SAUMYA
SUN PHARMA
lenvatinibe
Antineoplásico. Inibidor da tirosina-quinase.
Apresentações.
Saumya 4 mg e 10 mg, embalagem com 30 cápsulas duras.
USO ORAL
USO ADULTO
Composição.
Cada cápsula dura de Saumya 4 mg contém: mesilato de lenvatinibe 4,9 mg (equivalente a 4 mg de lenvatinibe), excipientes q.s.p 1 cápsula dura
Cada cápsula dura de Saumya 10 mg contém: mesilato de lenvatibibe 12,25 mg (equivalente a 10 mg de lenvatinibe), excipientes q.s.p 1 cápsula dura
Excipientes: manitol, óxido de magnésio, celulose microcristalina, hiprolose de baixa substituição (LH-21), hiprolose, talco, hipromelose, dióxido de titânio, óxido de ferro vermelho e óxido de ferro amarelo, goma laca, óxido de ferro preto, propilenoglicol e hidróxido de potássio.
Informações técnicas.
1. INDICAÇÕES
Saumya é indicado para o tratamento de pacientes adultos com carcinoma diferenciado da tireoide (CDT) (papilífero, folicular ou célula de Hürthle) localmente avançado ou metastático, progressivo, refratário a radioiodoterapia (RIT).
Saumya é indicado em combinação com everolimo para o tratamento de pacientes com carcinoma de células renais avançado (CCR) após tratamento prévio com terapia anti-angiogênica.
Saumya é indicado para o tratamento de pacientes com carcinoma hepatocelular (CHC), que não receberam terapia sistêmica anterior para doença avançada ou não ressecável.
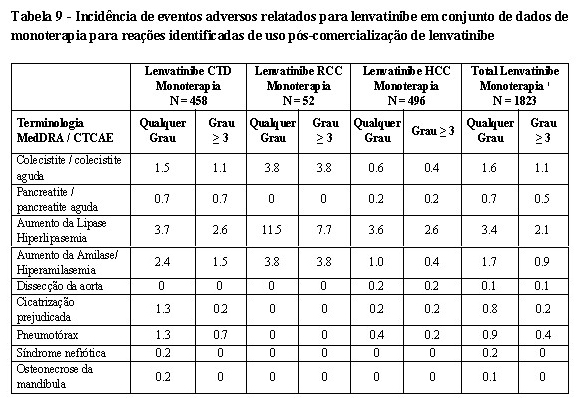
2. RESULTADOS DE EFICÁCIA
Eficácia e segurança clínica
Câncer de Tireoide
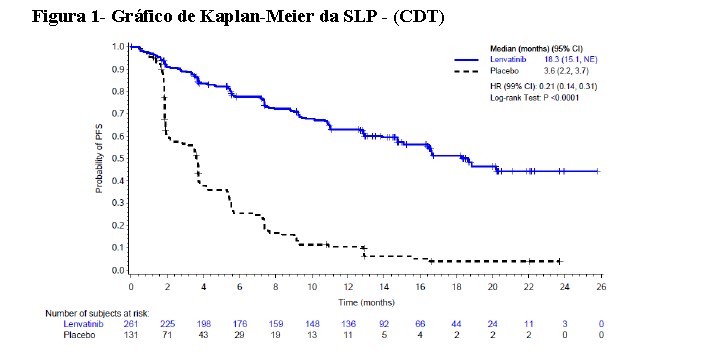
Um estudo multicêntrico, randomizado, duplo-cego, controlado com placebo (SELECT)* foi conduzido em 392 pacientes com câncer diferenciado de tireoide, progressivo e refratário à iodoterapia (iodo radioativo) com evidência radiográfica de progressão da doença dentro de 13 meses antes da admissão. Estado refratário à iodoterapia foi definido como uma ou mais lesões mensuráveis sem nenhuma captação de iodo durante a radioterapia ou que progrediram nos últimos 12 meses, apesar de terem apresentado captação de iodo durante a radioiodoterapia, ou apresentando uma atividade cumulativa de radioiodo > 600 mCi ou 22 GBq, com a última dose administrada pelo menos 6 meses antes da entrada no estudo. A randomização foi estratificada por região geográfica (Europa, América do Norte, e outros), terapia anterior direcionada para VEGF/VEGFR (pacientes poderiam ter recebido 0 ou 1 terapia anterior direcionada para VEGF/VEGFR), e idade (≤ 65 anos ou > 65 anos). O desfecho primário de eficácia medido foi a sobrevida livre de progressão conforme determinado pela revisão radiológica independente cega utilizando os Critérios de Avaliação de Resposta em Tumores Sólidos (RECIST) 1.1. Os desfechos de eficácia secundários medidos incluíram a taxa de resposta objetiva e sobrevida global. Os pacientes do braço placebo poderiam receber tratamento com lenvatinibe no momento da progressão confirmada da doença. Os pacientes elegíveis com doença mensurável de acordo com RECIST versão 1.1 foram randomizados na proporção de 2:1 para receber lenvatinibe 24 mg uma vez ao dia (n=261) ou placebo (n=131). Os dados demográficos e as características da doença foram bem balanceados para ambos os grupos de tratamento. Dos 392 pacientes randomizados, 76,3% não tinham recebido terapias anteriores direcionadas para VEGF/VEGFR, 49,0% eram mulheres, 49,7% eram europeus, e a idade média era 63 anos. Histologicamente, 66,1% tinham diagnóstico confirmado de câncer de tireoide papilar e 33,9% tinham câncer de tireoide folicular que incluiu célula de Hürthle 14,8% e célula clara 3,8%. Metástases estavam presentes em 99% dos pacientes: pulmões em 89,3%, linfonodos em 51,5%, ossos em 38,8%, fígado em 18,1%, pleura em 16,3%, e cérebro em 4,1%. A maioria dos pacientes tinha uma condição de desempenho ECOG de 0; 42,1% tinham uma condição de 1; 3,9% tinham uma condição acima de 1. A mediana da atividade cumulativa de radioiodo administrado antes da entrada no estudo foi de 350 mCi (12,95 GBq). Um prolongamento estatisticamente significativo da SLP foi demonstrado em pacientes tratados com lenvatinibe em comparação com os que receberam placebo. O efeito positivo sobre a SLP foi similar nos subgrupos que receberam 0 ou 1 terapia anterior direcionada para VEGF/VEGFR (ver Tabela 1). Além disso, o efeito positivo sobre a SLP foi observado através dos subgrupos de idade (abaixo ou acima de 65 anos), sexo, raça, subtipo histológico e região geográfica. Após confirmação de progressão da doença por revisão independente, 109 (83,2%) pacientes randomizados para placebo migraram para o tratamento com lenvatinibe na fase aberta.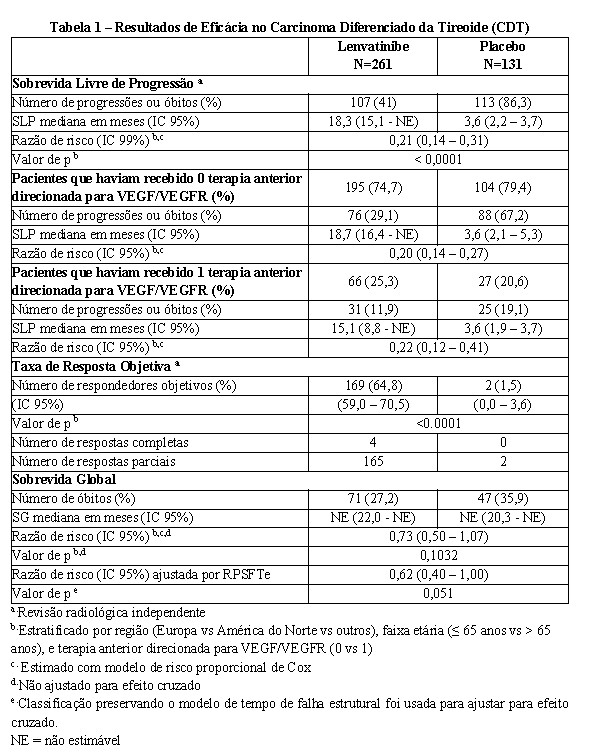
Câncer de células renais (CCR)
Um estudo multicêntrico, randomizado, aberto para determinar a segurança e eficácia do lenvatinibe administrado sozinho ou em combinação com everolimo em indivíduos com CCR não ressecáveis, avançado ou metastático. O estudo consistiu numa Fase 1b para definição de dose e uma Fase 2. O estudo de Fase 2 envolveu um total de 153 pacientes com carcinoma de células renais avançado ou metastático (CCR) após 1 tratamento previamente direcionado para VEGF. Os pacientes tiveram confirmação histológica do CCR predominante de células claras, evidência radiográfica de progressão da doença de acordo com RECIST versão 1.1, um tratamento prévio com VEGF e avaliação de 0 ou 1 de ECOG.
