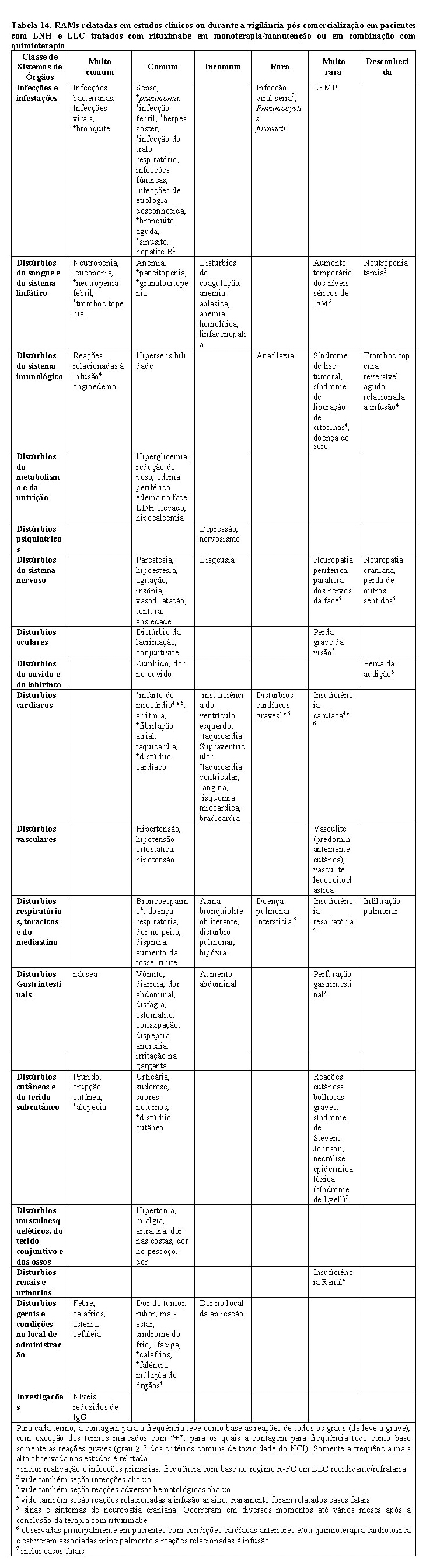
RUXIENCE
PFIZER
rituximabe
Antineoplásico.
Apresentações.
Ruxience® solução para diluição para infusão em embalagens contendo 1 frasco-ampola com 10 mL (100 mg/10 mL) ou 1 frasco-ampola com 50 mL (500 mg/50 mL).
VIA DE ADMINISTRAÇÃO: VIA INTRAVENOSA
USO ADULTO
CUIDADO: AGENTE CITOTÓXICO
Composição.
Ruxience® 100 mg concentrado para solução para infusão
Cada mL contém 10 mg de rituximabe.
Cada frasco-ampola de Ruxience® com 10 mL contém 100 mg de rituximabe.
Ruxience® 500 mg concentrado para solução para infusão
Cada mL contém 10 mg de rituximabe.
Cada frasco-ampola de Ruxience® com 50 mL contém 500 mg de rituximabe.
Excipientes: L-histidina, cloridrato de L-histidina monoidratada, EDTA, polissorbato 80, sacarose, água para injetáveis.
Informações técnicas.
As informações disponíveis nesta bula aplicam-se exclusivamente a Ruxience® (via intravenosa).
1. INDICAÇÕES
Ruxience® é indicado para o tratamento de:
Linfoma não Hodgkin
- pacientes com linfoma não Hodgkin de células B, baixo grau ou folicular, CD20 positivo, recidivado ou resistente à quimioterapia;
- pacientes com linfoma não Hodgkin difuso de grandes células B, CD20 positivo, em combinação à quimioterapia CHOP;
- pacientes com linfoma não Hodgkin de células B, folicular, CD20 positivo, não tratados previamente, em combinação com quimioterapia;
- pacientes com linfoma folicular, como tratamento de manutenção, após resposta à terapia de indução.
