VENCLEXTA
ABBVIE
venetoclax
Antineoplásico.
Apresentações.
-Tratamento inicial 1° mês: caixa com 42 comprimidos revestidos divididos em 4 embalagens semanais contendo:
Semana 01(cartela amarela): 14 comprimidos revestidos com 10 mg de venetoclax
Semana 02 (cartela rosa): 7 comprimidos revestidos com 50 mg de venetoclax
Semana 03 (cartela verde): 7 comprimidos revestidos com 100 mg de venetoclax
Semana 04 (cartela roxa): 14 comprimidos revestidos com 100 mg de venetoclax
- Tratamento semanal avulso de 10 mg: cartela amarela contendo 14 comprimidos revestidos com 10 mg de venetoclax
- Tratamento semanal avulso de 50 mg: cartela rosa contendo 7 comprimidos revestidos com 50 mg de venetoclax
- Tratamento mensal de manutenção: embalagem contendo 120 comprimidos revestidos com 100 mg de venetoclax
USO ORAL
USO ADULTO
Composição.
Cada comprimido revestido de VENCLEXTA® (venetoclax) de 10 mg contém: venetoclax 10 mg. Excipientes: copovidona, polissorbato 80, dióxido de silício, fosfato de cálcio dibásico, estearilfumarato de sódio, água purificada, álcool polivinílico, dióxido de titânio, macrogol, talco e óxido de ferro amarelo.
Cada comprimido revestido de VENCLEXTA® (venetoclax) de 50 mg contém: venetoclax 50 mg. Excipientes: copovidona, polissorbato 80, dióxido de silício, fosfato de cálcio dibásico, estearilfumarato de sódio, água purificada, álcool polivinílico, dióxido de titânio, macrogol, talco, óxido de ferro amarelo, óxido de ferro vermelho e óxido de ferro preto.
Cada comprimido revestido de VENCLEXTA® (venetoclax) de 100 mg contém: venetoclax 100 mg. Excipientes: copovidona, polissorbato 80, dióxido de silício, fosfato de cálcio dibásico, estearilfumarato de sódio, água purificada, álcool polivinílico, dióxido de titânio, macrogol, talco e óxido de ferro amarelo.
Informações técnicas.
1. INDICAÇÕES
Leucemia Linfocítica Crônica
VENCLEXTA® (venetoclax) em monoterapia está indicado para o tratamento da Leucemia Linfocítica Crônica (LLC) em pacientes adultos:
· na presença de deleção 17p e/ou mutações no TP53 e que receberam tratamento prévio com inibidor de receptor de célula B (BCRi), ou que a critério médico, não sejam elegíveis ao inibidor de receptor de célula B;
· na ausência de deleção 17p e/ou mutações no TP53 e que receberam tratamento prévio com imuno-quimioterapia e inibidor de receptor de célula B.
Leucemia Mielóide Aguda
VENCLEXTA® (venetoclax), em combinação com um agente hipometilante, ou em combinação com citarabina em baixa dose, é indicado para pacientes recém-diagnosticados com leucemia mielóide aguda (LMA) e que são inelegíveis para quimioterapia intensiva, a critério do médico.
2. RESULTADOS DE EFICÁCIA
Leucemia Linfocítica Crônica
Estudo M13-982
O estudo clínico M13-9821 foi um estudo multicêntrico, aberto de braço único de 107 pacientes com LLC previamente tratados e com a deleção 17p. Dos pacientes, 65,4% eram do sexo masculino e 97,2% eram brancos. A idade média foi de 67 anos (variação: 37 a 85 anos), e o tempo médio desde o diagnóstico foi de 6,8 anos (variação: 0,1 a 32 anos; N=106). O número médio de tratamentos anteriores para LLC foi de 2 (intervalo: 1a 10 tratamentos). No início do estudo, 53,3% dos pacientes tinham um ou mais nódulos ≥ 5 cm, e 50,5% dos pacientes apresentaram ALC ≥ 25 x 109 / L. O estado de performance baseline do ECOG foi zero para 39,3%, um para 52,3%, e 2 para 8,4% dos pacientes.
Dos pacientes, 37,4% (34/91) eram refratários à fludarabina, 81,1% (30/37) apresentavam o gene IGHV não mutado, e 23,8% (19/80) tiveram deleção 11q.
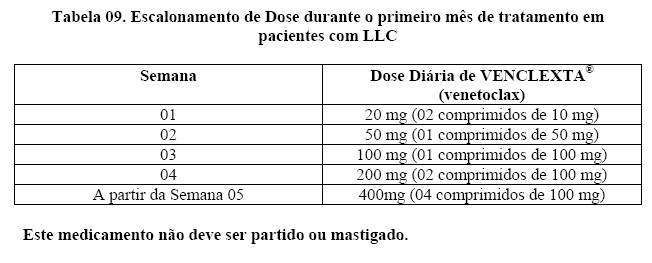
Os pacientes receberam VENCLEXTA® (venetoclax) através de uma programação de escalonamento de dose semanal a partir de 20 mg e aumentando até 50 mg, 100 mg, 200 mg e, finalmente, 400 mg uma vez por dia. Os pacientes continuaram a receber 400 mg de venetoclax por via oral, uma vez ao dia, até a progressão da doença ou toxicidade inaceitável. O tempo médio de tratamento no momento da avaliação foi de 12,1 meses (variação: 0 a 21,5 meses).
O desfecho primário de eficácia foi a taxa de resposta global (ORR) como avaliado por uma Comissão de Revisão Independente (IRC) usando o Workshop Internacional para Leucemia Linfocítica Crônica (IWCLL) atualizadas pelas diretrizes do Grupo de Trabalho patrocinado pela National Cancer Institute (NCI-WG) (2008). Os resultados de eficácia estão apresentados na Tabela 01.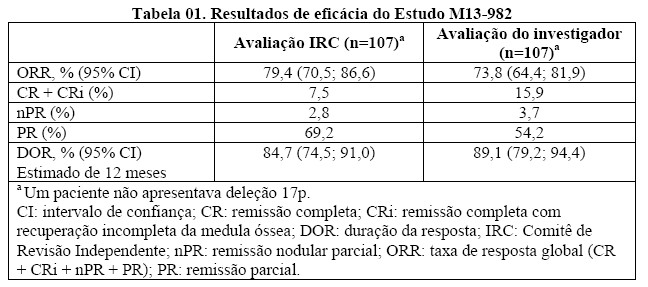
A doença residual mínima foi avaliada por citometria de fluxo em 45 dos 107 pacientes que alcançaram a remissão completa (CR), a remissão completa com a recuperação incompleta da medula óssea (CRi), ou remissão parcial (PR) com a doença permanecendo limitada com o tratamento de VENCLEXTA® (venetoclax). O ponto de corte para um estado negativo era uma célula de LLC por 104 leucócitos na amostra (isto é, um valor de MRD de < 10-4 foi considerado MRD negativo). Dezessete por cento (18/107) dos pacientes estavam com MRD negativo no sangue periférico, incluindo 06 pacientes que eram também MRD negativo na medula óssea.
Houve 73 pacientes que completaram a avaliação do estado de saúde global (GHS) e 76 pacientes que completaram as avaliações de Funcionamento emocional (FE) e Social (FS) no questionário EORTC QLQ-C30, tanto no baseline quanto na semana 24. Houve 74 e 77 pacientes, respectivamente, que completaram as avaliações do papel funcional (RF) e a da escala de sintomas de fadiga, tanto no baseline quanto na semana 24. Após o tratamento com VENCLEXTA® (venetoclax), os pacientes apresentaram melhora no GHS (16%), FE (10,6%), FS(17,1%), RF (16,2%), e a pontuação de sintomas de fadiga (17,5%) na semana 24. As melhoras nestas medidas já foram observadas a partir da semana 04.
