma PIO < 18 mmHg consistente em todos os 9 pontos de tempo de eficácia medidos (Fig. 2) (diferença, 11,6%; 95% IC, 4,3-18,9; P = 0,005). Da mesma forma, uma porcentagem maior de indivíduos no grupo LBN 0,024% (34,9%) em comparação com o grupo timolol 0,5% (19,5%) teve uma redução de PIO > 25% consistentemente em todos os 9 pontos de tempo medidos (Figura 2) (diferença, 15,3%; 95% IC, 6,6-24,0; P = 0.001). Resultados similares foram observados na população PP.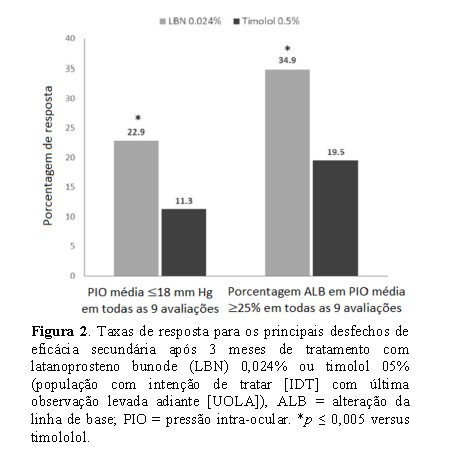
Uma análise adicional da ANCOVA mostrou que a média ALB em PIO médio foi maior no grupo LBN 0,024% (faixa, -7,7 a -9,1 mmHg) do que no grupo timolol 0,5% (faixa, -6,6 a -8,0 mmHg) em todos os 9 pontos de tempo de eficácia (Figura 3).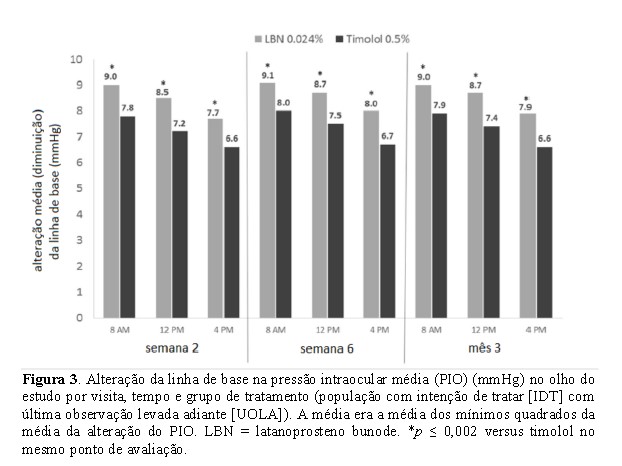
A diferença no ALB na PIO média entre os grupos de tratamento foi estatisticamente significativa em todos os 9 pontos de tempo de eficácia (P _ < 0,002). Como no caso dos pontos de tempo individuais, a PIO média diurna (média da PIO às 8h, 12h e 16h) foi significativamente menor no grupo LBN 0,024% em comparação com o grupo timolol 0,5% a cada visita (18,2 vs. 19,5 mmHg na semana 2, 18,1 vs. 19,3 mmHg na semana 6, e 18,2 vs. 19,4 mmHg no mês 3; P < 0,001 para todos). Semelhante ao ALB em pontos de tempo individuais, a cada visita de estudo, houve uma ALB estatisticamente significante (P < 0,001) maior no PIO diurno médio no grupo LBN 0,024% (faixa, -8,4 a -8,6 mmHg) do que no grupo timolol 0,5% (faixa, -7,1 a -7,3 mmHg).
O estudo LUNAR foi um estudo clínico prospectivo de grupo paralelo, randomizado, multicêntrico, duplo mascarado, realizado em 46 locais de investigação nos Estados Unidos (40), Reino Unido (3), Alemanha (2) e Itália (1)
O objetivo principal do estudo foi avaliar a não-inferioridade do efeito médio de redução da PIO de LBN 0,024% durante 3 meses de tratamento ao de timolol 0,5%. Se a não-inferioridade da LBN 0,024% fosse alcançada, o objetivo secundário era avaliar a superioridade estatística da LBN 0,024% para o timolol 0,5%. O estudo foi composto de 2 fases: uma fase de 3 meses de eficácia controlada ativa seguida por uma fase de extensão de segurança de 3 meses em estudo aberto.
