TRAZIMERA
PFIZER
150mg
trastuzumabe
Anticorpo monoclonal. Antineoplásico.
Apresentações.
Trazimera pó liofilizado para solução injetável em embalagens contendo 1 frasco-ampola de 150 mg de pó liofilizado.
VIA DE ADMINISTRAÇÃO: USO INTRAVENOSO
USO ADULTO
Composição.
Cada frasco-ampola de Trazimera pó liofilizado para solução injetável contém 150 mg de trastuzumabe. Cada mL da solução reconstituída contém 21 mg de trastuzumabe.
Excipientes: Frasco-ampola de Trazimera 150 mg: sacarose, cloridrato de histidina monoidratado, histidina e polissorbato 20.
Informações técnicas.
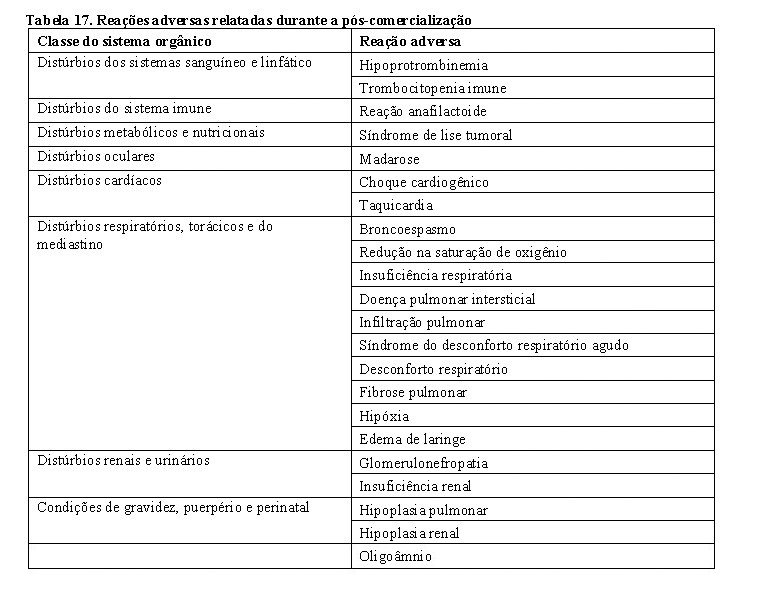
1. INDICAÇÕES
Câncer de mama metastático
Trazimera é indicado para o tratamento de pacientes com câncer de mama metastático que apresentam tumores com superexpressão do HER2:
• em monoterapia para o tratamento de pacientes que já tenham recebido um ou mais tratamentos quimioterápicos para suas doenças metastáticas;
• em combinação com paclitaxel ou docetaxel para o tratamento de pacientes que ainda não tenham recebido quimioterapia para suas doenças metastáticas.
Câncer de mama inicial
Trazimera é indicado para o tratamento de pacientes com câncer de mama inicial HER2-positivo:
• após cirurgia, quimioterapia (neoadjuvante ou adjuvante) e radioterapia (quando aplicável);
• após quimioterapia adjuvante com doxorrubicina e ciclofosfamida, em combinação com paclitaxel ou docetaxel;
• em combinação com quimioterapia adjuvante de docetaxel e carboplatina;
• em combinação com quimioterapia neoadjuvante seguida por terapia adjuvante com Trazimera para câncer de mama localmente avançado (inclusive inflamatório) ou tumores > 2 cm de diâmetro.
Câncer gástrico avançado
Trazimera em associação com capecitabina ou 5-fluorouracil (5-FU) intravenoso e um agente de platina é indicado para o tratamento de pacientes com adenocarcinoma inoperável, localmente avançado, recorrente ou metastático do estômago ou da junção gastroesofágica, HER2-positivo, que não receberam tratamento prévio contra o câncer para sua doença metastática.
2. RESULTADOS DE EFICÁCIA
Trazimera é um medicamento biológico desenvolvido pela via de comparabilidade (biossimilar). O programa de desenvolvimento do produto foi projetado para demonstrar a comparabilidade entre Trazimera e Herceptin®.
Resultados de Eficácia de Trazimera
O programa de desenvolvimento clínico do biossimilar para Trazimera incluiu um total de dois estudos randomizados, multicêntricos, duplos-cegos e controlados que foram conduzidos em pacientes adultos (n=933) com uso de trastuzumabe intravenoso em combinação com quimioterapia.
O estudo B3271002 foi um estudo comparando Trazimera com Herceptin® quando administrado em associação com paclitaxel em pacientes com câncer de mama metastático HER2-positivo. O endpoint primário para este estudo foi a taxa de resposta objetiva (TRO) alcançada na Semana 25, de acordo com o RECIST 1.1 baseada nas avaliações de uma revisão radiológica central. A análise do endpoint primário atingiu o critério de equivalência pré-especificado.
O estudo B3271004 foi um ensaio que compara Trazimera com Herceptin® quando administrado em associação com docetaxel e carboplatina em pacientes com câncer de mama HER2-positivo operável na configuração neoadjuvante. Os endpoint secundários do estudo incluíram taxa de resposta patológica completa (RPC) definida como ausência de células neoplásicas invasivas na mama e nódulos linfáticos, segurança e imunogenicidade. A porcentagem de pacientes que alcançaram o RPC, com base em um patologista local qualificado, foi comparável nos 2 grupos de tratamento.
Em ambos os estudos, os resultados de segurança e imunogenicidade confirmam perfis de segurança comparáveis para Trazimera e Herceptin®. Não há diferença clinicamente significativa na eficácia ou segurança entre o produto comparador Herceptin® e Trazimera quando administrado por via intravenosa em indivíduos com câncer de mama HER-2 positivo.
Resultados de Eficácia do Comparador - Herceptin®
Câncer de mama metastático
Herceptin® como monoterapia foi utilizado em estudos clínicos para pacientes com câncer de mama metastático que apresentavam tumores com superexpressão do HER2 tratados sem sucesso com um ou mais esquemas quimioterápicos prévios para essas doenças metastáticas.1
Herceptin® também foi utilizado em estudos clínicos, em combinação com paclitaxel ou com uma antraciclina (doxorrubicina ou epirrubicina) mais ciclofosfamida (AC), como terapia de primeira linha para pacientes com câncer de mama metastático que apresentavam tumores com superexpressão HER2.2
Pacientes que tinham recebido previamente quimioterapia adjuvante à base de antraciclina foram tratados com paclitaxel (175 mg/m2, com infusão durante três horas) com ou sem Herceptin®. Os pacientes poderiam ser tratados com Herceptin® até a progressão da doença.2
A monoterapia com Herceptin®, utilizada no tratamento de segunda ou terceira linha de mulheres com câncer de mama metastático com superexpressão do HER2, resultou em taxa de resposta tumoral global de 15% e sobrevida mediana de 13 meses.1
A utilização de Herceptin® em combinação com paclitaxel, como tratamento de primeira linha de mulheres com câncer de mama metastático com superexpressão do HER2, prolonga significativamente o tempo mediano até a progressão da doença, em comparação com paclitaxel em monoterapia. O aumento no tempo mediano até a progressão da doença para os pacientes tratados com Herceptin® e paclitaxel é de 3,9 meses (6,9 meses versus 3,0 meses). A resposta tumoral e a taxa de sobrevida em um ano também aumentaram com Herceptin® em combinação com paclitaxel versus paclitaxel isolado.2
Herceptin® também foi avaliado em estudo randomizado, controlado, em combinação com docetaxel, como tratamento de primeira linha de mulheres com câncer de mama metastático. A combinação de Herceptin® com docetaxel aumentou significativamente o índice de resposta (61% versus 34%) e prolongou a mediana de tempo até a progressão da doença (em 5,6 meses), em comparação com pacientes tratados apenas com docetaxel. A sobrevida mediana também aumentou de forma significativa em pacientes tratados com a combinação, em comparação com aqueles que receberam docetaxel isoladamente (31,2 meses versus 22,7 meses).3
Câncer de mama inicial
No tratamento adjuvante, Herceptin® foi investigado em quatro grandes estudos de Fase III, multicêntricos e randomizados:
O estudo BO16348 foi desenhado para comparar um e dois anos de tratamento com Herceptin® a cada três semanas versus observação em pacientes com câncer de mama inicial HER2-positivo após cirurgia, quimioterapia e radioterapia (se aplicável). Adicionalmente, uma comparação de tratamento com Herceptin® por dois anos versus um ano foi realizado. Pacientes designados para Herceptin® receberam uma dose de ataque inicial de 8 mg/kg, seguida por 6 mg/kg, a cada três semanas, durante um4 ou dois8 anos.
