VEMLIDY
GILEAD
tenofovir alafenamida, hemifumarato de
Antiviral.
Apresentações.
Vemlidy® é apresentado em frascos contendo 30 comprimidos revestidos. Cada comprimido revestido contém hemifumarato de tenofovir alafenamida que equivale a 25 mg de tenofovir alafenamida. Cada frasco contém um dissecante de sílica gel.
USO ORAL
USO ADULTO
Composição.
Cada comprimido contém 28,04 mg de hemifumarato de tenofovir alafenamida que equivale a 25 mg de tenofovir alafenamida.
Excipientes: lactose monoidratada, celulose microcristalina, croscarmelose sódica, estearato de magnésio, álcool polivinílico, dióxido de titânio, macrogol, talco e óxido de ferro amarelo.
Informações técnicas.
1. INDICAÇÕES
Vemlidy® é indicado para o tratamento da infecção crônica pelo vírus da hepatite B (VHB) em adultos com doença hepática compensada.
2. RESULTADOS DE EFICÁCIA
A eficácia e a segurança de tenofovir alafenamida em pacientes com hepatite B crônica baseiam-se em dados referentes a 48 e 96 semanas de dois estudos randomizados, duplo-cegos, controlados com comparador ativo, "Estudo 108"e"Estudo 110". A segurança do tenofovir alafenamida é também suportada por dados agrupados dos Estudos 108 e 110, de pacientes que permaneceram no tratamento cego a partir da semana 96 até a semana 144 e adicionalmente dos pacientes na fase aberta dos estudos 108 e 110 a partir da semana 96 até a semana 144 (N=360 permaneceram com tenofovir alafenamida; N=180 trocaram de tenofovir desoproxila para tenofovir alafenamida na semana 96).
No Estudo 108, pacientes AgHBe-negativos sem experiência prévia ao tratamento e com experiência prévia ao tratamento, com função hepática compensada foram randomizados numa razão de 2:1 para receberem tenofovir alafenamida (25 mg; N = 285) uma vez por dia ou tenofovir desoproxila (245 mg; N = 140) uma vez por dia. A idade média foi de 46 anos, 61% eram do sexo masculino, 72% eram asiáticos, 25% eram caucasianos e 2% (8 indivíduos) eram negros; 24%, 38% e 31% tinham VHB de genótipo B, C e D, respetivamente. 21% tinham experiência prévia ao tratamento (tratamento anterior com antivirais orais, incluindo entecavir (N = 41), lamivudina (N = 42), tenofovir desoproxila (N = 21), ou outro (N = 18)). No início do estudo, o DNA-VHB plasmático médio era 5,8 log10 UI/mL, a ALT sérica média era 94 U/L, e 9% dos pacientes tinham histórico de cirrose.
No Estudo 110, pacientes AgHBe-positivos sem experiência prévia ao tratamento e com experiência prévia ao tratamento, com função hepática compensada foram randomizados numa razão de 2:1 para receberem tenofovir alafenamida (25 mg; N = 581) uma vez por dia ou tenofovir desoproxila (245 mg; N = 292) uma vez por dia. A idade média foi de 38 anos, 64% eram do sexo masculino, 82% eram asiáticos, 17% eram caucasianos e < 1% (5 indivíduos) eram negros. 17%, 52% e 23% tinham VHB de genótipo B, C e D, respetivamente. 26% tinham experiência prévia ao tratamento (tratamento anterior com antivirais orais, incluindo adefovir (N = 42), entecavir (N = 117), lamivudina (N = 84), telbivudina (N = 25), tenofovir desoproxila (N = 70), ou outro (N = 17)). No início do estudo, o DNA-VHB plasmático médio era 7,6 log10 UI/mL, a ALT sérica média era 120 U/L, e 7% dos pacientes tinham história de cirrose.
O desfecho primário de eficácia em ambos os estudos era a proporção de pacientes com níveis de DNA-VHB plasmático inferiores a 29 UI/mL na semana 48. A não-inferioridade foi avaliada usando uma abordagem de intervalo de confiança de 95% com uma margem de não inferioridade pré-especificada de 10%. A determinação do tamanho da amostra baseou-se nos resultados da Semana 48 de ensaios clínicos de fase 3 anteriores em pacientes AgHBe-positivos e AgHBe-negativos com infecção crônica pelo VHB comparando tenofovir desoproxila com adefovir dipivoxila (Estudos GS-US-174-0103 e GS-US-174-0102) e comparando adefovir dipivoxila com placebo (Estudos GS-98-437 e GS-98-438). Vemlidy® foi determinado para atingir os critérios de não-inferioridade se o limite inferior do intervalo de confiança bilateral de 95% da diferença de tratamento (grupo Vemlidy® -grupo Viread®) na proporção de pacientes que atingiram DNA-VHB inferior a 29 UI/mL na Semana 48 for maior que -10%.
