SOMAVERT®
PFIZER
pegvisomanto
Análogo do hormônio de crescimento humano.
Apresentações.
Forma farmacêutica: pó liofilizado injetável
Via de administração: subcutânea
Somavert® 10 mg ou 15 mg, pó liofilizado injetável, em embalagens contendo 30 frascos-ampola + 30 frascos-ampola de diluente.
USO ADULTO
Composição.
Cada frasco-ampola de Somavert® 10 mg ou 15 mg contém 10 mg ou 15 mg de pegvisomanto, respectivamente.
Após a reconstituição de Somavert® 10 mg ou 15 mg, 1 mL da solução contém 10 mg ou 15 mg de pegvisomanto, respectivamente.
Excipientes: glicina, manitol, fosfato de sódio dibásico (anidro) e fosfato de sódio monobásico (monoidratado).
Cada frasco-ampola de diluente contém 8 mL de água para injetáveis.
Indicações.
Somavert® (pegvisomanto) é indicado para o tratamento da acromegalia em pacientes que apresentaram resposta inadequada à cirurgia e/ou à radioterapia e para aqueles pacientes cujo tratamento médico com análogos da somatostatina não normalizou as concentrações séricas de IGF-I ou não foi tolerado.
O objetivo do tratamento com Somavert® é normalizar os níveis séricos de IGF-I.
Resultados de eficácia.
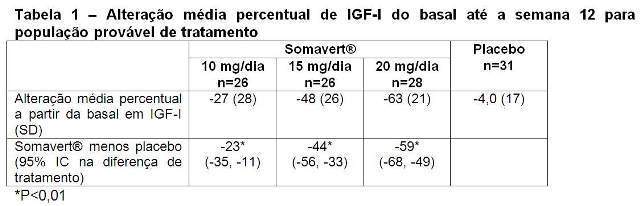
Pacientes acromegálicos (n=112) previamente tratados com cirurgia, radiação, e/ou terapias medicamentosas participaram em um estudo multicêntrico, randomizado, duplo-cego, durante 12 semanas, comparando placebo com pegvisomanto. Após suspensão do tratamento médico prévio, os 80 pacientes randomizados para o tratamento com pegvisomanto receberam uma dose de ataque subcutânea, seguida por 10 mg/dia, 15 mg/dia ou 20 mg/dia por via subcutânea. Estes 3 grupos que receberam pegvisomanto demonstraram reduções estatisticamente significativas e dose-dependentes dos níveis séricos de IGF-I (p < 0,0001), de IGF-I livre (p < 0,05), de IGFBP-3 (p < 0,05) e de ALS (p < 0,05) em todas as visitas após a visita basal nos grupos de tratamento que receberam pegvisomanto (vide figura 2 e tabela 1).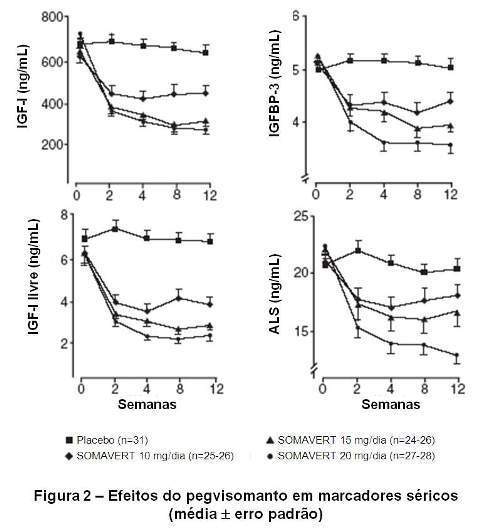
O nível sérico de IGF-I foi normalizado no final do estudo (semana 12) em 10%, 39%, 75% e 82% dos indivíduos tratados com placebo, Somavert® (pegvisomanto) 10 mg/dia, 15 mg/dia ou 20 mg/dia, respectivamente (vide figura 3).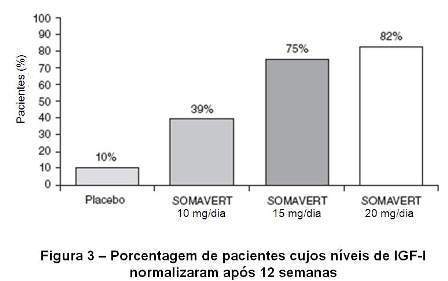
Diferenças estatisticamente significantes em relação ao placebo (p < 0,05) foram observadas quanto a melhora da pontuação total no escore de sinais e sintomas para todos os grupos de dose comparados ao placebo.
