TYKERB
NOVARTIS
lapatinibe
Inibidor da tirosina-quinase.
Apresentações.
Comprimidos revestidos de 250 mg em embalagens com 70 comprimidos.
VIA ORAL
USO ADULTO
Composição.
Cada comprimido revestido contém 250 mg de lapatinibe equivalente a 405 mg de ditosilato de lapatinibe monohidratado. Excipientes: celulose microcristalina, povidona, amidoglicolato de sódio, estearato de magnésio, hipromelose, dióxido de titânio, macrogol, polissorbato 80, óxido de ferro amarelo, óxido de ferro vermelho
Informações técnicas.
1. INDICAÇÕES
Câncer de mama metastático com superexpressão do HER2
Tykerb®, em combinação com capecitabina, é indicado no tratamento de pacientes com câncer de mama, avançado ou metastático, cujos tumores apresentem superexpressão da proteína HER2/neu (ErbB2) e que tenham progredido com terapia prévia inclusive com trastuzumabe, em tumores com metástase (ver Estudos Clínicos, em Resultados de Eficácia).
Tykerb®, em combinação com trastuzumabe, é indicado para o tratamento de pacientes com câncer de mama metastático negativo para receptores de hormônios, cujos tumores superexpressem HER2/neu (ErB2) e que tenham progredido em terapia prévia com trastuzumabe em combinação com quimioterapia, em tumores com metástase (ver Estudos Clínicos, em Resultados de Eficácia).
Câncer de mama metastático hormônio sensível Tykerb®, em combinação com letrozol, é indicado para mulheres na pós-menopausa, com câncer de mama avançado ou metastático positivo para receptores de hormônios, cujos tumores superexpressem HER2/neu (ErbB2) e para as quais a terapia hormonal é recomendada.
2. RESULTADOS DE EFICÁCIA
Estudos Clínicos
Dados de dois estudos randomizados em cenário metastático (EGF111438 (CEREBREL) e EGF108919 (COMPLETE)) mostraram que Tykerb® em combinação com quimioterapia é menos efetivo que trastuzumabe combinado a quimioterapia.
A combinação de Tykerb® com trastuzumab foi avaliada no estudo clínico randomizado EGF104900 e demonstrou eficácia superior versus Tykerb® sozinho em pacientes com câncer de mama metastático que progrediram em um regime anterior contendo trastuzumabe.
Tykerb® também foi estudado em combinação com letrozol e teve eficácia superior versus letrozol sozinho em pacientes com câncer de mama HER2-positivo, hormônio receptor-positivo avançado ou metastático.
Veja abaixo para detalhes.
O Tykerb® não é indicado em cenário de adjuvância.
Tratamento combinado com Tykerb® e capecitabina
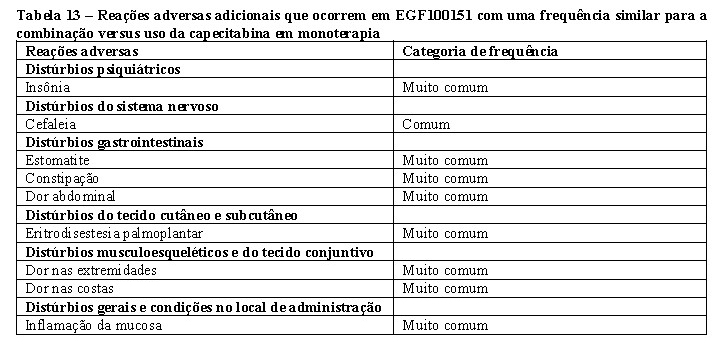
Estudo EGF100151 A eficácia e a segurança de Tykerb® em combinação com capecitabina no tratamento de câncer de mama foram avaliadas em um estudo EGF100151 randomizado de Fase III. Participaram do estudo pacientes com câncer de mama metastático ou localmente avançado, com superexpressão de ErbB2 (IHC 3+ ou IHC 2+ e FISH positivo) e em progressão após tratamento anterior com taxanos, antraciclinas e trastuzumabe. A fração de ejeção do ventrículo esquerdo (FEVE) foi avaliada em todas as pacientes por meio de ecocardiograma [ECG] ou de cintilografia de perfusão do miocárdio (MUGA, na sigla em inglês) antes do início do tratamento com Tykerb® para assegurar que a FEVE basal estivesse dentro dos limites normais. Em estudos clínicos, a FEVE foi monitorizada em intervalos de aproximadamente oito semanas durante o tratamento com lapatinibe para assegurar que não fique abaixo da fração para níveis menores que o limite inferior de normalidade. Observou-se a maioria dos casos de declínio da FEVE ( > 60% dos eventos) durante as primeiras nove semanas de tratamento, mas os dados disponíveis sobre a exposição a longo prazo eram limitados.
A distribuição das pacientes foi aleatória para receber Tykerb® em regime de 1.250 mg uma vez por dia (continuamente) em combinação com capecitabina (2.000 mg/m2/dia nos dias 1 a 14 a cada 21 dias) ou somente capecitabina (2.500 mg/m2/dia nos dias 1 a 14 a cada 21 dias). O tratamento do estudo foi administrado até a progressão da doença ou abandono do paciente por alguma outra razão. O objetivo primário (primary endpoint) foi o Tempo Para Progressão (TTP, na sigla em inglês) da doença, e os resultados abaixo se basearam na revisão conduzida por um comitê de revisão independente.