Os resultados de tratamento do Estudo 108 e do Estudo 110 até a semana 48 são apresentados na Tabela 1 e Tabela 2.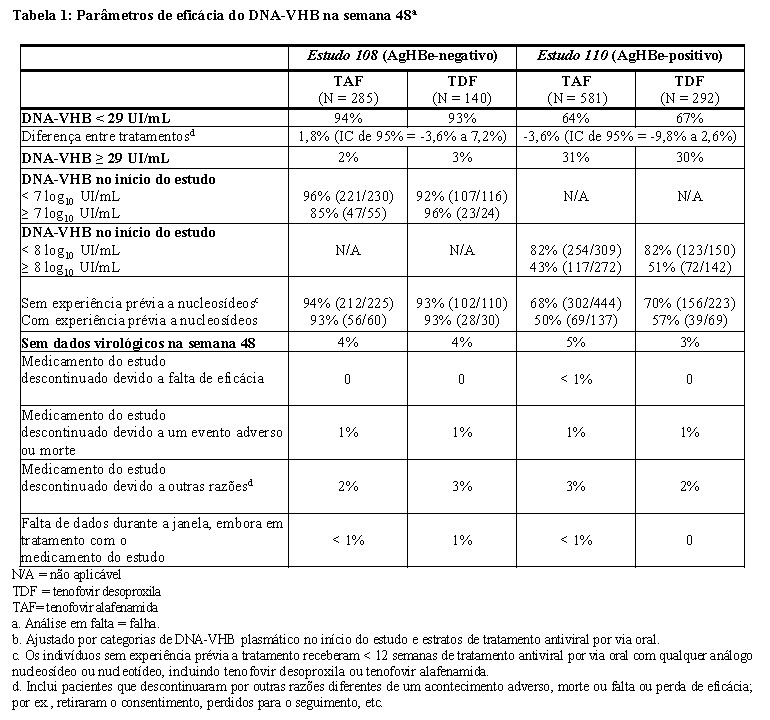
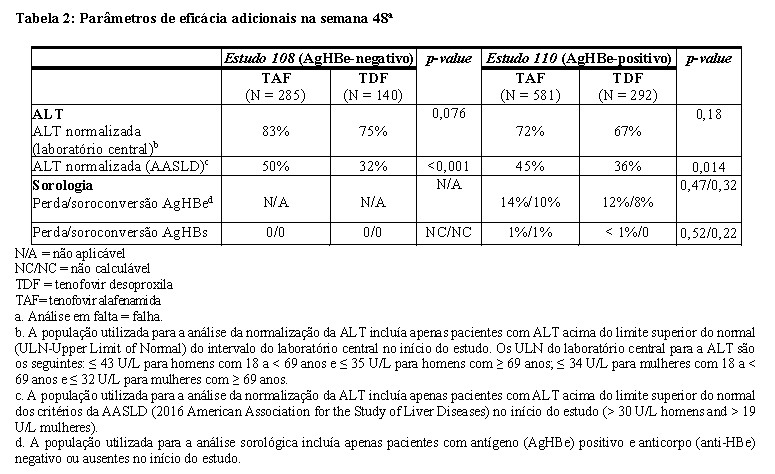
Experiência após 48 semanas no Estudo 108 e no Estudo 110
Na semana 96, manteve-se a supressão viral, bem como as respostas bioquímicas e sorológicas com a continuação do tratamento com tenofovir alafenamida (ver Tabela 3).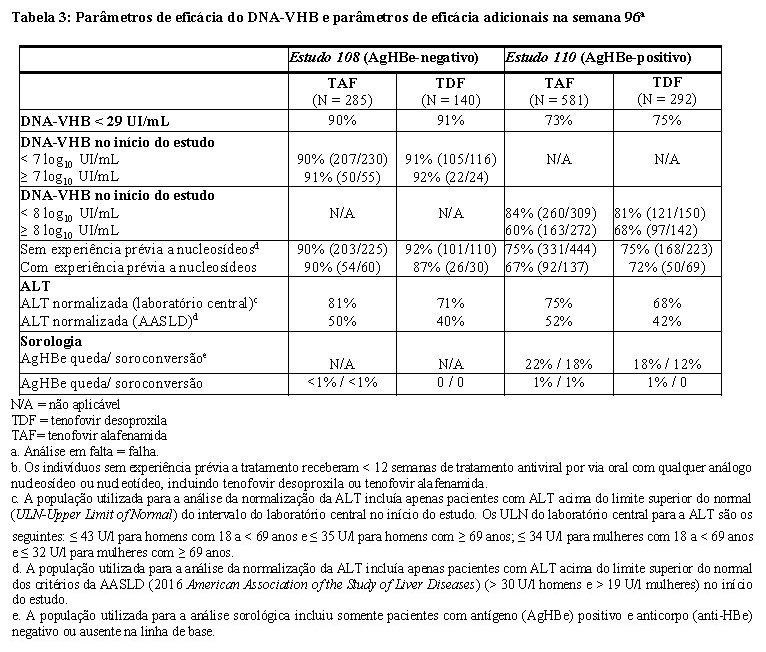
Alterações na determinação da densidade mineral óssea nos estudos 108 e 110
Em ambos os estudos, o tenofovir alafenamida foi associado a menores percentagens médias de redução da densidade mineral óssea (DMO; determinada por absorciometria por duplo feixe de raio X [DXA] do quadril e da coluna lombar) em comparação com tenofovir desoproxila após 96 semanas de tratamento (p < 0,001). Em pacientes que permaneceram em tratamento cego além da semana 96, a variação percentual média da DMO, em cada grupo na 144 semana foi similar àquela da semana 96. Na fase aberta de ambos os estudos, a variação percentual média da DMO da semana 96 para a semana 144 nos pacientes que permaneceram com tenofovir alafenamida foi +0,4% na coluna lombar e -0,3% no quadril, em comparação com +2,0% na coluna lombar e +0,9% no quadril naqueles que trocaram de tenofovir desoproxila para tenofovir alafenamida na semana 96.
Alterações na determinação da função renal nos estudos 108 e 110
Em ambos os estudos, o tenofovir alafenamida foi associado a menores alterações dos parâmetros de segurança renal (menores reduções mediana da ClCr estimada por Cockcroft-Gault e menores percentagens mediana de aumento da razão entre a ligação da proteína retinol na urina e a creatinina e a razão entre a beta-2-microglobulina na urina e a creatinina) em comparação com tenofovir desoproxila após 96 semanas de tratamento (p < 0,001) (ver também item 5. Advertências e Precauções). Em pacientes que permaneceram em tratamento cego além da semana 96 nos Estudos 108 e 110, a alteração na linha de base nos valores de parâmetro laboratorial renal em cada grupo na semana 144 foram similares aqueles na semana 96. Na fase aberta dos Estudos 108 e 110, a alteração de média (±SD) de creatinina sérica da semana 96 para a semana 144 foi de +0,002 (0,0924) mg/dL naqueles que permaneceram com tenofovir alafenamida, em comparação com -0,018 (0,0691) mg/dL com aqueles que trocaram de tenofovir desoproxila para tenofovir alafenamida na semana 96. Na fase aberta, a alteração mediana de eGFR da semana 96 para a semana 144 foi -1,2 mL/min nos pacientes que permaneceram com tenofovir alafenamida, comparado à +4,2 mL/min nos pacientes que trocaram de tenofovir desoproxila para tenofovir alafenamida na semana 96.
