SAPHNELO
ASTRAZENECA
anifrolumabe
Tratamento do lúpus eritematoso sistêmico.
Apresentações.
Solução para diluição para infusão de 300 mg/2 mL (150 mg/mL) em embalagem com 1 frasco-ampola contendo 2,0 mL da solução.
VIA INTRAVENOSA
USO ADULTO
Composição.
Cada mL da solução para diluição para infusão contém 150 mg de anifrolumabe. Cada frasco-ampola de 2mL contém 300 mg de anifrolumabe.
Excipientes: histidina, cloridrato de histidina monoidratado, cloridato de lisina, trealose di-hidratada, polissorbato 80, água para injetáveis.
Informações técnicas.
1. INDICAÇÕES
SAPHNELO® é indicado para o tratamento de pacientes adultos com lúpus eritematoso sistêmico (LES) moderado a grave, positivo para autoanticorpos, em adição à terapia padrão.
2. RESULTADOS DE EFICÁCIA
A segurança e a eficácia de SAPHNELO® foram avaliadas em três estudos clínicos multicêntricos, randomizados, duplo-cegos, controlados por placebo com duração de 52 semanas - Estudo 1 (MUSE), Estudo 2 (TULIP 1) e Estudo 3 (TULIP 2). Os pacientes foram diagnosticados com LES de acordo com os critérios de classificação do American College of Rheumatology (revisados em 1997).
Todos os pacientes eram ≥ 18 anos de idade e apresentavam doença moderada a grave, com uma pontuação no Índice de Atividade do LES 2000 (Systemic Lupus Erythematosus Disease Activity Index 2000 - SLEDAI-2K) ≥ 6 pontos, nível de acometimento de órgãos com base na avaliação pelo British Isles Lupus Assessment Group (BILAG) e uma pontuação de Avaliação Global do Médico (Physician's Global Assessment - PGA) ≥ 1, independentemente de na linha de base receberem terapia padrão para LES consistindo de uma ou qualquer combinação de corticosteroides orais (OCS), medicamentos antimaláricos e/ou imunossupressores. Os pacientes continuaram a receber a sua terapia para LES existente em doses estáveis durante os estudos clínicos, com exceção de OCS (prednisona ou equivalente), cuja redução gradual era um componente do protocolo do estudo. Os pacientes que tinham nefrite lúpica ativa grave e pacientes com lúpus do sistema nervoso central ativo grave foram excluídos. O uso de outros agentes biológicos e ciclofosfamida não foi permitido durante os estudos clínicos; pacientes recebendo outras terapias biológicas deveriam concluir o período de descontinuação (wash-out) de pelo menos 5 meias-vidas antes da inclusão. Todos os três estudos foram realizados na América do Norte, Europa, América do Sul e Ásia. Os pacientes receberam anifrolumabe ou placebo, administrado por infusão IV, a cada 4 semanas.
Fase II
No Estudo 1 (MUSE - NCT01438489), 305 pacientes foram randomizados (1:1:1) e receberam anifrolumabe 300 mg ou 1000 mg ou placebo. O desfecho primário foi uma avaliação combinada do Índice de Resposta ao LES - SLE Responder Index (SRI-4, um desfecho composto) e a redução sustentada de OCS ( < 10 mg/dia e uma dose de OCS ≤ na semana 1, sustentada por 12 semanas) medida na Semana 24; uma proporção significantemente maior de pacientes tratados com anifrolumabe 300 mg atingiu resposta SRI-4 e redução sustentada de OCS (34,3% no grupo anifrolumabe vs. 17,6% no grupo placebo). A análise pré-especificada de atividade da doença medida pelo British Isles Lupus Assessment Group-based Composite Lupus Assessment (BICLA) foi 53,5% para anifrolumabe e 25,1% para placebo na Semana 52. O modelo de dose-resposta e perfil de benefício-risco suportou a avaliação da dose de 300 mg nos estudos subsequentes; a dose de 1000 mg não é recomendada.
Fase III
Os Estudos 2 e 3 tiveram desenho similares: o desfecho primário foi a melhora na atividade da doença avaliada na Semana 52, medida por SRI-4 e BICLA, respectivamente. Os desfechos secundários de eficácia comuns incluídos em ambos estudos foram a taxa de resposta no subgrupo com alta assinatura de interferon- (usando a medida do desfecho primário), manutenção da redução de OCS, melhora na atividade de LES cutânea pelo Índice de Gravidade e Área de Doença no Lúpus Eritematoso Cutâneo (Cutaneous Lupus Erythematosus Disease Area and Severity Index - CLASI) e taxa anualizada de exacerbações. Uma avaliação de melhora na atividade em articulações foi incluída como um desfecho secundário no Estudo 3. Ambos os estudos avaliaram a eficácia de anifrolumabe 300 mg versus placebo; a dose de anifrolumabe 150 mg também foi avaliada para a dose-resposta no Estudo 2.