Os pacientes foram randomizados em um dos três braços: lenvatinibe 18 mg + everolimo 5 mg, lenvatinibe 24 mg, ou everolimo 10 mg usando uma relação 1: 1: 1. Os pacientes foram estratificados pelo nível de hemoglobina (≤ 13 g/dL vs > 13 g/dL para os machos e ≤ 11,5 g/dL vs > 11,5 g/dL para as fêmeas) e cálcio sérico corrigido (≥ 10 mg/dL < 10 vs. mg/dL). O desfecho primário de eficácia, com base na resposta tumoral avaliada pelo investigador, foi a sobrevida livre de progressão (SLP) do braço lenvatinibe + everolimo vs o braço everolimo e do braço lenvatinibe vs o braço everolimo. Outros desfechos de eficácia incluíram a sobrevivência global (SG) avaliada pelo investigador e a taxa de resposta objetiva (TRO).
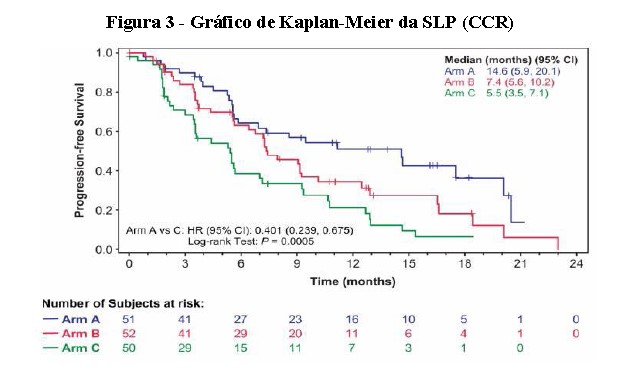
As avaliações dos tumores foram avaliadas de acordo com os Critérios de Avaliação da Resposta em Tumores Sólidos Versão 1.1 (RECIST). O braço de lenvatinibe + everolimo mostrou uma melhora estatisticamente significativa e clinicamente significativa na SLP em comparação com o braço everolimo (Figura 3). O braço de lenvatinibe mostrou também uma melhora na SLP em comparação com o braço everolimo. A sobrevida global foi maior no braço lenvatinibe + everolimo (Figura 4).
O braço de lenvatinibe + everolimo apresentou melhora estatisticamente significativa e clinicamente significativa na SLP (Hazard ratio [HR] = 0,50, [IC95%: 0,26, 0,79], P = 0,003) em comparação com o braço everolimo. Resultados para TRO foram consistentes com os das avaliações dos investigadores, 35,3% no braço lenvatinibe + everolimo, com 1 resposta completa e 17 respostas parciais; nenhum paciente teve uma resposta objetiva no braço everolimo (valor P < 0,0001) em favor do braço lenvatinibe + everolimo.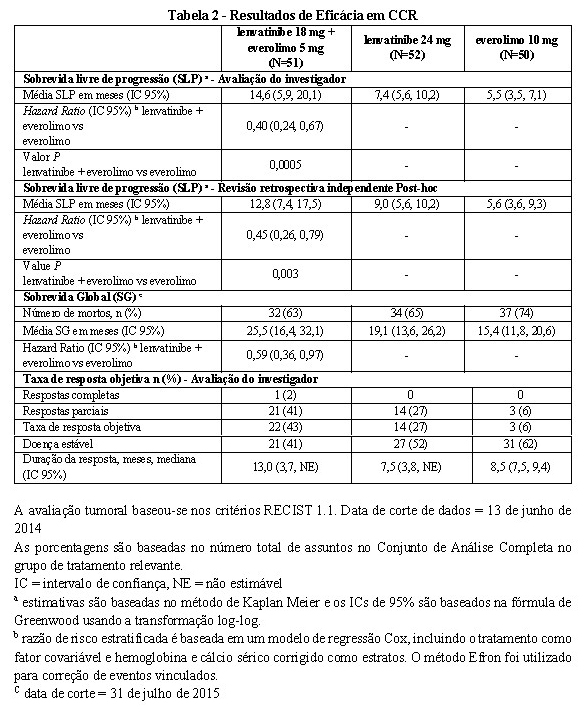
Experiência Clínica - Carcinoma Hepatocelular
Um estudo multicêntrico aberto foi conduzido em 954 pacientes com carcinoma hepatocelular (CHC) não ressecável que foram randomizados para mesilato de lenvatinibe (12 mg [peso corporal basal ≥ 60 kg] ou 8 mg [peso corporal basal < 60 kg]) administrado por via oral uma vez ao dia ou sorafenibe 400 mg administrado por via oral duas vezes ao dia.