Alterações nos exames laboratoriais lipídicos nos estudos 108 e 110
Para pacientes que mudaram para tenofovir alafenamida na fase aberta do estudo na semana 96, as alterações no valor basal na fase duplo-cega para os pacientes randomizados inicialmente para tenofovir alafenamida e tenofovir desoproxila nas semanas 96 e semana 144 em colesterol total, colesterol HDL, colesterol HDL, colesterol LDL, triglicérides e a relação colesterol total/HDL são apresentadas na Tabela 4.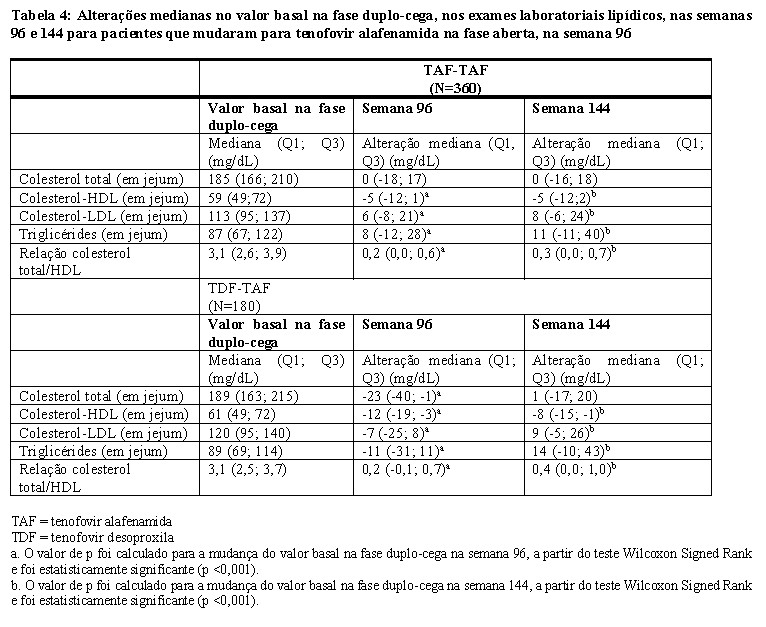
Pacientes adultos virologicamente suprimidos no Estudo 4018
A eficácia e segurança do Vemlidy® em adultos virologicamente suprimidos com hepatite B crônica é baseada em dados de 48 semanas de um estudo randomizado, duplo-cego e ativo-controlado, Estudo 4018 (N=243 em tenofovir alafenamida; N=245 em tenofovir desoproxila), incluindo dados de pacientes que participaram da fase em regime aberto do Estudo 4018 da semana 48 até a semana 96 (N=235 permaneceram tratados com tenofovir alafenamida [TAF-TAF]; N= 237 ou mudaram de tenofovir desoproxila para tenofovir alafenamida na semana 48 [TDF-TAF]).
No estudo 4018, foram incluídos adultos virologicamente suprimidos com hepatite B crônica (N = 488) que haviam sido previamente mantidos em 245 mg de tenofovir desoproxila uma vez ao dia, por pelo menos 12 meses, com DNA do VHB < limite inferior de quantificação (LLOQ) por avaliação laboratorial local pelo menos 12 semanas antes da triagem e HBV DNA < 20 UI/mL na triagem. Os pacientes foram estratificados pela situação de AgHBe (AgHBe positivo ou AgHBe negativo) e idade (≥ 50 ou < 50 anos) e randomizados em uma proporção de 1: 1 para mudar para 25 mg de tenofovir alafenamida (N = 243) ou permanecer em 245 mg tenofovir desoproxila uma vez ao dia (N = 245). A idade média foi de 51 anos (22% eram ≥ 60 anos), 71% eram homens, 82% eram asiáticos, 14% eram brancos e 68% eram negativos para AgHBe. No início do estudo, a duração média do tratamento prévio com tenofovir desoproxila foi de 220 e 224 semanas nos grupos tenofovir alafenamida e tenofovir desoproxila, respectivamente. O tratamento anterior com antivirais também incluiu interferon (N = 63), lamivudina (N = 191), adefovir dipivoxila (N = 185), entecavir (N = 99), telbivudina (N = 48) ou outro (N = 23). No início, a ALT sérica média foi de 27 U/L, a TFGe média por Cockcroft-Gault foi de 90,5 mL/min; 16% dos pacientes tinham histórico de cirrose.
O desfecho primário da eficácia foi a proporção de pacientes com níveis plasmáticos de DNA HBV ≥ 20 UI/mL na semana 48 (conforme determinado pelo algoritmo Snapshot da FDA EUA modificado). Os parâmetros de eficácia adicionais incluíram a proporção de pacientes com níveis de DNA do HBV < 20 UI/mL, normalização da ALT e normalização da ALT, perda e soroconversão de AgHBs e perda e soroconversão de AgHBe. O tenofovir alafenamida não foi inferior na proporção de indivíduos com DNA do VHB ≥ 20 UI / mL na semana 48, quando comparado ao tenofovir desoproxila, conforme avaliado pelo algoritmo Snapshot da FDA EUA modificado. Os resultados do tratamento (DNA do HBV < 20 UI / mL por falta = falha) na semana 48 entre os grupos de tratamento foram semelhantes entre os subgrupos por idade, sexo, raça, status basal do AgHBe e ALT.
Os resultados do tratamento do Estudo 4018 na semana 48 e na semana 96 são apresentados na Tabela 5 e na Tabela 6.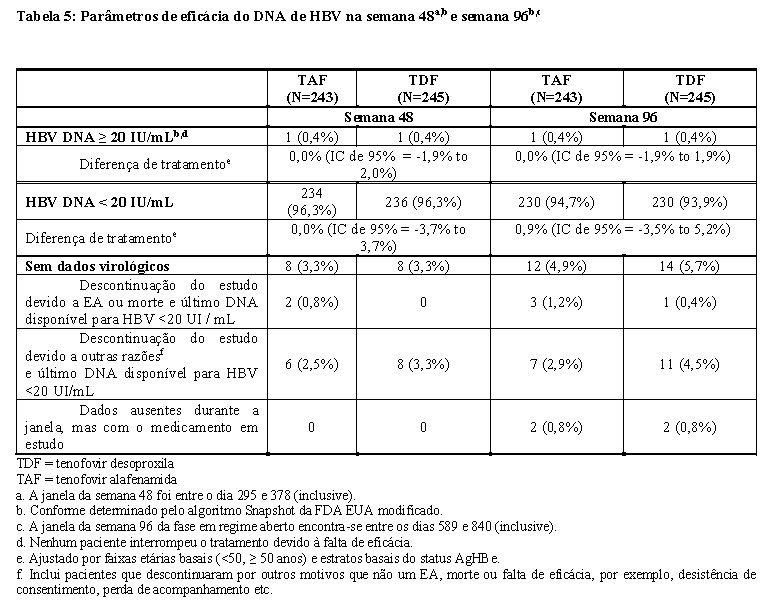
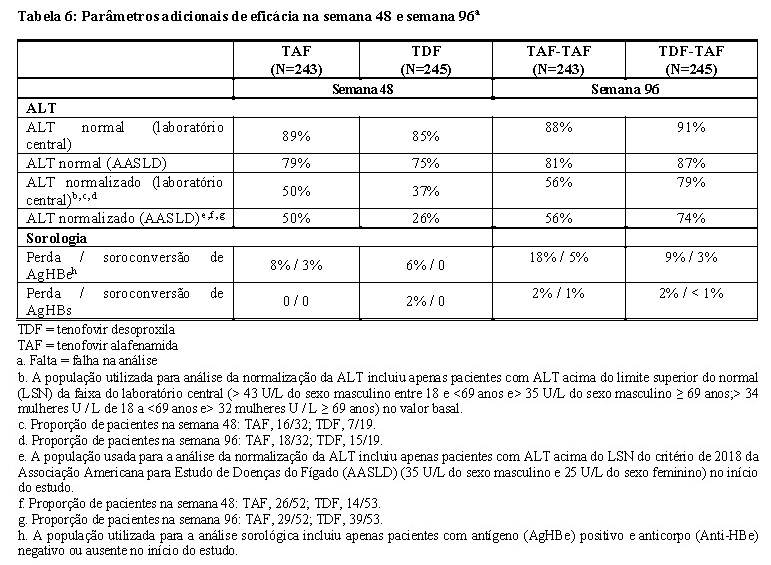
Alterações na densidade mineral óssea no Estudo 4018
A alteração percentual média na DMO no valor basal para a Semana 48, avaliada pelo DXA, foi de + 1,7% com tenofovir alafenamida em comparação com -0,1% com tenofovir desoproxila na coluna lombar e + 0,7% em comparação com -0,5% no quadril total. Declínios de DMO acima de 3% na coluna lombar foram observados por 4% dos pacientes com tenofovir alafenamida e 17% dos pacientes com tenofovir desoproxila na semana 48. Declínios de DMO superior a 3% no quadril total foram observados por 2% de pacientes com tenofovir alafenamida e 12% dos pacientes com tenofovir desoproxila na semana 48.