Os dados demográficos foram em geral similares em ambos os estudos; nestes estudos, 92% e 93% eram mulheres, 71% e 60% eram brancos, 14% e 12% eram negros/afrodescendentes e 5% e 17% eram asiáticos, respectivamente. Em ambos estudos, 72% dos pacientes tiveram atividade da doença alta (SLEDAI-2K pontuação ≥ 10). Nos Estudos 2 e 3, respectivamente, 47% e 49% tiveram doença grave (BILAG-A) em pelo menos 1 sistema de órgãos e 46% e 47% dos pacientes tiveram doença moderada (BILAG-B) em pelo menos 2 sistemas de órgãos. Os sistemas de órgãos mais comumente afetados (BILAG-A ou B na linha de base) foram sistemas mucocutâneo (Estudo 2: 87%, Estudo 3: 85%) e musculoesquelético (Estudo 2: 89%, Estudo 3: 88%); 7,4% e 8,8% dos pacientes tiveram manifestações cardiorrespiratórias e 7,9% e 7,5% manifestações renais na linha de base, nos Estudos 2 e 3, respectivamente.
Nos Estudos 2 e 3, 90% dos pacientes (em ambos estudos) foram soropositivos para anticorpos antinucleares (ANA) e 45% e 44% para anticorpos anti-DNA de fita dupla (anti-double-stranded DNA - anti-dsDNA); 34% e 40% dos pacientes tiveram C3 baixo e 21% e 26% tiveram C4 baixo.
Os pacientes foram testados prospectivamente para super expressão de quatro genes induzíveis por interferon tipo I nas amostras de sangue total em um laboratório central usando um método baseado em reação em cadeia de polimerase (polymerase chain reaction - PCR) (QIAGEN therascreen® IFIGx RGQ RT-PCR). Em ambos estudos, a maioria dos pacientes foi classificada como com alta assinatura para interferon (interferon gene signature test-high) na linha de base (Estudo 2: 82%, Estudo 3: 83%).
Na linha de base, os medicamentos da terapia padrão concomitante incluíram corticosteroide oral (Estudo 2: 83%, Estudo 3: 81%), antimaláricos (Estudo 2: 73%, Estudo 3: 70%) e imunossupressores (Estudo 2: 47%, Estudo 3: 48%; incluindo azatioprina, metotrexato, micofenolato e mizoribina). Para aqueles pacientes recebendo OCS
(prednisona ou equivalente) no basal, a dose média diária foi 12,3 mg no Estudo 2 e 10,7 mg no Estudo 3. Durante as Semanas 8-40, pacientes em uso de OCS na linha de base ≥ 10 mg/dia deveriam ter a dose reduzida para ≤ 7,5 mg/dia, a menos que houvesse piora da atividade da doença.
A randomização foi estratificada por atividade da doença (pontuação SLEDAI-2K na linha de base, < 10 vs. ≥ 10 pontos), dose de OCS no Dia 1 ( < 10 mg/dia vs. ≥ 10 mg/dia de prednisona ou equivalente) e resultados do teste de assinatura para interferon (alto vs. baixo).
Estudo 2 (TULIP-1 - NCT02446912)
No Estudo 2, 457 pacientes foram randomizados (1:2:2) e receberam anifrolumabe 150 mg ou 300 mg ou placebo. O desfecho primário, resposta SRI-4, foi definido pelo atingimento de cada um dos seguintes critérios na Semana 52 em comparação com o basal:
3. Redução em relação a linha de base de ≥ 4 pontos no SLEDAI-2K;
4. Nenhum novo sistema de órgão afetado, conforme definido por 1 ou mais itens BILAG-A ou 2 ou mais itens BILAG-B em comparação com a linha de base;
• Sem piora em relação à linha de base na atividade do lúpus do sujeito de pesquisa, definida por um aumento ≥ 0,30 pontos em escala visual análoga (visual analogue scale - VAS) do PGA de 3 pontos;
• Sem descontinuação de tratamento;
• Sem uso de medicamentos restritos além dos limites permitidos pelo protocolo do estudo.
Para o desfecho primário (SRI-4 na Semana 52), o tratamento com anifrolumabe não resultou em melhora estatisticamente significativa em comparação com placebo (valor de p=0,455). Os desfechos secundários não foram formalmente testados; no entanto, melhoras clinicamente significativas na resposta BICLA, redução de dose de OCS sustentada, resposta CLASI, taxa de exacerbação e resposta de articulação foram observados para pacientes recebendo anifrolumabe 300 mg em comparação com aqueles recebendo placebo. A taxa de responsivos BICLA foi 47,1% (85/180) para anifrolumabe 300 mg versus 30,2% (55/184) para placebo (diferença de 17,0%, IC 95% 7,2, 26,8, valor de p nominal < 0,001). Não houve padrão consistente de eficácia entre os desfechos ou ao longo do tempo em pacientes recebendo 150 mg de anifrolumabe. A dose de 150 mg não é recomendada.
Estudo 3 (TULIP-2 - NCT02446899)
No Estudo 3, 362 pacientes foram randomizados (1:1) e receberam anifrolumabe 300 mg ou placebo. O desfecho primário, resposta BICLA na Semana 52, foi definido como melhora em relação à linha de base em todos os domínios referentes aos órgãos com atividade moderada ou grave:
• Redução de todos os BILAG-A na linha de base para B/C/D e BILAG-B na linha de base para C/D e sem piora do BILAG em outros sistemas de órgãos, conforme definido por ≥ 1 BILAG-A novo ou ≥ 2 BILAG-B novos;
• Sem piora em relação à linha de base no SLEDAI-2K, em que a piora é definida como um aumento em relação à linha de base de > 0 pontos no SLEDAI-2K;
• Sem piora em relação à linha de base na atividade do lúpus do sujeito de pesquisa, em que a piora é definida por um aumento ≥ 0,30 pontos em uma VAS do PGA de 3 pontos;
• Sem descontinuação de tratamento;
• Sem uso de medicamentos restritos além dos limites permitidos pelo protocolo do estudo.