Indivíduos
O estudo inscreveu indivíduos masculinos e femininos > 18 anos de idade com um diagnóstico de GAA (incluindo pigmentar ou pseudoexfoliativo) ou HO em 1 ou ambos os olhos. A pressão intraocular foi avaliada uma vez na visita 1 (triagem) e às 8h, 12h e 16h durante a visita 3 (elegibilidade, Dia 0) para estabelecer valores de linha de base e elegibilidade. Os indivíduos que já estavam recebendo tratamento com um medicamento que diminui a PIO foram obrigados a passar por um período de washout antes da visita 3, cuja duração variou dependendo do tipo de medicamento que diminui a PIO utilizado (período máximo de washout, 28 þ 5 dias). Na visita 3, e após o washout, se necessário, os participantes do estudo eram obrigados a ter uma PIO > 26 mm Hg em um mínimo de 1 de 3 pontos de tempo (8 AM, 12 PM, e 4 PM), > 24 mm Hg em um mínimo de 1 ponto de tempo, e > 22 mm Hg em 1 ponto de tempo, tudo no mesmo olho, e PIO < 36 mm Hg em todos os 3 pontos de tempo de medição em ambos os olhos. Os possíveis indivíduos submetidos a washout foram excluídos da participação no estudo se a PIO excedesse 36 mm Hg em qualquer dos olhos em qualquer ponto durante o período de washout.
Tratamentos e Avaliações do Estudo
O produto investigativo LBN 0,024%, seu veículo e o comparador timolol 0,5% foram fabricados pela Bausch & Lomb Inc (Tampa, Flórida, EUA). Dados demográficos e clínicos de base (ou seja, histórico médico e ocular relevante, medicamentos concomitantes, avaliações oculares) foram registrados na visita 1 (triagem). O olho do estudo foi definido como o olho que se qualificou por critério de inclusão na visita 3 (elegibilidade, Dia 0). Se ambos os olhos foram qualificados, o olho de estudo foi o olho com o maior valor médio de PIO diurno (definido como a média de PIO registradas às 8h, 12h e 16h) na visita 3, ou o olho direito se ambos os olhos tivessem o mesmo valor médio da PIO diurna. Se ambos os olhos de um sujeito tivessem um diagnóstico de GAA ou HO, então ambos os olhos foram tratados durante a duração do estudo, mesmo se apenas 1 olho atendesse aos critérios de inclusão no estudo na visita 3.
Os indivíduos elegíveis foram randomizados na visita 3 em uma proporção de 2:1 para receber 3 meses de tratamento com LBN 0,024% uma vez por dia à noite (aproximadamente 8 PM) e uma vez por dia do veículo pela manhã (aproximadamente 8 AM) ou timolol 0,5% duas vezes ao dia.
Cada indivíduo recebeu kits de estudo contendo 4 frascos de colírios com rótulos investigacionais gerados por computador (isto é, para evitar a rotulagem comercial) e foi instruído a instilar 1 gota de medicamento de estudo do frasco dosador ''Noturno'' no(s) olho(s) afetado(s) por volta das 20h cada noite e 1 gota do frasco dosador ''Dia'' por volta das 8h cada manhã. Um cronograma de randomização foi criado antes de qualquer inscrição no estudo por um estatístico não envolvido de outra forma no estudo usando o SAS (SAS Institute,Cary, Carolina do Norte, EUA; Versão 9.2). A alocação do medicamento em estudo foi concluída através do uso da IRT (Interactive Response Technology), que determinou qual kit atribuir a cada indivíduo. Os indivíduos e o pessoal envolvido no estudo foram totalmente mascarados para tarefas de tratamento. A conformidade foi determinada como o número real de instilações, conforme registrado em um diário do paciente, dividido pelo número de instilações esperadas.
Os indivíduos completaram 3 visitas de estudo (visita 4 [semana 2 ± 2 dias]; visita 5 [semana 6 ± 3 dias]; visita 6 [mês 3 ± 10 dias]) após a randomização. A pressão intraocular foi avaliada a cada visita em ambos os olhos às 8h, 12h e 16h, utilizando um tonômetro de aplanação de Goldmann. Nesses dias, a dose diurna do medicamento em estudo foi instilada após a primeira avaliação PIO do dia. A pressão intraocular foi medida duas vezes consecutivas e a PIO média foi registrada para medições consecutivas dentro de 2 mm Hg. Em casos de medidas consecutivas que diferiam por > 2 mm Hg, uma terceira medida foi tomada e a PIO mediana foi registrada. Sempre que possível, a PIO foi medida pelo mesmo operador usando o mesmo tonômetro em cada visita para um determinado indivíduo.