Na fase em regime aberto, a alteração percentual média na DMO do valor basal até a Semana 96 em pacientes que permaneceram tratados com tenofovir alafenamida foi de +2,3% na coluna lombar e +1,2% no quadril total, comparado à +1,7% na coluna lombar e +0,2% no quadril total naqueles que trocaram de tenofovir desoproxila para tenofovir alafenamida na semana 48.
Alterações nos exames laboratoriais renais no Estudo 4018
A mudança mediana no valor basal para a Semana 48 no TFGe pelo método de Cockcroft-Gault foi de +2,2 mL por minuto no grupo tenofovir alafenamida e -1,7 mL por minuto naqueles que receberam tenofovir desoproxila. Na semana 48, houve um aumento mediano em relação ao valor basal na creatinina sérica entre os pacientes randomizados para continuar o tratamento com tenofovir desoproxila (0,01 mg/dL) em comparação com uma dimunuição mediana no valor basal entre os pacientes que foram alterados para tenofovir alafenamida (0,01 mg/dL)
Na fase em regime aberto do estudo, a alteração mediana na TFGe, desde o valor basal até a Semana 96 foi 1,6 mL/min nos pacientes que permaneceram com tenofovir alafenamida, comparado à +0,5 mL/min em pacientes que mudaram do tenofovir desoproxila para o tenofovir alafenamida na semana 48. A mudança mediana em relação ao valor basal para a semana 96 na creatinina sérica foi de -0,02 mg/dL naqueles que permaneceram com tenofovir alafenamida, comparado à ?0,01 mg/dL naqueles que alteraram de tenofovir desoproxila para o tenofovir alafenamida na semana 48.
Alterações nos exames laboratoriais lipídicos no Estudo 4018
Alterações desde o valor basal em duplo-cego até a Semana 48 e Semana 96 em colesterol total, colesterol HDL, colesterol LDL, triglicérides e relação colesterol total/HDL tenofovir desoproxila são apresentados na Tabela 7.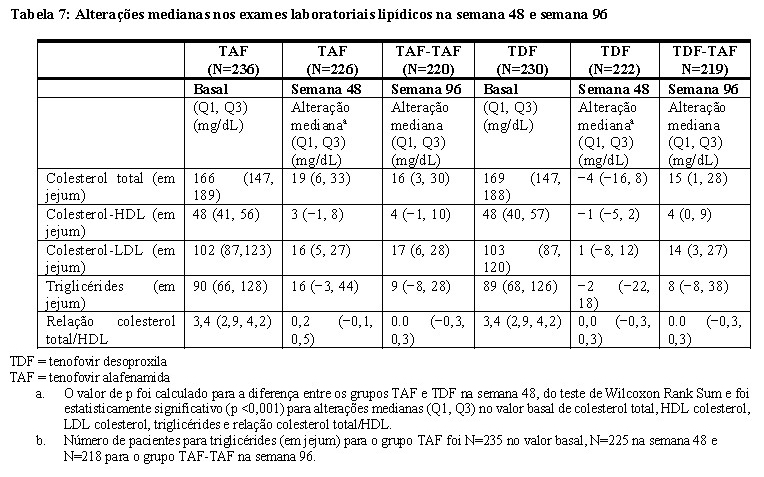
Referências
1 Buti M, Gane E, Seto WK, Chan HL, Chuang W-L, Stepanova T, et al. Tenofovir alafenamide tenofovir disoproxil fumarate vs. for the treatment of patients with HBeAg-negative chronic hepatitis B virus infection: a randomised, double-blind, phase 3, non-inferiority trial. Lancet Gastroenterol Hepatol 2016;1:196-206.
2 Chan HL, Fung S, Seto WK, Chuang WL, Chen CY, Kim HJ, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate for the treatment of HBeAg-positive chronic hepatitis B virus infection: a randomised, double-blind, phase 3, noninferiority trial. Lancet Gastroenterol Hepatol 2016;1:185-195.
3 Agarwal K, Brunetto M, Seto WK, et al. 96 weeks treatment of Tenofovir alafenamide vs. tenofovir disoproxil fumarate for hepatitis B virus infection. J Hepatol. 2018;68:672-681.
4 Lampertico P, Buti M, Fung S, Ahn SH, Chuang WL, Tak WY, et al. Switching from tenofovir disoproxil fumarate to tenofovir alafenamide in virologically suppressed patients with chronic hepatitis B: a randomised, double-blind, phase 3, multicentre non inferiority study. Lancet Gastroenterol Hepatol, 2020;5(5):441-453.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades farmacodinâmicas
Grupo farmacoterapêutico: Antivirais para uso sistêmico, inibidores nucleosídeos e nucleotídeos da transcriptase reversa, código ATC: J05AF13.
Mecanismo de ação
O tenofovir alafenamida é um pró-fármaco fosfonoamidato do tenofovir (análogo 2'-deoxiadenosina monofosfato). O tenofovir alafenamida penetra nos hepatócitos primários através de difusão passiva e dos transportadores hepáticos OATP1B1 e OATP1B3. O tenofovir alafenamida é em primeiramente hidrolisado para formar o tenofovir pela carboxilesterase-1 nos hepatócitos primários. O tenofovir intracelular é subsequentemente fosforilado dando origem ao metabólito farmacologicamente ativo tenofovir difosfato. O tenofovir difosfato inibe a replicação do VHB por incorporação no DNA viral através da transcriptase reversa do VHB, o que resulta na terminação da cadeia de DNA.