Os estudos NCCTG N9831 e NSAPB-B31, que incluem a análise conjunta, foram desenhados para investigar o uso clínico do tratamento combinado de Herceptin® IV com paclitaxel após quimioterapia AC (adriamicina e ciclofosfamida). Adicionalmente o estudo NCCTG N9831 investigou a adição de Herceptin® após a quimioterapia de AC-paclitaxel em pacientes com câncer de mama inicial HER2-positivo após cirurgia.
O estudo BCIRG 006 foi desenhado para investigar o tratamento combinado de Herceptin® IV com docetaxel após a quimioterapia AC ou em combinação com docetaxel e carboplatina em pacientes com câncer de mama inicial HER2-positivo após cirurgia.
No estudo BO16348, o câncer de mama inicial foi limitado a operável, primário, adenocarcinoma invasivo da mama, com tumores de nódulos axilares positivos ou negativos de, pelo menos, 1 cm de diâmetro.
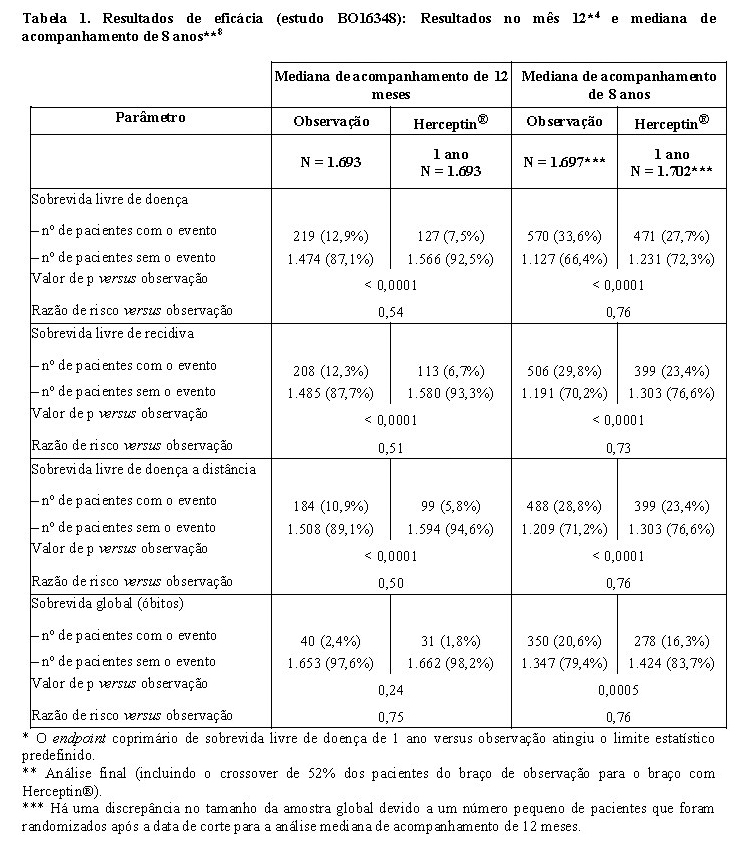
Os resultados de eficácia do estudo BO16348 estão resumidos na tabela a seguir: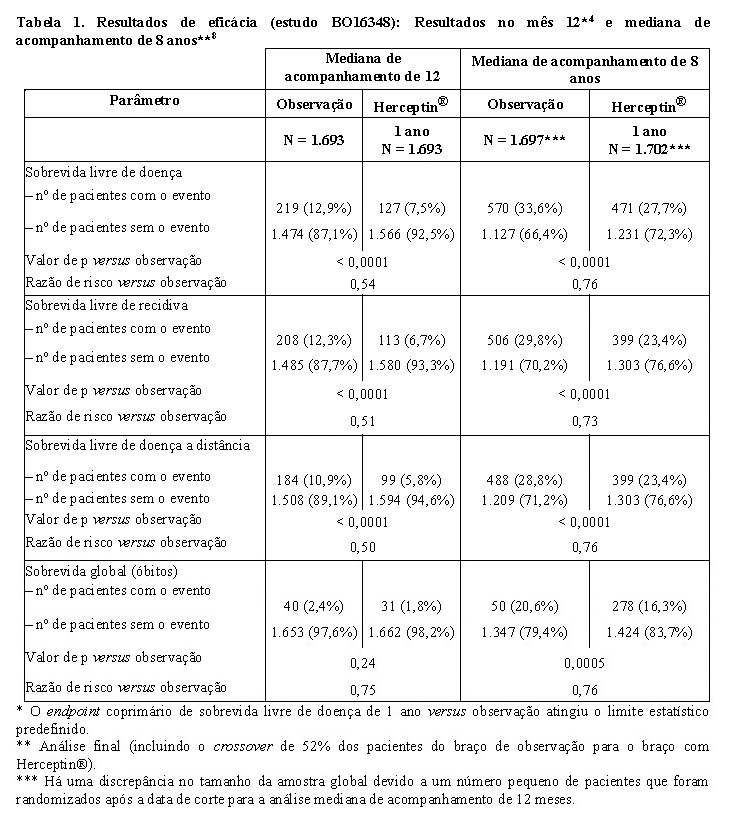
Os resultados de eficácia da análise interina cruzaram o limite estatístico predeterminado no protocolo para a comparação estatística de um ano de Herceptin® versus observação. Após a mediana de acompanhamento de 12 meses, a razão de risco (HR) para a sobrevida livre de doença (SLD) foi de 0,54 (IC 95% 0,44, 0,67), que se traduz em um benefício absoluto, em termos de taxa de sobrevida livre de doença durante dois anos, de 7,6 pontos percentuais (85,8% versus 78,2%) favoráveis ao braço com Herceptin®.