Os pacientes deveriam ter um diagnóstico confirmado histologicamente ou citologicamente de CHC não ressecável ou um diagnóstico clinicamente confirmado de CHC de acordo com os critérios da Associação Americana para o Estudo de Doenças Hepáticas, incluindo cirrose de qualquer etiologia ou com infecção crônica de hepatite B ou C. Os pacientes apresentavam pelo menos 1 lesão alvo hepática ou não-hepática mensurável de acordo com o mRECIST, e função hepática, da medula óssea, de coagulação sanguínea, renal e pancreática adequadas. Os pacientes foram estratificados por região, presença ou ausência de invasão macroscópica da veia porta (MPVI) ou disseminação extra-hepática (EHS) ou ambos, Eastern Cooperative Oncology Group Performance Status (ECOG PS) 0 ou 1 e Peso Corporal ( < 60 kg ou ≥ 60 kg). A maioria dos pacientes em ambos os braços de tratamento apresentava ECOG PS de 0 no basal (63%), classificação Child- Pugh score de 5 (76%) e peso ≥ 60 kg (69%). A idade mediana foi de 62 anos, 84% eram do sexo masculino, 16% eram do sexo feminino, 69% eram asiáticos, 1% era de negros e 29% eram brancos.
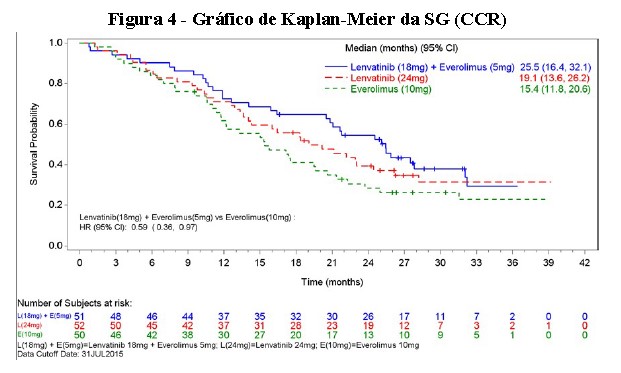
O lenvatinibe não foi inferior para Sobrevida Global (SG) ao sorafenibe 400 mg duas vezes ao dia. A SG mediana foi de 13,6 meses em comparação com 12,3 meses para o sorafenibe com HR = 0,92 [IC 95% de (0,79, 1,06)].
Com base na avaliação do investigador avaliada de acordo com o mRECIST, o tratamento com lenvatinibe resultou em melhora estatisticamente significativa (P < 0,00001) e clinicamente significativa em relação ao sorafenibe nos desfechos secundários de SLP e TRO. O tratamento com lenvatinibe prolongou significativamente a TPP em comparação com o sorafenibe, com uma TPP mediana que foi mais do que o dobro que a de sorafenibe. A revisão retrospectiva independente da imagem corroborou os desfechos secundários de SLP, TPP e TRO. Estes resultados de eficácia estão resumidos na Tabela 3 e nas Figuras 5, 6 e 7.
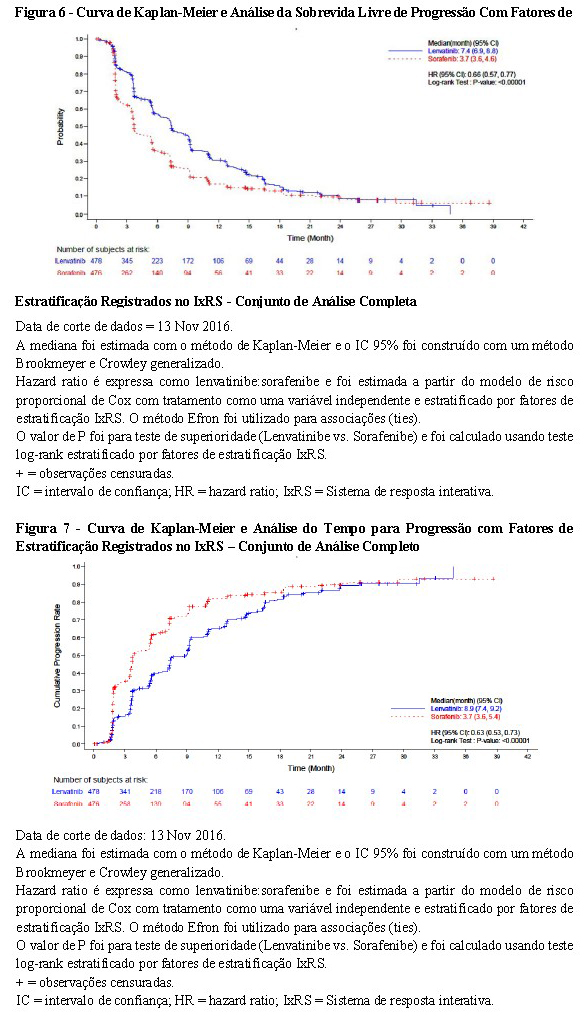
Avaliação da Qualidade de Vida (QV) em Pacientes com CHC
Foram administrados três questionários de QV, EORTC QLQ-C30, EORTC QLQ-CHC18 e o EQ-5D-3L. Em comparação com os pacientes tratados com lenvatinibe, aqueles tratados com sorafenibe experimentaram maior risco de tempo mais rápido para piora clinicamente significativa dos sintomas e função para os domínios de Nutrição (p nominal = 0,0113) e Imagem Corporal (p nominal = 0,0051) do EORTC QLQ-CHC18 bem como Dor (p nominal = 0,0105), Diarreia (p nominal < 0,0001) e Funcionamento do Papel (p nominal = 0,0193) do EORTC QLQ-C30.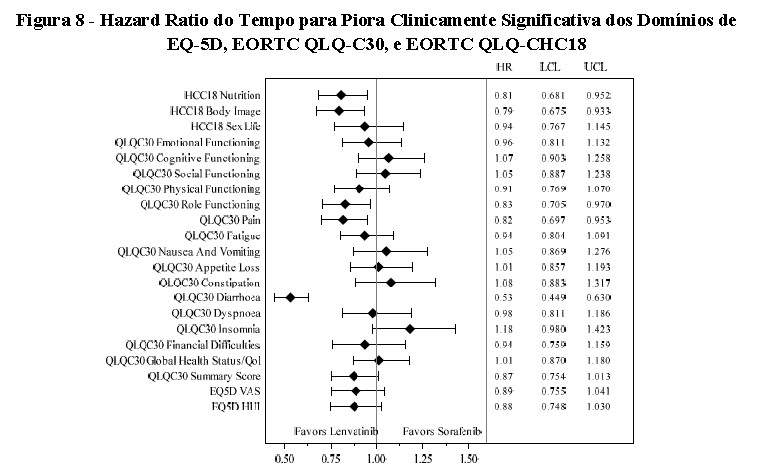
3. CARACTERÍSTICAS FARMACOLÓGICAS
Mecanismo de Ação
O lenvatinibe é um inibidor de múltiplos receptores de tirosina quinase (RTK) que inibe seletivamente as atividades dos receptores de fator de crescimento endotelial vascular (VEGF), receptores VEGFR1 (FLT1), VEGFR2 (KDR), e VEGFR3 (FLT4), além de outros RTKs relacionados à via pró-angiogênica e oncogênica, incluindo os receptores de fator de crescimento de fibroblastos (FGF), receptores FGFR1, 2, 3, e 4; o receptor de fator de crescimento derivado de plaquetas (PDGF) receptor PDGFRa, KIT e RET.