A tabela 2 mostra os efeitos do tratamento com Somavert® no tamanho do anel (tamanho padrão de joalheiro convertido para um escore numérico variando de 1 a 63) e em ambos os escores total e individual para sinais e sintomas de acromegalia. Cada escore individual foi baseado em uma escala ordinal de nove pontos (0=ausente e 8=grave e incapacitante) e o escore total foi derivado da soma dos escores individuais. Os escores basais médios foram os seguintes: tamanho do anel=47,1; sinais e sintomas totais=15,2; edema de tecidos moles=2,5; artralgia=3,2; cefaleia=2,4; transpiração=3,3 e fadiga =3,7.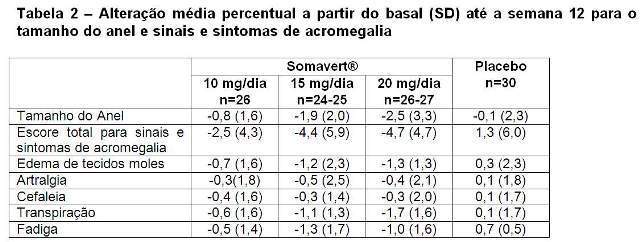
Quando pacientes com acromegalia receberam uma dose de ataque de Somavert® seguida por uma dose fixa diária, a elevação do hormônio do crescimento foi inversamente proporcional à queda em IGF-I e geralmente estabilizou na semana 2. As concentrações séricas do hormônio do crescimento (GH) também permaneceram estáveis em pacientes tratados com Somavert® por até 18 meses (vide figura 4).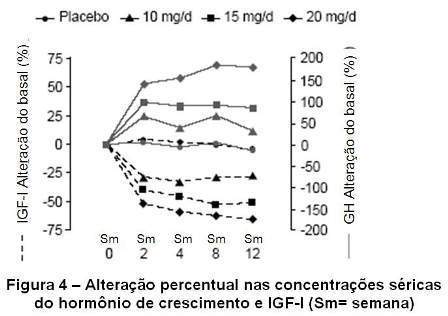
Foi realizado um estudo de corte aberto de longa duração com 38 pacientes acromegálicos, com titulação da dose, durante pelo menos 12 meses consecutivos de administração diária de pegvisomanto (média = 55 semanas). A concentração média de IGF-I neste estudo caiu de 917 ng/mL (± 356) para 299 ng/mL (± 134) no grupo tratado com pegvisomanto, com 92% atingindo uma concentração normal de IGF-I (ajustada para a idade).
Caract. farmacológicas.
Propriedades Farmacodinâmicas
O pegvisomanto é produzido em E. Coli por tecnologia de DNA recombinante. É uma proteína contendo 191 resíduos de aminoácidos para os quais vários polímeros de polietilenoglicol (PEG) estão covalentemente ligados (predominantemente 4 a 6 PEG/molécula de proteína). O pegvisomanto é um análogo do hormônio de crescimento humano (GH) geneticamente modificado para agir como antagonista do receptor do hormônio de crescimento.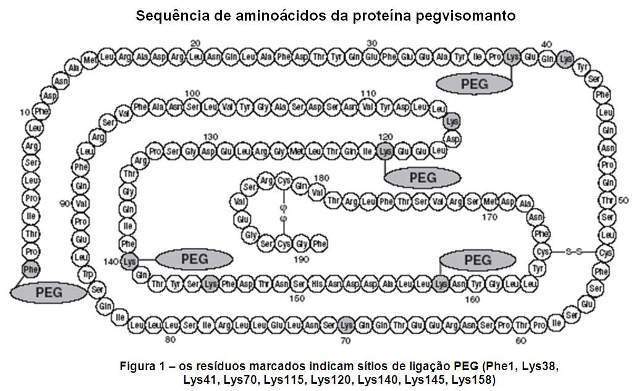
O pegvisomanto liga-se seletivamente aos receptores do hormônio de crescimento na superfície das células, bloqueando a ligação do hormônio de crescimento endógeno, interferindo, dessa forma, na transdução do sinal intracelular do hormônio de crescimento. O pegvisomanto é altamente seletivo para o receptor de GH, e não apresenta reação cruzada com outros receptores de citocina, incluindo a prolactina. A inibição da ação do hormônio de crescimento pelo pegvisomanto leva à redução das concentrações séricas do fator do crescimento semelhante à insulina-I (IGF-I), bem como das outras proteínas séricas responsivas ao hormônio de crescimento, como o IGF-I livre, a subunidade ácido-lábil do IGF-I (ALS) e a proteína de ligação do fator de crescimento semelhante à insulina-3 (IGFBP-3).