Estudo M14-032
O estudo M14-0322,3 foi um estudo aberto, multicêntrico que avaliou a eficácia de VENCLEXTA® (venetoclax) em pacientes com LLC previamente tratados e que haviam progredido durante ou após o uso de ibrutinibe (Braço A) ou idelalisibe (Braço B). Os pacientes receberam uma dose diária de 400 mg de VENCLEXTA® (venetoclax) após a fase de escalonamento de dose. Os pacientes continuaram a receber VENCLEXTA® (venetoclax) 400 mg, uma vez ao dia, até que a progressão da doença ou toxicidade inaceitável fosse observada.
A eficácia foi avaliada por uma Comissão de Revisão Independente (IRC) utilizando o Workshop Internacional para Leucemia Linfocítica Crônica (IWCLL) atualizadas pelas diretrizes do Grupo de Trabalho patrocinado pela National Cancer Institute (NCI-WG). As avaliações de resposta foram realizadas na Semana 08, Semana 24 e posteriormente, a cada 12 semanas para os 64 pacientes da coorte principal, enquanto os pacientes recrutados na coorte de expansão tiveram avaliação da doença nas semanas 12 e 36.
Um total de 127 pacientes foram recrutados no estudo: 64 pacientes no coorte principal (43 previamente com ibrutinibe, 21 previamente com idelalisibe) e 63 pacientes no coorte de expansão (48 previamente com ibrutinibe, 15 previamente com idelalisibe). A idade média dos pacientes era 66 anos (intervalo: 28 a 85 anos), 70% eram do sexo masculino e 92% eram brancos. O tempo médio desde o diagnóstico foi de 8,3 anos (intervalo: 0,3-18,5 anos; N = 96). O número médio de tratamentos anteriores para LCC foi de 4 (intervalo: 1 a 15 tratamentos). No baseline, 41% dos pacientes tinham um ou mais nódulos ≥ 5 cm e 31% dos pacientes apresentavam CAL ≥25 x 109 / L. Os dados de eficácia são apresentados com data de corte de 31 de janeiro de 2017. A eficácia avaliada pelo investigador (N = 108) incluiu todos os 64 pacientes na coorte principal com mais de 24 semanas de avaliação, 37 pacientes da coorte de expansão com 36 semanas de avaliação e 7 pacientes que haviam progredido antes da avaliação de 36 semanas. Os resultados de eficácia pelo IRC (N = 97) incluíram 64 pacientes da coorte principal e 33 pacientes da coorte de expansão.
Os resultados de eficácia para 108 pacientes avaliados pelo investigador e 97 pacientes avaliados pelo IRC são apresentados na Tabela 02.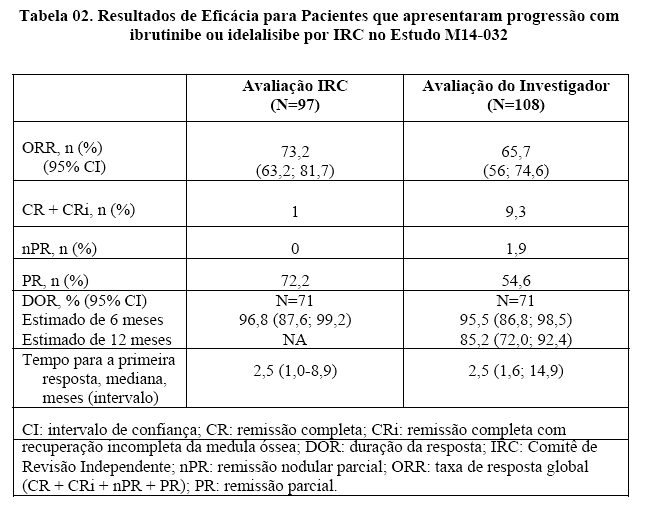
A duração média do tratamento com VENCLEXTA® (venetoclax) para 127 pacientes com avaliação do investigador foi de 10,2 meses (intervalo: 0,1 a 25,6 meses). A duração média do tratamento com VENCLEXTA® (venetoclax) para 97 pacientes com avaliação do IRC foi de 12,3 meses (intervalo: 0,1 a 25,6 meses). A taxa de DRM negativo no sangue periférico para todos os 127 pacientes foi de 30% (29/127), incluindo 5 pacientes que alcançaram DRM negativo na medula óssea.
Leucemia Mielóide Aguda
A eficácia de VENCLEXTA® (venetoclax) foi estudada em dois ensaios não randomizados em pacientes com LMA recém-diagnosticados que não eram elegíveis para quimioterapia intensiva. A eficácia foi estabelecida com base na taxa de remissão completa (CR) / remissão completa com recuperação hematológica parcial (CRh), a duração de CR / CRh e a taxa de conversão de dependência transfusional à independência transfusional. A independência transfusional foi baseada na ausência de qualquer transfusão de hemácias ou plaquetas durante qualquer período consecutivo de 56 dias durante o período de tratamento do estudo e foi avaliada em todos os pacientes.