As avaliações de segurança registradas em cada visita incluíram os eventos adversos emergentes do tratamento, oculares e sistêmicos.
Desfechos
O desfecho primário de eficácia foi a PIO no olho do estudo medido nos pontos de tempo especificados das 8h, 12h e 16h em cada visita pós-randomização (semana 2, semana 6, e mês 3). Os desfechos secundários de eficácia incluíram a proporção de olhos com PIO < 18 mmHg; a proporção com redução da PIO > 25% da linha de base consistentemente em todas as 9 avaliações pós-randomização; a alteração da linha de base na PIO às 8h, 12h e 16h; e a alteração da linha de base na PIO diurna na semana 2, semana 6 e mês 3. Os desfechos de segurança incluíram a incidência de eventos adversos oculares e sistêmicos (EA), sinais vitais, MAVC e avaliações de hiperemia conjuntival.
Análise Estatística
Um tamanho de amostra de 300 indivíduos na população por protocolo (PP) foi calculado como fornecendo potência adequada (90%) para detectar uma diferença PIO (margem de não inferioridade) de 1,5 mm Hg, assumindo um desvio padrão (DP) de 3,75 mmHg e um desvio de 2 lados a=0,05. O DP foi obtido através do agrupamento do DP de um estudo LBN 0,024% fase 2b e o braço timolol de um estudo fase 3. Um total de 393 indivíduos deveriam ser randomizados em uma proporção de 2:1 para os grupos de tratamento LBN 0,024% e timolol 0,5%, respectivamente, para responder por potenciais violações de protocolo e desistências.
As análises de eficácia primária foram realizadas utilizando a análise de covariância (ANCOVA) na população com intenção de tratar (IDT) (ou seja, todos os indivíduos randomizados que instilaram pelo menos 1 dose do medicamento em estudo e tiveram uma PIO de base e pelo menos 1 PIO pós-randomização), com o grupo de tratamento randomizado como uma variável de classificação e PIO média compatível com o tempo como uma covariância. Os dados faltantes foram imputados usando o método de última observação levada adiante (UOLA) para a PIO no olho de estudo medido às 8h, 12h e 16h em cada visita pós-randomização. Os 2 tratamentos, LBN 0,024% e timolol 0,5%, foram comparados para cada ponto de tempo em cada visita, determinando-se a média dos mínimos quadrados (MQ) de cada grupo de tratamento, a diferença na média MQ (LBN 0,024% - timolol 0,5%), e o intervalo de confiança de 95% (IC) dos 2 lados para a diferença. A não-inferioridade deveria ter sido estabelecida se o limite superior das ICs para a diferença não excedesse 1,5 mmHg em todas as 9 avaliações da PIO e não excedesse 1,0 mm Hg durante pelo menos 5 dos 9 pontos de tempo. Se a não-inferioridade foi determinada, a superioridade foi concluída se o limite superior do IC 95% não excedesse 0 mm Hg em nenhum dos 9 pontos de tempo de medição da PIO. Estas análises foram repetidas na população PP a fim de complementar as análises primárias.
As análises dos desfechos secundários foram realizadas após a demonstração de não-inferioridade do LBN 0,024% para o timolol 0,5%. A proporção de indivíduos com PIO < 18 mm Hg em todos os 9 pontos de tempo de medição da PIO pós-randomização e a proporção com redução de PIO > 25% em todos os 9 pontos de tempo foram resumidas categoricamente.
A porcentagem de redução a partir da linha de base em PIO foi calculada como 100 x (linha de base média de PIO - PIO pós-base média) / PIO média de linha de base média. Para cada desfecho secundário, foram apresentados os 95% IC de 2 lados em torno da diferença nas proporções (LBN 0,024% -timolol 0,5%) e o valor P do teste qui-quadrado (c2) de Pearson. Uma ANCOVA de alteração da linha de base em PIO foi realizada com termos de efeito fixo para tratamento e linha de base para os 3 pontos de tempo pós-randomização (8h, 12h e 16h) na visita 4/semana 2, visita 5/semana 6 e visita 6/mês 3. Uma ANCOVA de alteração da linha de base em PIO média diurna também foi realizada com termos de efeito fixo para tratamento e PIO diurna de linha de base em cada visita pós randomização.