Propriedades Farmacocinéticas
Absorção: a absorção do pegvisomanto após administração subcutânea é lenta e prolongada e, em geral, concentrações séricas máximas de pegvisomanto são atingidas em 33-77 horas após a administração. O nível médio de absorção de uma dose subcutânea de 20 mg foi de 57% em relação a uma dose intravenosa.
Distribuição: o volume aparente médio de distribuição do pegvisomanto é de 7 L (com coeficiente de variação de 12%), o que sugere que o pegvisomanto não seja extensivamente distribuído entre os tecidos. Após administração subcutânea única, a exposição ao pegvisomanto (Cmax, AUC) aumenta desproporcionalmente com o aumento da dose. As concentrações sanguíneas médias (± SEM) de pegvisomanto após 12 semanas de tratamento com doses diárias de 10, 15 e 20 mg foram de, respectivamente, 6600 ± 1330; 16000 ± 2200; e 27000 ± 3100 ng/mL.
A média do clearance sistêmico corporal total após doses múltiplas é estimada como sendo de 28 mL/h para doses subcutâneas, variando de 10 mg/dia a 20 mg/dia. O clearance renal do pegvisomanto é desprezível correspondendo a menos de 1% do clearance corporal total. O pegvisomanto é lentamente eliminado do soro, com estimativas médias de meia-vida variando geralmente de 74 a 172 horas após doses únicas ou múltiplas. O metabolismo deste fármaco não foi estudado.
A farmacocinética do pegvisomanto é semelhante em voluntários saudáveis normais e em pacientes com acromegalia, apesar de indivíduos com maior massa corpórea terem a tendência a apresentar clearance corporal total maior do que indivíduos com menor massa corpórea. Por este motivo, pode ser necessário administrar doses maiores de pegvisomanto nestes indivíduos.
Metabolismo e eliminação: a molécula de pegvisomanto contém ligações covalentes a polímeros de polietilenoglicol, o que torna a velocidade de clearance reduzida. O clearance de pegvisomanto observado após doses múltiplas é mais baixo do que o observado após uma única dose. O clearance sistêmico corporal médio de pegvisomanto após doses múltiplas subcutâneas de 10 a 20 mg/dia varia de 36 a 28 ml/h respectivamente. O clearance de pegvisomanto costuma aumentar de acordo com o peso corporal. O pegvisomanto é eliminado do soro sanguíneo com uma meia-vida de aproximadamente 6 dias, tanto após dose única como doses múltiplas. Menos de 1% da dose administrada é recuperada na urina após 96 horas. A rota de eliminação do pegvisomanto não foi estudada em humanos.
Populações especiais:
Renal: nenhum estudo farmacocinético foi conduzido em pacientes com insuficiência renal.
Hepático: nenhum estudo farmacocinético foi conduzido em pacientes com insuficiência hepática.
Geriátrico: nenhum estudo farmacocinético foi conduzido em pacientes idosos.
Pediátrico: nenhum estudo farmacocinético foi conduzido em pacientes pediátricos.
Gênero: nenhum efeito farmacocinético relacionado ao gênero (sexo masculino ou feminino) dos pacientes foi observado durante a análise farmacocinética de uma população.
Raça: efeitos farmacocinéticos do pegvisomanto relacionados à raça não foram estudados.
Dados de Segurança Pré-Clínicos
Os dados pré-clínicos não revelaram riscos especiais para humanos com base nos estudos convencionais de toxicidade de dose repetida em ratos e macacos e de potencial carcinogênico em ratos. No entanto, devido à resposta farmacológica acentuada em macacos, não foram estudadas exposições sistêmicas mais elevadas do que aquelas atingidas em pacientes nas doses terapêuticas. Com exceção de um teste em coelhos, nenhum outro estudo de toxicidade reprodutiva foi conduzido.
Contraindicações.
Somavert® (pegvisomanto) é contraindicado a pacientes com hipersensibilidade ao princípio ativo ou a qualquer componente da fórmula.
Advertências e precauções.
Somavert® (pegvisomanto) só deve ser administrado por via subcutânea.
Os tumores hipofisários secretores de hormônio de crescimento algumas vezes podem se expandir causando sérias complicações (por exemplo, defeitos de campo visual). O tratamento com Somavert® não reduz o tamanho do tumor na hipófise. Todos os pacientes que apresentarem esses tumores devem ser monitorados rigorosamente a fim de evitar qualquer progressão eventual do tamanho do tumor durante o tratamento.