O desfecho primário foi atingido; anifrolumabe 300 mg demonstra eficácia estatística e clinicamente significativa na atividade geral da doença em comparação com placebo. Melhoras maiores em todos os componentes do desfecho composto de BICLA foram observadas para anifrolumabe em comparação com placebo (Tabela 1).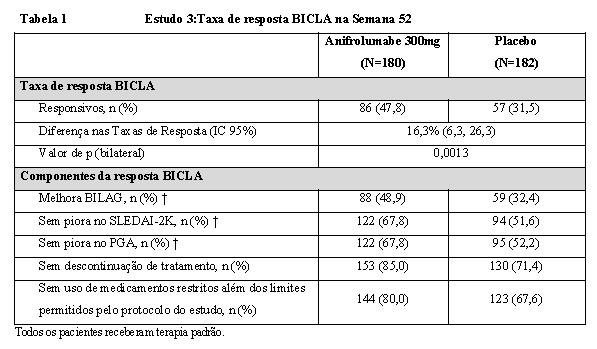
BICLA: British Isles Lupus Assessment Group-based Composite Lupus Assessment, BILAG: British Isles Lupus Assessment Group, PGA: Avaliação Global do Médico, SLEDAI-2K: Systemic Lupus Erythematosus Disease Activity Index 2000.
† Pacientes que descontinuaram o tratamento ou usaram medicamentos restritos além dos limites permitido pelo protocolo do Estudo são considerados como não responsivos.
As diferenças clinicamente significativas na taxa de resposta BICLA foram observadas a partir da Semana 8 e foram mantidas até a Semana 52 (Figura 1).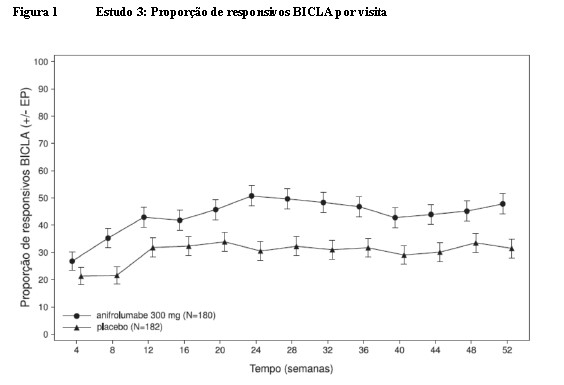
O tratamento com anifrolumabe reduziu o tempo até a primeira visita na qual a resposta BICLA foi obtida e subsequente mantida até, e incluindo, a Semana 52. Em qualquer momento durante o estudo, pacientes tratados com anifrolumabe tiveram uma probabilidade 55% maior de atingir uma resposta BICLA sustentada, em relação aos pacientes recebendo placebo (razão de risco = 1,55, IC 95% 1,11; 2,18). A separação entre os braços de tratamento começou aproximadamente na Semana 4 (Figura 2).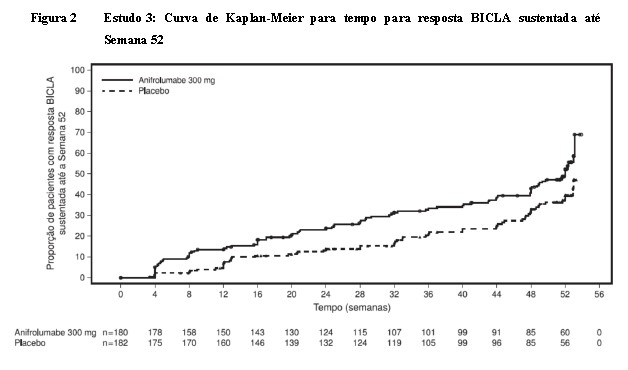
Pacientes sem uma resposta BICLA sustentada até a Semana 52 foram censurados ou na data da descontinuação do tratamento ou na Semana 52, o que houver ocorrido primeiro.
O efeito do tratamento de anifrolumabe em relação ao placebo foi consistente entre os subgrupos (por idade, gênero, raça, etnia, intensidade da doença [SLEDAI-2K na linha de base] e uso de OCS na linha de base).
A análise pré-especificada de atividade da doença medida por SRI-4 foi consistente com a resposta medida por BICLA (taxa de responsivos SRI-4; anifrolumabe 55,5% vs. placebo 37,3%; diferença de 18,2% [IC 95% 8,1, 28,3]).
Efeito no tratamento concomitante com esteroides: Nos 47% dos pacientes com um uso de OCS na linha de base ≥ 10 mg/dia, anifrolumabe demonstrou uma redução estatística e clinicamente significativa no uso de OCS, de pelo menos 25% para ≤ 7,5 mg/dia na Semana 40 mantida até a Semana 52 (valor de p=0,004); 51,5% (45/87) dos pacientes no grupo anifrolumabe versus 30,2% (25/83) no placebo atingiram esse nível de redução de esteroide (diferença de 21,2% [IC 95% 6,8; 35,7]). Em pacientes com o uso de OCS na linha de base ≥ 10 mg/dia, a dose cumulativa de OCS foi menor em pacientes tratados com anifrolumabe em comparação com placebo. A mediana (min, max) da dose cumulativa de OCS na Semana 52 foi 3197 mg (309, 13265) no grupo anifrolumabe em comparação com 3640 mg (1745, 10920) no grupo placebo.