Os resultados da data de corte de 03 de Abril de 2006 (a data em que o recrutamento para o estudo foi encerrado), mostrou um aumento no TTP para pacientes tratados com Tykerb® em combinação com capecitabina (representando uma redução de 43% no risco de progressão da doença ou morte devido ao câncer de mama versus monoterapia com capecitabina, conforme avaliado pelo comitê de revisão independente). Ver Tabela 01.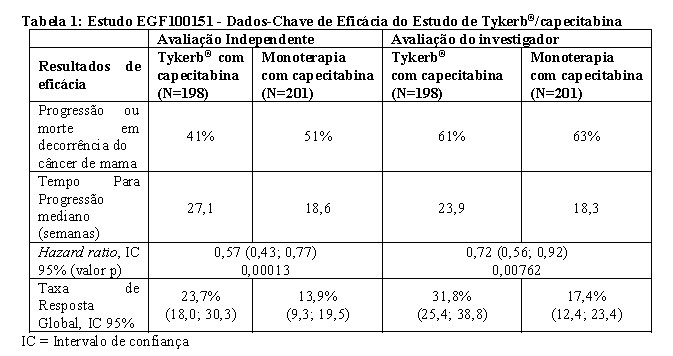
A taxa de resposta global avaliada pelo comitê de revisão independente foi de 23,7% para pacientes que receberam Tykerb® em combinação com capecitabina e 13.9% para pacientes que receberam capecitabina. A duração de resposta mediana foi de 32,1 e 30,6 semanas para Tykerb®, respectivamente.
No grupo de tratamento combinado houve 4 (2%) progressões da doença no sistema nervoso central, em comparação a 13 (6%) progressões observadas no grupo tratado com capecitabina em monoterapia, conforme avaliado pelo comitê de revisão independente (ver Efeito do lapatinibe em metástase no SNC, em Resultados de Eficácia).
Na ocasião em que o recrutamento foi encerrado para o estudo (03 de abril de 2006), 399 pacientes foram randomizados para inclusão no estudo e outros 9 pacientes estavam sendo submetidos à triagem. O tratamento combinado foi oferecido a 9 pacientes em triagem e a todos aqueles que já estavam recebendo monoterapia com capecitabina. No total, 207 pacientes foram designadas para o tratamento combinado e 201 pacientes para a monoterapia com capecitabina.
Um resumo da análise dos dados de sobrevida até 1° de Outubro é exposto na Tabela 2.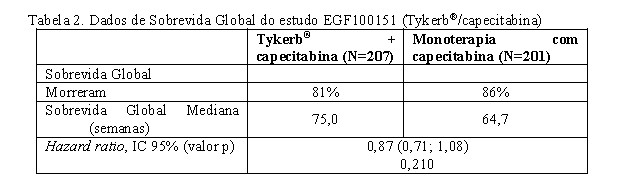
Depois que o estudo foi encerrado, 36 pacientes foram transferidos do tratamento com capecitabina para o tratamento com lapatinibe + capecitabina. Desses pacientes, 26 foram transferidos antes da progressão da doença, enquanto recebiam a monoterapia com capecitabina. Para isolar o efeito do tratamento em presença de cross-over, foi conduzida uma análise de regressão de Cox, considerando o cross-over como uma covariada dependente do tempo e o efeito do tratamento. Os resultados dessa análise indicam uma redução clinicamente relevante de 20% no risco de morte, com um Hazard ratio de 0.80 [intervalo de confiança (IC) de 95%: 0,64; 0,99; p=0,043].
Estudo EGF111438 (CEREBEL)
Este Estudo de Fase III randomizado (EGF111438) (N=540) comparou o efeito de Tykerb® em combinação com capecitabina em relação à trastuzumabe em combinação com capecitabina na incidência de SNC como local da primeira recidiva em mulheres com câncer de mama metastático com superexpressão do HER2. As pacientes foram randomizadas para receber Tykerb® 1250 mg uma vez ao dia (de forma contínua) mais capecitabina (2000 mg/m2/dia nos dias 1-14 a cada 21 dias) ou trastuzumabe (dose de ataque de 8 mg/kg seguida por infusões de 6 mg/kg a cada 3 semanas) mais capecitabina (2500 mg/m2/dia, nos dias 1-14, a cada 21 dias). A randomização foi estratificada por tratamento anterior com trastuzumabe e número de tratamentos anteriores para doença metastática (nenhum versus ≥1ª linha). O estudo foi interrompido quando uma análise interina pré-planejada (N=475) revelou eficácia superior do braço de trastuzumabe mais capecitabina e baixa incidência de eventos do SNC.
A análise final confirmou que os resultados do desfecho primário foram inconclusivos devido ao baixo número de eventos de SNC [8 pacientes (3,2%) no braço de Tykerb® mais capecitabina apresentaram metástase no SNC como local da primeira progressão versus 12 pacientes (4,8%) no braço de trastuzumabe mais capecitabina] (ver Efeito do Tykerb® em metástase no SNC, em Resultados de Eficácia). Os resultados finais da sobrevida livre de progressão e sobrevida global são apresentados na tabela 3. A análise final confirmou a eficácia superior do braço de trastuzumabe mais capecitabina.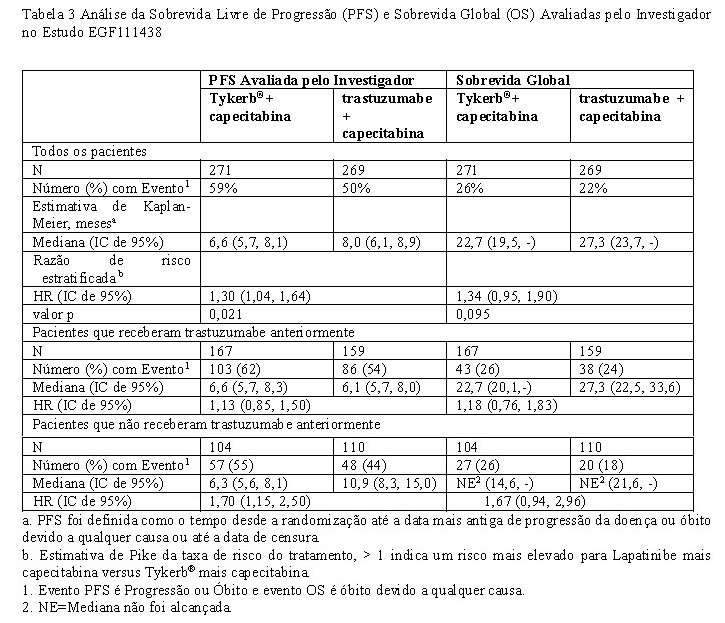
Efeito do Tykerb® em metástase no SNC
Em termos de resposta objetiva, a monoterapia com Tykerb® demonstrou atividade mínima no tratamento de metástases no SNC bem estabelecidas. Tykerb® não é recomendado para a prevenção de metástases no SNC.