Além disso, o lenvatinibe apresentou atividade seletiva, direta antiproliferativa nas linhagens de células hepatocelulares dependentes da sinalização de FGFR ativado, a qual é atribuída à inibição da sinalização de FGFR pelo lenvatinibe.
A combinação de lenvatinibe e everolimo demonstrou atividade anti-angiogênica e antitumoral aumentada, em comparação com cada fármaco isolado, como demonstrado pela diminuição da proliferação de células endoteliais humanas, formação de tubo e sinalização VEGF in vitro e volume tumoral em modelos de xenoenxerto de hamster de câncer de células renais humanas.
Embora não tenha sido estudado diretamente com lenvatinibe, o mecanismo de ação para hipertensão é postulado para ser mediado pela inibição de VEGFR2 em células endoteliais vasculares. Da mesma forma, embora não seja estudado diretamente, o mecanismo de ação para proteinúria é mediado por VEGFR1 e VEGFR2 nos podócitos do glomérulo.
O mecanismo de ação para o hipotireoidismo não está totalmente elucidado.
O mecanismo de ação para o agravamento da hipercolesterolemia com a combinação não foi estudado diretamente e não está totalmente elucidado.
Embora não seja estudado diretamente, o mecanismo de ação para o agravamento da diarreia com a combinação é confirmado pelo comprometimento da função intestinal relacionada aos mecanismos de ação para os agentes individuais - VEGF / VEGFR e inibição c-KIT pela ligação de lenvatinibe com Inibição mTOR / NHE3 por everolimo.
Propriedades Farmacocinéticas
Absorção
O lenvatinibe é rapidamente absorvido após administração oral com tmáx geralmente observado 1 a 4 horas após a dose. Os alimentos não afetam o grau de absorção, mas diminuem a taxa de absorção. Quando administrado com alimentos a indivíduos saudáveis, o pico das concentrações plasmáticas é retardado em 2 horas.
Em indivíduos com tumores sólidos que receberam doses únicas e múltiplas de lenvatinibe uma vez ao dia, a exposição ao lenvatinibe (Cmáx e AUC) aumentou de forma diretamente proporcional à dose administrada no intervalo de 3,2 a 32 mg. O lenvatinibe apresentou acúmulo mínimo no estado de equilíbrio. No estudo SELECT, a AUC mediana de lenvatinibe de uma dose normalizada para 24 mg foi 3490 ng*h/mL (faixa 1410 a 10700 ng*h/mL) e mostrou variabilidade moderada (CV de 38 %). Lenvatinibe mostra acumulação mínima no estado de equilíbrio. Neste intervalo, o índice mediano de acúmulo (Rac) variou de 0,96 (20 mg) a 1,54 (6,4 mg).
Distribuição
A taxa de ligação in vitro de lenvatinibe a proteínas plasmáticas humanas foi alta e variou de 98% a 99% (0,3 - 30 mg/mL, mesilato). Esta ligação ocorreu principalmente com a albumina e houve mínima ligação à glicoproteína ácida a1 e à c-globulina. Em indivíduos com câncer de tireoide o valor da população para o clearance aparente médio de lenvatinibe foi estimado ser 6,56 L/h, que foi independente da dose (3,2 a 32 mg) e tempo. Volumes de distribuição aparentes dos compartimentos central e 2 periféricos foram estimados serem 49,3, 30,7 e 37,1 L, respectivamente. In vitro, a razão de concentração de sangue-para-plasma de lenvatinibe variou de 0,589 a 0,608 (0,1 - 10 mg/mL, mesilato). O lenvatinibe é um substrato para a P-gp e para a BCRP. O lenvatinibe não é um substrato para a OAT1, OAT3 (transportador de ânions orgânicos), OATP1B1, OATP1B3 (polipeptídeo transportador de ânions orgânicos), OCT1, OCT2 (transportador de cátions orgânicos), MATE1, MATE2-K nem para a BSEP (bomba de transporte de sais biliares).
Biotransformação
In vitro, o citocromo P450 3A4 foi a isoforma de citocromo predominante ( > 80%) envolvida no metabolismo do lenvatinibe mediado por P450. Porém, dados in vivo indicam que vias metabólicas não mediadas pelo citocromo P450 contribuem com uma porção significante do metabolismo geral do lenvatinibe. Consequentemente, in vivo, os indutores e inibidores da CYP3A4 tiveram um efeito mínimo sobre a exposição ao lenvatinibe. Em microssomas hepáticos humanos, a forma desmetilada de lenvatinibe (M2) foi identificada como o metabólito principal. M2´e M3´, os principais metabólitos excretados nas fezes humanas, foram formados a partir do M2 e do lenvatinibe, respectivamente, por aldeído oxidase (AO). Nas amostras de plasma coletadas até 24 horas após a administração, o lenvatinibe constituiu 97% da radioatividade nos radiocromatogramas de plasma enquanto o metabólito M2 respondeu por mais 2,5%. Baseado na AUC0-inf, o lenvatinibe respondeu por 60% e 64% da radioatividade total no plasma e sangue, respectivamente. Os dados de um estudo de balanço de massa/excreção humana indicam que o lenvatinibe é amplamente metabolizado em humanos. As principais vias metabólicas em humanos foram identificadas como oxidação por aldeído oxidase (AO), desmetilação via CYP3A4, conjugação com glutationa com eliminação do grupo O-arila (porção clorbenzila), e combinações destas vias seguidas de biotransformações adicionais (p.ex., glucuronidação, hidrólise da porção glutationa, degradação da porção cisteína, e rearranjo intramolecular de cisteinilglicina e conjugados cisteína com dimerização subsequente). Estas vias metabólicas in vivo se alinham com os dados fornecidos nos estudos in vitro utilizando biomateriais humanos.
Eliminação
As concentrações plasmáticas decaem biexponencialmente após a Cmáx. A meia-vida exponencial terminal de lenvatinibe é de aproximadamente 28 horas. Após a administração de lenvatinibe radiomarcado a 6 indivíduos com tumores sólidos, aproximadamente dois terços e um quarto do composto radiomarcado foram eliminados nas fezes e urina, respectivamente. O metabolito M2 foi o analito predominante nas excreções (~ 5% da dose), sendo o lenvatinibe o segundo mais proeminente (~ 2,5%).