Efeito na atividade de LES cutânea: Em pacientes com doença cutânea moderada a grave na linha de base (pontuação de atividade CLASI ≥ 10; n=89), anifrolumabe demonstrou melhoras estatística e clinicamente significativas na atividade de lúpus cutâneo (resposta CLASI definida como redução de pelo menos 50% na pontuação de atividade CLASI em comparação com basal) na Semana 12 (taxa de responsivos 49,0% [24/49] e 25,0% [10/40] para o grupo anifrolumabe e placebo, respectivamente; diferença observada de 24,0% [IC 95% 4,3; 43,6], valor de p=0,017). Em comparação com placebo, o benefício do tratamento de anifrolumabe foi mantido até a Semana 52. Em qualquer momento durante o estudo, pacientes tratados com anifrolumabe tiveram uma probabilidade 55% maior de atingir uma resposta CLASI sustentada (definida como uma resposta CLASI obtida em qualquer momento durante o estudo e subsequentemente sustentada em todos as visitas até a Semana 52), em relação aos pacientes recebendo placebo (razão de risco=1,55, IC 95% 0,87; 2,85).
Efeito nas manifestações de LES: A manifestação da doença foi definida como atividade da doença grave (BILAG-A) em um ou mais sistemas de órgão novos ou atividade de doença moderada (BILAG-B) em 2 ou mais sistemas de órgãos novos em comparação com a visita anterior. No Estudo 3, 68,9% [124/180] dos pacientes recebendo anifrolumabe não apresentaram manifestações de LES em comparação com 57,7% [105/182] dos pacientes recebendo placebo, durante o período de tratamento de 52 semanas. O tratamento com anifrolumabe levou a uma redução clinicamente significativa de 33% na taxa anual de manifestações versus placebo (taxa anual de 0,43 e 0,64 para o grupo anifrolumabe e placebo, respectivamente; razão da taxa 0,67 [IC 95% 0,48, 0,94], valor de p=0,020); essa diferença não foi estatisticamente significativa após ajuste para comparações múltiplas. O tempo para primeira manifestação foi maior no grupo anifrolumabe, em qualquer momento durante o estudo e os pacientes tiveram um risco 35% menor de apresentar uma primeira manifestação em relação a pacientes recebendo placebo (razão de risco=0,65; IC 95% 0,46, 0,91).
Efeito na atividade de articulações: Na linha de base, 44% dos pacientes tinham ≥ 6 articulações edemaciadas e ≥ 6 articulações sensíveis. A resposta foi definida como melhora ≥ 50% na contagem de articulações edemaciadas/sensíveis na Semana 52. Não houve diferença notável na resposta das articulações entre os grupos de tratamento (taxa de resposta de 42,2% [30/71] e 37,5% [34/90] para o grupo anifrolumabe e placebo, diferença observada 4,7% [IC 95% -10,6, 20,0], valor de p= 0,547).
Resultados relacionados com a saúde: A proporção de pacientes que relataram melhora na fadiga, conforme medido pela taxa de responsivos FACIT-F (melhora em relação à linha de base na Semana 52 > 3 pontos), foi 33,2% para anifrolumabe e 24,7% para placebo (diferença de 8,5%, IC 95% -0,9, 17,9).
REFERÊNCIAS BIBLIOGRÁFICAS
Estudo MUSE (Estudo 1 - NCT01438489): Furie R, et al. CD1013 Study Investigators. Anifrolumab, an Anti-Interferon-a Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017;69(2):376-386.
Estudo TULIP-1 (Estudo 2 - NCT02446912): Furie R, et al. Type I interferon inhibitor anifrolumab in active systemic lupus erythematosus (TULIP-1): a randomised, controlled, phase 3 trial. The Lancet. 2019;1(4):208-219.
Estudo TULIP-2 (Estudo 3 - NCT02446899): Morand EF, et al. TULIP-2 Trial Investigators. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N Engl J Med. 2020;382(3):211-221.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Mecanismo de ação
Anifrolumabe é um anticorpo monoclonal de cadeia kappa do tipo imunoglobulina G1 humana, que se liga à subunidade 1 do receptor interferon tipo I (IFNAR1) com alta especificidade e afinidade. Essa ligação inibe a sinalização de IFN tipo I bloqueando assim a atividade biológica dos IFNs tipo I. O anifrolumabe também induz a internalização de IFNAR1, reduzindo assim os níveis de IFNAR1 na superfície celular disponíveis para a fabricação do receptor. O bloqueio do receptor mediado pela sinalização de IFN tipo I inibe a expressão genética responsiva de IFN, bem como diminui os processos inflamatórios e imunológicos. A inibição de IFN tipo I bloqueia a diferenciação de plasmócitos e normaliza os subconjuntos periféricos de célula T, restaurando o equilíbrio entre imunidade adaptativa e inata que está desregulada em diversos distúrbios autoimunes.
Os IFNs tipo I desempenham um papel importante na patogênese de LES. A maioria dos pacientes adultos com LES (aproximadamente 60-80%) expressa níveis elevados de genes induzíveis de IFN tipo I, que são associados com o aumento da atividade e gravidade da doença.