1. A Phase III, Randomized, Open-Label, Multicenter Study Comparing GW572016 and Capecitabine (Xeloda) versus Capecitabine in Women with Refractory Advanced or Metastatic Breast Cancer. Study EGF100151. Report UM2004/00001/00.
Tratamento combinado com Tykerb® e trastuzumabe
Estudo EGF104900
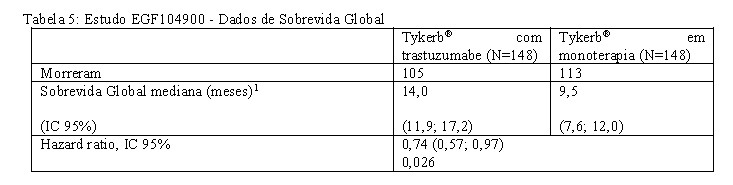
A eficácia e a segurança de Tykerb® em combinação com trastuzumabe em câncer de mama metastático foram avaliadas no estudo randomizado EGF104900. As pacientes elegíveis eram mulheres com câncer de mama metastático em estágio IV, com amplificação do gene ErbB2 (ou com superexpressão de proteína), expostas a tratamento com antraciclinas ou taxanos. De acordo com o protocolo, os investigadores deviam atestar que as pacientes mostraram progressão no esquema de tratamento mais recente contendo trastuzumabe em condições metastáticas. O número mediano de esquemas de tratamento anteriores contendo trastuzumabe em condições de metástase foram três. As pacientes foram randomizadas para receber Tykerb® 1000 mg por via oral uma vez ao dia com trastuzumabe 4 mg/kg, administrado como uma dose de ataque intravenosa (IV), seguida de 2 mg/kg IV semanalmente (N=148), ou Tykerb® 1500 mg por via oral uma vez ao dia (N=148). As pacientes com progressão objetiva da doença após pelo menos 4 semanas com Tykerb® como monoterapia eram elegíveis para passar para o tratamento combinado. Das 148 pacientes que foram tratadas com a monoterapia, 77 (52%) escolheram receber o tratamento combinado na ocasião da progressão da doença. O objetivo primário desse estudo foi avaliar e comparar a Sobrevida Livre de Progressão (PFS) em pacientes com câncer de mama metastático tratadas com Tykerb® e trastuzumabe, em comparação com a monoterapia com Tykerb®. Outros objetivos secundários foram avaliar e comparar os dois grupos de tratamento com relação à Sobrevida Global (OS), à Taxa de Resposta Tumoral Global (ORR), à Taxa Benefício Clínico (CBR) e ao Tempo Para Resposta. A média de idade foi de 51 anos, e 13% das pacientes tinham 65 anos ou mais. 94% das pacientes eram brancas. A maioria das pacientes nos dois grupos de tratamento tinha doença visceral [215 (73%) pacientes, no total]. Metade das pacientes na população do estudo mostrava receptor de estrógeno negativo e receptor de progesterona negativo [150 (51%) no total]. Um resumo dos pontos de avaliação de eficácia é apresentado na Tabela 4, e os dados de sobrevida total são apresentados na Tabela 5. Resultados da análise de subgrupos com base no fator de estratificação pré-definido (status do receptor hormonal) são apresentados na Tabela 06.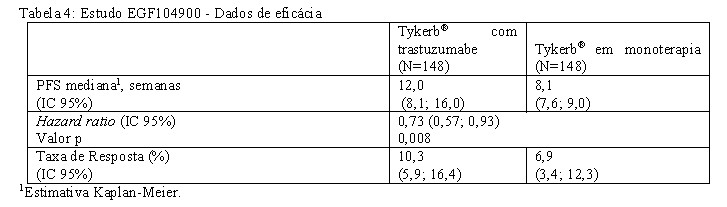

Tratamento combinado com Tykerb® e inibidor de aromatase -letrozol
Estudo EGF30008
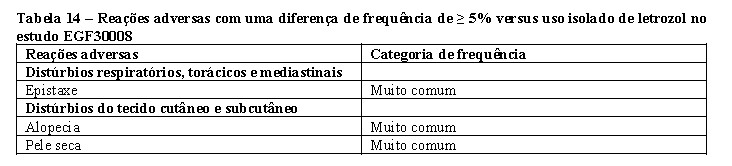
Tykerb® foi estudado em combinação com letrozol para o tratamento de câncer de mama avançado ou metastático em mulheres pós-menopáusicas positivas para receptores de hormônios (positivas para receptores de estrogênio [ER] e/ou positivas para receptores de progesterona [PgR]).
O estudo EGF30008 foi um estudo randomizado, duplo-cego e controlado em pacientes com câncer de mama (CM) localmente avançado ou metastático positivo para receptores de hormônios (RH+), que não haviam recebido tratamento sistêmico anterior para a doença metastática. Mil duzentas e oitenta e seis pacientes foram randomizadas para receber letrozol 2,5 mg uma vez ao dia combinado a Tykerb® 1500 mg uma vez ao dia (N=642), ou letrozol mais placebo (N=644). A randomização foi estratificada por locais da doença e terapia adjuvante antiestrogênio anterior. O status do receptor HER2 foi determinado de maneira retrospectiva por testes conduzidos em laboratório central. De todas as pacientes randomizadas para tratamento, 219 tinham tumores que apresentavam superexpressão do receptor HER2 que foi a população primária previamente especificada para a análise da eficácia.
Na população positiva para HER2, a Sobrevida Livre de Progressão da doença determinada pelo investigador foi significativamente mais alta com letrozol combinado com Tykerb® do que com letrozol combinado com placebo (ver Tabela 7).