M14-358
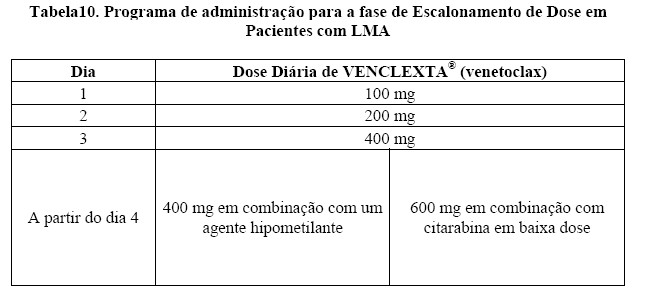
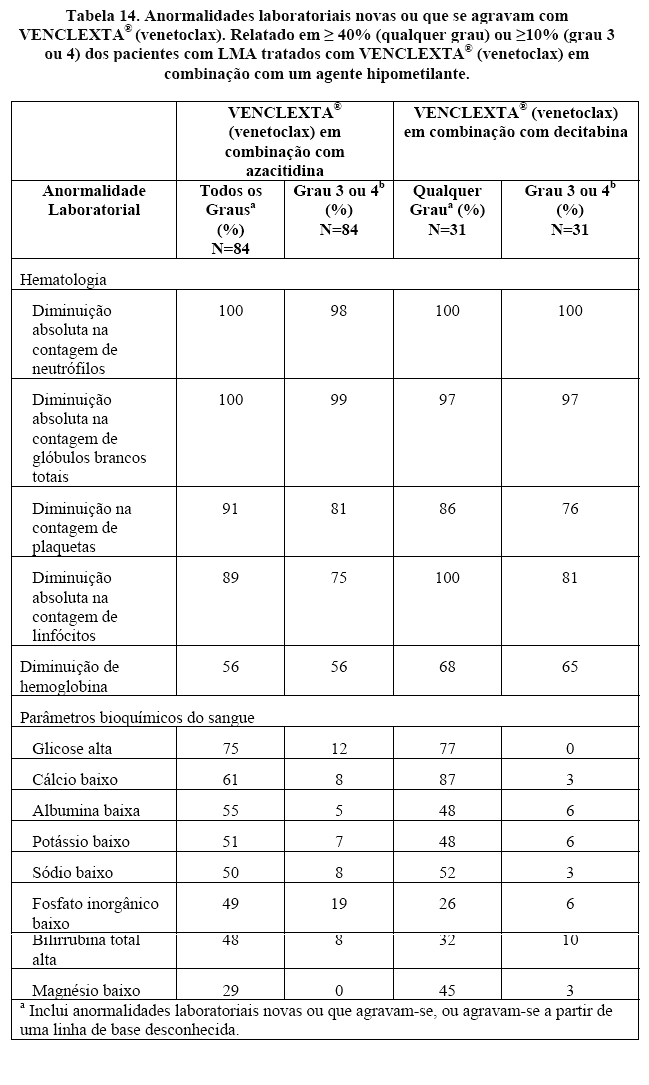
A eficácia de VENCLEXTA® (venetoclax) foi estabelecida em um ensaio clínico não randomizado de VENCLEXTA® (venetoclax) em combinação com azacitidina (n = 84) ou decitabina (n = 31) em pacientes recém-diagnosticados com LMA que não eram elegíveis para quimioterapia intensiva. Os doentes receberam [venetoclax] através de um aumento diário até uma dose final de 400 mg uma vez por dia. Durante o período de escalonamento da dose, os pacientes receberam profilaxia à SLT e foram hospitalizados para monitoramento. Azacitidina a 75 mg / m2 foi administrada por via intravenosa ou subcutânea nos Dias 1-7 de cada ciclo de 28 dias começando no Ciclo 1 Dia 1. A decitabina a 20 mg / m2 foi administrada por via intravenosa nos Dias 1-5 de cada ciclo de 28 dias começando em Ciclo 1 Dia 1. Os pacientes continuaram a receber ciclos de tratamento até a progressão da doença ou toxicidade inaceitável. A redução da dose de azacitidina foi implementada no ensaio clínico para o controle da toxicidade hematológica. Consulte a informação completa sobre prescrição da azacitidina. Reduções de dose para decitabina não foram implementadas no ensaio clínico.
A Tabela a seguir resume as características demográficas e da doença da população do estudo.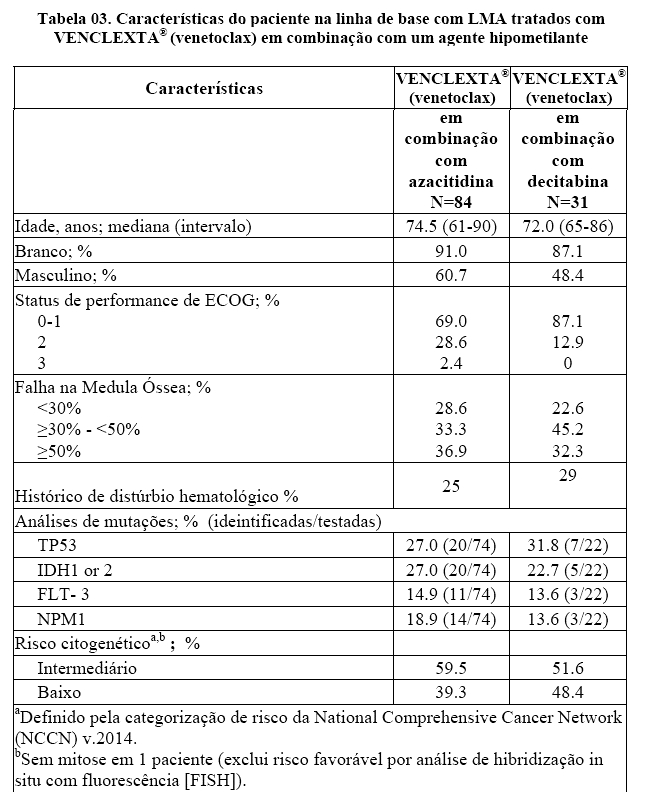
A mediana do seguimento foi de 8,2 meses (intervalo: 0,4 a 35,5 meses) para VENCLEXTA® (venetoclax) em combinação com azacitidina e 16,2 meses (intervalo: 0,7 a 36,7 meses) para VENCLEXTA® (venetoclax) em combinação com decitabina.
Os resultados de eficácia são mostrados na Tabela 04 e 05 e foram semelhantes para ambas as combinações.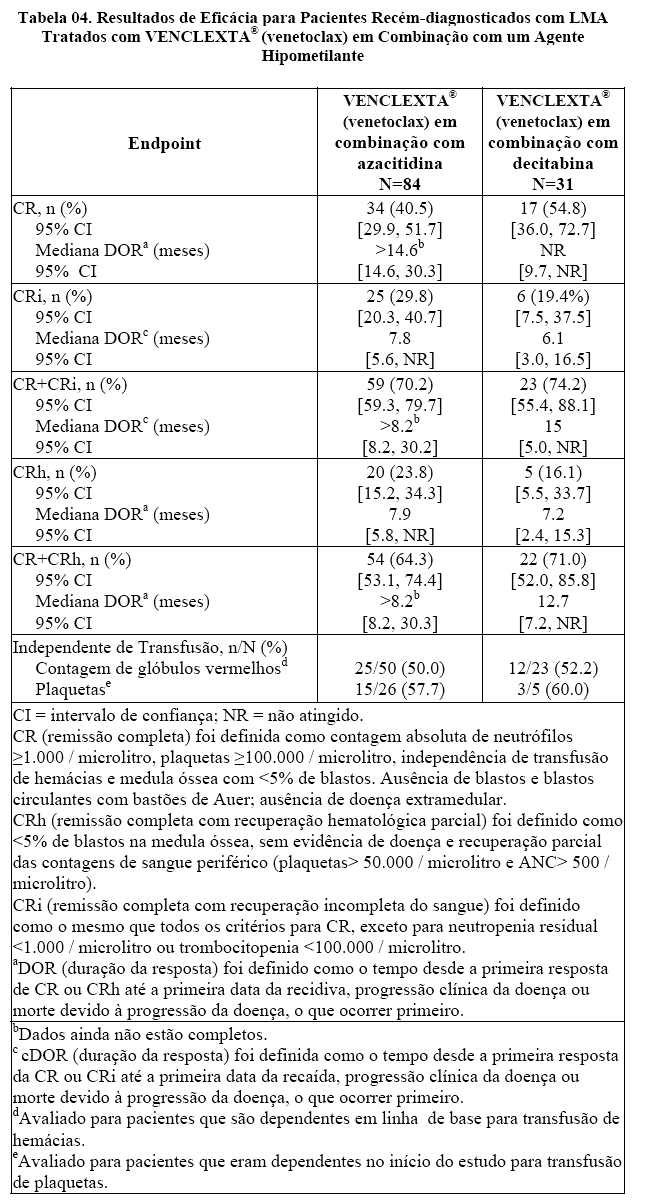

VENCLEXTA® (venetoclax) em combinação com azacitidina
Os resultados de eficácia são mostrados na Tabela 04 e 05.
A sobrevida global mediana (OS) para pacientes em tratamento com VENCLEXTA® (venetoclax) em combinação com azacitidina foi de 14,9 meses (95% CI: 10,2, NR).