Propriedades Farmacodinâmicas
Em pacientes adultos com LES, a administração de anifrolumabe em doses ≥ 300 mg, via infusão IV a cada 4 semanas demonstrou neutralização consistente (≥ 80%) na assinatura farmacodinâmica (PD) sanguínea de interferon tipo I de 21 genes. Essa supressão ocorreu a partir de 4 semanas após o tratamento e foi ou mantida ou ainda mais suprimida ao longo do período de tratamento de 52 semanas. O anifrolumabe 150 mg IV demonstrou supressão < 20% da assinatura gênica em pontos de tempo iniciais, que atingiu um máximo de < 60% no final do período de tratamento.
A neutralização (≥ 70%) da assinatura farmacodinâmica de IFN tipo I também foi observada no tecido cutâneo, conforme demonstrado no estudo de Fase I em pacientes com escleroderma (anifrolumabe ≥1.0 mg/kg).
Após a descontinuação de anifrolumabe no final do período de tratamento de 52 semanas nos estudos clínicos com LES, a assinatura farmacodinâmica do IFN tipo I nas amostras de sangue retornaram aos níveis basais dentro de 8 a 12 semanas.
Nos estudos de Fase III em pacientes com LES positivos para anticorpos anti-dsDNA na linha de base, o tratamento com anifrolumabe 300 mg levou a reduções numéricas nos anticorpos anti-dsDNA ao longo do tempo (mediana da alteração em relação à linha de base na Semana 52: -14,82 U/mL no grupo anifrolumabe vs. -5,37 U/mL no grupo placebo). Na Semana 52, 7,8% (13/167) dos pacientes tratados com anifrolumabe e 5,8% (9/155) dos pacientes recebendo placebo tinham convertido para anti-dsDNA negativo.
Em pacientes com níveis baixos de C3 na linha de base, o tratamento com anifrolumabe 300 mg levou a aumentos numéricos maiores no C3 ao longo do período de tratamento de 52 semanas (média da alteração em relação à linha de base na Semana 52: 0,13 g/L no grupo anifrolumabe vs. 0,04 g/L no grupo placebo). Em pacientes com nível anormal de C4 na linha de base, pequenos aumentos foram observados ao longo de 52 semanas em ambos os grupos de tratamento (média da alteração em relação à linha de base na Semana 52: 0,02 g/L anifrolumabe vs. 0,02 g/L placebo). Em pacientes com níveis baixos de complemento na linha de base, a normalização de C3 e C4 foi observada em 16,2% (21/130) e 22,6% (19/84) dos pacientes recebendo anifrolumabe e em 9,5% (13/137) e 7,1% (6/85) dos pacientes recebendo placebo, respectivamente, na Semana 52.
O tratamento com anifrolumabe 300 mg levou a números significantemente aumentados (p < 0,05) dos subconjuntos de célula T em pacientes com assinatura gênica de interferon alta. As contagens normalizadas do subconjunto de células T foram observadas a partir da Semana 12 e até a Semana 52.
Em comparação com o placebo, anifrolumabe 300 mg inibiu a produção de proteínas envolvidas na sobrevivência e recrutamento de célula B (CXCL13, BAFF). A inibição foi observada a partir da Semana 12 e se manteve até a Semana 52, que é consistente com a diminuição de determinados níveis de auto anticorpo pelo anifrolumabe.
Imunogenicidade
Nos estudos de Fase III, os anticorpos antidroga emergentes do tratamento foram detectados em 6 de 352 (1,7%) pacientes tratados com SAPHNELO® no esquema posológico recomendado durante o período de estudo de 60 semanas. Um total de 0,3% (1/351) dos pacientes tratados com SAPHNELO® desenvolveu anticorpos neutralizantes detectados in-vitro. Os anticorpos anti-anifrolumabe não foram associados com aumento na depuração de anifrolumabe ou perda da atividade farmacodinâmica em comparação com pacientes negativos para anticorpo antidroga. Não foi observada evidência de associação de anticorpos antidroga com a eficácia ou segurança.
Propriedades Farmacocinéticas
A farmacocinética (PK) do anifrolumabe foi estudada em pacientes adultos com LES após doses IV variando de 100 a 1000 mg, uma vez a cada 4 semanas e em voluntários sadios após dose única.
O anifrolumabe exibe PK não linear na faixa de dose de 100 mg a 1000 mg. A exposição PK reduziu mais rapidamente em doses menores do que 300 mg a cada 4 semanas (posologia recomendada).
Absorção
SAPHNELO® é administrado por infusão intravenosa.
Distribuição
Com base na análise farmacocinética populacional, o volume de distribuição periférico e central para anifrolumabe foi respectivamente 2,93 L (com variabilidade interindividual CV 26,9%) e 3,3 L para um paciente com 69,1 kg.
Biotransformação
O anifrolumabe é uma proteína, portanto estudos específicos de metabolismo não foram realizados.
SAPHNELO® é eliminado por uma via de eliminação mediada por IFNAR alvo e pelo sistema reticuloendotelial, onde espera-se que SAPHNELO® seja degradado em pequenos peptídeos ou aminoácidos individuais, por enzimas proteolíticas que são amplamente distribuídas no corpo.
Eliminação
Houve um aumento maior que proporcional à dose na exposição ao medicamento devido à depuração do medicamento mediada por IFNAR1.