O benefício de Tykerb® com letrozol na Sobrevida Livre de Progressão da doença na população positiva para HER2 foi confirmado em uma análise de regressão de Cox previamente planejada (HR=0,65 [IC 95%: 0,47; 0,89]; p=0,008). Alémdo benefício da Sobrevida LivredeProgressão dadoençaobservado nessapopulação, o tratamento combinado com Tykerb® e letrozol melhorou no Objetivo de Taxa de Resposta (27,9% e 14,8%, respectivamente) e na Taxa de Benefício Clínico (47,7% e 28,7%, respectivamente), em comparação com a monoterapia com letrozol. No momento da análise final da Sobrevida Livre de Progressão (com período de acompanhamento médio de 2,64 anos), os dados de Sobrevida Global não eram maduros e não houve diferença significativa entre os grupos de tratamento na população HER2-positiva. Isto não alterou com o acompanhamento adicional (tempo médio de acompanhamento de > 7,5 anos; Tabela 8).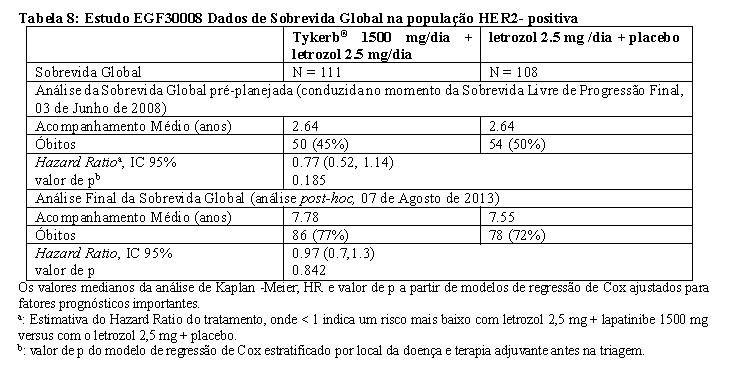
Estudo LAP016A2307 -EGF114299 (ALTERNATIVE) -Qualquer inibidor de aromatase
A eficácia e segurança de Tykerb® em combinação com um inibidor de aromatase foram confirmadas em um estudo de Fase III. Os pacientes inscritos eram mulheres na pós-menopausa que tinham câncer de mama metastático HR-positivo/HER2-positivo, que havia progredido após regime de quimioterapia contendo trastuzumabe e terapias endócrinas. O estudo foi projetado principalmente para avaliar um benefício potencial de PFS do bloqueio duplo de HER2 (Tykerb® + trastuzumabe) versus bloqueio único de HER2 (Tykerb® ou trastuzumabe). Todos os 3 braços do estudo continham um inibidor de aromatase (IA). Um total de 355 pacientes foram randomizados em uma proporção de 1:1:1 para Tykerb® 1000 mg + trastuzumabe (dose de ataque: 8 mg/kg; dose de manutenção: 6 mg/kg IV a cada 3 semanas) + IA (N = 120), ou trastuzumabe (dose de ataque: 8 mg/kg; dose de manutenção: 6 mg/kg IV a cada 3 semanas) + IA (N=117), ou Tykerb® 1500 mg + IA (N=118). O endpoint primário foi PFS com base na avaliação radiológica local comparando Tykerb® + trastuzumabe + IA versus trastuzumabe + IA. O estudo atingiu seu objetivo principal, demonstrando uma redução de risco estatisticamente significativa e clinicamente significativa de 38% na PFS e um prolongamento de 5,4 meses da PFS mediana em favor do tratamento com Tykerb® + trastuzumabe + IA. A PFS mediana (IC 95%) foi de 11,0 meses (8,3, 13,8) para Tykerb® + trastuzumabe + IA e 5,6 meses (5,4, 8,3) para trastuzumabe + IA (ver Tabela 9).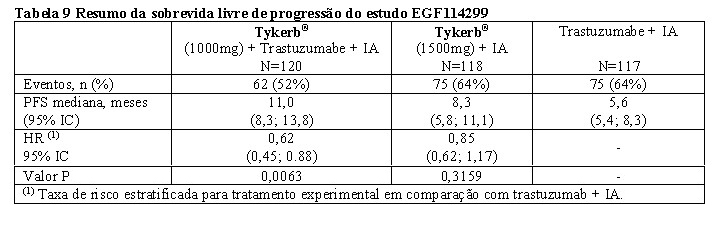
Neste estudo, o perfil de segurança dos grupos de tratamento foi consistente com a segurança conhecida desses agentes.
Referências Bibliográficas
Clinical Overview. COMPLETE and CEREBEL, clinical studies final results, EGF111438, EGF108919. GSK, 2013 (v17-EGF111438-EGF108919-FR, COMPLETE).
Clinical Overview. Without lapatinib + paclitaxel indication D2012-6157, GSK, 2012.
Clinical Overview. With lapatinib + paclitaxel indication, D2012-6158, GSK, 2012.
Clinical Overview. Lapatinib in combination with capecitabine, GSK, 2006 (v1-2.5 Seq000). Summary of Clinical Efficacy. GSK, 2006 v1-2.7.3 Seq000). [Study EGF100151]. Specific obligation SO-01: Report of an updated analysis of survival to 01-Oct-2008 for Study EGF100151. GSK, November 2008 (v7-D2008-6163, Seq020).
Clinical Overview. Clinical studies (TTP and OS EGF100151). D2009-5293. GSK, 26-Aug-2009. (v7-D20095293).
Clinical Overview. Clinical studies (CNS metastases), GSK, 2015.
Clinical Overview. GSK, 2012 (v13-2.5 Seq098).
Summary of Clinical Pharmacology. GSK, 2012 (v13-2.7.2 Seq098).
Summary of Clinical Efficacy. GSK, 2012 (v13-2.7.3 Seq098). [Study EGF104900] A Randomized, Multicenter, Open-Label, Phase III Study of Lapatinib in Combination with Trastuzumab versus Lapatinib Monotherapy in Subjects with Metastatic Breast Cancer whose Disease has Progressed on a Trastuzumab-Containing Regimen. GSK, 2-Feb-2012 (v13 EGF104900/2011N114550_00, Seq098).
Clinical Overview. 'Clinical studies (subgroup analysis)', GSK, 2013.
Clinical Overview. GSK, 2009 (v6-2.5 Seq026). Summary of Clinical Pharmacology. GSK, 2009 (v6-2.7.2 Seq026)]. Summary of Clinical Efficacy. GSK, 2009 (v6-2.7.3 Seq026).