Populações Especiais
Insuficiência Hepática
A farmacocinética do lenvatinibe após uma dose única de 10 mg foi avaliada em 6 indivíduos com insuficiência hepática leve e 6 indivíduos com insuficiência hepática moderada (Child-Pugh A e Child-Pugh B, respectivamente). Uma dose de 5 mg foi avaliada em 6 indivíduos com insuficiência hepática grave (Child-Pugh C). Oito indivíduos saudáveis, demograficamente pareados serviram como controles e receberam uma dose de 10 mg. A meia-vida mediana foi comparável em indivíduos com insuficiência hepática leve, moderada, e grave, assim como naqueles com função hepática normal e variou de 26 h a 31 h. A porcentagem da dose de lenvatinibe excretada na urina foi baixa em todas as coortes ( < 2,16% entre as coortes de tratamento). A exposição ao lenvatinibe baseada na AUC0-t, não ligado e AUC0-inf, não ligado ajustada pela dose foi de 119%, 107%, e 180% do normal para indivíduos com insuficiência hepática leve, moderada, ou grave, respectivamente. Não se sabe se ocorre mudança na ligação a proteínas plasmáticas em pacientes com insuficiência hepática. Indivíduos com insuficiência hepática grave (Child Pugh C) tem habilidade reduzida para eliminar lenvatinibe como demonstrado pela AUC que aumenta (aproximadamente 2,7 vezes) (não ligada) e 1,8 vezes maior (total) comparada a indivíduos e requerem ajuste de dose (vide item 8. POSOLOGIA E MODO DE USAR).
Insuficiência renal
A farmacocinética do lenvatinibe após uma dose única de 24 mg foi avaliada em 6 indivíduos cada com insuficiência renal leve, moderada ou grave e comparada com a de 8 indivíduos saudáveis, demograficamente pareados. Indivíduos com doença em estágio terminal não foram estudados.
A exposição ao lenvatinibe, baseada em dados de AUC0-inf, foi de 101%, 90% e 122% para indivíduos normais com insuficiência renal, leve, moderada e grave, respectivamente. Não se sabe se ocorre mudança na ligação a proteínas plasmáticas em pacientes com insuficiência renal (vide item 8. POSOLOGIA E MODO DE USAR).
Sexo
Com base em uma análise de farmacocinética populacional de pacientes recebendo até 24 mg de lenvatinibe uma vez ao dia, o sexo não apresentou nenhum efeito significativo sobre o clearance aparente (CL/F) do lenvatinibe.
Idosos (65 anos de idade ou mais)
Com base em uma análise de farmacocinética populacional de pacientes recebendo até 24 mg de lenvatinibe uma vez ao dia, a idade não apresentou nenhum efeito significativo sobre o clearance aparente (CL/F) do lenvatinibe.
Raça
Com base em uma análise de farmacocinética populacional de pacientes recebendo até 24 mg de lenvatinibe uma vez ao dia, a raça (japoneses vs outros, caucasianos vs outros) não apresentou nenhum efeito significativo sobre o clearance aparente (CL/F) do lenvatinibe.
População Pediátrica
A segurança e eficácia de lenvatinibe em pacientes pediátricos (menores de 18 anos) ainda não foram estabelecidas.
Avaliação genômica dos parâmetros farmacocinéticos do lenvatinibe
Devido ao amplo metabolismo do lenvatinibe, o efeito de fenótipos de enzimas metabolizadoras do medicamento sobre o clearance do lenvatinibe foi investigado utilizando dados derivados da plataforma de genotipagem microarray do transportador e enzima metabolizadora de medicamento Affymetrix (DMET Plus). Nenhum dos fenótipos para CYP3A5, CYP1A2, CYP2A6, e CYP2C19 apresentou um impacto significativo sobre o clearance do lenvatinibe.
4. CONTRAINDICAÇÕES
Hipersensibilidade a substância ativa ou a qualquer um dos excipientes do produto.
Este medicamento não deve ser usado durante a amamentação (ver item 5. ADVERTÊNCIAS E PRECAUÇÕES - Lactação).
5. ADVERTÊNCIAS E PRECAUÇÕES
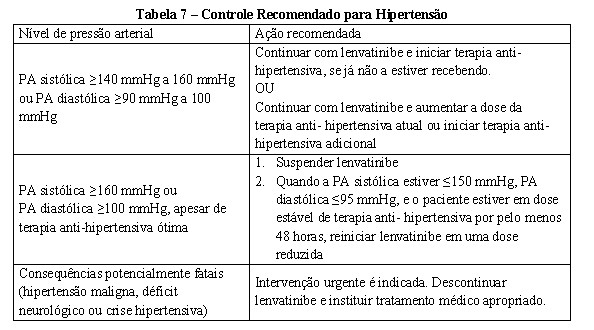
Hipertensão
A hipertensão foi relatada em pacientes tratados com lenvatinibe (ver item 9). O tempo mediano de início foi de 16 dias no estudo CDT, 34 dias no estudo CCR e 26 dias no estudo CHC.
A pressão arterial deve ser bem controlada antes do tratamento com lenvatinibe. A detecção precoce e o gerenciamento efetivo da hipertensão são importantes para minimizar a necessidade de interrupções e reduções da dose de lenvatinibe. Foram relatadas complicações graves de hipertensão mal controlada, incluindo dissecção aórtica. A pressão sanguínea deve ser monitorada após 1 semana de tratamento com lenvatinibe, a cada 2 semanas durante os primeiros 2 meses e mensalmente depois durante o tratamento. Se o paciente desenvolver uma PA sistólica ≥ 140 mmHg ou uma PA diastólica ≥ 90 mmHg, o gerenciamento ativo é indicado (veja Tabela 4 no item 8. POSOLOGIA E MODO DE USAR).
Proteinúria
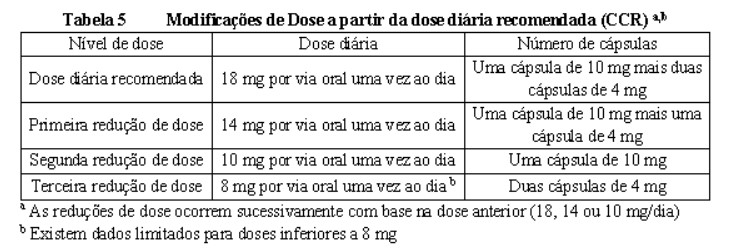
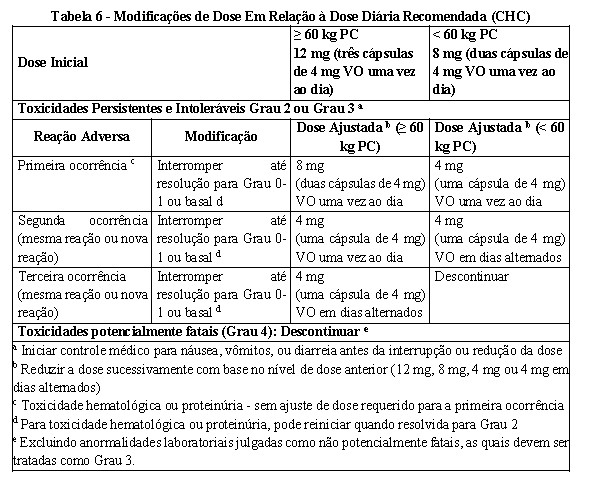
Proteinúria foi relatada em pacientes tratados com lenvatinibe. Monitorar proteínas na urina regularmente. Se for detectada proteinúria em fita reagente ≥ 2+, interrupções, ajustes ou descontinuação da dose podem ser necessários (veja Tabela 4 Câncer de tireoide, Tabela 5 CCR e Tabela 6 CHC no item 8. POSOLOGIA E MODO DE USAR). Descontinuar o tratamento com mesilato de lenvatinibe se ocorrer síndrome nefrótica
Insuficiência e Comprometimento Renal/Toxicidade Gastrointestinal
A insuficiência renal (incluindo falência renal) foi relatada em pacientes tratados com lenvatinibe (vide Reações adversas). O principal fator de risco identificado foi a desidratação/hipovolemia devido à toxicidade gastrointestinal. A toxicidade gastrointestinal deve ser ativamente gerenciada para reduzir o risco de desenvolvimento de insuficiência renal ou falência renal. Podem ser necessárias interrupções, ajustes da dose ou descontinuação (veja Tabela 4 Câncer de tireoide, Tabela 5 CCR e Tabela 6 CHC no item 8. POSOLOGIA E MODO DE USAR).