As remissões (CR ou CRh) foram observadas em subgrupos com diferentes características basais. Para os pacientes com citogenética de risco baixo ou intermediário, taxas de remissão semelhantes foram observadas, a taxa foi de 57,6% ou 70,0%, respectivamente. Para pacientes com as seguintes mutações identificadas, as remissões foram as seguintes: TP53: 65,0%; IDH1 / 2: 75,0%; FLT-3: 72,7% e NPM1: 71.4%.
A doença residual mínima (DRM) foi avaliada a partir de amostras de aspirado de medula óssea para pacientes que atingiram CR ou CRh após tratamento com VENCLEXTA® (venetoclax) em associação com azacitidina. Desses pacientes, 50% (27/54) atingiram MRD menor que uma célula AML por 103 leucócitos na medula óssea.
Dos pacientes tratados com VENCLEXTA® (venetoclax) em associação com azacitidina, 9,5% (8/84) atingiram um CR / CRi e subsequentemente receberam transplante de células tronco.
VENCLEXTA® (venetoclax) em combinação com Decitabina
Os resultados de eficácia são mostrados na Tabela 04 e 05.
A sobrevida global mediana (OS) para pacientes em tratamento com VENCLEXTA® (venetoclax) em combinação com decitabina foi de 16,2 meses (95% CI: 9,1, NR).
As remissões (CR ou CRh) foram observadas em subgrupos com diferentes características basais. Para os pacientes com citogenética de risco baixo ou intermediário, taxas semelhantes de remissão foram observadas, a taxa foi de 73,3% ou 68,8%, respectivamente. Para pacientes com as seguintes mutações identificadas, as remissões foram as seguintes: TP53: 5/7; IDH1 / 2: 5/5; FLT-3: 1/3 e NPM1: 3/3.
A doença residual mínima (DRM) foi avaliada a partir de amostras de aspirado de medula óssea para pacientes que alcançaram CR ou CRh após tratamento com VENCLEXTA® (venetoclax) em combinação com decitabina. Desses pacientes, 36,4% (8/22) atingiram MRD menor que uma célula AML por 103 leucócitos na medula óssea.
Dos pacientes tratados com VENCLEXTA® (venetoclax) em combinação com decitabina, 9,7% (3/31) atingiram um CR / CRi e subsequentemente receberam transplante de células tronco.
M14-387
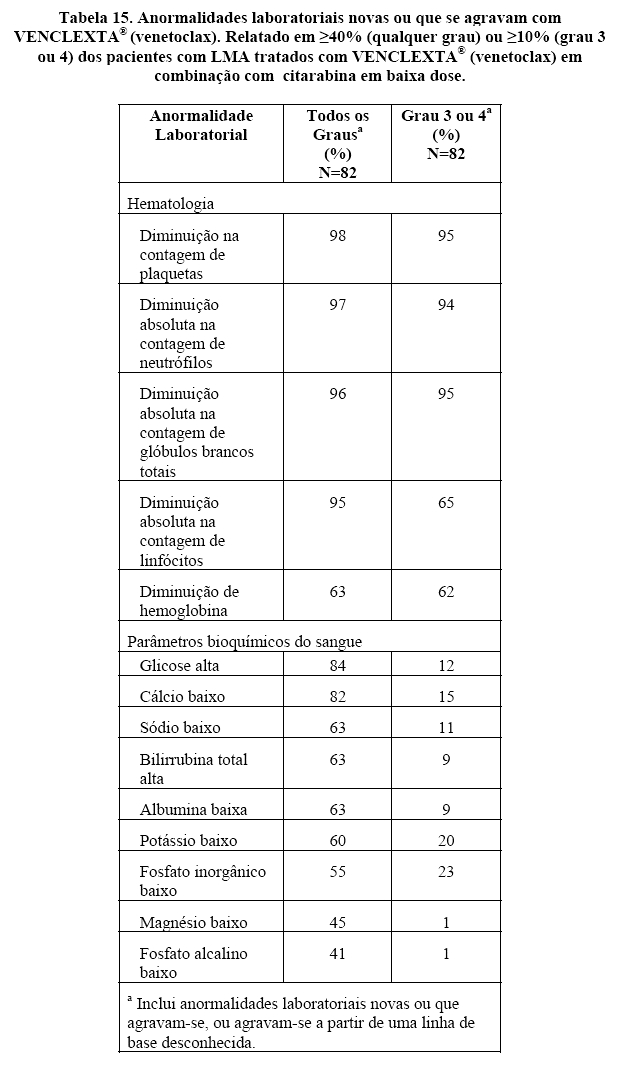
A eficácia de VENCLEXTA® (venetoclax) foi estabelecida em um estudo clínico não randomizado de VENCLEXTA® (venetoclax) em combinação com citarabina em baixa dose (n = 82) em pacientes recém-diagnosticados com LMA não elegíveis para quimioterapia intensiva, incluindo pacientes com exposição anterior a agente hipometilante para um distúrbio hematológico antecedente. Os pacientes iniciaram VENCLEXTA® (venetoclax) através de um escalonamento de dose até uma dose final de 600 mg uma vez por dia. Durante a fase de escalonamento de dose, os pacientes receberam profilaxia da SLT e foram hospitalizados para monitoramento. A citarabina a uma dose de 20 mg / m2 foi administrada por via subcutânea, uma vez ao dia nos Dias 1-10 de cada ciclo de 28 dias começando no Ciclo 1 Dia 1. Os pacientes continuaram a receber ciclos de tratamento até progressão da doença ou toxicidade inaceitável. A redução da dose para citarabina em baixa dose não foi implementada nos estudos clínicos.
A Tabela 06 resume as características demográficas e de doença da população de estudo.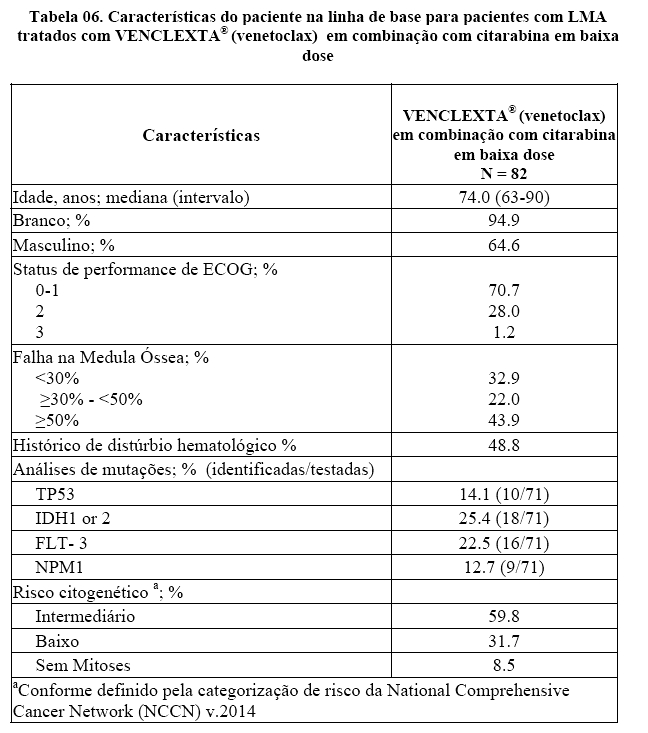
A mediana de acompanhamento foi de 7,1 meses (intervalo: 0,3 a 34,3 meses). Os resultados de eficácia são mostrados nas Tabelas 07 e 08.

A sobrevida global mediana (OS) para pacientes em VENCLEXTA® (venetoclax) em combinação com citarabina em baixa dose foi de 10,1 meses (95% CI: 5,7, 14,2).