[Study EGF30008] A Randomized, Double-Blind, Placebo-Controlled, Multicenter, Phase III Study Comparing Lapatinib and Letrozole versus Letrozole in Subjects with Estrogen/Progesterone Receptor-Positive Advanced or Metastatic Breast Cancer. GSK, 20-Feb-2009 (v6-EGF30008/ YM2007/00007/00, Seq026). Clinical Overview. Clinical studies (AIs on OS-DFS), GSK, 2015. Summary of Clinical Efficacy. Clinical studies GSK, 2014 (v-18-2.7.3 Seq143).
Clinical Overview in hormone receptor positive, HER2-positive metastatic breast cancer in postmenopausal women who have received prior trastuzumab and endocrine therapies (Final analysis of the primary efficacy endpoint Study EGF114299) / LAP016A2307). Novartis. 06-Dec-2018.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Grupo farmacoterapêutico:
Receptor de fator de crescimento epidérmico humano 2 (HER)2 inibidor de tirosina quinase (código ATC: L01EH01)
Propriedades Farmacodinâmicas
Mecanismo de Ação
O lapatinibe é um novo inibidor da quinase 4-anilinoquinazolina. Tem mecanismo de ação peculiar, pois representa um inibidor potente, reversível e seletivo dos domínios da tirosina quinase dos receptores EGFR (ErbB1) e de HER2/neu (ErbB2) (valores Kiapp estimados de 3nM e 13nM respectivamente), com dissociação lenta desses receptores (meia-vida ≥300 minutos). Demonstrou-se que esse índice de dissociação é mais lento que os de outros inibidores da quinase 4-anilinoquinazolina estudados. O lapatinibe inibe, in vitro e em vários modelos animais, o crescimento de células tumorais orientado pelos receptores ErbB. Além de sua atividade como agente único, demonstrou-se um efeito adicional em estudo in vitro quando lapatinibe e 5-FU (o metabólito ativo da capecitabina) foram usados em combinação nas quatro linhagens de células tumorais testadas. O significado clínico desses dados in vitro ainda é desconhecido. A combinação de Tykerb® e trastuzumabe pode oferecer mecanismos de ação complementares e possíveis mecanismos de resistência que não se sobrepõem. Os efeitos de inibição de crescimento demonstrados pelo lapatinibe foram avaliados em linhagens celulares pré-condicionadas com trastuzumabe. O lapatinibe demonstrou atividade significativa in vitro contra linhagens celulares de câncer de mama com amplificação de HER2 selecionadas para crescimento de longo prazo em meio que continha trastuzumabe e mostrou sinergia em combinação com trastuzumabe nessas linhagens celulares. Essas descobertas sugerem ausência de resistência cruzada entre esses dois agentes específicos para HER2/neu (ErbB2).
Células de câncer de mama sensíveis a hormônios (positivas para receptores de estrogênio [ER] e/ou positivas para receptores de progesterona [PgR]) que coexpressam ErbB2 tendem a tornar-se resistentes a tratamentos endócrinos estabelecidos. As células de câncer de mama sensíveis a hormônios que inicialmente não contêm ErbB1 e ErbB2 regularão para cima esses receptores à medida que o tumor se tornar resistente ao tratamento endócrino. Estudos randomizados em câncer de mama metastático sensível a hormônios indicam que um inibidor de tirosina quinase de ErbB1 ou ErbB2 potencialmente aumenta a eficácia clínica, quando acrescentado ao tratamento endócrino.
Efeitos Farmacodinâmicos
Eletrofisiologia cardíaca
Prolongamento QT
Estudo EGF114271: O efeito de Tykerb® no intervalo QT foi avaliado em um estudo único, controlado com placebo, com sequência única (tratamento com placebo e substância ativa) e com cross-over em pacientes com tumor sólido avançado (n=58). Durante o período de quatro dias de tratamento, três doses de placebo combinadas foram administradas com 12 horas de intervalo na manhã e na noite do dia 01 e na manhã do dia 02. Estas doses foram seguidas por três doses de Tykerb® 2000 mg administradas do mesmo modo. Foram realizadas medições, incluindo ECGs e amostras farmacocinéticas na linha de base e nos mesmos momentos no dia 02 e no dia 04. Na população avaliável (n=37), a média máxima do prolongamento do intervalo QT corrigido pelo método de Fridericia (ddQTcF) (IC de 90%) de 8.75 ms (4,08; 13,42) foi observada 10 horas após a ingestão da terceira dose de Tykerb®de 2000 mg. O ddQTcF excedeu o limiar de 5 ms e o limite superior ICs de 90% excedeu o limiar de 10 ms em múltiplos pontos. Os resultados para a população farmacodinâmica (n=52) foi consistente com aqueles da população avaliável [ddQTcF máximo (IC de 90%) de 7,91 ms (4,13; 11,68)] observado 10 horas após a ingestão da terceira dose de lapatinibe. A análise da farmacocinética/farmacodinâmica confirmou uma relação
positiva entre a concentração plasmática de lapatinibe a ddQTcF.
Propriedades Farmacocinéticas
Absorção
Após a administração oral do lapatinibe, a absorção é incompleta e variável (coeficiente de variação de aproximadamente 50% a 100% da área sob a curva (AUC, na sigla em inglês)). As concentrações séricas aparecem após intervalo médio de 0,25 hora (faixa de 0 a 1,5 hora). As concentrações máximas no plasma (Cmáx) são atingidas cerca de 4 horas após a administração. A dosagem diária de 1.250 mg produz média geométrica estável (intervalo de confiança de 95%) dos valores de Cmáx de 2,43 (de 1,57 a 3,77) mg/mL e dos valores de AUC de 36,2 (de 23,4 a 56,0) mg.h/mL. A exposição sistêmica ao lapatinibe aumenta quando a substância é administrada às refeições (ver Posologia e Modo de usar e Interações Medicamentosas). Os valores de AUC são cerca de três a quatro vezes mais altos (Cmáx aproximadamente 2,5 a 3 vezes mais alta) quando o medicamento é administrado com refeições de baixo teor de gordura (5% [500 calorias]) ou de alto teor (50% [1.000 calorias]) respectivamente.