A análise final foi realizada após a mediana de acompanhamento de 8 anos e demonstrou que o tratamento com Herceptin® por um ano está associado a uma redução do risco de 24% em relação à observação somente (HR = 0,76, IC 95% 0,67, 0,86). Isso se traduz em um benefício absoluto em termos de taxa de sobrevida livre de doença durante 8 anos, de 6,4 pontos percentuais a favor de um ano de tratamento com Herceptin®.8
Nessa análise final, a extensão do tratamento com Herceptin®
por um período de dois anos não mostrou benefício adicional sobre o tratamento por um ano [SLD HR na população com intenção de tratamento (ITT) de dois anos versus um ano = 0,99 (IC 95% 0,87, 1,13), valor de p = 0,90 e SG HR = 0,98 (0,83, 1,15), valor de p = 0,78]. A taxa de disfunção cardíaca assintomática foi maior no grupo de tratamento de dois anos (8,1% versus 4,6% no grupo de tratamento de um ano). Mais pacientes tiveram pelo menos um evento adverso de grau 3 ou 4 no grupo de tratamento de dois anos (20,4%), em comparação com o grupo de tratamento de 1 ano (16,3%).8
Na análise conjunta dos estudos NCCTG N9831 e NSAPB-B31, o câncer de mama inicial foi limitado a mulheres com câncer de mama operável de alto risco, definido como HER2-positivo e linfonodo axilar positivo ou HER2-positivo e linfonodo negativo com características de alto risco (tamanho do tumor > 1 cm e receptor hormonal negativo ou tamanho do tumor > 2 cm, independentemente do status hormonal). Herceptin® foi administrado em combinação com paclitaxel após quimioterapia AC. O paclitaxel foi administrado conforme segue:
- paclitaxel intravenoso: 80 mg/m2, na forma de infusão intravenosa contínua, administrada toda semana, por um período de 12 semanas; ou
- paclitaxel intravenoso: 175 mg/m2, na forma de infusão intravenosa contínua, administrada a cada três semanas, por um período de quatro ciclos (dia 1 de cada ciclo).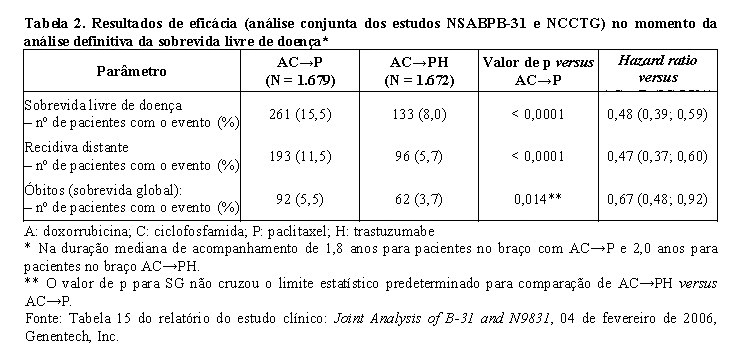
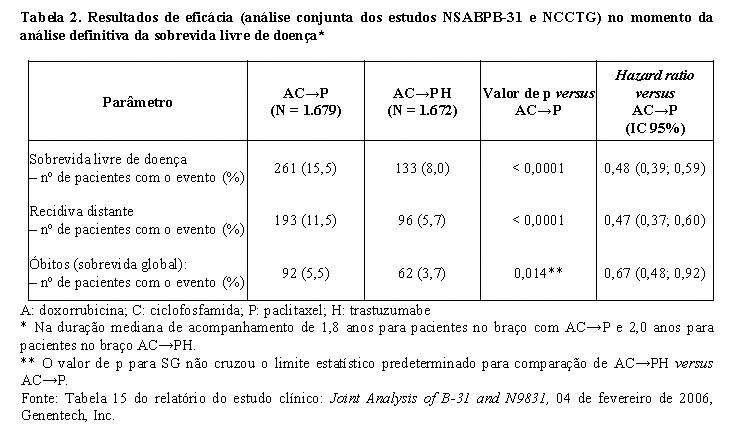
Para o endpoint primário, sobrevida livre de doença, a adição de Herceptin® à quimioterapia com paclitaxel resultou em redução de 52% no risco de recidiva da doença. O hazard ratio transforma-se em um benefício absoluto, em termos de taxa de sobrevida livre de doença durante três anos, de 11,8 pontos percentuais (87,2% versus 75,4%) favoráveis ao braço de AC→PH (Herceptin®).
A análise final pré-planejada da SG a partir da análise conjunta dos estudos NSABPB-31 e NCCTG N9831 foi realizada quando 707 mortes ocorreram (acompanhamento mediano de 8,3 anos no grupo AC→PH). O tratamento com AC→PH resultou em uma melhora significativa da SG comparada com AC→P (estratificado HR=0,64%; IC 95% [0,55, 0,74]; valor de p log-rank < 0,0001). Em 8 anos, a taxa de sobrevivência foi estimada em 86,9% para o braço AC→PH e 79,4% para o braço AC→P, um benefício absoluto de 7,4% (IC 95% 4,9, 10,0%).
A análise final de SG a partir da análise conjunta dos estudos NSABPB-31 e NCCTG N9831 foi resumida na tabela a seguir.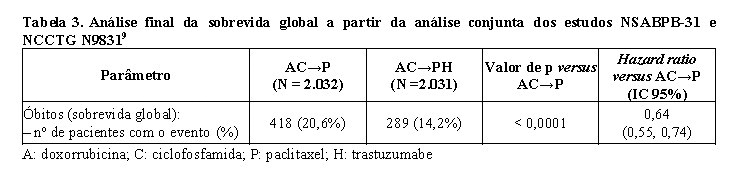
No estudo BCIRG 006, o câncer de mama inicial HER2-positivo foi limitado a pacientes com linfonodo positivo ou com nódulo negativo de alto risco, definido como envolvimento de linfonodo negativo (pN0) e com, pelo menos, um dos seguintes fatores: tamanho do tumor maior que 2 cm, receptor de estrógeno e progestágeno negativo, grau histológico e/ou nuclear 2 - 3 ou idade < 35 anos. Herceptin® foi administrado em combinação com docetaxel, após quimioterapia AC (AC-DH) ou em combinação com docetaxel e carboplatina (DCarbH).
O docetaxel foi administrado conforme segue:
- docetaxel intravenoso: 100 mg/m2, na forma de infusão intravenosa, durante uma hora, administrada a cada três semanas, por um período de quatro ciclos (dia 2 do primeiro ciclo de docetaxel e dia 1 de cada ciclo subsequente);
ou
- docetaxel intravenoso: 75 mg/m2, na forma de infusão intravenosa, durante uma hora, administrada a cada três semanas, por um período de seis ciclos (dia 2 do ciclo 1 e dia 1 de cada ciclo subsequente);
que foi seguido por:
- carboplatina: objetivo de AUC = 6 mg/mL/min administrada por infusão intravenosa durante 30 - 60 minutos, repetida a cada três semanas para um total de seis ciclos.
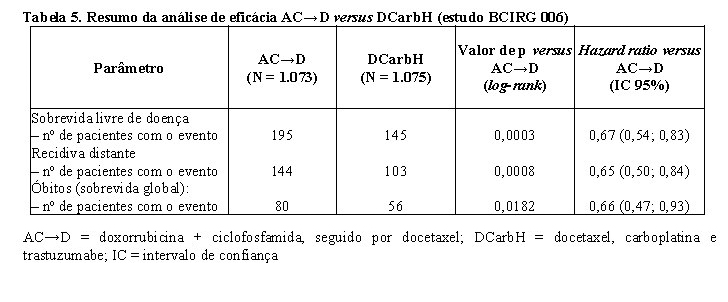
Os resultados de eficácia do estudo BCIRG 006 estão resumidos nas tabelas a seguir:

No estudo BCIRG 006, para o endpoint primário, sobrevida livre de doença, o hazard ratio transforma-se em um benefício absoluto, em termos de taxa de sobrevida livre de doença durante três anos, de 5,8 pontos percentuais (86,7% versus 80,9%) favoráveis ao braço de AC→DH (Herceptin®) e 4,6 pontos percentuais (85,5% versus 80,9%) favoráveis ao braço de DCarbH (Herceptin®) comparados a AC→D.
Para o endpoint secundário, sobrevida global, o tratamento com AC→DH reduziu o risco de óbito em 42% quando comparado a AC→D [hazard ratio 0,58 (IC 95%: 0,40; 0,83); p = 0,0024; teste log-rank], e o risco de óbito foi reduzido em 34% em pacientes tratados com DCarbH quando comparado aos pacientes tratados com AC→D [hazard ratio 0,66 (IC 95%: 0,47; 0,93); p = 0,0182]. Na segunda análise interina do estudo BCIRG 006, 185 pacientes randomizados foram a óbito: 80 pacientes (7,5%) no braço AC→D, 49 (4,6%) no braço AC→DH e 56 pacientes (5,2%) no braço DCarbH. A duração mediana do acompanhamento foi 2,9 anos para o braço AC→D e 3,0 anos para os braços AC→DH e DCarbH.