As remissões (CR ou CRh) foram observadas em subgrupos com diferentes características basais. Para os pacientes com citogenética de risco baixo ou intermediário, taxas semelhantes de remissão foram observadas, a taxa foi de 34,6% ou 57,1%, respectivamente. Para pacientes com as seguintes mutações identificadas, as remissões foram as seguintes: TP53: 20,0%, IDH1 / 2: 66,7%, FLT-3: 31,3% e NPM1: 88,9%. A doença residual mínima (DRM) foi avaliada na medula óssea para pacientes que alcançaram CR ou CRh após tratamento com VENCLEXTA® (venetoclax) em combinação com citarabina em baixa dose. Desses pacientes, 34,2% (13/38) atingiram MRD menor que uma célula AML por 103 leucócitos na medula óssea. Dos pacientes tratados com VENCLEXTA® (venetoclax) em combinação com citarabina em baixa dose, 1,2% (1/82) atingiram uma CR / CRh e subsequentemente receberam transplante de células tronco.
Referências Bibliográficas
1. Stilgenbauer S, Eichhorst B, Schetelig J, et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, openlabel, phase 2 study.Lancet Oncol. 2016 Jun;17(6):768-778
2. Jones JA, Mato AR, Wierda WG, et al. Venetoclax for chronic lymphocytic leukaemia progressing after ibrutinib: an interim analysis of a multicentre, openlabel, phase 2 trial. Lancet Oncol. 2018 Jan;19(1):65-75.
3. Coutre S, Choi M, Furman RR, et al. Venetoclax for patients with chronic lymphocytic leukemia who progressed during or after idelalisib therapy. Blood. 2018 Apr 12;131(15):1704-1711.
4. DiNardo CD, Pratz KW, Letai A, et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-randomised, open-label, phase 1b study. Lancet Oncol. 2018 Feb;19(2):216-228.
5. Wei A, Strickland S, Roboz G, et al. Safety and efficacy of venetoclax plus lowdose cytarabine in treatment-naive patients aged ≥65 years with acute myeloid leukemia. Blood 2016 128:102.
3. CARACTERÍSTICAS FARMACOLÓGICAS
O ativo de VENCLEXTA® (venetoclax) é um sólido amarelo claro a amarelo escuro com fórmula empírica C45H50ClN7O7S e peso molecular de 868,44. VENCLEXTA® (venetoclax) tem solubilidade muito baixa em água. O nome químico de VENCLEXTA® (venetoclax) é definido como 4-(4-{[2-(4-clorofenil)-4,4-dimetilciclohex-1-em-1-il]metil}piperazina-1-il)-N-({3-nitro-4-[(tetrahidro-2H-piran-4-ilmetil)amino]fenil}sulfonil)-2-(1H-pirrolo[2,3-b]piridina-5-iloxi)benzamida) e possui a seguinte estrutura química: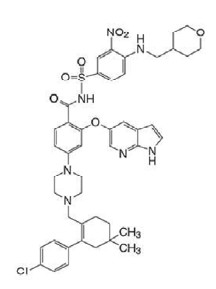
Mecanismo de ação
VENCLEXTA® (venetoclax) é um potente, seletivo e oralmente biodisponível inibidor de pequena molécula da célula de linfoma B (Bcl-2), uma proteína antiapoptótica. A superexpressão da Bcl-2 tem sido demonstrada em várias doenças malignas hematológicas e tumores sólidos, e tem sido implicada como um fator de resistência para determinados agentes terapêuticos. VENCLEXTA® (venetoclax) se liga diretamente ao canal de ligação-BH3 da Bcl-2, deslocando a proteína pro-apoptótica BH3 motif-containing como BIM, para iniciar a permeabilização da membrana mitocondrial externa (MOMP), a ativação de caspase e, a morte celular programada. Em estudos não clínicos, venetoclax demonstrou atividade citotóxica em uma grande variedade de células B e outras doenças malignas hematológicas.
Farmacodinâmica
- Eletrofisiologia Cardíaca
O efeito no intervalo QTc de doses múltiplas de VENCLEXTA® (venetoclax) de até 1200 mg uma vez por dia foi avaliado em um estudo aberto de braço único de 176 pacientes com LLC ou Linfoma não-Hodgkin (NHL) tratados previamente. VENCLEXTA® (venetoclax) não teve efeito sobre o intervalo QTc e não houve relação entre a exposição de venetoclax e mudanças no intervalo QTc.
Farmacocinética
- Absorção
Após múltiplas administrações orais, a concentração plasmática máxima de venetoclax foi atingida em 5-8 horas após a dose. A AUC no estado estacionário de venetoclax aumentou proporcionalmente no intervalo de doses de 150-800mg. Em condições de refeição com baixo teor de gordura, a média (± desvio padrão) da Cmax no estado estacionário foi de 2,1 ± 1,1 mg / mL e AUC24 foi de 32,8 ± 16,9 mg • h/mL na dose de 400 mg uma vez por dia.
- Efeito dos Alimentos
A administração com uma refeição de baixo teor de gordura aumentou a exposição de venetoclax em cerca de 3,4 vezes e a administração com uma refeição rica em gorduras aumentou a exposição de venetoclax de 5,1 para 5,3 vezes em comparação com condições de jejum. VENCLEXTA® (venetoclax) deve ser administrado durante as refeições [veja Posologia].
- Distribuição
VENCLEXTA® (venetoclax) é fortemente ligado às proteínas do plasma humano com fração livre no plasma < 0,01 em uma faixa de concentração de 1-30 mM (0,87-26 ug / mL). A proporção média de sangue para plasma foi de 0,57. A estimativa da população para o volume aparente de distribuição (Vdss / F) de venetoclax variou de 256 - 321 L em pacientes.
- Metabolismo
Estudos in vitro demonstraram que venetoclax é predominantemente metabolizado pelo CYP3A4.O M27 foi identificado como o principal metabólito no plasma, com uma atividade inibidora da Bcl-2, de pelo menos 58 vezes menor do que venetoclax in vitro.
- Eliminação
A estimativa da população para a meia-vida de eliminação da fase terminal de venetoclax foi de aproximadamente 26 horas. Após uma única administração oral de 200 mg de venetoclax radiomarcado (14C) em indivíduos saudáveis, > 99,9% da dose foi recuperada nas fezes e < 0,1% da dose administrada foi excretada na urina em 9 dias. O venetoclax inalterado representou 20,8% na dose radioativa administrada e excretada nas fezes. A farmacocinética de venetoclax não muda ao longo do tempo.
- Populações especiais
Idade, Raça, Gênero e Peso: baseando-se na análise farmacocinética populacional, idade, raça, gênero e peso não afetaram a liberação de venetoclax.
Uso pediátrico: a farmacocinética de venetoclax não foi avaliada em pacientes < 18 anos (veja em "5. ADVERTÊNCIAS E PRECAUÇÕES - Populações especiais").
Insuficiência renal: com base em uma análise farmacocinética da população, que incluiu 211 pacientes com insuficiência renal leve (CrCl ≥ 60 e < 90 mL/min), 83 pacientes com insuficiência renal moderada (CrCl ≥30 e < 60 mL / min) e 210 pacientes com função renal normal (CrCl ≥90 mL / min), as exposições de venetoclax em pacientes com insuficiência renal leve ou moderada foram semelhantes àqueles com função renal normal. A farmacocinética de venetoclax não foi estudada em pacientes com insuficiência renal grave (CrCl < 30 mL / min) ou indivíduos em diálise [veja em "5. ADVERTÊNCIAS E PRECAUÇÕES - Populações especiais"].