Distribuição
O lapatinibe apresenta forte ligação ( > 99%) com a albumina e com a glicoproteína ácida alfa1. Estudos in vitro indicam que ele representa um substrato para as proteínas transportadoras BCRP (ABCG1) e a glicoproteína P (ABCB1). Olapatinibetambémdemonstrou inibiraPgp (IC502.3mg/mL), BCRP(IC500.014 mg/mL), assimcomo
o transportador de captação hepática OATP1B1(IC50 2.3mg/mL), in vitro, em concentrações clinicamente relevantes. O significado clínico desses efeitos sobre a farmacocinética de outros medicamentos ou sobre a atividade farmacológica de outros agentes antineoplásicos ainda é desconhecido. O lapatinibe não inibe significativamente o transportador renal OAT ou OCT (valores de IC50 in vitro foram ≥ 6.9mg/mL).
Biotransformação/Metabolismo
O lapatinibe sofre metabolismo extenso, principalmente pelas enzimas CYP3A4 e CYP3A5, com contribuições menores de CYP2C19 e CYP2C8, para vários metabólitos oxidados, nenhum dos quais é responsável por mais de 14% da dose recuperada nas fezes nem por mais de 10% da concentração da substância no plasma.
Eliminação
A meia-vida do lapatinibe medida após doses únicas aumenta conforme a elevação das doses. Entretanto, a dosagem diária de Tykerb®resulta em um estado de equilíbrio dentro de seis a sete dias, o que indica meia-vida efetiva cerca de 1 dia. O lapatinibe é eliminado predominantemente por meio do metabolismo efetuado pela enzima CYP3A4/5. A via primária de eliminação do lapatinibe e seus metabólitos são as fezes, e menos de 2% da dose (como lapatinibe e metabólitos) é excretada na urina. A recuperação dessa substância nas fezes representa a média de 27% (faixa de 3% a 67%) de uma dose oral do agente.
Avaliação in vitro do potencial de interação medicamentosa
O lapatinibe inibe a ação da CYP3A (Ki de 0,6 a 2,3 mg/mL) e da CYP2C8 (0,3 mg/mL) in vitro em concentrações clinicamente relevantes. Não demonstrou ação inibidora significativa das enzimas CYP1A2, CYP2C9, CYP2C19 e CYP2D6, existentes em microssomos do fígado humano, nem das enzimas UGT (os valores de IC50 in vitro se mostraram ≥ 6,9 mg/mL).
Populações Especiais
Pacientes pediátricos (abaixo de 18 anos) A farmacocinética de Tykerb® em pacientes pediátricos não foi estabelecida.
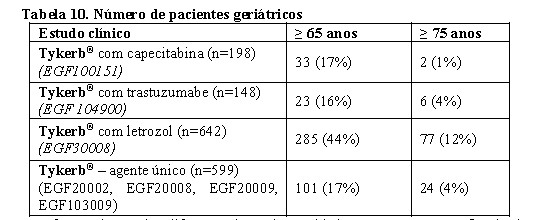
Pacientes geriátricos (65 anos ou mais)
A idade não parece afetar a farmacocinética do lapatinibe, com base na análise dos resultados dos estudos individuais. Um exame de dados combinados, abrangendo um intervalo de 18 a 82 anos, não sugere nenhum efeito.
Gênero
O sexo não parece afetar a farmacocinética do lapatinibe. Um exame de dados combinados, incluindo > 300 mulheres e > 450 homens, não sugere nenhuma diferença.
Raça/etnia
Os dados disponíveis do estudo não indicam distinção relacionada à raça/etnia.
Insuficiência renal
A farmacocinética do lapatinibe não foi estudada especificamente em pacientes com disfunção renal nem nos que se submetem a hemodiálise. Entretanto, é pouco provável que a disfunção renal afete a farmacocinética do lapatinibe, uma vez que menos de 2% da dose administrada (como lapatinibe inalterado e metabólitos) é eliminada pelos rins.
Insuficiência hepática
A farmacocinética do lapatinibe foi examinada em pacientes com disfunção hepática moderada (n=8) ou grave (n=4) e em oito pacientes sadios de controle. A exposição sistêmica (AUC) ao lapatinibe após uma única dose oral de 100 mg aumentou cerca de 56% e 85%, respectivamente, em pacientes com insuficiência hepática moderada e grave. Portanto, a administração de Tykerb® a pacientes com disfunção hepática requere cautela devido ao aumento da exposição ao fármaco. O médico deve reduzir a dose para pacientes com insuficiência hepática grave preexistente. Caso se desenvolva hepatotoxicidade grave durante o tratamento, Tykerb® deve ser descontinuado permanentemente (ver Posologia e Modo de Usar e Advertências e Precauções).
Farmacogenômica
Variações polimórficas nas enzimas metabolizadoras de fármacos, transportadores, receptores e outras proteínas que podem afetar a farmacocinética do lapatinibe não foram exploradas.
Os alelos HLA DQA1*02:01 e DRB1*07:01 foram associados à hepatotoxicidade em um subestudo genético de um estudo de monoterapia com Tykerb® (ver Advertências e precauções -Hepatotoxicidade).
4. CONTRAINDICAÇÕES
Tykerb® é contraindicado para pacientes com hipersensibilidade a qualquer componente da formulação (ver Reações Adversas).