Artrite reumatoide
Ruxience® em combinação com metotrexato está indicado para o tratamento de pacientes adultos com artrite reumatoide ativa que tiveram resposta inadequada ou intolerância a uma ou mais terapias de inibição do fator de necrose tumoral (TNF).
Leucemia linfoide crônica
Ruxience® em combinação com quimioterapia é indicado para o tratamento de pacientes com leucemia linfoide crônica (LLC) não tratados previamente e com recaída / refratária ao tratamento.
Granulomatose com poliangiite (Granulomatose de Wegener) e poliangiite microscópica (PAM)
Ruxience® em combinação com glicocorticoides é indicado para o tratamento das seguintes vasculites ativas graves: granulomatose com poliangiite (GPA, conhecida também como Granulomatose de Wegener) e poliangiite microscópica (PAM).
2. RESULTADOS DE EFICÁCIA
Ruxience® é um medicamento biológico desenvolvido pela via de comparabilidade (biossimilar). O programa de desenvolvimento do produto foi projetado para demonstrar a comparabilidade entre Ruxience® e o medicamento comparador MabThera®.
Resultados de Eficácia de Ruxience®
Estudos clínicos comparativos de Ruxience®
O programa de desenvolvimento clínico biossimilar para Ruxience® incluiu um estudo randomizado, duplo-cego em pacientes com CD20 positivo, baixa carga tumoral de linfoma folicular no tratamento de primeira linha (Estudo B3281006), um estudo de grupo paralelo duplo-cego randomizado em pacientes com artrite reumatoide (Estudo B3281001), e estudo de extensão duplo-cego randomizado de extensão conduzido para fornecer acesso contínuo ao tratamento para pacientes do estudo B3281001 (Estudo B3281004).
B3281006
O estudo B3281006 foi um estudo randomizado, duplo-cego, comparando a eficácia e segurança de Ruxience® (n = 196) vs. MabThera® (n = 198) em pacientes com CD20 positivo, com baixa carga tumoral e linfoma folicular no tratamento de primeira linha. Os pacientes foram randomizados na proporção de 1:1 para receber Ruxience® ou MabThera® administrado como infusão IV à dose de 375 mg/m2 nas Visitas 2, 3, 4 e 5 (Dias 1, 8, 15 e 22). A dose máxima de rituximabe administrada em um dia foi de 1.125 mg via infusão IV.
O objetivo primário deste estudo foi comparar a eficácia de Ruxience® a MabThera® quando administrados como tratamento de primeira linha em pacientes com CD20 positivo, com baixa carga tumoral e linfoma folicular. O parâmetro de avaliação primário de eficácia foi a taxa de resposta global (TRO) de Ruxience® e MabThera® na Semana 26, e é definido como a proporção de pacientes que obtiveram resposta completa (RC) ou resposta parcial (RP), de acordo com os critérios de resposta revistos para o linfoma maligno. Os parâmetros de avaliação secundários incluíram eficácia, segurança, farmacocinética, farmacodinâmica e imunogenicidade adicionais.
A semelhança entre o Ruxience® e o MabThera® foi demonstrada estatisticamente para o parâmetro de avaliação primário de eficácia, TRO, com base em critérios pré-especificados de -16,0% a 16,0%. As TROs foram n = 148 (75,5%) para Ruxience® e n = 140 (70,7%) para MabThera®. A análise da TRO derivada de avaliações de revisão central mostrou uma diferença estimada de 4,66%, com um IC 95% de (-4,16%, 13,47%), que caiu inteiramente dentro da margem de equivalência. Os resultados de outros parâmetros de avaliação secundários foram comparáveis entre os 2 grupos de tratamento.
Não houve diferenças clinicamente significativas na eficácia, na segurança ou na imunogenicidade entre o Ruxience® e o MabThera® em pacientes com CD20 positivo, com baixa carga tumoral e linfoma folicular.