Insuficiência hepática: com base em uma análise farmacocinética da população que incluiu 69 pacientes com insuficiência hepática leve, 7 pacientes com insuficiência hepática moderada e 429 pacientes com função hepática normal, as exposições de venetoclax foram semelhantes em pacientes com insuficiência hepática leve e moderada e função hepática normal. A insuficiência hepática leve foi definida como bilirrubina total normal total e aspartato transaminase (AST) > que o limite superior normal (ULN) ou bilirrubina total > 1,0 a 1,5 vezes de ULN, insuficiência hepática moderada como bilirrubina total > 1,5 a 3,0 vezes da ULN, e insuficiência hepática grave como bilirrubina total > 3,0 ULN. Em um estudo dedicado a insuficiência hepática, a Cmax e a AUC de venetoclax em indivíduos com insuficiência hepática leve (Child-Pugh A) ou moderada (Child-Pugh B) foram semelhantes aos indivíduos com função hepática normal. Em indivíduos com insuficiência hepática grave (Child-Pugh C), a média da Cmax do venetoclax foi semelhante à dos indivíduos com função hepática normal, mas a AUC do venetoclax foi 2,3 a 2,7 vezes superior à dos indivíduos com função hepática normal. [veja em "8. POSOLOGIA E MODO DE USAR"].
- Interação medicamentosa
Inibidores da CYP3A: a coadministração de 400mg de cetoconazol uma vez ao dia, um forte inibidor da CYP3A, P-gp e inibidor da BCRP, por 07 dias em 11 pacientes com linfoma não-Hodgkin tratados previamente aumentou a Cmax de venetoclax em 130 % e AUC∞ em 540%.
A coadministração de 50 mg de ritonavir uma vez ao dia, um forte inibidor da CYP3A, Pgp e inibidor da OATP1/B3B1, por 14 dias em 6 pacientes sádios aumentou a Cmax de venetoclax em 140% e AUC∞ em 690%.
Comparado com 400 mg de venetoclax administrado isoladamente, a administração concomitante de 300 mg de posaconazol, um forte inibidor da CYP3A e da P-gp, com 50 mg e 100 mg de venetoclax durante 7 dias em 12 pacientes com LMA recém-diagnosticados resultou em um aumento de 61% e 86% na Cmax de venetoclax, respectivamente. A AUC24 do venetoclax foi 90% e 144% maior, respectivamente.
Indutores da CYP3A: a coadministração de 600 mg de rifampicina uma vez ao dia, um forte indutor da CYP3A, por 13 dias em 10 indivíduos saudáveis diminuiu a Cmax de venetoclax em 42% e AUC∞ em 71%.
Inibidores da OATP1B1/1B3 e P-gp: a coadministração de 600 mg de rifampicina em dose única, um inibidor da OATP1B1/1B3 e P-gp, em 11 indivíduos saudáveis aumentou a Cmax de venetoclax em 106% e AUC∞ em 78%.
Azitromicina: A co-administração de 500 mg de azitromicina, no primeiro dia seguida de 250 mg de azitromicina durante 4 dias em 12 indivíduos saudávei diminuiu a Cmax de venetoclax em 25% e a AUC∞ em 35%.
Agentes redutores da acidez gástrica: com base na análise farmacocinética populacional, agentes redutores da acidez gástrica (por exemplo, inibidores da bomba de prótons, antagonistas do receptor H2 e antiácidos) não afetou a biodisponibilidade de venetoclax.
Varfarina: em um estudo de interação medicamento-medicamento em três voluntários saudáveis, a administração de 400 mg de venetoclax em dose única com 5 mg de varfarina resultou em um aumento de 18% para 28% na Cmax e AUC∞ da R-varfarina e S-varfarina.
Digoxina: em um estudo de interação medicamento-mediamento em 10 voluntários saudáveis, a administração de uma dose única de 100 mg de venetoclax com 0,5 mg de digoxina, um substrato da P-gp, resultou em um aumento de 35% na Cmax da digoxina e um aumento de 9% na AUC∞ da digoxina.
- Estudos in vitro
Os estudos in vitro indicaram que venetoclax não é um inibidor ou indutor da CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 ou CYP3A4 em concentrações clinicamente relevantes. O venetoclax é um inibidor fraco da UGT1A1 in vitro mas, não é previsto causar uma inibição clinicamente relevante da UGT1A1. O venetoclax não é um inibidor da UGT1A4, UGT1A6, UGT1A9 e UGT2B7.
O venetoclax é um substrato de P-gp e BCRP, bem como um inibidor da P-gp e BCRP e fraco inibidor da OATP1B1 in vitro. Não é esperado que venetoclax iniba OATP1B3, OCT1, OCT2, OAT1, OAT3, MATE1 ou MATE2K em concentrações clinicamente relevantes.
Dados de segurança pré-clínicos
- Carcinogenicidade. Mutagênese e Alterações na Fertilidade
Não foram conduzidos estudos de carcinogenicidade com VENCLEXTA® (venetoclax). VENCLEXTA® (venetoclax) não foi mutagênico em um ensaio in vitro de mutagenicidade bacteriana (Ames), não induziu anormalidades numéricas ou estruturais em um ensaio in vitro de anormalidade cromossômicas utilizando linfócitos periféricos de sangue humano, e não foi clastogênico em um ensaio in vivo de micronúcleos da medula óssea de ratos em doses de até 835 mg /kg. O metabolito M27 foi negativo para a atividade genotóxica nos ensaios in vitro de Ames e de alterações cromossômicas e em camundongos tiveram efeitos semelhantes ao venetoclax (diminuição de linfócitos e massa de hemácias), mas de menor magnitude, consistente com sua baixa potência farmacológica in vitro.
Estudos de desenvolvimento embrionário precoce e de fertilidade foram realizados em camundongos machos e fêmeas. Estes estudos avaliaram o acasalamento, a fertilização e o desenvolvimento embrionário por meio de implantação. Não houve efeitos de venetoclax nos ciclos estrais, de acasalamento, fertilidade, corpo lúteo, implantes uterinos ou embriões vivos por ninhada em doses de até 600 mg/kg/dia (em camundongos machos e fêmeas, com aproximadamente 2,8 e 3,2 vezes da exposição AUC humana na dose recomendada, respectivamente). No entanto, um risco para a fertilidade masculina humana existe com base na toxicidade testicular (perda de células germinativas) observada em cães em todas as doses examinadas (exposições de 0,5 a 18 vezes da exposição AUC humana na dose recomendada). A reversibilidade deste achado não foi demonstrada.