No tratamento neoadjuvante-adjuvante, Herceptin® foi avaliado em um estudo Fase III:
• O estudo MO16432 investigou um total de 10 ciclos de quimioterapia neoadjuvante [uma antraciclina e um taxano (AP+H) seguido por P+H, seguido por CMF+H] concomitantemente com terapia neoadjuvante-adjuvante com Herceptin®, ou quimioterapia neoadjuvante isolada seguida por tratamento adjuvante com Herceptin®, até a duração total de um ano de tratamento em pacientes com diagnóstico recente de câncer de mama HER2-positivo localmente avançado (estágio III) ou inflamatório.
O MO16432 é um estudo de Fase III, aberto e randomizado, de comparação de um ano de tratamento neoadjuvante e adjuvante de Herceptin® com observação em 231 pacientes com câncer de mama HER2-positivo localmente avançado ou inflamatório, tratados com um regime de quimioterapia neoadjuvante sequencial que incluiu doxorrubicina, paclitaxel, ciclofosfamida, metotrexato e 5-fluorouracil. A população alvo para o estudo MO16432 consistia em mulheres ≥ 18 anos que foram recentemente diagnosticadas com câncer de mama localmente avançado e que não haviam recebido qualquer tratamento anterior para uma doença invasiva. O tumor primário deveria ser T3N1 ou T4 (invasão do mamilo ou da pele, peau d'orange, extensão para a parede torácica ou carcinoma inflamatório); qualquer T mais N2 ou N3; ou qualquer T mais envolvimento dos nódulos supraclaviculares ipsilaterais. As pacientes precisavam ter doença HER2-positivo, definida como doença com superexpressão de HER2 por imuno-histoquímica IHC 3+ e/ou amplificação de HER2 de acordo com a hibridização fluorescente in situ (FISH), com base na confirmação do laboratório central (entretanto, permitiu-se que as pacientes entrassem no estudo com base em um resultado IHC 3+/FISH central negativo).
Os resultados de eficácia do estudo MO16432 estão resumidos na tabela a seguir. A mediana de duração do acompanhamento no braço de Herceptin® foi 3,8 anos.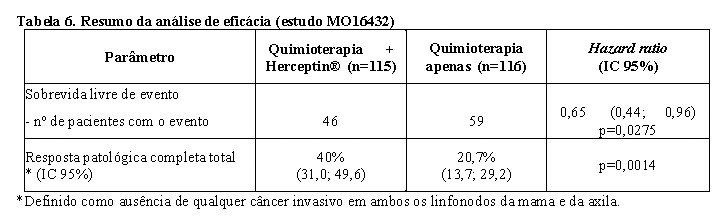
Para o endpoint primário, sobrevida livre de evento, a adição de Herceptin® à quimioterapia neoadjuvante, seguida pelo tratamento adjuvante com Herceptin® para uma duração total de 52 semanas, resultou em redução de 35% no risco de recidiva/progressão da doença. O hazard ratio traduz-se em um benefício absoluto, em termos de taxa de sobrevida livre de evento de três anos, estimada em 13 pontos percentuais (65% versus 52%) favoráveis ao braço com Herceptin®.5
Câncer gástrico avançado
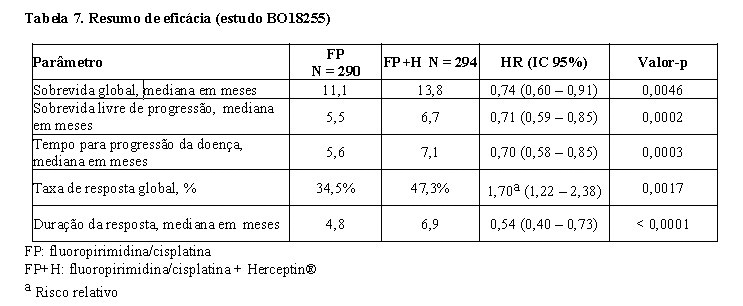
Os resultados de eficácia do estudo BO18255 estão resumidos na tabela 7. Os pacientes com adenocarcinoma localmente avançado inoperável ou metastático e/ou recorrente do estômago ou da junção gastroesofágica, HER2-positivo sem possibilidade de terapia curativa e não tratados previamente foram recrutados para o estudo. O endpoint primário foi a sobrevida global, a qual foi definida como o tempo a partir da data de randomização até o dia do óbito por qualquer causa. No momento da análise, um total de 349 pacientes randomizados foi a óbito: 182 pacientes (62,8%) no braço controle e 167 pacientes (56,8%) no braço tratamento. A maioria dos óbitos foi devida a eventos relacionados com o câncer subjacente.6
A sobrevida global foi significativamente maior no braço Herceptin® + capecitabina/5-FU e cisplatina comparada ao braço capecitabina/5-FU e cisplatina (p = 0,0046, teste log-rank). O tempo mediano da sobrevida foi de 11,1 meses com capecitabina/5-FU e cisplatina e 13,8 meses com Herceptin® + capecitabina/5-FU e cisplatina. O risco de óbito diminuiu em 26% [hazard ratio 0,74 IC 95% (0,60-0,91)] para pacientes no braço com Herceptin®, comparado ao braço com capecitabina/5-FU.6
Análises de subgrupo post-hoc indicam que ter como alvo tumores com níveis mais elevados da proteína HER2 (IHQ 2+/FISH+ e IHQ 3+/independentemente do status FISH) resulta em melhor efeito terapêutico. A mediana de sobrevida global para o grupo com alta expressão de HER2 foi de 11,8 meses versus 16 meses, HR 0,65 (IC 95% 0,51-0,83), e a mediana de sobrevida livre de progressão foi de 5,5 meses versus 7,6 meses, HR 0,64 (IC 95% 0,51-0,79) para capecitabina/5-FU e cisplatina e Herceptin® + capecitabina/5-FU e cisplatina, respectivamente.6
Em estudo de comparação de método, um alto grau de concordância ( > 95%) foi observado para as técnicas SISH e FISH para a detecção da amplificação do gene HER2 em pacientes com câncer gástrico.7 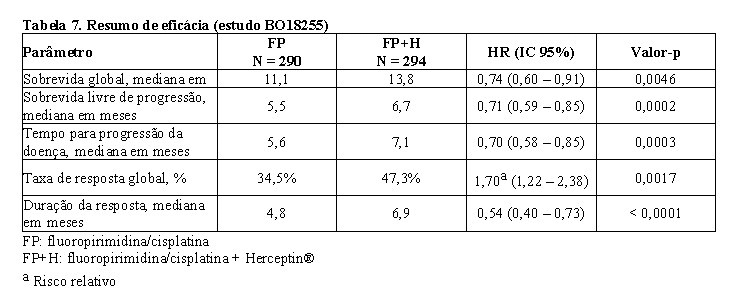
Referências do Herceptin®
1 Cobleigh MA, Vogel CL, Tripathy D, et al. Multinational Study of the Efficacy and Safety of Humanized Anti-HER2 Monoclonal Antibody in Women Who Have HER2-Overexpressing Metastatic Breast Cancer That Has Progressed After Chemotherapy for Metastatic Disease. Journal of Clinical Oncology; 17 (9):2639-2648, 1999.
2 Slamon DJ, Leyland-Jones B, Hak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresss HER2. The New England Journal of Medicine; 344 (11): 783, 2001.
3 Marty M, Cognetti F, Maraninchi D,et al. Efficacy and Safety of Trastuzumab Combined With Docetaxel in Patients With Human Epidermal Growth Factor Receptor 2-Positive Metastatic Breast Cancer Administered as First-Line Treatment: Results of a Randomized Phase II Trial by the M77001 Study Group. Journal of Clinical Oncology; 23(19): 1, 2005.