Categoria D de risco na gravidez.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
5. ADVERTÊNCIAS E PRECAUÇÕES
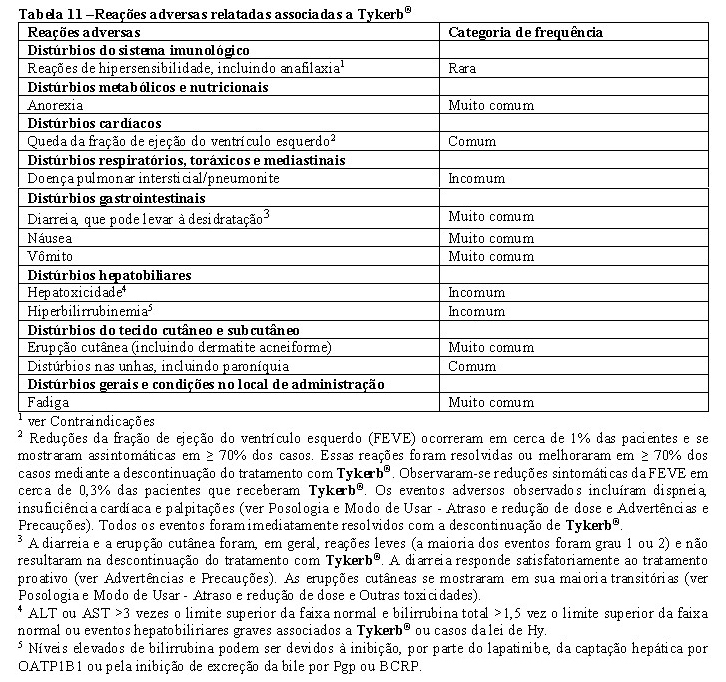
Toxicidade cardíaca: Tykerb® associa-se à redução da fração de ejeção do ventrículo esquerdo (FEVE) (ver Reações Adversas). Dessa forma, este medicamento deve ser administrado com cautela a pacientes que apresentem condições clínicas que possam prejudicar a função do ventrículo esquerdo. A FEVE deve ser avaliada em todos os pacientes antes de iniciar o tratamento com Tykerb® para assegurar que esteja dentro dos limites normais. A FEVE deve ser continuamente avaliada durante o uso de Tykerb® para assegurar que não caia a níveis inaceitáveis (ver Atraso e redução de dose e Reações cardíacas, em Posologia e Modo de Usar, e Estudos Clínicos, em Resultados de Eficácia). Em estudos conduzidos durante o programa de desenvolvimento clínico para Tykerb® , eventos cardíacos, incluindo reduções da FEVE, foram relatados em aproximadamente 1% das pacientes. Reduções sintomáticas da FEVE foram observadas em aproximadamente 0,3% das pacientes tratadas com Tykerb®. No entanto, quando Tykerb® foi administrado em combinação com trastuzumabe em condições de metástase, a incidência de eventos cardíacos, incluindo reduções da FEVE, foi mais alta (7%), quando comparada ao grupo tratado com Tykerb® como monoterapia (2%) no estudo principal. Os eventos cardíacos observados nesse estudo mostraram natureza e gravidade comparáveis àquelas observadas anteriormente com Tykerb® .
Uma elevação do intervalo QTc, concentração-dependente, foi observada em um estudo dedicado, controlado com placebo, com cross-over em pacientes com tumor sólido avançado. Devem ser tomadas precauções se Tykerb® for administrado a pacientes que tenham ou possam desenvolver prolongamento do intervalo QTc. Isso pode incluir pacientes com hipocalemia ou hipomagnesemia, síndrome congênita do QT longo e pacientes em uso de antiarrítmicos ou outros medicamentos que possam causar prolongamento do intervalo QT. Hipocalemia, hipocalcemia e hipomagnesemia devem ser tratados antes do início do tratamento com Tykerb® .
Doença pulmonar intersticial e pneumonite: Tykerb® associa-se também com doença pulmonar intersticial e pneumonite (ver Reações Adversas). As pacientes devem ser monitorizadas para detecção de sintomas pulmonares indicativos de doença pulmonar intersticial e pneumonite (ver Posologia e Modo de Usar).
Hepatotoxicidade: Observou-se hepatotoxicidade (ALT ou AST > 3 vezes o limite superior da faixa normal e bilirrubina total > 1,5 vez o limite superior da faixa normal) em estudos clínicos ( < 1% das pacientes) e na prática pós-comercialização. Hepatotoxicidade pode ser grave, e houve relatos de morte, embora a relação com Tykerb® seja incerta. Hepatotoxicidade pode ocorrer depois de dias a meses após o início do tratamento.
Os testes de função hepática (transaminases, bilirrubina e fosfatase alcalina) devem ser monitorizados antes do início do tratamento, a cada 4 a 6 semanas durante o tratamento e conforme indicação clínica. Se as alterações da função hepática forem graves, deve-se descontinuar permanentemente o tratamento com Tykerb® (ver Reações Adversas).
Pacientes apresentando os alelos HLA como DQA1*02:01 e DRB*07:01 possuem um maior risco de hepatotoxicidade associada ao uso de Tykerb® . Em um grande estudo clínico randomizado de Tykerb® em monoterapia (EGF114471; n=1.194), o risco global de dano hepático grave (ALT > 5 x o limite máximo normal, grau 3 CTCAE NCI) foi 2% (1:50), o risco nos portadores de DQA1*02:01 e DRB*07:01 foi de 8% (1:12) e o risco de não portadores foi de 0,5% (1:200). A presença dos alelos de risco HLA é comum (15 a 25%) em caucasianos, asiáticos, africanos e na população hispânica. No entanto, é baixa (1%) na população japonesa.
Se for necessário administrar Tykerb® a pacientes com insuficiência hepática grave, recomenda-se a redução da dose. Se houver desenvolvimento de hepatotoxicidade grave durante o tratamento, deve-se descontinuar permanentemente a administração de Tykerb® (ver Posologia e Modo de Usar; e Populações Especiais, em Propriedades Farmacocinéticas - Características Farmacológicas).
Diarreia: A diarreia induzida pelo uso de Tykerb® é normalmente de baixo grau, com diarreia grave de grau 3 ou 4 CTCAE NCI ocorrendo em < 10% e < 1% dos pacientes, respectivamente. A identificação e a intervenção precoces são essenciais no tratamento ideal da diarreia. O uso de Tykerb® tem sido associado à diarreia, incluindo diarreia grave (ver Reações Adversas). Diarreia pode ser grave e casos de óbito foram relatados. A diarreia normalmente ocorre no início do tratamento com Tykerb® e, em aproximadamente metade desses pacientes a diarreia inicia-se nos primeiros seis dias. Esta dura, geralmente, 4 a 5 dias. Os pacientes devem ser instruídos a relatar imediatamente qualquer alteração dos padrões intestinais. Recomenda-se o tratamento imediato da diarreia com antidiarreicos, como loperamida, depois da primeira evacuação sem formação de bolo fecal. O tratamento imediato dessa reação com antidiarreicos, como loperamida, após a primeira alteração nas fezes é recomendado. Quadros graves de diarreia podem exigir fluidos e eletrólitos orais ou intravenosos, antibióticos como fluoroquinolona (especialmente se a diarreia persistir por mais de 24 horas, com febre ou neutropenia grau 3 ou 4) ou interrupção ou descontinuação de Tykerb® (ver Atraso e Redução de dose e Outras toxicidades, em Posologia e Modo de Usar).