O tenofovir apresenta atividade específica contra o vírus da hepatite B e o vírus da imunodeficiência humana (HIV-1 e HIV-2). O tenofovir difosfato é um inibidor fraco das polimerases do DNA dos mamíferos que incluem a polimerase c do DNA mitocondrial, não existindo evidência de toxicidade mitocondrial in vitro com base em diversos ensaios que incluíram análises de DNA mitocondrial.
Atividade antiviral
A atividade antiviral do tenofovir alafenamida contra um painel de isolados clínicos do VHB representando os genótipos A-H foi avaliada em células HepG2. Os valores da CE 50 (concentração eficaz a 50%) para o tenofovir alafenamida encontravam-se no intervalo de 34,7 a 134,4 nM, com uma CE 50 média global de 86,6 nM. A CC50 (concentração citotóxica a 50%) em células HepG2 foi > 44.400 nM.
Resistência
Em pacientes recebendo tenofovir alafenamida, foi realizada uma análise sequencial em isolados de VHB emparelhados no início do estudo e durante o tratamento de pacientes que apresentaram recidiva virológica (2 consultas consecutivas com DNA-VHB ≥ 69 UI/mL depois de terem apresentado < 69 UI/mL, ou 1,0 log10 ou mais de DNA-VHB acima do nadir) ou pacientes com DNA-VHB ≥ 69 UI/mL na semana 48 ou semana 96 ou na descontinuação precoce na semana 24 ou após.
Na análise agrupada de pacientes recebendo tenofovir alafenamida nos Estudos 108 e 110, na semana 48 (n=20) e na semana 96 (n=72), não foram identificadas substituições de aminoácidos associadas à resistência à tenofovir alafenamida nesses isolados (análise genotípica e fenotípica).
Em pacientes virologicamente suprimidos que receberam tenofovir alafenamida após a troca do tratamento com tenofovir desoproxila no Estudo 4018, durante 96 semanas do tratamento com tenofovir alafenamida um paciente do grupo TAFTAF apresentou viremia detectável transitória (uma consulta com HBV DNA ≥ 69 UI/mL) e um paciente do grupo TDFTAF apresentou uma recidiva virológica. Não foram detectadas quaisquer substituições de aminoácidos do VHB associadas a resistência a TAF ou TDF durante 96 semanas de tratamento.
Resistência cruzada
A atividade antiviral de tenofovir alafenamida foi avaliada contra um painel de isolados contendo mutações associadas a resistência a inibidores nucleosídeos e nucleotídeos da transcriptase reversa em células HepG2. Os isolados de VHB que expressavam as substituições de rtV173L, rtL180M e rtM204V/I associadas à resistência à lamivudina permaneceram com sensibilidade ao tenofovir alafenamida (alteração < 2 vezes na CE50). Os isolados de VHB que expressavam as substituições de rtL180M, rtM204V mais rtT184G, rtS202G ou rtM250V associadas à resistência ao entecavir permaneceram com sensibilidade ao tenofovir alafenamida. Os isolados de VHB que expressavam as substituições individuais de rtA181T, rtA181V ou rtN236T associadas à resistência ao adefovir permaneceram com sensibilidade ao tenofovir alafenamida; no entanto, o isolado de VHB expressando rtA181V mais rtN236T apresentou sensibilidade reduzida ao tenofovir alafenamida (alteração de 3,7 vezes na CE50). A relevância clínica destas mutações é desconhecida.
Propriedades farmacocinéticas
Absorção
Após administração oral de tenofovir alafenamida, em jejum, em pacientes adultos com hepatite B crônica, observaram-se concentrações plasmáticas máximas de tenofovir alafenamida aproximadamente 0,48 horas após a administração. Com base na análise farmacocinética populacional de Fase 3 em indivíduos com hepatite B crônica, a AUC0-24 média no estado estacionário do tenofovir alafenamida (N = 698) e do tenofovir (N = 856) foi respetivamente de 0,22 mg•hr/mL e 0,32 mg•hr/mL. As Cmax no estado estacionário do tenofovir alafenamida e do tenofovir foram, respetivamente, 0,18 e 0,02 mg /mL. Em relação ao estado de jejum, a administração de uma dose única de tenofovir alafenamida com uma refeição com teor alto em gordura resultou num aumento de 65% da exposição ao tenofovir alafenamida.
Distribuição
A ligação do tenofovir alafenamida às proteínas plasmáticas humanas, em amostras recolhidas durante os ensaios clínicos, foi de aproximadamente 80%. A ligação do tenofovir às proteínas plasmáticas humanas foi inferior a 0,7% e é independente da concentração no intervalo 0,01 a 25 mg/mL.
Biotransformação
O metabolismo é uma importante via de eliminação do tenofovir alafenamida no ser humano, sendo responsável por > 80% de uma dose oral. Estudos in vitro demonstraram que o tenofovir alafenamida é metabolizado, dando origem ao tenofovir (metabolito principal) pela carboxilesterase-1 nos hepatócitos e pela catepsina A nas CsMSP e nos macrófagos. In vivo,o tenofovir alafenamida é hidrolisado nas células de modo a formar tenofovir (metabólito principal), o qual é fosforilado dando origem ao metabólito ativo tenofovir difosfato.
In vitro, o tenofovir alafenamida não é metabolizado pelo CYP1A2, CYP2C8, CYP2C9, CYP2C19, ou CYP2D6. O tenofovir alafenamida é minimamente metabolizado pelo CYP3A4.
Eliminação
A excreção renal do tenofovir alafenamida intacto é uma via menor, em que < 1% da dose é eliminada na urina. O tenofovir alafenamida é eliminado principalmente após o metabolismo de tenofovir. O tenofovir alafenamida e o tenofovir têm uma meia-vida plasmática mediana de 0,51 e 32,37 horas, respetivamente. O tenofovir é eliminado do organismo pelos rins, tanto por filtração glomerular como por secreção tubular ativa.
Linearidade/não linearidade
As exposições ao tenofovir alafenamida são proporcionais à dose num intervalo de doses de 8 a 125 mg.
Farmacocinética em populações especiais
Idade, gênero e etnia
Não foram identificadas diferenças clinicamente relevantes na farmacocinética de acordo com a idade ou etnia. As diferenças na farmacocinética de acordo com o gênero não foram consideradas clinicamente relevantes.