4 Piccart-Gebhart MJ, Procter M, Leyland-Jones B, et al. Trastuzumab after Adjuvant Chemotherapy in HER2-Positive Breast Cancer. The New England Journal of Medicine; 353 (16): 1659, 2005.
5 Gianni L, Eiermann W, Semiglazov V, Manikhas A, Lluch A, Tjulandin S, Zambetti M, Vazquez F, Byakhow M, Lichinitser M, Climent MA, Ciruelos B, Mansutti M, Bozhok A, Baronio R, Feyereislova A, Barton C, Valagussa P, Baselga J: Neoadjuvant chemotherapy with trastuzumab followed by adjuvant trastuzumab versus neoadjuvant chemotherapy alone, in patients with HER2-positive locally advanced breast cancer (the NOAH trial): a randomised controlled superiority trial with a parallel HER2-negative cohort. Lancet 2010, 375:377-384.
6 Bang Y-J, Van Cutsem E, Feyereislova A, et al; for the ToGA Trial Investigators. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687-697.
7 Method Comparison Study of CONFIRM anti-HER2/neu(4B5) Primary Antibody and INFORM HER2 DNA Probe VS Hercep Test and HER2 FISH PharmDx on human gastric cancer. Dated: 27th July 2009.
8 Update Clinical Study Report BO16348 (HERA): A randomized three-arm, multicenter comparison of 1 year and 2 years of Herceptin versus no Herceptin in women with HER2-positive primary breast cancer who have completed adjuvant chemotherapy. Report No. 1044055. March 2013
9 Joint Analysis (B-31 & N9831) Clinical Study Report 2013.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades Farmacodinâmicas
O trastuzumabe é um anticorpo monoclonal IgG1 humanizado recombinante contra o receptor tipo 2 do fator de crescimento epidérmico humano (HER2). A superexpressão do HER2 é observada em 20% - 30% dos cânceres de mama primários. Estudos das taxas do HER2-positivo no câncer gástrico (CG) usando imuno-histoquímica (IHQ) e hibridação in situ fluorescente (FISH) ou hibridização in situ cromogênica (CISH) demonstraram que existe uma ampla da classificação de positividade do HER2, variando entre 6,8% - 34,0% para IHQ e 7,1% - 42,6% para FISH. Estudos indicam que pacientes com câncer de mama cujos tumores superexpressam HER2 têm uma sobrevida livre de doença reduzida em comparação com pacientes cujos tumores não superexpressam HER2. O domínio extracelular do receptor (ECD, p105) pode estar presente na corrente sanguínea e mensurado em amostras de soro.
Mecanismo de ação
O trastuzumabe liga-se com alta afinidade e especificidade ao subdomínio IV, uma região justamembranar do domínio extracelular do HER2. A ligação do trastuzumabe ao HER2 inibe a sinalização independente do ligante do HER2 e previne a clivagem proteolítica do seu domínio extracelular, mecanismo de ativação do HER2. Como resultado, o trastuzumabe demonstrou, tanto em estudos in vitro quanto em animais, inibir a proliferação de células tumorais humanas com superexpressão do HER2. Além disso, o trastuzumabe é um potente mediador da citotoxicidade mediada por células dependente de anticorpos (ADCC). In vitro, a ADCC mediada pelo trastuzumabe demonstrou ser exercida preferencialmente nas células cancerosas que superexpressam HER2 em comparação com células cancerosas que não superexpressam HER2.
Detecção de superexpressão do HER2 ou amplificação do gene HER2
Detecção de superexpressão do HER2 ou amplificação do gene HER2 em câncer de mama
O trastuzumabe só deve ser utilizado em pacientes cujos tumores possuem superexpressão do HER2 ou amplificação do gene HER2 conforme determinado por um ensaio preciso e validado. A superexpressão do HER2 deve ser detectada usando uma avaliação baseada em imuno-histoquímica (IHQ) de amostras de tumor fixos. A amplificação do gene HER2 deve ser detectada usando hibridação in situ fluorescente (FISH) ou hibridização in situ cromogênica (CISH) de amostras tumorais fixas. Os pacientes são elegíveis para o tratamento com trastuzumabe se apresentarem uma forte superexpressão do HER2, descrito por uma classificação de 3+ por IHQ ou um resultado positivo de FISH ou CISH.
Para garantir resultados precisos e reproduzíveis, o teste deve ser realizado em um laboratório especializado, que pode garantir a validação dos procedimentos de teste.
O sistema de classificação recomendado para avaliar os padrões de coloração de IHQ é como indicado na Tabela 8:
No geral, o teste de FISH é considerado positivo se a razão entre o número de cópias do gene HER2 por célula tumoral e o número de cópias do cromossomo 17 for maior ou igual a 2, ou se houver mais de 4 cópias do gene HER2 por célula tumoral, caso o cromossomo 17 não seja usado como controle.
No geral, o teste de CISH é considerado positivo se houver mais de 5 cópias do gene HER2 por núcleo em mais de 50% das células tumorais.
Para instruções completas sobre a realização e a interpretação dos testes, consulte os folhetos de instruções nas embalagens dos testes de FISH e CISH validados. Recomendações oficiais sobre o teste com HER2 também podem se aplicar.
Para qualquer outro método que possa ser utilizado na determinação da proteína ou da expressão gênica do HER2, as análises devem ser realizadas somente por laboratórios que utilizem métodos validados, com desempenho adequado. Tais métodos devem claramente ser precisos e exatos o suficiente para demonstrar a superexpressão de HER2 e devem ser capazes de distinguir entre a superexpressão de HER2 moderada (classificado como 2+) e forte (classificado como 3+).
Detecção da superexpressão de HER2 ou amplificação do gene HER2 no câncer gástrico
Somente um teste preciso e validado deve ser utilizado para detectar a superexpressão do HER2 ou a amplificação do gene HER2. A IHQ é recomendada como a primeira modalidade de teste e, nos casos em que o estado de amplificação do gene HER2 também é necessário, deve ser aplicado um teste de hibridação in situ por prata (SISH) ou um teste de FISH. No entanto, a tecnologia SISH é recomendada para permitir a avaliação paralela da histologia e morfologia do tumor. Para garantir a validação dos procedimentos dos testes e a geração de resultados precisos e reproduzíveis, o teste do HER2 deve ser realizado em um laboratório com pessoal treinado. As instruções completas sobre a realização do teste e a interpretação dos resultados devem ser obtidas nos folhetos de instruções fornecidos com os testes do HER2 utilizados.
No estudo ToGA (BO18255), os pacientes cujos tumores eram IHQ3+ ou FISH positivos foram definidos como HER2-positivos e, portanto, incluídos no estudo. Com base nos resultados dos estudos clínicos, os efeitos benéficos foram limitados aos pacientes com maior nível de superexpressão da proteína HER2, definido pela classificação de 3+ pela IHQ, ou uma classificação 2+ pela IHQ e um resultado positivo de FISH.
Em um estudo de comparação de métodos (estudo D008548) foi observado um alto grau de concordância ( > 95%) para os testes SISH e FISH na detecção da amplificação do gene HER2 em pacientes com câncer gástrico.
A superexpressão do HER2 deve ser detectada utilizando uma avaliação baseada na imuno-histoquímica (IHQ) de amostras tumorais fixas; A amplificação do gene HER2 deve ser detectada utilizando hibridação in situ, ou SISH ou FISH, em amostras tumorais fixas.