- Toxicidade e/ou Farmacologia Animal
Além da perda de células germinativas testiculares, outras toxicidades foram observadas em estudos em animais com venetoclax que incluíram reduções dose-dependentes de linfócitos e massa de glóbulos vermelhos. Ambos os efeitos foram reversíveis após a interrupção da administração de venetoclax, com recuperação dos linfócitos ocorrendo em 18 semanas após o tratamento. As células-B e células-T foram afetadas, mas as perdas mais significativas ocorreram com células-B. A diminuição de linfócitos não foi associada com infecções oportunistas. VENCLEXTA® (venetoclax) também causou necrose de única célula em vários tecidos, incluindo a vesícula biliar e pâncreas exócrino, sem evidência de ruptura da integridade do tecido ou disfunção dos órgãos; estes achados foram de mínimos a leves em magnitude. Após um período de dose de quatro semanas e subsequente período de recuperação de quatro semanas, a necrose mínima de única célula ainda estava presente em alguns tecidos e a reversibilidade não foi avaliada por com períodos mais longos de dosagem ou de recuperação. Além disso, depois de aproximadamente três meses de administração diária em cães, venetoclax causou descoloração branca e progressiva do pêlo, devido à perda de pigmento melanina no cabelo. Não foram observadas alterações na qualidade do revestimento do pêlo ou da pele, nem em outros tecidos pigmentados examinados (por exemplo, a íris e o fundo ocular do olho). A reversibilidade das alterações do pêlo não tem sido avaliada em cães. Em ratos fêmeas grávidas, a exposição sistêmica materna (AUC) ao venetoclax foi aproximadamente 14 vezes superior à exposição em humanos na dose de 400 mg. Níveis mensuráveis de radioatividade nos tecidos fetais (fígado, trato GI) foram > 15 vezes menores que os níveis maternos nos mesmos tecidos. A radioatividade derivada de venetoclax não foi detectada no sangue fetal, cérebro, olhos, coração, rim, pulmão, músculo ou medula espinhal. Venetoclax foi administrado (dose única; 150 mg / kg por via oral) em ratos lactentes 8-10 dias de parto. O venetoclax identificado no leite foi 1,6 vezes menor do que no plasma. A droga principal (venetoclax) representou a maior parte do total de material relacionado ao medicamento no leite, com níveis vestigiais de três metabólitos.
4. CONTRAINDICAÇÕES
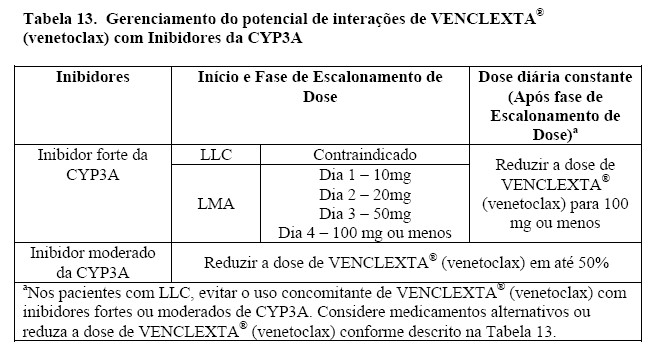
Em pacientes com LLC, o uso concomitante de VENCLEXTA® (venetoclax) e um inibidor forte da CYP3A é contraindicado no início do tratamento e durante a fase de escalonamento de dose (veja em "8. POSOLOGIA E MODO DE USAR e "6. INTERAÇÕES MEDICAMENTOSAS").
5. ADVERTÊNCIAS E PRECAUÇÕES
Síndrome da Lise Tumoral (SLT)
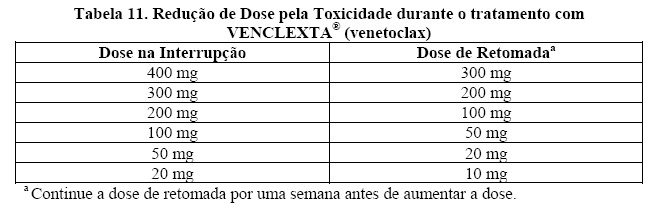
Pacientes com elevada carga tumoral, apresentaram Síndrome da Lise Tumoral quando tratados com VENCLEXTA® (venetoclax), um evento adverso grave que pode levar à morte (veja em "9. REAÇÕES ADVERSAS"). VENCLEXTA® (venetoclax) pode causar uma rápida redução do tumor e, portanto, representa um risco para SLT no início e durante a fase de escalonamento de dose. Podem ocorrer alterações nos eletrólitos consistentes à SLT que exigir rápido gerenciamento logo nas primeiras 6-8 horas após a primeira dose de VENCLEXTA® (venetoclax) e em cada aumento de dose. O risco de SLT é constante com base em vários fatores, incluindo comorbidades. Pacientes com uma elevada carga tumoral (por exemplo, qualquer linfonodo com diâmetro maior ou igual a 5 cm ou uma contagem absoluta de linfócitos [ALC] maior ou igual a 25X109/L]) estão em maior risco de SLT quando o tratamento com VENCLEXTA® (venetoclax) é iniciado. Uma função renal reduzida aumenta ainda mais este risco. Os pacientes devem ser avaliados quando o risco de SLT e devem receber profilaxia apropriada para SLT, incluindo hidratação e uso de agentes anti-hiperuricemicos. Deve-se monitorar a composição sanguínea e gerenciar prontamente as anormalidades. Interrompa a dose se for necessário. Empregue medidas mais intensivas (hidratação intravenosa, monitoramento frequente e hospitalização) com o aumento global do risco (veja em "8. POSOLOGIA E MODO DE USAR"). O uso concomitante de VENCLEXTA® (venetoclax) e um inibidor forte ou moderado da CYP3A aumentam a exposição de venetoclax podem aumentar o risco de SLT no início e durante a fase de escalonamento de dose (veja em "8. POSOLOGIA E MODO DE USAR" e "6. INTERAÇÕES MEDICAMENTOSAS"). Além disso, os inibidores da Pgp podem aumentar a exposição ao venetoclax (veja em "6. INTERAÇÕES MEDICAMENTOSAS").
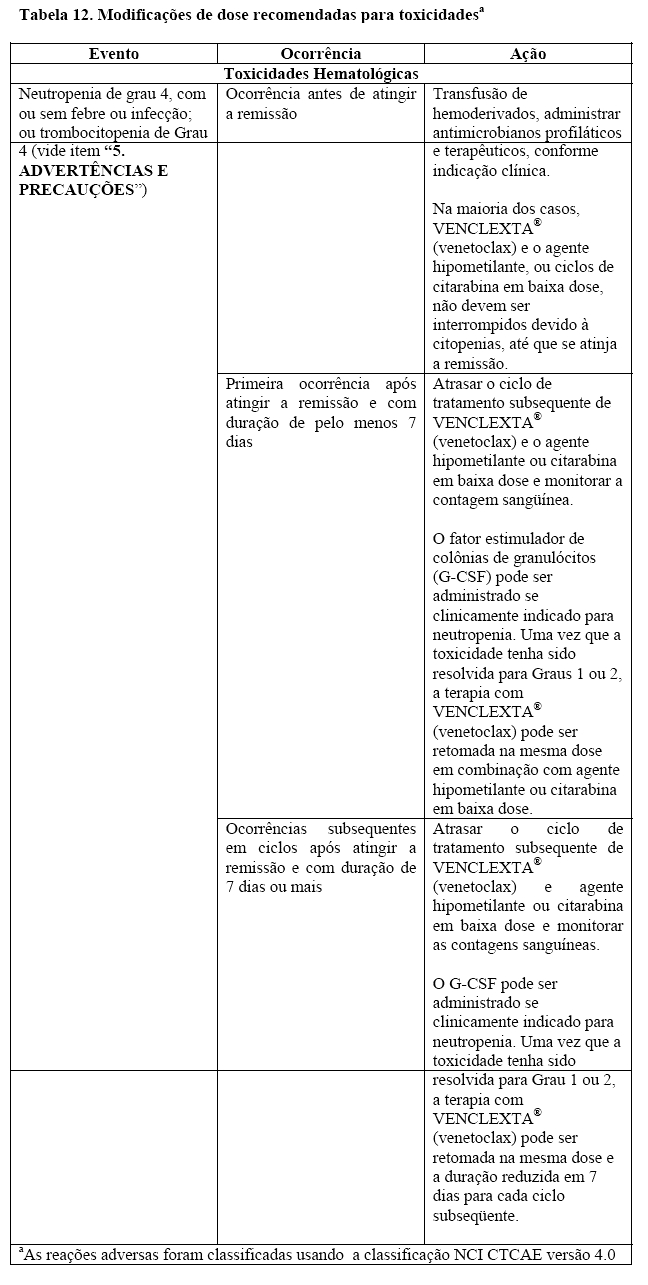
Neutropenia
Foi apresentada neutropenia de grau 3 ou 4 em pacientes com LLC tratados com VENCLEXTA® (venetoclax) (veja em "9. REAÇÕES ADVERSAS"). Em pacientes com LMA, neutropenia de Grau 3 ou 4 são comuns antes do início do tratamento. A contagem de neutrófilos pode piorar com VENCLEXTA® (venetoclax) em combinação com um agente hipometilante ou com citarabina em baixa dose. A neutropenia pode recorrer com ciclos subseqüentes da terapia. Durante todo o período do tratamento, os hemogramas completos devem ser monitorados. Interrupções da dose ou reduções da dose são recomendadas no caso de neutropenia grave. Devem ser consideradas medidas de suporte, incluindo antimicrobianos para qualquer sinal de infeção e o uso profilático de fatores de crescimento (por exemplo, G-CSF) (veja em "8. POSOLOGIA", "4.CONTRAINDICAÇÕES", "6. INTERAÇÕES MEDICAMENTOSAS" e "3. CARACTERÍSTICAS FARMACOLÓGICAS").