B3281001 e B3281004
O estudo B3281001 foi um estudo de grupo paralelo, duplo-cego, randomizado, de 1:1:1 em pacientes com artrite reumatoide, que comparou a farmacocinética, a farmacodinâmica e a segurança (inclusive a imunogenicidade) de Ruxience®, MabThera® ou Rituxan. Os desfechos secundários incluíram avaliações da atividade clínica da doença.
O estudo B3281004 foi um estudo de extensão, duplo-cego, randomizado, conduzido em pacientes com artrite reumatoide que participaram por, pelo menos, 16 semanas no estudo B3281001. Os pacientes em tratamento com Ruxience® no estudo B3281001 continuaram a receber Ruxience® e os pacientes que receberam MabThera® ou Rituxan trocaram para Ruxience®. Os desfechos secundários incluíram avaliações da atividade clínica da doença.
O estudo B3281001 e o estudo B3281004 não foram concebidos para comparação estatística formal de desfecho de eficácia. Os resultados de eficácia desses estudos foram comparáveis entre Ruxience®, MabThera® e Rituxan. Não houve diferenças clinicamente significativas na segurança ou na imunogenicidade entre o Ruxience®, o MabThera® e o Rituxan em pacientes com artrite reumatoide.
Resultados de Eficácia do comparador - MabThera®
1. Linfoma não Hodgkin (LNH) de baixo grau ou folicular
Monoterapia
Tratamento inicial, semanal, em quatro doses:
No estudo pivotal, 166 pacientes com LNH de baixo grau ou folicular de células B, recidivado ou resistente à quimioterapia receberam quatro doses de 375 mg/m2 de MabThera® em infusão IV, uma vez por semana. A taxa de resposta global (TRG) na população ITT (intenção de tratamento) foi de 48% (IC95% 41% - 56%), com 6% de respostas completas (RC) e 42% de respostas parciais (RP). A mediana projetada do tempo para progressão da doença nos pacientes responsivos foi de 13 meses.
Em uma análise de subgrupo, a TRG foi maior em pacientes com subtipos histológicos da "International Working Formulation" B, C e D, em comparação com o subtipo A (58% versus 12%), foi maior em pacientes cuja maior lesão era < 5 cm versus > 7 cm no seu maior diâmetro (53% versus 38%) e foi maior em pacientes que apresentaram recidiva quimiossensível versus recidiva quimiorresistente (definida como duração de resposta < três meses) (50% versus 22%). A TRG em pacientes previamente tratados com transplante de medula óssea autólogo foi de 78% contra 43% em pacientes não submetidos a transplante de medula óssea autóloga. Idade, gênero, grau do linfoma, diagnóstico inicial, presença ou ausência de doença volumosa, desidrogenase láctica (LDH) alta ou normal ou presença de doença extranodal não apresentaram efeito estatisticamente significativo (teste exato de Fisher) sobre a resposta a MabThera®.
Relação estatisticamente significativa foi encontrada entre taxas de resposta e comprometimento da medula óssea. Quarenta por cento dos pacientes com comprometimento da medula óssea responderam, em comparação a 59% dos pacientes sem comprometimento da medula óssea (p = 0,0186). Esse achado não foi suportado por uma análise de regressão logística passo a passo, na qual os seguintes fatores foram identificados como prognósticos: tipo histológico, positividade bcl-2 no quadro inicial, resistência à última quimioterapia e doença volumosa.
Tratamento inicial, semanal, em oito doses:
Em um estudo multicêntrico de braço único, 37 pacientes com LNH de células B, baixo grau ou folicular, recidivado ou resistente à quimioterapia receberam oito doses de 375 mg/m2 de MabThera® em infusão IV, uma vez por semana. A TRG foi 57% (IC 95% 41% - 73%; RC 14%, RP 43%), com uma mediana projetada do tempo até a progressão da doença de 19,4 meses (variando de 5,3 até 38,9 meses).