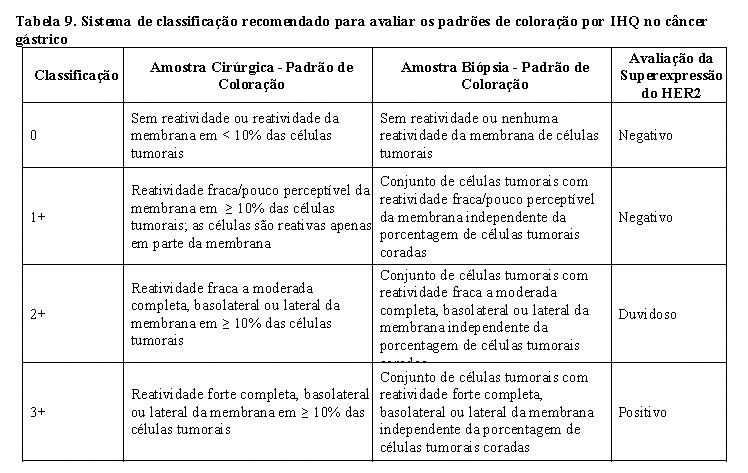
O sistema de classificação recomendado para avaliar os padrões de coloração de IHQ é como indicado na Tabela 9:
No geral, SISH ou FISH são considerados positivos se a razão entre o número de cópias do gene HER2 por célula tumoral e o número de cópias do cromossomo 17 for maior ou igual a 2.
Propriedades Farmacocinéticas
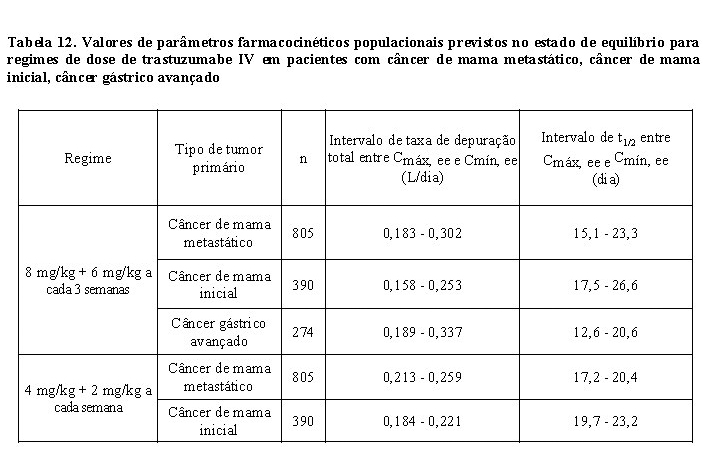
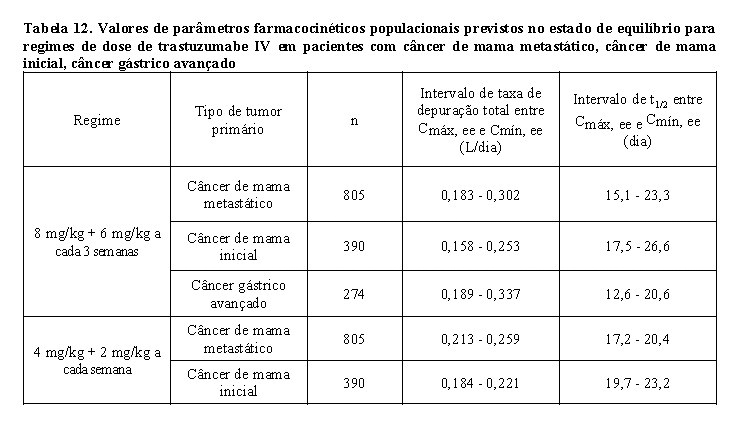
A farmacocinética de trastuzumabe foi avaliada em uma análise de modelo de farmacocinética populacional utilizando dados agrupados de 1.582 indivíduos, incluindo pacientes com câncer de mama metastático HER2-positivo, câncer de mama inicial, câncer gástrico avançado ou outros tipos de tumores, e voluntários saudáveis, em 18 estudos de Fase I, II e III que receberam trastuzumabe IV. Um modelo de dois compartimentos com eliminação linear e não linear do compartimento central descreveu o perfil de concentração-tempo de trastuzumabe. Devido à eliminação não linear, a depuração total aumentou com a concentração decrescente. Portanto, nenhum valor constante para a meia-vida de trastuzumabe pode ser deduzido. O t1/2 diminui com as concentrações decrescentes dentro de um intervalo de dose (ver Tabela 12). Os pacientes com câncer de mama metastático e câncer de mama inicial apresentaram parâmetros farmacocinéticos similares (por exemplo, depuração (clearance - CL), volume do compartimento central (Vc)) e exposição farmacocinética populacional prevista no estado de equilíbrio (Cmín, Cmáx e AUC). A depuração linear foi 0,136 L/dia para câncer de mama metastático, 0,112 L/dia para câncer de mama inicial e 0,176 L/dia para câncer gástrico avançado. Os valores dos parâmetros de eliminação não linear foram 8,81 mg/dia para a taxa de eliminação máxima (Vmáx) e 8,92 mg/mL para a constante de Michaelis-Menten (Km) para pacientes com câncer de mama metastático, câncer de mama inicial e câncer gástrico avançado. O volume do compartimento central foi de 2,62 L para pacientes com câncer de mama metastático e câncer de mama inicial e 3,63 L para pacientes com câncer gástrico avançado.
No modelo farmacocinético da população final, além do tipo de tumor primário, o peso corporal, a aspartato aminotransferase e a albumina no soro foram identificados como covariáveis estatisticamente significativas que afetam a exposição do trastuzumabe. No entanto, a magnitude do efeito destas covariáveis na exposição ao trastuzumabe sugere que estas covariáveis provavelmente não terão um efeito clinicamente significativo nas concentrações de trastuzumabe.
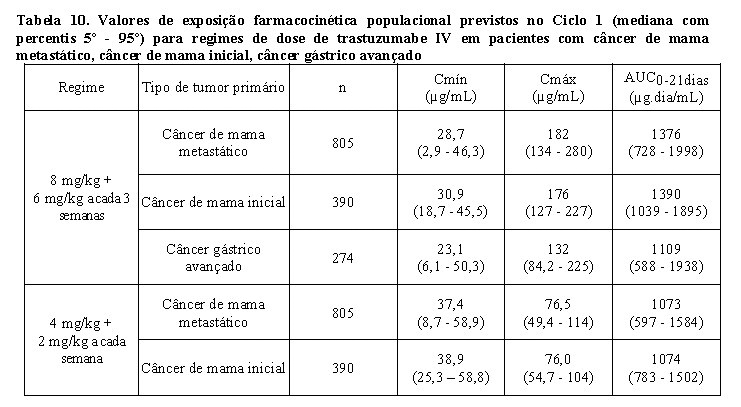
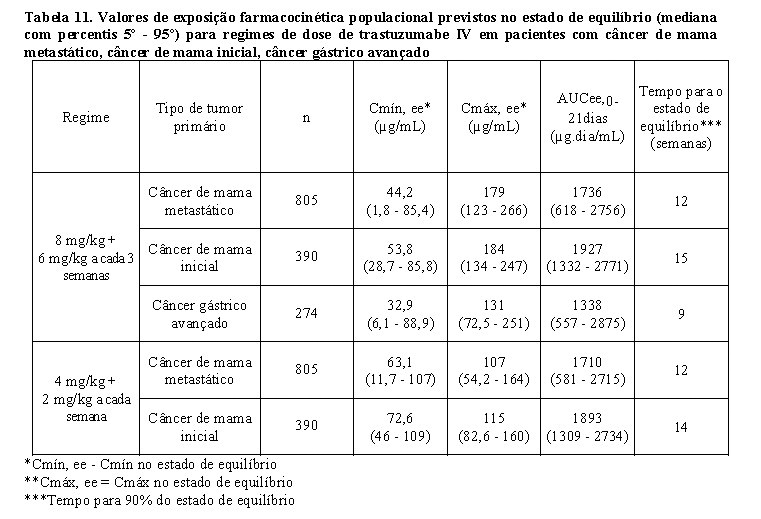
Os valores de exposição farmacocinética populacional prevista (mediana com percentis 5° - 95°) e os valores de parâmetros farmacocinéticos de concentrações clinicamente relevantes (Cmáx e Cmín) para pacientes com câncer de mama metastático, câncer de mama inicial e câncer gástrico avançado tratados com os regimes de dose aprovados a cada 1 semana e a cada 3 semanas são mostrados na Tabela 10 (Ciclo 1), Tabela 11 (estado de equilíbrio) e Tabela 12 (parâmetros farmacocinéticos).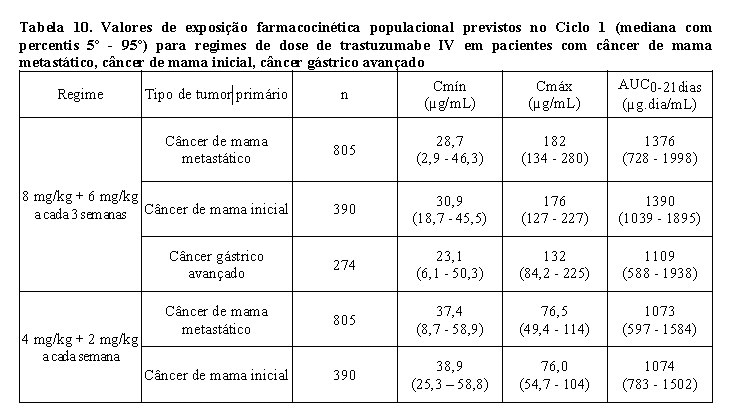
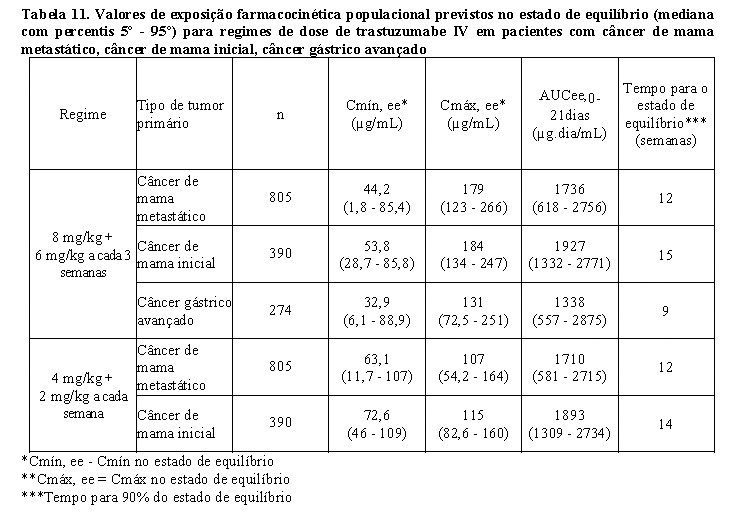
Washout de trastuzumabe
O washout do trastuzumabe foi avaliado após administração intravenosa a cada 1 semana ou a cada 3 semanas usando o modelo da farmacocinética populacional. Os resultados dessas simulações indicam que pelo menos 95% dos pacientes irão alcançar concentrações < 1 mg/mL (aproximadamente 3% da população prevista Cmín, ee, ou cerca de 97% de washout) por 7 meses.
Segurança não clínica
Carcinogenicidade
Não foram realizados estudos de carcinogenicidade para estabelecer o potencial carcinogênico de trastuzumabe.
Diminuição da fertilidade
Os estudos de reprodução foram realizados em macacas Cynomolgus com doses de até 25 vezes a dose semanal humana de manutenção de 2 mg/kg de trastuzumabe IV, e não revelaram evidência de diminuição da fertilidade.