Todos os ICs, testes estatísticos e os valores P resultantes foram relatados como dois lados e foram avaliados no nível de significância de 5%. Os dados contínuos foram resumidos com estatísticas descritivas (número, média, DP, mediana, mínimo e máximo). As análises estatísticas foram realizadas utilizando o software SAS (SAS Institute, Cary, Carolina do Norte, EUA) versão 9.2 ou superior.
Desfechos:
Desfecho primário
A PIO média no olho do estudo foi significativamente menor no grupo LBN 0,024% do que no grupo timolol 0,5% na maioria dos pontos de tempo medidos (12 PM, 16 PM na semana 2, 8 AM, 12 PM, 16 PM na semana 6 e mês 3) (Tabela 2). A não-inferioridade do LBN 0,024% ao timolol 0,5% foi demonstrada com base nos resultados da ANCOVA (o limite superior dos 95% ICs não excederam 1,0 mm Hg em nenhum dos 9 pontos de tempo) (Tabela 2).
LBN 0,024% também satisfez os critérios de superioridade estatística sobre o timolol 0,5% em todos os pontos de tempo, exceto o ponto de tempo 8 AM na semana 2 (limite superior do IC 95% excedeu 0 mm Hg neste ponto de avaliação único) (Tabela 2).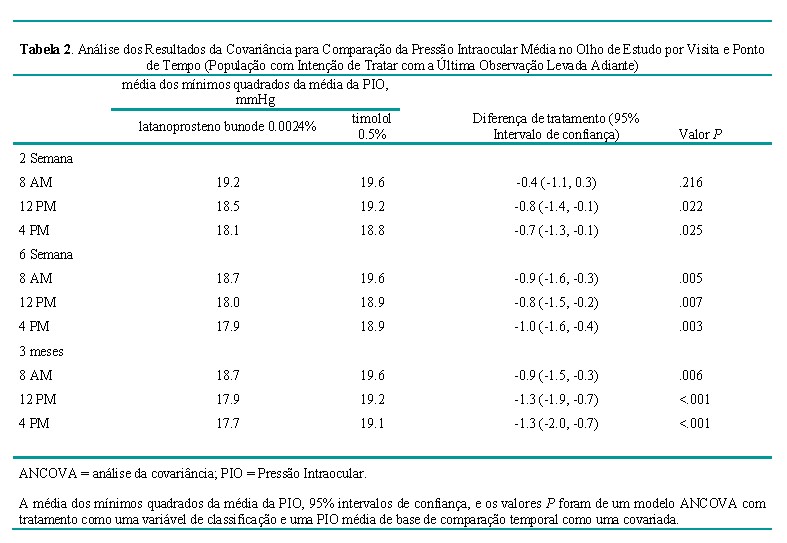
Os resultados na população PP foram consistentes com estas descobertas. Outras análises exploratórias indicaram que os resultados do desfecho primário não diferiram com base no status anterior do tratamento com medicação que reduziu a PIO no momento da inscrição ou se foram ou não incluídos na análise indivíduos com betabloqueadores sistêmicos concorrentes (15,1% de LBN e 13,2% de indivíduos com timolol).
Desfechos secundários
A porcentagem de indivíduos com uma redução do PIO > 25% consistentemente em todos os 9 pontos de tempo foi significativamente maior no grupo LBN (31,0%) em comparação com o grupo timolol (18,5%; diferença de proporções 12,5%; IC 95% da diferença: 4,0%, 21,1%; P ¼.007) (Figura 4). A porcentagem de indivíduos com PIO médio < 18 mm Hg consistente em todos os 9 pontos de tempo não diferiu no grupo LBN em comparação com o grupo timolol (17,7% vs 11,1%, respectivamente; diferença de proporções, 6,6%; IC 95%: -0,4%, 13,5%; P =.084) (Figura 4).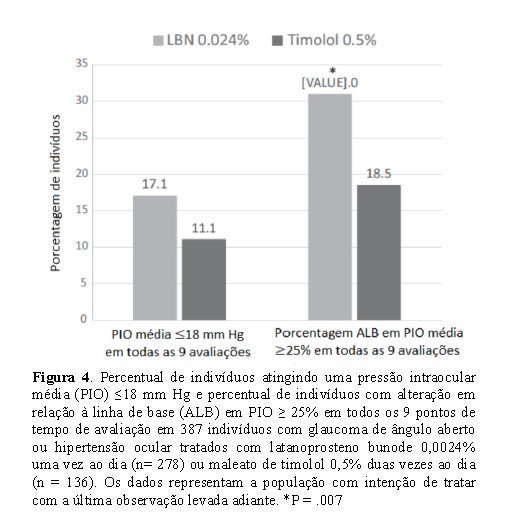
Além disso, a ANCOVA mostrou que a diferença de tratamento para a alteração da linha de base na PIO foi significativamente maior no grupo LBN 0,024% do que no grupo timolol 0,05% em todos os pontos do tempo (faixa PIO -7,5-8,8 mm Hg para LBN 0,024%; -6,6 a -7,9 mm Hg para timolol 0,5%; P < .025) exceto para a medição de 8 AM na semana 2 (-8,3 mm Hg para LBN 0,024% vs -7,9 mm Hg para timolol 0,5%; P < .216). Estas alterações corresponderam a uma redução da linha de base variando de 29,1% a 32,1% no grupo LBN 0,024% e variando de 25,2% a 28,7% no grupo de tratamento com timolol.