Imunizações
A segurança e eficácia de imunizações com vacinas de vírus atenuado durante e após o uso de VENCLEXTA® (venetoclax) não foram estudadas. Vacinas vivas não devem ser administradas durante e após o tratamento com VENCLEXTA® (venetoclax) até a recuperação das células B.
Cuidados e advertências em populações especiais
- Reprodução, Gravidez e Lactação
Estudos em animais demonstraram toxicidade embriofetal.
Dados em animais: em estudos de desenvolvimento embriofetal, VENCLEXTA® (venetoclax) foi administrado em camundongos fêmeas e coelhas grávidas para avaliar os efeitos potenciais após a implantação e subsequente desenvolvimento embrionário e fetal durante os respectivos períodos de organogênese. Em camundongos, venetoclax foi associado com o aumento da perda pós-implantação e diminuição do peso corpóreo fetal em 150 mg/kg/dia (exposição materna de aproximadamente de 1,2 vezes à exposição AUC humana na dose recomendada). Em coelhas, 300 mg/kg/dia de venetoclax produziu toxicidade materna, mas nenhuma toxicidade fetal (exposição materna de aproximadamente 0,2 vezes à exposição AUC humana na dose recomendada). Não foi observada teratogenicidade nos camundongos ou coelhas.
Reprodução: mulheres com potencial reprodutivo devem fazer o teste de gravidez antes do início de VENCLEXTA® (venetoclax). Mulheres com potencial reprodutivo devem usar um contraceptivo efetivo durante todo o tratamento com VENCLEXTA® (venetoclax) e no mínimo 30 dias após a última dose. Com base nos resultados obtidos em animais, a fertilidade masculina pode ser comprometida pelo tratamento com VENCLEXTA® (venetoclax) (veja em "Dados de segurança pré-clínicos").
Gravidez: VENCLEXTA® (venetoclax) não deve ser utilizado durante a gravidez. Não existem dados adequados e bem controlados sobre o uso de VENCLEXTA® (venetoclax) em mulheres grávidas. Os estudos com animais mostraram toxicidade embrionária e fetal.
Categoria de risco: C Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião dentista.
Lactação: a excreção no leite humano de VENCLEXTA® (venetoclax) ou seus metabólitos é desconhecida. Dados disponíveis em estudos com animais mostraram excreção de venetoclax / metabólitos de venetoclax no leite (vide item "2. RESULTADOS DE EFICÁCIA"). O risco para recém nascidos e/ou lactentes não pode ser excluído. A amamentação deve ser descontinuada durante o tratamento com VENCLEXTA® (venetoclax).
- Uso em crianças
A segurança e eficácia de VENCLEXTA® (venetoclax) em crianças e adolescentes com menos de 18 anos de idade não foi estabelecida.
Em um estudo de toxicologia juvenil, VENCLEXTA® (venetoclax) foi administrado em camundongos de 07 a 60 dias de idade em 10, 30 ou 100 mg/kg/dia via oral. Os sinais clínicos de toxicidade incluíram diminuição da atividade, desidratação, palidez da pele e postura arqueada com ≥ 30 mg/kg/dia. Além disso, os efeitos da mortalidade e do peso corporal ocorreram em 100 mg/kg/dia. Outros efeitos relacionados ao venetoclax foram reduções reversíveis nos linfócitos a ≥10 mg/kg/dia, que foram consistentes com os camundongos adultos e considerados não adversos.
O nível de evento adverso não observado (NOAEL) para VENCLEXTA® (venetoclax) e 10 mg/kg/dia em camundongos é aproximadamente 0,06 vezes a dose clínica de 400 mg em uma base de mg/m2 para uma criança de 20 kg.
- Uso em idosos
Não é necessário ajuste específico de dose para os pacientes idosos (≥65 anos).
- Insuficiência renal
Não foram conduzidos estudos clínicos específicos em pacientes com insuficiência renal. Não é necessário qualquer ajuste de dose para pacientes com insuficiência renal leve ou moderada (CrCl ≥ 30 mL/min). A dose recomendada não foi determinada para pacientes com insuficiência renal grave (CrCl < 30 mL/min) ou em pacientes em diálise (veja Propriedades Farmacológicas).
Pacientes com função renal diminuída (CrCl < 80 mL/min) podem exigir a profilaxia e monitoramento mais intensos para reduzir o risco de SLT quando o tratamento com VENCLEXTA® (venetoclax) for iniciado (veja posologia e administração).
- Insuficiência hepática
Não é necessário ajuste de dose em pacientes com insuficiência hepática leve ou moderada (veja propriedades farmacológicas). Recomenda-se uma redução da dose de 50% durante o tratamento em pacientes com insuficiência hepática grave. O monitoramento desses pacientes deve ser realizado mais de perto quanto aos sinais de toxicidade.
Efeito na habilidade de dirigir ou operar máquinas
Não foram conduzidos estudos sobre os efeitos de VENCLEXTA® (venetoclax) na habilidade de dirigir ou operar máquinas. VENCLEXTA® (venetoclax) tem pouca ou nenhuma influência na habilidade de dirigir ou operar máquinas.
Abuso de drogas e dependência
Não há dados disponíveis quanto ao uso de VENCLEXTA® (venetoclax) e o abuso ou dependência de drogas.
6. INTERAÇÕES MEDICAMENTOSAS
Efeitos de outros medicamentos em VENCLEXTA® (venetoclax)
VENCLEXTA® (venetoclax) é predominantemente metabolizado pela CYP3A4.
- Inibidores da CYP3A
A coadministração de cetoconazol aumentou a Cmax de venetoclax em 130% e a AUC∞ em 540%.
A co-administração de ritonavir aumentou a Cmax de venetoc