Insuficiência hepática
Em pacientes com insuficiência hepática grave, as concentrações plasmáticas totais de tenofovir alafenamida e tenofovir são inferiores às observadas em indivíduos com função hepática normal. Quando corrigidas para a ligação às proteínas, as concentrações plasmáticas não ligadas (livres) de tenofovir alafenamida na insuficiência hepática grave e na função hepática normal são semelhantes.
Insuficiência renal
Não se observaram diferenças clinicamente relevantes na farmacocinética do tenofovir alafenamida ou tenofovir entre indivíduos saudáveis e indivíduos com insuficiência renal grave (ClCr estimada > 15 mas < 30 ml/min) em estudos com o tenofovir alafenamida.
Dados de segurança pré-clínica
Os dados não clínicos em ratos e cães revelaram que o osso e o rim são os órgãos alvo primários de toxicidade. A toxicidade óssea foi observada como redução da DMO em ratos e cães, com exposições de tenofovir pelo menos quatro vezes superiores às que são esperadas após a administração de tenofovir alafenamida. Observou-se a presença de uma infiltração mínima de histiócitos no olho em cães, com exposições de tenofovir alafenamida e tenofovir aproximadamente 4 e 17 vezes superiores, respectivamente, aquelas esperadas após a administração de tenofovir alafenamida.
O tenofovir alafenamida não foi mutagênico nem clastogênico em estudos convencionais de genotoxicidade.
Apenas foram realizados estudos de carcinogenicidade e um estudo peri/pós-natal em ratos com o tenofovir desoproxila, uma vez que a exposição de tenofovir é menor em ratos e camundongos após a administração de tenofovir alafenamida, em comparação com o tenofovir desoproxila. Não se demonstraram riscos especiais para o ser humano segundo estudos convencionais de potencial carcinogênico com tenofovir desoproxila (como fumarato), e de toxicidade reprodutiva e de desenvolvimento com tenofovir desoproxila (como fumarato) ou tenofovir alafenamida. Os estudos de toxicidade reprodutiva em ratos e coelhos não demonstraram alterações nos parâmetros de acasalamento, fertilidade, gravidez ou nos parâmetros fetais. No entanto, o tenofovir desoproxila reduziu o índice de viabilidade e o peso das crias num estudo de toxicidade peri/pós-natal com doses tóxicas maternas. Um estudo a longo prazo de carcinogenicidade de administração por via oral em camundongos demonstrou uma baixa incidência de tumores do duodeno, considerados provavelmente relacionados com as altas concentrações locais no trato gastrointestinal na dose elevada de 600 mg/kg/dia. O mecanismo de formação dos tumores nos camundongos e a potencial relevância para o ser humano são incertos.
4. CONTRAINDICAÇÕES
Este medicamento é contraindicado em pacientes com histórico de hipersensibilidade à substância ativa ou a qualquer um dos excipientes listados no item Composição.
5. ADVERTÊNCIAS E PRECAUÇÕES
Transmissão de VHB
Os pacientes devem ser advertidos que Vemlidy® não previne o risco de transmissão de VHB a outras pessoas, através do contato sexual ou contaminação com sangue. As precauções adequadas devem continuar sendo utilizadas.
Pacientes com doença hepática descompensada
A segurança e eficácia de Vemlidy® em pacientes infectados pelo VHB com doença hepática descompensada (Child-Pugh-Turcotte (CPT) classe B ou C) não foram estabelecidas. Portanto, Vemlidy® não é recomendado para pacientes com doença hepática descompensada (CPT Classe B ou C).
Exacerbação da hepatite B
Exacerbações durante o tratamento
As exacerbações espontâneas na hepatite B crônica são relativamente frequentes e caracterizam-se por aumentos transitórios da alanina aminotransferase (ALT) sérica. Após o início da terapêutica antiviral, os níveis séricos de ALT podem aumentar em alguns pacientes. Em pacientes com doença hepática compensada, estes aumentos da ALT sérica não são geralmente acompanhados de aumento da bilirrubina sérica ou descompensação hepática. Os pacientes com cirrose podem estar em maior risco de descompensação hepática após exacerbação da hepatite, e devem ser cuidadosamente monitorados durante o tratamento.
Exacerbações após interrupção do tratamento
Tem sido notificada exacerbação aguda da hepatite, em pacientes que interromperam o tratamento da hepatite B, normalmente em associação com um aumento dos níveis plasmáticos de DNA-VHB. A maioria dos casos é autolimitada, mas podem ocorrer exacerbações graves, inclusive resultados fatais, após a descontinuação do tratamento da hepatite B. A função hepática deve ser monitorada em intervalos regulares, com acompanhamento clínico e laboratorial durante pelo menos 6 meses após interrupção do tratamento da hepatite B. Se apropriado, pode justificar-se o recomeço da terapêutica da hepatite B.
Em pacientes com doença hepática avançada ou cirrose, a interrupção do tratamento não é recomendada uma vez que a exacerbação da hepatite após a interrupção do tratamento pode levar a descompensação hepática. As exacerbações hepáticas são particularmente graves, e por vezes fatais em pacientes com doença hepática descompensada.
Insuficiência renal
Pacientes com depuração de creatinina < 30 mL/min
A utilização de Vemlidy® uma vez por dia em pacientes com ClCr ≥ 15 mL/min mas < 30 mL/min e em pacientes com ClCr < 15 mL/min que estejam fazendo hemodiálise baseia-se em dados farmacocinéticos bastante limitados e em modelos e simulações. Não existem dados de segurança sobre a utilização de Vemlidy® para o tratamento de pacientes infectados pelo VHB com ClCr < 30 mL/min.
A utilização de Vemlidy® não é recomendada em pacientes com ClCr < 15 mL/min que não estejam fazendo hemodiálise (ver item 8. Posologia e modo de usar).
Nefrotoxicidade
Não se pode excluir um risco potencial de nefrotoxicidade resultante da exposição crônica a níveis baixos de tenofovir resultantes da administração de tenofovir alafenamida (ver item 3. Características farmacológicas).
Pacientes coinfectados pelo VHB e pelo vírus da hepatite C ou D
Não existem dados sobre a segurança e eficácia de Vemlidy® em pacientes coinfectados pelo vírus da hepatite C ou D. Devem ser seguidas as orientações sobre a coadministração para o tratamento da hepatite C (ver item 6. Interações medicamentosas).
Hepatite B e coinfecção pelo HIV
Devem ser disponibilizados testes anti-HIV a todos os pacientes infectados pelo VHB cujo estado de infecção por HIV-1 seja desconhecido antes de iniciarem o tratamento com Vemlidy®. Nos pacientes coinfectados pelo VHB e HIV, Vemlidy® deve ser coadministrado com outros antirretrovirais para garantir que o paciente receba um regime apropriado para o tratamento do HIV (ver item 6. Interações medicamentosas).