A partir da modelagem de PK populacional, a depuração sistêmica típica estimada (CL) foi 0,193 L/dia com uma variabilidade interindividual CV de 33,0%. A mediana da CL reduz lentamente ao longo do tempo, com 8,4% após 1 ano de tratamento.
Com base na análise de PK populacional, as concentrações séricas estavam abaixo do nível de detecção em 95% dos pacientes, aproximadamente 16 semanas após a última dose de anifrolumabe, quando anifrolumabe foi administrado por um ano.
Populações especiais
Não há diferença clinicamente significativa na depuração sistêmica com base na idade, raça, etnia, região, gênero, condição IFN ou peso corporal que necessite de ajuste de dose.
Pacientes idosos (≥ 65 anos de idade)
Com base na análise de PK populacional, a idade (intervalo de 18 a 69 anos) não impactou a depuração de anifrolumabe; houve 20 (3%) pacientes ≥65 anos de idade. Nenhuma diferença geral na segurança ou efetividade foi observada entre pacientes idosos e mais jovens que receberam anifrolumabe em estudos clínicos.
Insuficiência renal
Nenhum estudo clínico específico foi realizado para investigar o efeito do comprometimento renal no anifrolumabe. Com base nas análises de PK populacional, a depuração de anifrolumabe foi comparável em pacientes com LES com leve (60-89 mL/min/1,73 m2) e moderada (30-59 mL/min/1,73 m2) redução nos valores da TFGe e pacientes com função renal normal (≥ 90 mL/min/1,73 m2). Pacientes com LES com uma redução grave na TFGe ou doença renal em estágio terminal ( < 30 mL/min/1,73 m2) foram excluídos dos estudos clínicos; o anifrolumabe não é depurado por via renal.
Pacientes com razão de proteína/creatinina urinária (UPCR) > 2 mg/mg foram excluídos de estudos clínicos. Com base nas análises de PK populacional, a UPCR não afetou significantemente a depuração de anifrolumabe.
Insuficiência hepática
Nenhum estudo clínico específico foi realizado para investigar o efeito do comprometimento hepático sobre o anifrolumabe.
Como um anticorpo monoclonal IgG1, o anifrolumabe é eliminado principalmente via catabolismo e não se espera que passe por metabolismo por enzimas hepáticas, uma vez que tais alterações na função hepática são improváveis de ter qualquer efeito na eliminação do anifrolumabe. Com base nas análises farmacocinéticas populacionais, os biomarcadores basais de função hepática (ALT e AST ≤2,0 × ULN e bilirrubina total) não tiveram efeito clinicamente relevante na depuração do anifrolumabe.
Interações medicamentosas
Nenhum estudo formal de interação medicamentosa foi realizado com anifrolumabe. Não se espera um efeito de anifrolumabe na farmacocinética dos medicamentos administrados concomitantemente.
Com base na análise de PK populacional, o uso concomitante de corticosteroides orais, antimaláricos, imunossupressores (incluindo azatioprina, metotrexato e mizoribina), AINEs, inibidores da ECA, inibidores da HMG-CoA redutase não influenciaram significantemente a PK do anifrolumabe.
Dados de segurança pré-clínica
Segurança não-clínica
Dados não clínicos não revelaram riscos especiais para humanos com base nos estudos convencionais de farmacologia de segurança ou estudos de toxicidade de dose repetida em macacos cinomolgo.
Mutagenicidade e carcinogenicidade
O anifrolumabe é um anticorpo monoclonal, desta forma, estudos de genotoxicidade e carcinogenicidade não foram realizados.
Foi observado o aumento do potencial carcinogênico em modelos de roedores do bloqueio de IFNAR1. A relevância clínica deste achado é desconhecida.
Toxicidade reprodutiva
Toxicidade do desenvolvimento
Em um estudo de desenvolvimento pré e pós-natal, realizado em macacos cinomolgo, não houve efeitos observados no desenvolvimento materno, embriofetal ou pós-natal para anifrolumabe nas doses de 30 ou 60 mg/kg administradas por via intravenosa (exposições de aproximadamente até 28 vezes a dose máxima recomendada para humanos [MRHD] com base na ASC) a partir do Dia de Gestação 20, uma vez a cada 2 semanas a partir de então, ao longo da gestação até 1 mês após o parto (aproximadamente Dia 28 de Lactação).
Fertilidade
Os efeitos na fertilidade masculina e feminina não foram avaliados diretamente nos estudos em animais. No estudo de dose repetida de 9 meses não houve efeitos adversos relacionados ao anifrolumabe sobre as medidas indiretas de fertilidade masculina ou feminina, com base na análise de sêmen, estadiamento de espermatogênese, ciclos menstruais, peso dos órgãos e achados histopatológicos nos órgãos reprodutores de macacos cinomolgo em doses até 50 mg/kg IV uma vez por semana (aproximadamente 18 vezes a MRHD com base na ASC).
4. CONTRAINDICAÇÕES
SAPHNELO® é contraindicado em pacientes com hipersensibilidade à substância ativa ou a qualquer um de seus excipientes listados em Identificação do Medicamento - COMPOSIÇÃO.
5. ADVERTÊNCIAS E PRECAUÇÕES
Hipersensibilidade
Reações de hipersensibilidade grave, incluindo anafilaxia, foram relatadas após a administração de SAPHNELO® (vide seção 9. REAÇÕES ADVERSAS).