A PIO diurna média foi significativamente menor no grupo LBN 0,024% em comparação com o grupo do timolol 0,5% a cada visita (18,6 mm Hg vs 19,2 mm Hg na semana 2, 18,2 mm Hg vs 19,1 mm Hg na semana 6, e 18,1 mm Hg vs 19,3 mm Hg no mês 3; P < .034 para todos). Semelhante à alteração da linha de base em pontos de tempo individuais, a alteração média da linha de base na PIO média diurna foi significativamente maior no grupo LBN 0,024% do que no grupo timolol 0,5% a cada visita de estudo pós-randomização (semana 2, diferença, -0,6 mm Hg; P =.034; semana 6, diferença, -0,9 mm Hg; P =.002; mês 3, diferença, -1,2 mm Hg; P < .001.
Características demográficas
ESTUDO LUNAR
Na população IDT, os indivíduos tinham em média 64,7 anos de idade (variação, 23-88 anos) e eram predominantemente do sexo feminino (58,2%), brancos (70,8%) e não hispânicos/não latinos (86,7%). Os indivíduos negros/afro-americanos representaram 27,8% da população total. As características demográficas e oftalmológicas (espessura média da córnea, esfera de refração e cilindro de refração) foram comparáveis entre os grupos de tratamento.
A maioria dos sujeitos (72%) foi tratada com medicamentos que reduzem a PIO nos 30 dias anteriores à sua inscrição.
Devido ao pequeno tamanho das amostras, a comparação estatística dentro dos grupos raciais além do grupo de "caucasianos" não foi forte o suficiente para ser conclusiva, porém não foi encontrada diferença significativa na resposta terapêutica em nenhum grupo racial.
ESTUDO APOLLO
Os indivíduos da população IDT tinham uma idade média de 64,2 anos, eram de ascendência predominantemente europeia ou africana, e de etnia não hispânica/não latina. Os grupos de tratamento eram comparáveis no que diz respeito à demografia e às características do olho na linha de base. Mais de dois terços dos sujeitos (71,9%) estavam recebendo medicação tópica para baixar a PIO na triagem ou usaram medicação para baixar a PIO 30 dias antes da triagem e participaram do washout.
O histórico médico não-ocular foi semelhante entre os grupos de tratamento.
Devido ao pequeno tamanho das amostras, a comparação estatística dentro dos grupos raciais além do grupo de "caucasianos" não foi forte o suficiente para ser conclusiva, porém não foi encontrada diferença significativa na resposta terapêutica em nenhum grupo racial.
1. Weinreb RN, Scassellati Sforzolini B, Vittitow J, Liebmann J. Latanoprostene bunod 0.024% versus timolol maleate 0.5% in subjects with open-angle glaucoma or ocular hypertension: the APOLLO study. Ophthalmology. 2016;123(5):965-973.
2. Medeiros FA, Martin KR, Peace J, Scassellati Sforzolini B, Vittitow JL, Weinreb RN. Comparison of latanoprostene bunod 0.024% and timolol maleate 0.5% in open-angle glaucoma or ocular hypertension: the LUNAR study. Am J Ophthalmol. 2016;168:250-259.