Coadministração com outros medicamentos
Vemlidy® não deve ser coadministrado com medicamentos contendo tenofovir alafenamida, tenofovir desoproxila ou adefovir dipivoxila.
A coadministração de Vemlidy® com determinados anticonvulsivantes (por ex., carbamazepina, oxcarbazepina, fenobarbital e fenitoína), antimicobacterianos (por ex., rifampicina, rifabutina e rifapentina) ou Erva-de-São João (Hypericum perforatum) não é recomendada, pois são indutores da glicoproteína P (gpP) e podem reduzir as concentrações plasmáticas de tenofovir alafenamida.
A coadministração de Vemlidy® com inibidores fortes da gpP (por ex., itraconazol e cetoconazol) pode aumentar as concentrações plasmáticas de tenofovir alafenamida. A coadministração não é recomendada.
Intolerância à lactose
Vemlidy® contém lactose monoidratada. Pacientes com problemas hereditários raros de intolerância à galactose, deficiência total de lactase ou má absorção de glucose-galactose não devem tomar este medicamento.
Este medicamento contém LACTOSE.
Uso em Populações Especiais
Gravidez
A quantidade de dados sobre a utilização de tenofovir alafenamida em mulheres grávidas é limitada (menos de 300 grávidas expostas) ou inexistente. Contudo, uma quantidade elevada de dados em mulheres grávidas (mais de 1.000 grávidas expostas) indica ausência de malformações ou toxicidade fetal/neonatal com tenofovir desoproxila.
Os estudos em animais não indicam efeitos nocivos diretos ou indiretos em relação à toxicidade reprodutiva (ver item 3. Características farmacológicas).
A utilização de tenofovir alafenamida pode ser considerada durante a gravidez, se necessário.
Categoria B de risco na gravidez: Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Amamentação
Desconhece-se se o tenofovir alafenamida é excretado no leite humano. No entanto, em estudos em animais demonstrouse que o tenofovir é excretado no leite. Existe informação insuficiente sobre os efeitos de tenofovir em recémnascidos/lactentes.
Não pode ser excluído qualquer risco para o lactente, portanto, tenofovir alafenamida não deve ser utilizado durante a amamentação.
Fertilidade
Não estão disponíveis dados no ser humano sobre o efeito de tenofovir alafenamida na fertilidade. Os estudos em animais não indicam efeitos nocivos do tenofovir alafenamida sobre a fertilidade.
Uso pediátrico
A segurança e eficácia de Vemlidy® em pacientes abaixo de 18 anos não foram estabelecidas.
Efeitos sobre a capacidade de conduzir e utilizar máquinas
Os efeitos de Vemlidy® sobre a capacidade de conduzir e utilizar máquinas são nulos ou desprezáveis. Os pacientes devem ser informados de que foram notificadas tonturas durante o tratamento com tenofovir alafenamida.
6. INTERAÇÕES MEDICAMENTOSAS
Os estudos de interação medicamentosa só foram realizados em adultos.
Vemlidy® não deve ser coadministrado com medicamentos contendo tenofovir desoproxila, tenofovir alafenamida ou adefovir dipivoxila.
Medicamentos que podem afetar o tenofovir alafenamida
O tenofovir alafenamida é transportado pela glicoproteína P (gpP) e pela proteína de resistência ao câncer de mama (BCRP). Prevê-se que os medicamentos que são indutores da gpP (por ex., rifampicina, rifabutina, carbamazepina, fenobarbital ou Erva-de-São-João (Hypericum perforatum)) diminuam as concentrações plasmáticas do tenofovir alafenamida, o que pode levar à perda do efeito terapêutico de Vemlidy®. A coadministração destes medicamentos com Vemlidy® não é recomendada.
A coadministração de tenofovir alafenamida com medicamentos que inibem a gpP e/ou a BCRP pode aumentar as concentrações plasmáticas do tenofovir alafenamida. A coadministração de inibidores fortes da gpP com tenofovir alafenamida não é recomendada.
O tenofovir alafenamida é um substrato do OATP1B1 e do OATP1B3 in vitro. A distribuição do tenofovir alafenamida no organismo pode ser afetada pela atividade do OATP1B1 e/ou do OATP1B3.
Efeito do tenofovir alafenamida em outros medicamentos
O tenofovir alafenamida não é um inibidor do CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 ou CYP2D6 in vitro. Não é inibidor do CYP3A in vivo.
O tenofovir alafenamida não é um inibidor da uridina difosfato glucuronosiltransferase (UGT) 1A1 humana in vitro. Não se sabe se o tenofovir alafenamida é inibidor de outras enzimas UGT.
A informação sobre as interações medicamentosas entre Vemlidy® e potenciais medicamentos concomitantes está resumida na Tabela 8 abaixo (um aumento é indicado como "↑", uma diminuição como "↓", sem alteração como "↔"; duas vezes por dia como "b.i.d.", dose única como "s.d.", uma vez por dia como "q.d."; e via intravenosa como "IV"). As interações medicamentosas descritas baseiam-se em estudos realizados com tenofovir alafenamida ou são interações medicamentosas potenciais que podem ocorrer com Vemlidy®.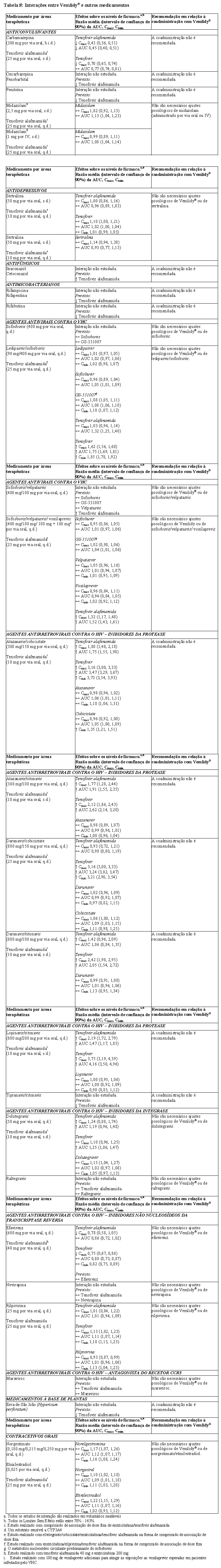
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Vemlidy® é um comprimido revestido amarelo, redondo, com a inscrição "GSI" de um lado e "25" do outro. Cada frasco contém 30 comprimidos, um dissecante de sílica gel, um chumaço de poliéster e é fechado com uma tampa com trava de segurança para crianças.
Armazenar em temperatura ambiente, entre 15°C e 30 °C. Proteger da umidade.
Dispensar apenas na embalagem original.