Terapias antineoplásicas anteriores
O lenvatinibe foi estudado em pacientes que receberam até 1 terapia anterior direcionada para VEGF/VEGFR, no entanto, não há dados sobre o uso de lenvatinibe imediatamente após o uso de sorafenibe ou outras terapias antineoplásicas. Pode haver um risco potencial de toxicidades aditivas a menos que haja um período de eliminação (washout) entre os tratamentos. O período de intervalo mínimo nos estudos clínicos foi de 4 semanas.
Insuficiência Cardíaca
Insuficiência cardíaca e fração de ejeção ventricular esquerda diminuída foram relatadas em pacientes tratados com lenvatinibe. Os pacientes devem ser monitorados quanto a sintomas ou sinais clínicos de descompensação cardíaca, uma vez que interrupções, ajustes, ou descontinuação de dose podem ser necessários (veja Tabela 4 Câncer de tireoide, Tabela 5 CCR e Tabela 6 CHC no item 8.
POSOLOGIA E MODO DE USAR).
Síndrome da Leucoencefalopatia Posterior Reversível (SLPR)
Foram relatados eventos de síndrome da leucoencefalopatia posterior reversível (SLPR) também conhecida como síndrome da encefalopatia reversível posterior (PRES) ( < 1%) em pacientes tratados com lenvatinibe (veja Reações adversas). O SLPR é um distúrbio neurológico que pode apresentar dor de cabeça, convulsão, letargia, confusão, função mental alterada, cegueira e outros distúrbios visuais ou neurológicos. A hipertensão leve a grave pode estar presente. A ressonância magnética é necessária para confirmar o diagnóstico de SLPR. Devem ser tomadas medidas adequadas para controlar a pressão arterial. Gerenciamento recomendado de hipertensão arterial). Em pacientes com sinais ou sintomas de SLPR, podem ser necessárias interrupções, ajustes de dose ou descontinuação (veja Tabela 4 Câncer de tireoide, Tabela 5 CCR e Tabela 6 CHC no item 8. POSOLOGIA E MODO DE USAR).
Hepatotoxicidade
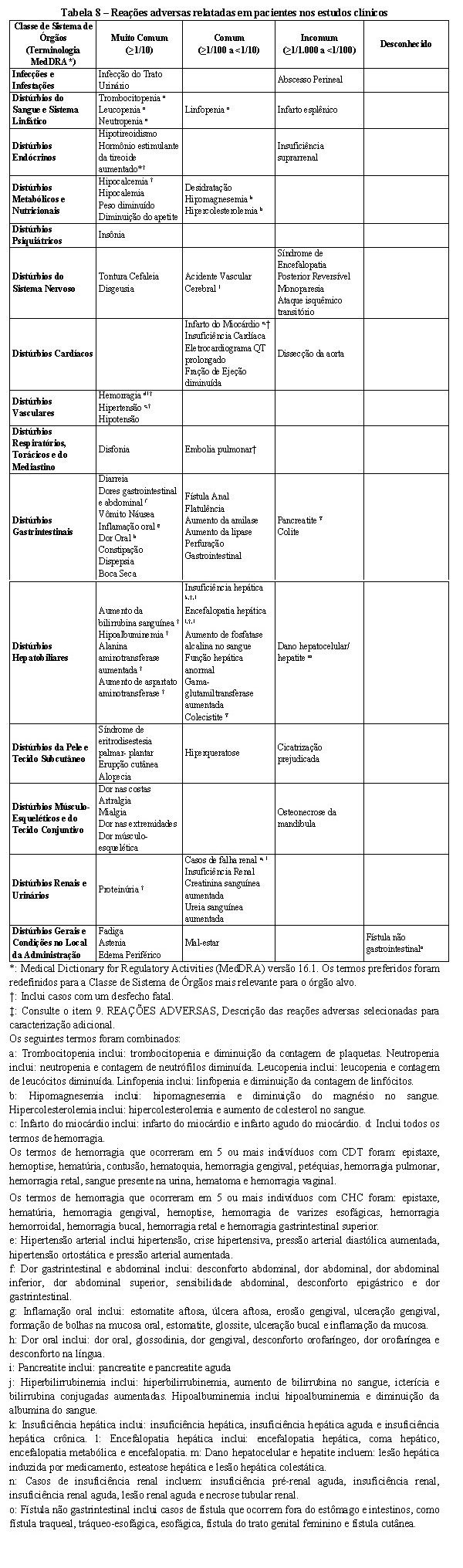
As reações adversas relacionadas ao fígado mais comumente relatadas em pacientes tratados com lenvatinibe incluíram aumentos de alanina aminotransferase (ALT), aspartato aminotransferase (AST), e bilirrubina no sangue. Insuficiência hepática e hepatite aguda ( < 1%) foram relatadas em pacientes com CDT e CCR tratados com lenvatinibe. Os eventos de insuficiência hepática foram em geral relatados em indivíduos com metástases hepáticas progressivas.
As reações adversas relacionadas ao fígado incluindo encefalopatia hepática e insuficiência hepática (incluindo reações fatais) foram relatadas em maior frequência em pacientes tratados com lenvatinibe com CHC (ver item 9. REAÇÕES ADVERSAS) do que com CDT e CCR. Pacientes com pior comprometimento hepático e/ou maior carga de tumor hepático no basal apresentaram maior risco de desenvolver encefalopatia hepática e insuficiência hepática. Encefalopatia hepática também ocorreu mais frequentemente em pacientes com 75 anos ou mais. Aproximadamente metade dos eventos de insuficiência hepática foi relatada em pacientes com progressão da doença.
Os testes de função hepática devem ser monitorados antes do início do tratamento, depois, a cada 2 semanas pelos primeiros 2 meses e mensalmente subsequentemente durante o tratamento. Pacientes com CHC devem ser monitorados para piora da função hepática incluindo encefalopatia hepática. Em caso de hepatotoxicidade, interrupções, ajustes ou descontinuação da dose podem ser necessários (veja Tabela 4 Câncer de tireoide, Tabela 5 CCR e Tabela 6 CHC no item 8. POSOLOGIA E MODO DE USAR).