Nos estudos clínicos controlados, os eventos graves de hipersensibilidade (incluindo angioedema) foram relatados por 0,6% (3/459) dos pacientes que receberam anifrolumabe. Houve um evento de reação anafilática no programa de desenvolvimento de LES após a administração de anifrolumabe.
Se ocorrer reação grave relacionada à infusão ou hipersensibilidade (por exemplo, anafilaxia), a administração de SAPHNELO® deve ser imediatamente interrompida e terapia adequada deve ser iniciada.
Infecções
SAPHNELO® aumenta o risco de infecções respiratórias e herpes zoster (foram observados eventos de herpes zoster disseminado) (vide seção 9. REAÇÕES ADVERSAS)
Nos estudos clínicos controlados, a taxa geral de infecções sérias em pacientes que receberam anifrolumabe foi 4,8% em comparação com 5,6% do grupo placebo (correspondendo às taxas de incidência ajustada à exposição [EAIR] de 5,4 e 6,6 por 100 pacientes-anos, respectivamente).
Não foram realizados estudos em pacientes com histórico de imunodeficiência primária.
Devido ao seu mecanismo de ação, SAPHNELO® deve ser usado com cautela em pacientes com infecções crônicas, histórico de infecções recorrentes ou fatores de risco conhecidos para infecção. O tratamento com SAPHNELO® não deve ser iniciado em pacientes com qualquer infecção clinicamente significante até a infecção ser resolvida ou adequadamente tratada. Instruir os pacientes a buscarem avaliação médica caso ocorram sinais ou sintomas de infecção clinicamente significantes. Se um paciente desenvolver uma infecção ou não estiver respondendo à terapia padrão, monitorar o paciente cautelosamente e considerar interromper a terapia com SAPHNELO® até a resolução da infecção.
Imunização
Não há dados disponíveis sobre a resposta a vacinas vivas ou atenuadas.
Antes de iniciar a terapia com SAPHNELO®, considerar a conclusão de todas as imunizações apropriadas de acordo com as diretrizes atuais de imunização. Evitar o uso concomitante de vacinas vivas ou atenuadas em pacientes tratados com SAPHNELO®.
Grupos de pacientes excluídos de estudos clínicos
O anifrolumabe não foi estudado em associação com outras terapêuticas biológicas, incluindo terapêuticas dirigidas às células B. Portanto, o tratamento com anifrolumabe não é recomendado em associação com terapêuticas biológicas.
O anifrolumabe não foi estudado em pacientes com lúpus ativo grave do sistema nervoso central ou com nefrite lúpica ativa grave.
Malignidades
O impacto do tratamento com SAPHNELO® no desenvolvimento potencial de malignidades é desconhecido. Estudos em pacientes com histórico de malignidades não foram realizados; no entanto, pacientes com câncer de pele basocelular ou de células escamosas e câncer de colo do útero completamente excisado ou adequadamente tratado foram elegíveis para a inclusão nos estudos clínicos em LES.
Nos estudos clínicos controlados, em qualquer dose, as malignidades (excluindo câncer de pele não melanoma) foram observadas em 0,7% (5/657) e 0,6% (3/466) dos pacientes recebendo SAPHNELO® e placebo, respectivamente. Neoplasias malignas (incluindo câncer de pele não melanoma) foram relatadas por 8/657 (1,2%) pacientes recebendo anifrolumabe, em comparação com 3/466 (0,6%) pacientes recebendo placebo (EAIR: 1,2 e 0,7 por 100 pacientes-anos, respectivamente). Em pacientes recebendo anifrolumabe, carcinomas de mama e de células escamosas foram as malignidades observadas em mais de um paciente.
Deve-se considerar o benefício-risco individual em pacientes com fatores de risco conhecidos para o desenvolvimento ou recidiva de malignidades. Deve-se ter cautela ao se considerar continuar a terapia com SAPHNELO® em pacientes que desenvolvem malignidades.
Efeito sobre a capacidade de dirigir veículos e operar máquinas
SAPHNELO® não tem ou tem influência desprezível sobre a capacidade de dirigir veículos e operar máquinas.
Gravidez
Há dados limitados sobre o uso de anifrolumabe em mulheres grávidas. Os dados são insuficientes para informar o risco associado ao medicamento.
Em um estudo de desenvolvimento pré e pós-natal, após a administração IV de anifrolumabe, não foram observados efeitos adversos nos animais maternos e em suas proles (vide seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
SAPHNELO® não deve ser usado durante a gravidez a menos que o potencial benefício justifique o potencial risco para o feto.
Categoria de risco na gravidez: B
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião dentista.
Lactação
Não se sabe se o anifrolumabe é excretado no leite materno. O anifrolumabe foi detectado no leite de macacos cinomolgo fêmeas administrados com 30 ou 60 mg/kg, por via intravenosa a cada 2 semanas (vide seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
O risco para o lactente não pode ser excluído. Deve-se tomar a decisão entre descontinuar ou a amamentação ou a terapia com SAPHNELO®, levando-se em consideração o benefício da amamentação para a criança e o benefício da terapia para a mãe.
Fertilidade
Não há dados sobre os efeitos de anifrolumabe na fertilidade humana.