Reações cutâneas graves
Reações cutâneas graves foram relatadas com o uso de Tykerb®. Em caso de suspeita de eritema multiforme ou reações de risco à vida, como Síndrome de Stevens-Johnson ou Necrólise Epidermal Tóxica (rash cutâneo progressivo com bolha ou lesão mucosa frequente), o tratamento com Tykerb® deve ser descontinuado (ver Posologia e Modo de Usar).
Tratamento concomitante com inibidores ou indutores de CYP3A4: Coadministração de inibidores ou indutores de CYP3A4 requer cautela devido ao risco de aumento ou de redução da exposição ao Tykerb® , respectivamente (ver Interações Medicamentosas).
Populações especiais
Pacientes pediátricos (abaixo de 18 anos) A segurança e a eficácia de Tykerb® em pacientes pediátricos ainda não foram devidamente estabelecidas.
Pacientes geriátricos (65 anos ou mais)
Existem dados limitados sobre o uso de Tykerb® em pacientes com 65 anos ou mais (ver Posologia e Modo de Usar).
Farmacologia de segurança
Nenhum efeito neurológico, respiratório ou cardiovascular foi identificado em um painel de estudos farmacológicos in vitro ou em estudos em animais in vivo com lapatinibe.
Toxicidade de dose repetida
O lapatinibe foi avaliado em estudos de toxicidade de dose repetida por até 6 meses em ratos e até 9 meses em cachorros. Os efeitos principais relacionados ao tratamento foram inflamação e atrofia da pele e estruturas anexiais, degeneração e inflamação do trato gastrointestinal e órgãos digestivos acessórios (incluindo fígado), glândula mamáriaepróstata. Osefeitosforamobservadosemdosde≥60 mg/kg/diaemratose≥40 mg/jg/dia em cachorros. O NOAEL em ratos machos e fêmeas foi 60 mg/kg/dia e 10 mg/kg/dia, respectivamente, com AUC estimada de 24,7 microgramas.h/mL e 25,1 microgramas.h/mL, respectivamente. O NOAEL em cachorros machos e fêmeas foi 10 mg/kg/dia com AUC estimada de 5,4 microgramas.h/mL e 8,2 microgramas.h/mL, respectivamente. A exposição sistêmica correspondente nesses níveis de doses foram 0,5 e 0,6 vezes a exposição clínica humana para ratos machos e fêmeas, respectivamente, e 0,1 e 0,2 vezes a exposição clínica humana para cachorros machos e fêmeas, respectivamente.
Carcinogenicidade e mutagenicidade
Em estudos de carcinogenicidade oral com lapatinibe, lesões cutâneas graves foram observadas com as doses mais altas testadas (150 e 300 mg/kg/dia em camundongos macho e 300 mg/kg/dia em camundongos fêmea, e 500 mg/kg/dia em ratos macho e 300 mg/kg/dia em ratos fêmea). Comparado às doses em humanos 1250 mg de Tykerb® e 2000 mg/m2 de capecitabina, estas doses produziram exposições com base na AUC até 1,7 vezes maior em camundongos e ratos do sexo masculino, e até 12 vezes maior em ratas. Não houve evidência de carcinogenicidade em ratos. Nestes, um aumento na incidência de hemangioma benigno dos linfonodos mesentéricos,ocorreu em machos que receberam 120 mg/kg/dia e fêmeas que receberam 180 mg/kg/dia, mas foi dentro do controle histórico da escala de fundo. Houve também um aumento de infartos e necrose papilar renal em ratos fêmeas com doses ≥ 60 mg/kg/dia e 180 mg/kg/dia, respectivamente (aproximadamente 5,8 a 8,2 vezes a exposição clínica em humanos recebendo 1250 mg de lapatinibe e 2000 mg/m2 de capecitabina, respectivamente). A relevância desses achados renais em humanos é incerta. O lapatinibe não foi clastogênico nem mutagênico em uma bateria de ensaios, incluindo o ensaio de aberrações cromossômicas em hamsters chineses, o ensaio de Ames,
o ensaio de aberrações cromossômicas em linfócitos periféricos humanos e um ensaio de aberrações cromossômicas em medula óssea de ratos in vivo.
Mutagenicidade e reprodução
Não houve nenhum efeito sobre a função gonadal, o acasalamento ou a fertilidade em machos ou fêmeas de ratos em doses até 120 mg/kg/dia (fêmeas) e até 180 mg/kg/dia (machos) (6,4 e 2,6 vezes a exposição clínica prevista em seres humanos tratados com 1250 mg de lapatinibe e 2000 mg/m² de capecitabina, respectivamente). O efeito sobre a fertilidade humana é desconhecido.
Gravidez, lactação e potencial reprodutivo em homens e mulheres Gravidez
Resumo do risco
Não existem dados suficientes em mulheres grávidas expostas ao lapatinibe para avaliar os riscos. Mulheres grávidas devem ser aconselhadas do risco potencial para o feto e Tykerb® deve ser usado durante a gravidez somente se os benefícios esperados para os pacientes justificarem o risco potencial ao feto. Tykerb® não se mostrou teratogênico quando estudado em fêmeas grávidas de ratos e coelhos, mas causou anormalidades menores com doses consideradas tóxicas para as mães (veja Dados em animais).
Dados em animais
Em estudos de desenvolvimento embrionário, ratas e coelhas, animais grávidas, receberam doses orais de 30, 60 e 120 mg/kg/dia durante a organogênese. Não foram observados efeitos teratogênicos, no entanto, anomalias menores (do lado esquerdo da artéria umbilical, costela cervical e ossificação precoce) ocorreram em ratas na dose tóxica