Diminuição da supressão do hormônio estimulante da tireoide (TSH)
O hipotireoidismo foi relatado em pacientes tratados com lenvatinibe (veja item 9. REAÇÕES ADVERSAS). A função da tireoide, T3, T4 e TSH devem ser monitorados antes do início e periodicamente ao longo do tratamento com lenvatinibe. O hipotireoidismo deve ser tratado de acordo com a prática médica padrão para manter o estado eutiroideo.
Complicações de Cicatrização de Feridas
Não foram realizados estudos formais sobre o efeito do lenvatinibe na cicatrização de feridas. O comprometimento na cicatrização de feridas foi relatado em pacientes que receberam lenvatinibe. A interrupção temporária do lenvatinibe deve ser considerada em pacientes submetidos a procedimentos cirúrgicos importantes. Há uma experiência clínica limitada em relação ao momento da reintrodução do lenvatinibe após um procedimento cirúrgico importante. Portanto, a decisão de reintroduzir o lenvatinibe após um procedimento cirúrgico importante deve basear-se no julgamento clínico de cicatrização adequada da ferida.
Eventos Hemorrágicos
Eventos hemorrágicos graves foram notificados em doentes tratados com lenvatinibe. O evento hemorrágico mais frequentemente relatado foi epistaxe leve. No entanto, sangramentos graves relacionados a tumores foram relatados, incluindo eventos hemorrágicos fatais em pacientes tratados com lenvatinibe. O grau de invasão do tumor / infiltração dos principais vasos sanguíneos (por exemplo, artéria carótida) deve ser considerado devido ao risco potencial de hemorragia grave associada à contração/ necrose tumoral após a terapêutica com lenvatinibe.
No caso de hemorragias, podem ser necessárias interrupções da dose, ajustes ou descontinuação (veja Tabela 4 Câncer de tireoide, Tabela 5 CCR e Tabela 6 CHC no item 8. POSOLOGIA E MODO DE USAR).
Eventos Tromboembólicos Arteriais (ETAs)
Eventos tromboembólicos arteriais foram relatados em pacientes tratados com lenvatinibe. Lenvatinibe não foi estudado em pacientes que tiveram um evento tromboembólico arterial nos últimos 6 meses.
Síndrome da Eritrodistesia Palmar-Plantar (EPP)
Eventos de EPP foram relatados em 32% dos pacientes tratados com lenvatinibe comparado com 1% dos pacientes no grupo placebo. A incidência de EPP de Grau 3 ou maior foi 3% nos pacientes tratados com lenvatinibe comparado com nenhum no grupo placebo. Se EPP ocorrer, interrupção, ajuste ou descontinuação das doses pode ser necessário (veja Tabela 5 no item 8. POSOLOGIA E MODO DE USAR).
Formação de Fístula e Perfuração Gastrointestinal
Eventos de formação de fístula ou perfuração gastrointestinal e suas sequelas foram relatados em pacientes tratados com lenvatinibe. Fístulas (por exemplo, fístula gastrointestinal, broncopleural, traqueoesofágica, esofágica, cutânea, faríngea, do trato genital feminino) foram notificadas em ensaios clínicos de lenvatinibe e na experiência pós-comercialização. Além disso, o pneumotórax tem sido relatado com e sem evidência clara de uma fístula broncopleural. Alguns relatos de perfuração gastrointestinal, fístula e pneumotórax ocorreram em associação com regressão tumoral ou necrose. Na maioria dos casos de formação de fístula ou perfuração gastrointestinal, fatores de risco como cirurgia prévia ou radioterapia estavam presentes. No caso de formação de fístula ou perfuração gastrointestinal, interrupções, ajustes ou descontinuação da dose podem ser necessários. Foram notificadas perfurações gastrointestinais ou fístulas em doentes tratados com lenvatinibe (ver seção 9. REAÇÕES ADVERSAS). Na maioria dos casos, perfuração gastrointestinal e fístulas ocorreram em pacientes com fatores de risco, como cirurgia prévia ou radioterapia. No caso de uma perfuração gastrointestinal ou fístula, podem ser necessárias interrupções, ajustes ou descontinuação da dose.
(veja Tabela 4 Câncer de tireoide, Tabela 5 CCR e Tabela 6 CHC no item 8. POSOLOGIA E MODO DE USAR).
Hipocalcemia
Foi notificada hipocalcemia em doentes tratados com lenvatinibe. Monitorar os níveis de cálcio no sangue periodicamente e substituir o cálcio conforme necessário durante o tratamento com lenvatinibe. Interromper e ajustar a dose de lenvatinibe conforme necessário, dependendo da gravidade, presença de alterações no ECG e persistência da hipocalcemia.
Prolongamento do Intervalo QT
O efeito de uma dose única de 32 mg de lenvatinibe no intervalo QT / QTc foi avaliado num estudo QT minucioso em indivíduos saudáveis. Neste estudo, o lenvatinibe não prolongou o intervalo QT / QTc. O prolongamento do intervalo QT / QTc foi notificado a uma taxa mais elevada em doentes tratados com lenvatinibe. Monitorar eletrocardiogramas em pacientes com síndrome do QT longo congênita, insuficiência cardíaca congestiva, bradiarritmias e medicamentos conhecidos por prolongar o intervalo QT, incluindo antiarrítmicos Classe Ia e III. Monitorar e corrigir anormalidades eletrolíticas em todos os pacientes.
Osteonecrose da mandíbula (ONJ)
Foram observados eventos de osteonecrose da mandíbula (ONJ) com lenvatinibe (ver Reações Adversas). Procedimentos odontológicos invasivos são um fator de risco identificado para o desenvolvimento de ONJ. Um exame odontológico oral e uma odontologia preventiva apropriada devem ser considerados antes do início do lenvatinibe. Os pacientes devem ser informados sobre exames dentários periódicos e práticas de higiene oral durante a terapia com lenvatinibe. Evite procedimentos odontológicos invasivos durante o tratamento com lenvatinibe, se possível. Tenha cuidado em pacientes que recebem agentes associados com ONJ, como bifosfonatos e denosumabe.
Óbito embrio-fetal ou defeitos graves ao nascimento
Baseado no seu mecanismo de ação e em dados de estudos de toxicidade reprodutiva em animais, mesilato de lenvatinibe pode provocar óbito embrio-fetal ou defeitos graves ao nascimento quando administrado a gestantes (vide item Dados de Segurança Pré-Clinica/Toxicidade Reprodutiva e ao Desenvolvimento). O profissional de saúde deverá orientar os pacientes do sexo feminino e masculino sobre o risco potencial ao feto e a necessidade de uso de métodos contraceptivos altamente efetivos (vide item Fertilidade, gravidez e lactação).