Não utilizar se o lacre estiver rompido ou quebrado ou faltando.
Prazo de validade: 36 meses após a data de fabricação impressa na embalagem.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
O tratamento deve ser iniciado por um médico com experiência no tratamento da hepatite B crônica.
Posologia
Adultos: Um comprimido uma vez por dia com alimento.
Dose esquecida
Se um paciente se esquecer de uma dose e tiverem passado menos de 18 horas após o horário de costume, o paciente deve tomar Vemlidy® logo que possível e continuar com o seu esquema posológico normal. Se tiverem passado mais de 18 horas após o horário de costume, o paciente não deve tomar a dose esquecida e deve continuar simplesmente com o esquema posológico normal.
Se o paciente vomitar no espaço de 1 hora após tomar Vemlidy®, deve tomar outro comprimido. Se o paciente vomitar mais de 1 hora após tomar Vemlidy®, não é necessário tomar outro comprimido.
Populações especiais
Idosos
Não é necessário ajuste posológico de Vemlidy® em pacientes com idade igual ou superior a 65 anos (ver Item 3. Características farmacológicas).
Insuficiência renal
Não é necessário ajuste posológico de Vemlidy® em pacientes com uma depuração de creatinina (ClCr) estimada ≥ 15 ml/min ou em pacientes com ClCr < 15 ml/min que estejam atualmente fazendo hemodiálise.
Nos dias da hemodiálise, Vemlidy® deve ser administrado após a conclusão do tratamento de hemodiálise (ver Item 3. Características farmacológicas).
Não é possível efetuar recomendações posológicas em pacientes com ClCr < 15 ml/min que não estejam fazendo hemodiálise (ver item 5. Advertências e precauções).
Insuficiência hepática
Não é necessário ajuste posológico de Vemlidy® em pacientes com insuficiência hepática leve (Child Pugh Turcotte [CPT] A). Vemlidy® não é recomendado para pacientes com insuficiência hepática descompensada (CPT B ou C).
População pediátrica
A segurança e eficácia de Vemlidy® em pacientes com menos de 18 anos de idade ainda não foram estabelecidas.
Modo de administração Via oral. Vemlidy® comprimido revestido deve ser tomado com alimentos.
Este medicamento não deve ser partido ou mastigado.
9. REAÇÕES ADVERSAS
A exacerbação aguda grave da Hepatite B é discutida no item "5. Advertências e Precauções" desta bula.
Resumo do perfil de segurança
A avaliação das reações adversas baseia-se nos dados de segurança agrupados, de 2 estudos controlados de Fase 3 (GS US-320-0108 e GS-US-320-0110; "Estudo 108" e "Estudo 110", respectivamente), nos quais 866 pacientes virêmicos infectados pelo VHB, com níveis séricos elevados de ALT, receberam 25 mg de tenofovir alafenamida,uma vez por dia, de forma duplo-cega durante 96 semanas (duração média da exposição ao medicamento no estudo cego em 104 semanas). As reações adversas notificadas mais frequentemente foram cefaleias (12%), náuseas (6%) e fadiga (6%). Após a Semana 96, os pacientes permaneceram em seu tratamento cego original ou receberam tenofovir alafenamida de forma aberta. Alterações nos exames laboratoriais lipídicos foram observadas no "Estudo 108" e no "Estudo 110". Não foram identificadas reações adversas adicionais ao tenofovir alafenamida desde a Semana 96 até a Semana 144 na fase duplo-cega e no subconjunto de pacientes que receberam tratamento com tenofovir alafenamida de forma aberta (ver item 3.Características farmacológicas).
Em um estudo duplo-cego, randomizado e ativo-controlado (GS-US-320-4018; "Estudo 4018") em indivíduos virologicamente suprimidos que trocaram tenofovir desoproxila para tenofovir alafenamida 25 mg (N = 243), foram observadas alterações nos exames laboratoriais lipídicos.
Resumo tabelar das reações adversas
Foram identificadas as seguintes reações adversas medicamentosas com tenofovir alafenamida em pacientes com hepatite B crônica (Tabela 9). As reações adversas estão indicadas abaixo por classes de sistemas de órgãos e frequência com base na análise da semana 96. As frequências são definidas como se segue: muito comum (≥ 1/10), comum (≥ 1/100, < 1/10), incomum (≥ 1/1.000, < 1/100), raros (≥ 1/10.000, < 1/ 1.000) ou muito raros ( < 1/10.000).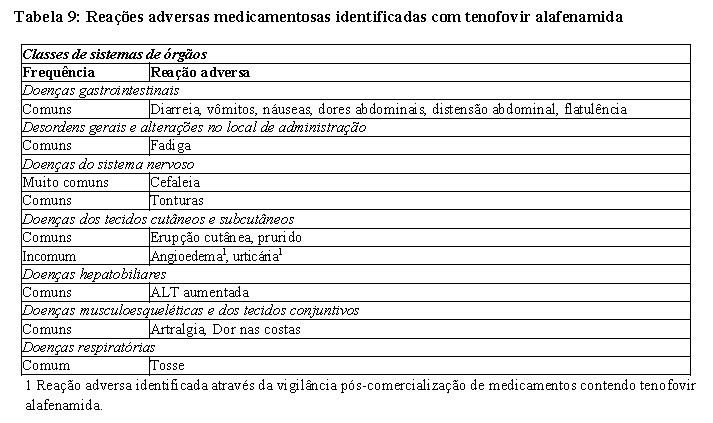
Alterações nos exames laboratoriais lipídicos
Numa análise conjunta dos Estudos 108 e 110, foram observadas alterações medianas nos parâmetros lipídicos em jejum, no valor basal, à Semana 96 em ambos os grupos de tratamento. No grupo tenofovir alafenamida, foram observadas reduções na mediana de colesterol total e HDL em jejum e aumentos na mediana de LDL direto e triglicérides em jejum, enquanto o grupo tenofovir desoproxila demonstrou reduções medianas em todos os parâmetros (vide Tabela 4). Em pacientes randomizados inicialmente para receber tenofovir alafenamida e trocados por tenofovir alafenamida em estudo aberto na semana 96, as alterações medianas (Q1; Q3) no valor basal na fase duplo-cega para a semana 144 foi a seguinte (mg/dL): colesterol total foi 0 (-16; 18); LDL foi 8 (-6; 24); HDL foi de -5 (-12; 2); triglicérides foram 11 (-11; 40); a relação colesterol total/HDL foi de 0,3 (0,0; 0,7). Em pacientes randomizados inicialmente para tenofovir desoproxila e trocados por tenofovir alafenamida em estudo aberto na Semana 96, as alterações medianas (Q1; Q3) no valor basal na fase duplo-cega par