Os estudos em animais não demonstram efeitos adversos do tratamento com anifrolumabe nas medidas indiretas de fertilidade (vide seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
6. INTERAÇÕES MEDICAMENTOSAS
Nenhum estudo formal de interação medicamentosa foi realizado com SAPHNELO®.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
SAPHNELO® deve ser conservado sob refrigeração (2°C a 8°C). Não congelar. Manter o frasco-ampola dentro da embalagem original para proteger da luz.
SAPHNELO® tem validade de 36 meses a partir da data de fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Após o preparo da solução diluída para infusão
Do ponto de vista microbiológico, a solução diluída para infusão deve ser utilizada imediatamente após o seu preparo. Se não for utilizada imediatamente, as suas condições de conservação são de responsabilidade do profissional de saúde que manipulará o medicamento. Foi comprovada a estabilidade química e física em uso da solução diluída para infusão a partir do momento da punção do frasco até o início da administração por no máximo 4 horas em temperatura ambiente de até 25°C e 24 horas sob refrigeração (2 a 8°C).
SAPHNELO® é apresentado como uma solução para diluição para infusão de 2,0 mL em um frasco-ampola de vidro transparente tipo I 2R fechado por uma tampa de elastômero revestida com Teflon e coberta por uma lâmina de alumínio. A solução para diluição para infusão é clara a opalescente, incolor a ligeiramente amarelada.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
Posologia
A dose recomendada de SAPHNELO® é 300 mg, administrada por infusão intravenosa ao longo de um período de 30 minutos, a cada 4 semanas.
Dose perdida
Se uma infusão planejada for perdida, administrar SAPHNELO® o mais rápido possível. Um intervalo mínimo de 14 dias deve ser mantido entre as doses.
Populações especiais
Idosos (≥ 65 anos de idade)
Nenhum ajuste de dose é necessário. Há informações limitadas em indivíduos ≥ 65 anos (vide seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Insuficiência renal
Nenhum ajuste de dose é necessário. Nenhum estudo específico com SAPHNELO® foi realizado em pacientes com insuficiência renal. Não há experiência em pacientes com insuficiência renal grave ou doença renal em estágio terminal (vide seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Insuficiência hepática
Nenhum ajuste de dose é necessário. Nenhum estudo específico foi realizado em pacientes com insuficiência hepática (vide seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
População pediátrica
A segurança e a eficácia de SAPHNELO® em crianças e adolescentes ( < 18 anos de idade) não foram estabelecidas. Não há dados disponíveis.
Método de administração
SAPHNELO® é destinado para uso intravenoso (IV).
Após a diluição com solução para injeção de cloreto de sódio (0,9%), SAPHNELO® é administrado como uma infusão IV ao longo de um período de 30 minutos. Não administrar como uma injeção intravenosa ou em bolus.
Para instruções sobre a diluição e armazenamento do medicamento antes da administração vide item Modo de Usar.
A velocidade da infusão pode ser retardada ou interrompida se o paciente desenvolver reação à infusão.
Incompatibilidades
Na ausência de estudos de compatibilidade, este medicamento não deve ser misturado com outros medicamentos.
Modo de usar
SAPHNELO® é fornecido como um frasco-ampola de dose única. A solução diluída para infusão deve ser preparada e administrada por um profissional da saúde, usando técnicas assépticas, conforme segue:
Preparação da solução
1. Inspecionar visualmente o frasco-ampola para verificar a presença de material particulado e alteração de cor. SAPHNELO® é uma solução clara a opalescente, incolor a ligeiramente amarelada. Descarte o frasco se a solução estiver turva, com alteração de cor ou se partículas visíveis forem observadas. Não agitar o frasco.
2. Retirar e descartar 2,0 mL da solução para injeção de cloreto de sódio a 9 mg/mL (0,9%) de uma bolsa para infusão de 100 mL.
3. Retire 2,0 mL do frasco-ampola de SAPHNELO® e adicione na bolsa de infusão. Misture a solução invertendo-a gentilmente. Não agitar.
4. O concentrado não contém qualquer conservante. Qualquer concentrado remanescente no frasco-ampola deve ser descartado.
Administração
1. Recomenda-se que a solução diluída para infusão seja administrada imediatamente após o preparo. Se a solução diluída para infusão for armazenada em refrigerador (vide seção 7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO), permita que atinja temperatura ambiente (15 a 25°C) antes da administração.
2. Administrar a solução diluída para infusão por via intravenosa ao longo de 30 minutos através de uma linha IV contendo um filtro em linha de 0,2 ou 0,22 mícron com baixa ligação à proteína e estéril.
3. Ao se concluir a infusão, limpe o equipo de infusão com 25 mL de solução de cloreto de sódio a 9 mg/mL (0,9%) para infusão para assegurar que toda a solução diluída para infusão foi administrada.
4. Não administre concomitantemente outros medicamentos através da mesma linha de infusão.
Descarte
Qualquer medicamento não utilizado ou resíduos devem ser descartados conforme os requerimentos locais.
9. REAÇÕES ADVERSAS
Resumo geral do perfil de segurança
A segurança de anifrolumabe foi avaliada em um total de 1029 indivíduos adultos, dos quais pelo menos 837 pacientes tinham LES e receberam anifrolumabe IV (150 mg, 300 mg ou 1000 mg) durante estudos clínicos. Destes, 688 pacientes com LES foram expostos ao anifrolumabe por pelo menos um ano e 263 pacientes foram expostos por pelo menos 3 anos.
Os dados descritos na Tabela 1 refletem a exposição a SAPHNELO®