Toxicidade reprodutiva
Os estudos de reprodução foram realizados em macacas Cynomolgus com doses de até 25 vezes a dose semanal humana de manutenção de 2 mg/kg de trastuzumabe IV, e não revelaram evidência de danos ao feto. No entanto, em relação à avaliação do risco de toxicidade reprodutiva em humanos, é importante considerar o significado do receptor HER2 dos roedores no desenvolvimento embrionário e na morte de embriões de ratos mutantes que não têm esse receptor. Foi observada transferência placentária de trastuzumabe durante o período de desenvolvimento fetal precoce (dias 20-50 de gestação) e tardio (dias 120-150 de gestação).
Lactação
Um estudo realizado em macacas Cynomolgus lactantes, com doses 25 vezes a dose semanal humana de manutenção de trastuzumabe IV, de 2 mg/kg, demonstrou que trastuzumabe é secretado no leite.
A presença de trastuzumabe no soro de macacos recém-nascidos não foi associada com qualquer efeito adverso no seu crescimento ou desenvolvimento desde seu nascimento até 1 mês de idade.
Farmacocinética em populações especiais
Não foram realizados estudos farmacocinéticos detalhados na população geriátrica ou em populações de pacientes com insuficiência renal ou hepática.
População geriátrica
Foi demonstrado que a idade não tem efeito sobre a disponibilidade do trastuzumabe (vide "Advertências e Precauções").
HER2-ECD livre circulante
As análises exploratórias de covariáveis com informação em apenas um subgrupo de pacientes sugeriram que os pacientes com maior nível do HER2-ECD livre circulante tiveram depuração não linear mais rápida (Km baixo) (P < 0,001). Houve correlação entre o antígeno livre circulante e os níveis de SGOT/AST; parte do impacto do antígeno livre circulante na depuração pode ter sido explicado pelos níveis de SGOT/AST.
Os níveis basais do HER2-ECD livre circulante observados em pacientes com câncer gástrico avançado foram comparáveis aos de pacientes com câncer de mama metastático e câncer de mama inicial e nenhum impacto aparente na depuração do trastuzumabe foi observado.
Estudos farmacocinéticos comparativos de Trazimera
A comparabilidade farmacocinética de Trazimera e Herceptin® foi avaliada no Estudo B3271001 em 105 indivíduos adultos saudáveis em um estudo clínico de três braços, duplo-cego, randomizado, de grupos paralelos (1:1:1), de dose única comparando Trazimera, Herceptin®-UE e Herceptin®-EUA administrado por via intravenosa.
Os IC de 90% para as razões teste-referência de Cmáx, AUCt e AUCinf estavam dentro dos intervalos de aceitação pré-especificados de 80% a 125% para as comparações de Trazimera com Herceptin®-EUA e Trazimera para Herceptin®-UE. As razões teste-referência (IC de 90% das razões) das médias geométricas ajustadas de Cmáx, AUCt e AUCinf foram 97,41% (90,71%, 104,62%), 99,94% (93,08%, 107,31%) e 99,83% (93,06%, 107,09%), respectivamente, para a comparação de Trazimera com Herceptin®-EUA; e 91,49% (85,32%, 98,09%), 92,66% (86,44%, 99,34%) e 92,15% (86,03%, 98,69%), respectivamente, para a comparação Trazimera com Herceptin®-UE.
No estudo B3271004 em pacientes com câncer de mama inicial tratados com Trazimera ou Herceptin®-UE, em associação com docetaxel e carboplatina, o endpoint primário do estudo foi a porcentagem de pacientes com Cvale no Ciclo 5 (concentração de trastuzumabe pré-dose no Ciclo 6) > 20 mg/mL. A análise do endpoint primário atendeu ao critério de não inferioridade. O estudo demonstrou uma porcentagem comparável de pacientes, no estado de equilíbrio (Ciclo 5), com Cvale > 20 mg/mL de Trazimera e Herceptin®-UE.
Dados de Segurança Pré-Clínicos
Não houve evidência de toxicidade aguda ou relacionada a múltiplas doses nos estudos até 6 meses, ou toxicidade reprodutiva em teratologia, fertilidade feminina ou toxicidade gestacional tardia/estudos de transferência placentária com trastuzumabe. O trastuzumabe não é genotóxico.