Somavert® é um antagonista potente da ação do hormônio de crescimento. Os pacientes devem ser monitorados quanto a sinais e sintomas de deficiência do hormônio do crescimento relativa, pois pode ocorrer um estado de deficiência funcional deste hormônio decorrente da administração de Somavert®, apesar da presença de níveis séricos elevados de hormônio de crescimento.
Somavert® produz uma reação cruzada com os testes disponíveis no mercado para dosagem dos níveis séricos de hormônio do crescimento, resultando em níveis séricos superestimados deste hormônio. Além disso, o próprio tratamento com Somavert® produz elevação nos níveis de hormônio do crescimento. Desta maneira, os níveis séricos de hormônio do crescimento não podem ser utilizados para avaliar o tratamento com Somavert®. Em contrapartida, as concentrações séricas de IGF-I devem ser monitoradas e mantidas dentro do intervalo normal ajustado para a idade.
Antes de iniciar o tratamento com Somavert® deve-se avaliar os níveis séricos basais de alanina aminotrasferase (ALT) e aspartato transaminase (AST), e após o início do tratamento, a cada 4 a 6 semanas, nos primeiros 6 meses de terapia. Deste ponto em diante, a avaliação deve ser realizada periodicamente ou a qualquer momento, caso o paciente apresente sintomas sugestivos de hepatite. Evidências de obstrução do trato biliar devem ser consideradas em pacientes com elevação nos níveis de ALT e AST ou em pacientes com história prévia de tratamento com análogo da somatostatina.
O tratamento com Somavert® não deve ser iniciado ou continuado caso o paciente apresente sinais de doenças no fígado, a menos que uma detalhada avaliação hepática seja realizada.
A tabela 3 lista as recomendações relacionadas ao início do tratamento com Somavert® baseando-se nos resultados destes testes hepáticos.
Se um paciente desenvolve elevações de testes hepáticos, ou qualquer outro sinal ou sintoma de disfunção hepática enquanto receber Somavert®, é recomendado o seguinte gerenciamento do paciente (vide tabela 4):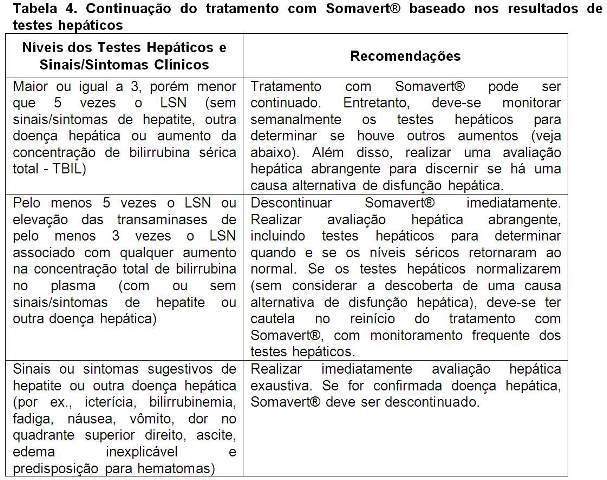
Durante o tratamento com Somavert®, pode haver a necessidade de redução das doses de insulina ou hipoglicemiantes orais em pacientes sob terapia anti-diabética, porque o pegvisomanto aumenta a sensibilidade à insulina e a tolerância à glicose (vide "Interações Medicamentosas").
Atenção: Somavert® contém açúcar, portanto, deve ser usado com cautela em diabéticos.
Uso durante a Gravidez
Estudos de reprodução realizados em coelhos revelaram não haver evidências de efeitos teratogênicos em doses de Somavert® até dez vezes maiores do que a dose recomendada para humanos.
Os estudos em animais são insuficientes em relação aos efeitos sobre a gravidez, desenvolvimento embrionário/fetal, parto ou desenvolvimento pós-natal (vide "Dados de Segurança Pré-Clínicos").
Não existem dados disponíveis para o uso de Somavert® em mulheres grávidas.
Somavert® só deve ser utilizado durante a gravidez se o benefício justificar o risco potencial ao feto.
Somavert® é um medicamento classificado na categoria B de risco de gravidez. Portanto, Somavert® não deve ser utilizado por mulheres grávidas sem orientação médica (vide "Advertências").