3. CARACTERÍSTICAS FARMACOLÓGICAS
VYZULTA® (solução oftálmica de 0,024% latanoprosteno bunode) é um análogo da prostaglandina formulado como uma solução oftálmica tópica estéril. VYZULTA® contém o princípio ativo latanoprosteno bunode 0,24 mg/mL, o conservante cloreto de benzalcônio 0,2 mg/mL e os seguintes excipientes: polissorbato 80, glicerina, EDTA e água. A formulação é tamponada a pH 5,5 com ácido cítrico/citrato de sódio.
Seu nome químico é 4-(Nitrooxi)butil(5Z)-7-{(1R, 2R, 3R, 5S)-3,5-dihidroxi-2-[(3R)-3-hidroxi-5- fenilpentil] ciclopentil}hept-5-enoato.
Mecanismo de Ação
O latanoprosteno bunode reduz a pressão intraocular, aumentando o fluxo de humor aquoso através das redes trabecular e uveoscleral. A pressão intraocular é um importante fator de risco modificável para a progressão do glaucoma e a sua redução diminui o risco de perda do campo visual glaucomatoso.
Presume-se que o latanoprosteno bunode reduza a pressão intraocular através de um mecanismo duplo de ação, uma vez que a medicação é metabolizada em duas moléculas relevantes após a administração: (1) ácido latanoprosta, e (2) mononitrato de butanodiol,2,1,3.
Como um análogo da prostaglandina F2-alfa2, o grupo ácido latanoprosta opera como um agonista seletivo do receptor PGF2-alfa (FP) 3. Como os receptores FP ocorrem no músculo ciliar, epitélio ciliar e esclera, a parcela de ácido latanoprosta atua primariamente na via uveoscleral, onde aumenta a expressão das metaloproteinases de matriz (MMPs) como MMP-1, -3 e -9, que promovem a degradação do colágeno tipos I, III e IV nos feixes longitudinais do músculo ciliar e esclera 5 circundante. O resultante remodelamento da matriz extracelular do músculo ciliar consequentemente produz redução da resistência ao escoamento por meio do aumento da permeabilidade e aumento do escoamento de humor aquoso pela via uveo-escleral3.
Por outro lado, o mononitrato de butanodiol sofre metabolismo adicional para NO e uma fracção inativa de 1,4-butanodiol. Como um gás que pode se difundir livremente através das membranas plasmáticas, propõe-se que o efeito relaxante do NO para induzir reduções no volume celular e contratilidade do músculo liso vascular como células é dependente da ativação da via em cascata sGC/cGMP/PKG. O NO liberado do mononitrato de butanodiol consequentemente entra nas células da MT e na parede interna do SC, causando diminuição da fosforilação da cadeia leve-2 da miosina, aumento da fosforilação dos canais de potássio ativado por cálcio de grande condutividade (BKCa), e um subsequente efluxo de íons de potássio através desses canais BKCa. Todas essas alterações servem para diminuir a contratilidade e o volume celular, bem como para reorganizar o citoesqueleto de actina das células TM e SC. Estas alterações biomecânicas permitem, em última análise, uma saída convencional melhorada de humor aquoso3.
1. Kawase K, Vittitow JL, Weinreb RN, Araie M: Long-term Safety and Efficacy of Latanoprostene Bunod 0.024% in Japanese Subjects with Open-Angle Glaucoma or Ocular Hypertension: The JUPITER Study. Adv Ther. 2016 Sep;33(9):1612-27. doi: 10.1007/s12325-016-0385-7. Epub 2016 Jul 25. [PubMed:27457469]
2. Kaufman PL: Latanoprostene bunod ophthalmic solution 0.024% for IOP lowering in glaucoma and ocular hypertension. Expert Opin Pharmacother. 2017 Mar;18(4):433-444. doi: 10.1080/14656566.2017.1293654. Epub 2017 Feb 20. [PubMed:28234563]
3. Garcia GA, Ngai P, Mosaed S, Lin KY: Critical evaluation of latanoprostene bunod in the treatment of glaucoma. Clin Ophthalmol. 2016 Oct 18;10:2035-2050. eCollection 2016. [PubMed:27799730]
Farmacodinâmica
A redução da pressão intraocular começa aproximadamente em 1 a 3 horas após a primeira administração, tendo seu efeito máximo alcançado após 11-13 horas em olhos com pressão intraocular elevada.