Não foram realizados estudos em animais de longo prazo para estabelecer o potencial carcinogênico do trastuzumabe, ou para determinar seus efeitos sobre a fertilidade em machos.
4. CONTRAINDICAÇÕES
Trazimera é contraindicado a pacientes com hipersensibilidade conhecida ao trastuzumabe ou a qualquer outro excipiente da fórmula.
5. ADVERTÊNCIAS E PRECAUÇÕES
A terapia com trastuzumabe deve ser iniciada somente sob a supervisão de um médico experiente no tratamento de pacientes com câncer.
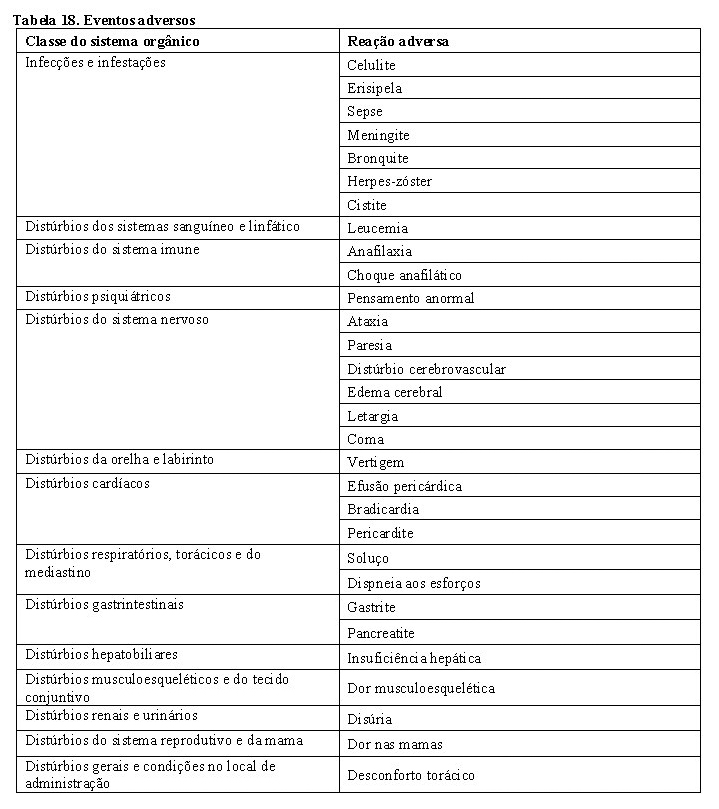
Reações relacionadas à infusão (RRI)
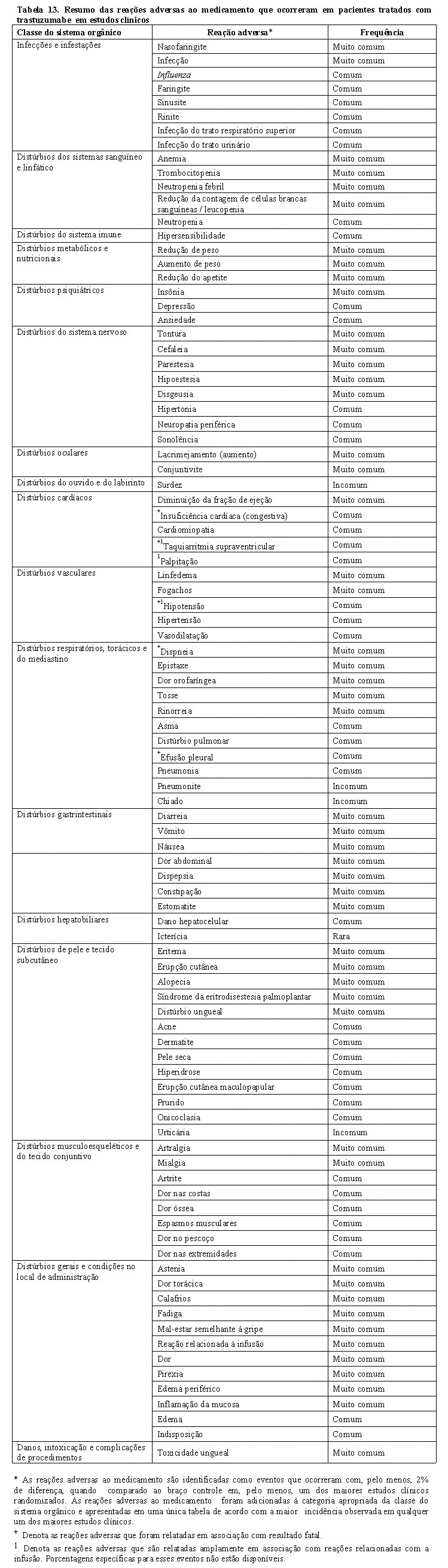
Sabe-se que reações relacionadas à infusão ocorrem com a administração de trastuzumabe (vide item "Reações adversas").
Pode ser difícil diferenciar, clinicamente, as reações relacionadas à infusão de reações de hipersensibilidade. Pré-medicação pode ser utilizada para reduzir o risco de ocorrência de reações relacionadas à infusão.
Reações graves relacionadas à infusão de trastuzumabe, que incluem dispneia, hipotensão, sibilância, broncoespasmo, taquicardia, redução na saturação de oxigênio e dificuldade respiratória, taquiarritmia supraventricular e urticária foram relatadas (vide item "Reações adversas"). O paciente deve ser monitorado em relação às reações relacionadas à infusão. A interrupção da infusão intravenosa pode ajudar no controle desses sintomas e a mesma poderá ser reinstituída assim que os sintomas forem controlados. Esses sintomas podem ser tratados com analgésico/antipirético, tais como a meperidina ou paracetamol, ou ainda com anti-histamínico, como a difenidramina. Reações graves têm sido tratadas, com sucesso, com terapias de suporte, tais como oxigenoterapia, beta-agonista e corticoides. Em casos raros, essas reações podem apresentar evolução fatal. Pacientes que apresentam dispneia de repouso decorrente de complicações de doença maligna avançada ou comorbidade podem ter risco aumentado para reação infusional fatal. Portanto, esses pacientes não devem ser tratados com trastuzumabe.
Reações pulmonares
Eventos adversos pulmonares graves com o uso de trastuzumabe foram relatados após sua comercialização. Esses eventos ocasionalmente resultaram em óbito e podem ocorrer como parte da reação relacionada à infusão ou serem de início tardio. Além disso, foram relatados casos de doença pulmonar intersticial, incluindo infiltrado pulmonar, síndrome do desconforto respiratório agudo, pneumonia, pneumonite, derrame pleural, dificuldade respiratória, edema pulmonar agudo e insuficiência respiratória.
Fatores de risco associados com a doença pulmonar intersticial incluem tratamento prévio ou concomitante com outras terapias antineoplásicas conhecidas por serem associadas a essa condição, como taxanos, gencitabina, vinorelbina e radioterapia. Pacientes com dispneia de repouso decorrente de complicações de doença maligna avançada ou comorbidade podem ter risco aumentado para reações pulmonares. Dessa forma, esses pacientes não devem ser tratados com trastuzumabe.
Disfunção cardíaca
Considerações gerais
Pacientes tratados com trastuzumabe apresentam maior risco de desenvolver insuficiência cardíaca congestiva (ICC) (New York Heart Association [NYHA] Classe II-IV) ou disfunção cardíaca assintomática. Esses eventos foram observados em pacientes que receberam trastuzumabe em monoterapia ou em combinação com taxano após regimes quimioterápicos com antraciclina (doxorrubicina ou epirrubicina). A insuficiência cardíaca pode ser de moderada a grave, e já houve casos de óbito (vide item "Reações adversas"). Além disso, deve-se ter cautela com pacientes em tratamento que apresentam risco cardíaco aumentado (por exemplo, hipertensão