Efeitos Sobre a Capacidade de Dirigir e Operar Máquinas
Não foram realizados estudos dos efeitos sobre a capacidade de conduzir veículos e operar máquinas. Mesilato de lenvatinibe pode ter uma influência mínima sobre a habilidade de dirigir ou operar máquinas devido a reações adversas como fadiga e tontura. Pacientes que experimentarem estes sintomas devem ser advertidos a não dirigir ou operar máquinas.
Dados de Segurança Pré-clínica
Estudos de Toxicidade de Doses Repetidas
Nos estudos de toxicidade de doses repetidas (até 39 semanas), o lenvatinibe causou alterações toxicológicas em vários órgãos e tecidos relacionados aos efeitos farmacológicos esperados do lenvatinibe incluindo glomerulopatia, hipocelularidade testicular, atresia folicular ovariana, alterações gastrointestinais, alterações ósseas, alterações adrenais (em ratos e cachorros) e lesões arteriais (necrose fibrinoide arterial, degeneração medial, ou hemorragia) em ratos, cães, e macacos cinomolgos. Níveis elevados de transaminase com sinais de hepatotoxicidade foram também observados em ratos, cães e macacos. A reversibilidade das alterações toxicológicas foi observada ao final de um período de recuperação de 4 semanas em todas as espécies animais investigadas.
Estudos de Toxicidade em Animais Jovens
Mortalidade foi a toxicidade dose-limitante em ratos jovens em que a dose foi iniciada no dia pós-natal 7 ou 21 e foi observada em exposições que foram respectivamente 125 ou 12 vezes menor, comparada com a exposição em que mortalidade foi observada em ratos adultos, sugerindo uma sensibilidade aumentada à toxicidade com idade decrescente. Portanto, mortalidade pode ser atribuída a complicações relacionadas a lesões primárias no duodeno com possível contribuição de toxicidades adicionais em órgãos-alvo imaturos. A toxicidade de lenvatinibe foi mais proeminente em ratos mais jovens (dose iniciada no dia pós-natal 7) comparada com aqueles em que a dose iniciou no dia pós-natal 21 e mortalidade e algumas toxicidades foram observadas mais cedo em ratos jovens a 10 mg/kg comparado com ratos adultos aos quais foi administrado o mesmo nível de dose. Retardo de crescimento, atraso secundário do desenvolvimento físico, e lesões atribuíveis aos efeitos farmacológicos (incisivos, fêmur [placa de crescimento], rins, adrenais e duodeno) também foram observados em ratos jovens.
Genotoxicidade
O lenvatinibe não foi mutagênico nos testes de mutação bacteriana reversa (Ames), e não foi clastogênico em um ensaio de linfoma de camundongos in vitro e um ensaio in vivo de micronúcleo de ratos.
Carcinogenicidade
Estudos de carcinogenicidade não foram conduzidos com lenvatinibe.
Toxicidade Reprodutiva e ao Desenvolvimento
Não foram conduzidos estudos específicos com lenvatinibe em animais para avaliar o efeito sobre a fertilidade. Entretanto, foram observadas alterações testiculares e ovarianas em estudos de toxicidade de doses repetidas em animais com exposições abaixo da exposição clínica prevista (baseada na AUC) na dose máxima recomendada para humanos. Estes efeitos foram reversíveis ao final de um período de recuperação de 4 semanas.
A administração de lenvatinibe durante a organogênese resultou em embrioletalidade e teratogenicidade tanto em ratos como em coelhos em exposições abaixo da exposição clínica (baseada na AUC) na dose máxima recomendada para humanos. Anomalias fetais externas e esqueléticas foram observadas em doses a partir de 0,1 mg/kg em ratos, e um NOAEL (No Observed Adverse Effect Level) fetal não foi identificado em ratos. Anomalias fetais externas, viscerais, ou esqueléticas foram observadas com 0,1 e 0,5 mg/kg em coelhos. O NOAEL fetal no estudo em coelhos foi de 0,03 mg/kg. Estes achados indicam que o lenvatinibe tem um potencial teratogênico, provavelmente relacionado à atividade farmacológica do lenvatinibe como um agente antiangiogênico.
Fertilidade, gravidez e lactação
Gravidez
Categoria D
Há informações insuficientes sobre o uso de lenvatinibe em mulheres grávidas. O lenvatinibe foi embriotóxico e teratogênico quando administrado a ratos e coelhos.
O lenvatinibe não deve ser usado durante a gravidez a menos que claramente necessário e após consideração cuidadosa das necessidades da mãe e do risco ao feto. As mulheres com potencial de engravidar devem evitar a gravidez e utilizar método contraceptivo altamente eficaz enquanto estiverem em tratamento com lenvatinibe ou até 30 dias após a última dose.
Uma mulher é considerada fértil a menos que esteja na pós-menopausa (amenorreica pelo menos 12 meses consecutivos, na faixa etária adequada e sem outra causa conhecida ou suspeita) ou que foi esterilizada cirurgicamente (ou seja, laqueadura bilateral, histerectomia total ou ooforectomia bilateral, todas com cirurgia realizada há pelo menos 1 mês antes de iniciar o tratamento com lenvatinibe).
É recomendável que mulheres com possibilidade de engravidar façam um teste de gravidez antes de iniciar o tratamento com lenvatinibe.
Como não se sabe se Saumya pode reduzir o efeito de contraceptivos orais, métodos contraceptivos de barreira devem ser adicionados durante o tratamento com lenvatinibe e por pelo menos 1 mês após a conclusão do tratamento. Atualmente não se sabe se lenvatinibe aumenta o risco de eventos tromboembólicos quando associado a contraceptivos orais [veja item 5. ADVERTÊNCIAS E PRECAUÇÕES - Eventos Tromboembólicos Arteriais (ETAs) e item 5. ADVERTÊNCIAS E PRECAUÇÕES - Toxicidade Reprodutiva e ao Desenvolvimento].
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica.
Informe imediatamente seu médico em caso de suspeita de gravidez.
Lactação
Não se sabe se o lenvatinibe é excretado no leite humano. O lenvatinibe e seus metabólitos são excretados no leite de ratas. Um risco ao recém-nascido ou aos bebês não pode ser descartado e, portanto, o lenvatinibe não deve ser usado enquanto a mulher estiver amamentando (veja item 4. CONTRAINDICAÇÕES).
Fertilidade
Saumya pode afetar a fertilidade masculina e feminina.
Os efeitos do lenvatinibe sobre a fertilidade em humanos são desconhecidos. No entanto, foi observada toxicidade testicular e ovariana em ratos, cães e macacos.
Doação de sangue e sêmen
Não há dados específicos que suportam sobre o tempo de doações de sangue e sêmen durante ou após o tratamento com lenvatinibe, portanto, os pacientes não devem doar sangue ou sêmen durante a terapia ou por até 4 semanas após a descontinuação de lenvatinibe.
Atenção: Contém os corantes dióxido de titânio, óxido de ferro vermelho, óxido de ferro amarelo e óxido de ferro preto que podem, eventualmente, causar reações alérgicas.
6. INTERAÇÕES MEDICAMENTOSA