Farmacocinética
Absorção
A exposição sistêmica de latanoprosteno bunode e seus metabólitos ácido de latanoprosta e mononitrato de butanodiol foi avaliada em um estudo com 22 indivíduos saudáveis após a administração ocular tópica de VYZULTA® 0,024% uma vez ao dia (uma gota bilateralmente pela manhã) por 28 dias. Não foi possível quantificar as concentrações plasmáticas de latanoprosteno bunode (limite inferior de quantificação, LLOQ, de 10,0 pg/mL) ou mononitrato de butanodiol (LLOQ de 200 pg/mL) após a dose no Dia 1 e Dia 28. A média das concentrações plasmáticas máximas (Cmax) do ácido de latanoprosta (LLOQ de 30 pg/mL) foi de 59,1 pg/mL e 51,1 pg/mL no Dia 1 e Dia 28, respectivamente. O tempo médio de concentração plasmática máxima (Tmax) para o ácido de latanoprosta foi de aproximadamente 5 minutos após a administração tanto no Dia 1 como no Dia 28.
Distribuição
Não foram realizados estudos de distribuição ocular em humanos.
Metabolismo
Após a administração ocular tópica, o latanoprosteno bunode é rapidamente metabolizado no olho para ácido de latanoprosta (parte ativa), um análogo da prostaglandina F2a, e mononitrato de butanodiol. Depois que o ácido de latanoprosta alcança a circulação sistêmica, ele é metabolizado principalmente pelo fígado nos metabólitos 1,2-dinor e 1,2,3,4-tetranor via b-oxidação de ácidos graxos.
O mononitrato de butanodiol é metabolizado em 1,4-butanodiol e óxido nítrico. O metabólito 1,4-butanodiol é ainda oxidado em ácido succínico e então primeiramente absorvido como um componente no ciclo do ácido tricarboxílico (TCA) na respiração aeróbia celular.
Eliminação
A eliminação do ácido de latanoprosta do plasma humano é rápida, uma vez que a concentração plasmática de ácido de latanoprosta caiu para abaixo do LLOQ (30 pg/mL) na maioria dos indivíduos em 15 minutos após a administração ocular de VYZULTA® 0,024% em humanos.
O componente ácido latanoprosta do latanoprosteno bunode é predominantemente metabolizado pelo fígado e excretado principalmente na urina.
Carcinogênese, Mutagênese, Diminuição da Fertilidade
Latanoprosteno bunode não se apresentou mutagênico em bactérias e não induziu a formação de micronúcleos no ensaio de micronúcleos de medula óssea em ratos in vivo. As aberrações cromossômicas foram observadas in vitro com linfócitos humanos na ausência de ativação metabólica.
Latanoprosteno bunode não foi testado para atividade carcinogênica em estudos de longo prazo em animais. O ácido de latanoprosta é um metabolito principal do latanoprosteno bunode. A exposição de ratos e camundongos ao ácido de latanoprosta, resultante da dosagem oral com latanoprosta em bioensaios com roedores ao longo da vida, não foi carcinogênica.
Não foram realizados os estudos de fertilidade com latanoprosteno bunode. O potencial de impacto na fertilidade pode ser parcialmente caracterizado pela exposição ao ácido de latanoprosta, um metabólito comum do latanoprosteno bunode e da latanoprosta. Não se descobriu qualquer efeito sobre a fertilidade masculina ou feminina do ácido de latanoprosta em estudos com animais.
Toxicologia Animal e/ou Farmacologia
Um estudo de toxicologia de 9 meses administrou doses oculares tópicas de latanoprosteno bunode a um olho de macacos cynomolgus: controle (somente veículo), uma gota de 0,024% duas vezes ao dia, uma gota de 0,04% duas vezes ao dia e duas gotas de 0,04% por dose, duas vezes ao dia. As exposições sistêmicas são equivalentes a 4,2 vezes, 7,9 vezes e 13,5 vezes a dose clínica, respectivamente, com base na superfície corporal (assumindo 100% de absorção). A avaliação microscópica dos pulmões após 9 meses observou fibrose/inflamação crônica pleural/subpleural nos grupos masculinos com dose de 0,04%, com incidência e gravidade crescentes em comparação aos controles. Não foi observada toxicidade pulmonar na dose de 0,024%.