Uso durante a Lactação
Não se sabe se o pegvisomanto é excretado no leite materno humano. Como vários medicamentos são excretados no leite materno, a administração de Somavert® em mulheres que estejam amamentando deve ser considerada com cautela.
Efeitos na Habilidade de Dirigir e Operar Máquinas
Não foram conduzidos estudos sobre os efeitos de Somavert® sobre a habilidade de conduzir veículos ou operar máquinas.
USO EM IDOSOS, CRIANÇAS E OUTROS GRUPOS DE RISCO
Vide "Posologia".
Interações medicamentosas.
Não foi realizado estudo de interação. Deve-se considerar se o tratamento com análogos da somatostatina deva ser mantido. O uso de Somavert® (pegvisomanto) em combinação com outros medicamentos para o tratamento da acromegalia não foi extensivamente investigado.
Os pacientes que estiverem recebendo insulina ou hipoglicemiantes orais podem requerer redução na dose dessas substâncias ativas devido ao efeito do pegvisomanto sobre a sensibilidade à insulina (vide "Advertências").
Somavert® apresenta estrutura significativamente semelhante a do hormônio de crescimento, causando assim reação cruzada com os testes de hormônio de crescimento comercialmente disponíveis. Como as concentrações séricas das doses terapêuticas eficazes de Somavert® são em geral 100 a 1.000 vezes maiores do que as concentrações séricas reais do hormônio de crescimento observadas em pacientes acromegálicos, a determinação das concentrações séricas de hormônio de crescimento apresentarão resultados falsos nos ensaios de hormônio de crescimento comercialmente disponíveis. Portanto, o tratamento com Somavert® não deve ser monitorado ou ajustado com base nas concentrações séricas de hormônio de crescimento relatadas por estes ensaios.
Cuidados de armazenamento.
Somavert® (pegvisomanto) deve ser armazenado sob refrigeração (entre 2 e 8°C). Não congelar. Manter o frasco dentro da embalagem original a fim de mantê-lo protegido da luz.
Utilizar Somavert® imediatamente após a reconstituição. Caso não seja possível, a solução reconstituída pode ser mantida em temperatura ambiente (entre 15 e 25°C) na seringa ou no frasco, porém deve ser utilizado dentro de, no máximo, 6 horas. Caso não seja utilizada neste período, a solução deve ser descartada.
Posologia e modo de usar.
MODO DE USAR E CUIDADOS DE CONSERVAÇÃO DEPOIS DE ABERTO
Instruções para Uso
Somavert® (pegvisomanto) deve ser utilizado por via subcutânea.
O diluente que acompanha Somavert® contém 8 mL de água para injetáveis, porém somente 1 mL é necessário para a diluição do medicamento. O restante deve ser descartado.
Para reconstituir Somavert®, injete 1 ml do diluente (água estéril para injeção) que encontra-se na mesma embalagem de Somavert®, no frasco que contém o pó liofilizado, direcionando o jato da água contra a parede do frasco. Segure o frasco entre as palmas das mãos e gire o frasco suavemente para dissolver o pó. Não agite vigorosamente o frasco, pois pode ocorrer a desnaturação da substância ativa pegvisomanto.
Após a reconstituição, cada frasco de Somavert conterá 10, 15 ou 20 mg de pegvisomanto em 1 ml de solução. A solução deve ser límpida após a reconstituição. Se a solução estiver turva ou contiver material particulado, o produto não deve ser utilizado. Apenas uma dose deve ser administrada por frasco e a solução deve ser administrada logo após a reconstituição.
Apenas para uso único. Descartar devidamente qualquer produto não utilizado.
Para maiores informações, vide "Folheto de Instruções" que acompanha a embalagem do produto.
Cuidados de Conservação Depois de Aberto
Somavert® deve ser armazenado sob refrigeração (entre 2 e 8°C). Não congelar. Manter o frasco dentro da embalagem original a fim de mantê-lo protegido da luz.
Utilizar Somavert® imediatamente após a reconstituição. Caso não seja possível, a solução reconstituída pode ser mantida em temperatura ambiente (entre 15 e 25°C) na seringa ou no frasco, porém deve ser utilizado dentro de, no máximo, 6 horas. Caso não seja utilizada neste período, a solução deve ser descartada.
POSOLOGIA
Administração
Somavert® (pegvisomanto) deve ser utilizado uma vez ao dia por via subcutânea.
Para os diferentes esquemas posológicos encontram-se disponíveis as seguintes apresentações: Somavert® 10 mg e Somavert® 15 mg.