Gravidez
Dados em Animais
Os estudos embriofetais foram realizados em coelhas prenhas que receberam latanoprosteno bunode diariamente por injeção intravenosa nos dias 7 a 19 da gestação, para atingir o período de organogênese. As doses administradas variaram de 0,24 a 80 mcg/kg/dia. O aborto ocorreu com doses de ≥ 0,24 mcg/kg/dia de latanoprosteno bunode (0,28 vezes a dose clínica, com base na superfície corporal, assumindo 100% de absorção). A letalidade embrionária (reabsorção) foi aumentada nos grupos de tratamento com latanoprosteno bunode, evidenciados por aumentos nas reabsorções iniciais em doses de ≥ 0,24 mcg/kg/dia e reabsorções tardias em doses de ≥ 6 mcg/kg/dia (aproximadamente 7 vezes a dose clínica). Nenhum feto sobreviveu em nenhuma gravidez de coelhas com doses de 20 mcg/kg/dia (23 vezes a dose clínica) ou maior. Latanoprosteno bunode produziu anormalidades estruturais em doses de ≥ 0,24 mcg/kg/dia (0,28 vezes a dose clínica). As malformações incluíram anomalias do esterno, coarctação da aorta com dilatação do tronco pulmonar, artéria subclávia retroesofágica com artéria braquiocefálica ausente, cabeça abobadada, hiperextensão da parte anterior e má-rotação do membro posterior, distensão/edema abdominal e vértebras caudais ausentes /fundidas.
Um estudo embriofetal foi realizado em ratas prenhas, que receberam latanoprosteno bunode diariamente por injeção intravenosa nos dias 7 a 17 da gestação, para atingir o período de organogênese. As doses administradas variaram de 150 a 1500 mcg/kg/dia. A toxicidade materna foi produzida a 1500 mcg/kg/dia (870 vezes a dose clínica, com base na superfície corporal, assumindo 100% de absorção), tal como evidenciado pela redução do ganho de peso materno. A letalidade embriofetal (reabsorção e morte fetal) e as anomalias estruturais foram produzidas em doses de ≥ 300 mcg/kg/dia (174 vezes a dose clínica). As malformações incluíam anomalias do esterno, cabeça abobadada, hiperextensão da pata dianteira e má-rotação do membro posterior, anomalias vertebrais e atraso na ossificação dos ossos do membro distal. Um nível de efeito adverso não observado (NOAEL | No Observed Adverse Effect Level) foi estabelecido em 150 mcg/kg/dia (87 vezes a dose clínica) neste estudo.
4. CONTRAINDICAÇÕES
Categoria de risco C.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
5. ADVERTÊNCIAS E PRECAUÇÕES
Pigmentação
VYZULTA® (solução oftálmica de 0,024% latanoprosteno bunode), pode causar alterações nos tecidos pigmentados. As alterações mais frequentemente relatadas com os análogos das prostaglandinas foram o aumento da pigmentação da íris e do tecido periorbital (pálpebra).
Espera-se que a pigmentação aumente enquanto a solução oftálmica de latanoprosteno bunode for administrada. A mudança na pigmentação se deve ao aumento do teor de melanina nos melanócitos, e não ao aumento do número de melanócitos. Com a interrupção no uso de VYZULTA®, a pigmentação da íris tende a ser permanente, enquanto que a pigmentação do tecido periorbital e as alterações dos cílios tendem a ser reversíveis na maioria dos pacientes. Deve-se informar aos pacientes que recebem análogos de prostaglandina, incluindo VYZULTA®, sobre a possibilidade de aumento da pigmentação, incluindo alterações permanentes. Os efeitos a longo prazo do aumento da pigmentação não são conhecidos.
A mudança de cor da íris pode não ser perceptível por vários meses ou anos. Normalmente, a pigmentação marrom ao redor da pupila se espalha concentricamente em direção à periferia da íris, e toda a íris, ou partes dela, torna-se mais acastanhada. O tratamento parece não afetar nem os nevos nem as sardas da íris. O tratamento com VYZULTA® (solução oftálmica de 0,024% latanoprosteno bunode), pode ser continuado em pacientes que desenvolvam uma pigmentação da íris visivelmente aumentada, porém, estes pacientes devem ser examinados regularmente. Os pacientes devem ser informados sobre o potencial aumento da pigmentação marrom da íris, que pode ser permanente. Os pacientes também devem ser informados sobre a possibilidade de escurecimento da pele das pálpebras, que geralmente é reversível após a interrupção do tratamento com VYZULTA®.
A utilização em pacientes pediátricos menores de 16 anos de idade não é recomendada devido as potenciais preocupações de segurança relacionadas com o aumento da pigmentação após utilização crônica a longo prazo.
Alterações nos Cílios
VYZULTA® pode mu