Somavert® 10 mg ou 15 mg contém 10 mg ou 15 mg de pegvisomanto em 1 mL da solução reconstituída, respectivamente.
O tratamento deve ser iniciado sob supervisão de um médico especializado no tratamento da acromegalia.
Os níveis séricos de IGF-I devem ser determinados antes do início da terapia.
Deve-se administrar uma dose de ataque de 80 mg de pegvisomanto por via subcutânea sob supervisão médica. Após esta dose inicial, Somavert® 10 mg reconstituído em 1 mL de diluente deve ser administrado uma vez por dia por via subcutânea. O local da administração deve ser revezado diariamente a fim de evitar lipohipertrofia.
Os ajustes de dose devem ser feitos com base nos níveis séricos de IGF-I. As concentrações séricas de IGF-I devem ser medidas a cada 4 a 6 semanas e ajustes de dose apropriados devem ser feitos aumentando-se 5 mg/dia a fim de manter a concentração sérica de IGF-I dentro do intervalo normal ajustado para a idade aliviando os sinais e sintomas da acromegalia.
A dose máxima não deve ser superior a 30 mg/dia.
Uso em Idosos
Não é necessário ajuste de doses em idosos.
Uso em Crianças
A segurança e a eficácia de Somavert® em crianças ainda não foram estabelecidas.
Uso em Pacientes com Insuficiência Hepática ou Renal
A segurança e a eficácia de Somavert® em pacientes com insuficiência renal ou hepática ainda não foram estabelecidas.
Uso em Pacientes Diabéticos
A sensibilidade à insulina pode aumentar após o início do tratamento com Somavert®. O risco de hipoglicemia foi observado em alguns pacientes diabéticos tratados com insulina ou com hipoglicemiantes orais durante o tratamento com Somavert®. Portanto, em pacientes com diabetes mellitus, pode ser necessário reduzir a dose da insulina ou do hipoglicemiante oral (vide "Advertências" e "Interações Medicamentosas").
Reações adversas.
Estudos clínicos:
Em um estudo clínico placebo-controlado, os eventos descritos a seguir foram reportados em pelo menos 2 pacientes e em uma frequência maior em pacientes tratados com Somavert® (pegvisomanto) do que naqueles pacientes tratados com placebo.
Reações adversas:
Gerais/condições do local da administração: dor, reação no local da injeção (incluindo hipersensibilidade no local da injeção), dor no peito, edema periférico, hipertrofia no local da injeção (exemplo: lipohipertrofia).
Sistema vascular: hipertensão.
Infecções e infestações: gripe, infecção e sinusite.
Dano, envenenamento e complicações de procedimento: ferimento.
Tecido conjuntivo e musculoesquelético: dor nas costas.
Sistema gastrintestinal: náusea, diarreia.
Testes laboratoriais: teste anormal das funções do fígado.
Sistema nervoso: tontura, parestesia.
O desenvolvimento de anticorpos anti-hormônio de crescimento isolados com baixa titulação foi observado em 16,9% dos pacientes tratados com Somavert®. O significado clínico destes anticorpos é desconhecido.
Período pós-comercialização
As seguintes reações adversas foram identificadas durante o período pós-comercialização de Somavert®. Essas reações são relatos voluntários de uma população de tamanho incerto, por isso, não é possível estimar sua frequência de forma confiável.
Sistema imune: reações de hipersensibilidade sistêmica incluindo reações anafilactoides/anafiláticas, laringoespasmo, angioedema, reações generalizadas da pele (rash, eritema, prurido, urticária). Alguns pacientes necessitaram de hospitalização. Sobre re-administração, os sintomas não voltaram a ocorrer em todos os pacientes.
ATENÇÃO: ESTE É UM MEDICAMENTO NOVO E, EMBORA AS PESQUISAS TENHAM INDICADO EFICÁCIA E SEGURANÇA ACEITÁVEIS PARA COMERCIALIZAÇÃO, EFEITOS INDESEJÁVEIS E NÃO CONHECIDOS PODEM OCORRER.
Superdose.
Um paciente que realizou a auto-administração de 80 mg/dia de Somavert® (pegvisomanto) por sete dias, não apresentou reações adversas clinicamente significativas relacionadas à superdose.
Em casos de superdose, a administração de Somavert® deve ser interrompida e não deve ser reiniciada até que os níveis de IGF-I estejam dentro ou acima do intervalo normal.
Dizeres legais.
MS - 1.0216.0178
VENDA SOB PRESCRIÇÃO MÉDICA.