Doença volumosa, tratamento inicial, semanal, em quatro doses:
Em dados compilados de três estudos, 39 pacientes com LNH de células B, baixo grau ou folicular, com doença volumosa (lesão única ≥ 10 cm de diâmetro), recidivado ou resistente à quimioterapia receberam quatro doses de 375 mg/m2 de MabThera®, em infusão IV, uma vez por semana. A TRG foi 36% (IC95% 21% - 51%, RC 3%, RP 33%), com a mediana do tempo até a progressão da doença de 9,6 meses (variando de 4,5 até 26,8 meses).
Retratamento, semanal, em quatro doses:
Em um estudo multicêntrico, de braço único, 58 pacientes com LNH de células B, baixo grau ou folicular, recidivado ou resistente à quimioterapia, que haviam apresentado resposta objetiva a um tratamento anterior com MabThera® foram novamente tratados com quatro doses de 375 mg/m2 de MabThera® em infusão IV, uma vez por semana. Três desses pacientes já haviam recebido dois ciclos anteriores de MabThera® antes do estudo; portanto, receberam o terceiro já após a inclusão. Dois pacientes foram retratados duas vezes durante o estudo. Para os 60 retratamentos, a TRG foi 38% (IC95% 26% - 51%; RC 10%; e RP 28%), com mediana projetada do tempo para progressão da doença de 17,8 meses (variando de 5,4 até 26,6). Esse dado é comparado favoravelmente com o tempo de 12,4 meses até a progressão da doença obtido após o primeiro tratamento com MabThera®.
Em associação à quimioterapia
Tratamento inicial:
Em um estudo randomizado, aberto, 322 pacientes com linfoma folicular sem tratamento prévio foram randomizados para receber quimioterapia CVP (ciclofosfamida 750 mg/m2, vincristina 1,4 mg/m2 até o máximo de 2 mg no dia 1 e prednisolona 40 mg/m2/dia nos dias 1 a 5) a cada três semanas, por oito ciclos, ou MabThera® 375 mg/m2 associado com CVP (R-CVP). MabThera® foi administrado no primeiro dia de cada ciclo de tratamento. No total, 321 pacientes (162 R-CVP, 159 CVP) receberam o tratamento e foram analisados quanto à eficácia.
O tempo mediano de acompanhamento foi de 53 meses. O esquema R-CVP levou a benefícios significativos, em comparação com CVP apenas em relação ao desfecho primário, tempo até a falha do tratamento (27 meses versus 6,6 meses, p < 0,0001, teste logrank). A proporção de pacientes com resposta tumoral (RC, RCu - não confirmada, RP) foi significantemente maior (p < 0,0001 teste do qui-quadrado) no grupo R-CVP (80,9%) que no grupo CVP (57,2%). O tratamento com R-CVP, em comparação ao CVP, prolongou significativamente o tempo até a progressão da doença ou óbito em 33,6 meses e 14,7 meses, respectivamente (p < 0,0001, teste logrank). A duração mediana da resposta foi de 37,7 meses no grupo R-CVP e de 13,5 meses no grupo CVP (p < 0,0001, teste logrank). A diferença entre os grupos de tratamento, em relação à sobrevida global, mostrou forte benefício clínico (p = 0,029, teste logrank estratificado por centro de estudo): a porcentagem de sobrevida em 53 meses foi de 80,9% para pacientes no grupo R-CVP, em comparação a 71,1% para pacientes no grupo CVP.
Os resultados de outros três estudos clínicos randomizados, usando MabThera® em combinação com outros regimes de quimioterapia, além do CVP, CHOP (ciclofosfamida, doxorrubicina, vincristina, prednisolona), MCP (mitoxantrona, clorambucil e prednisona), CHVP (ciclofosfamida, doxorrubicina, teniposídeo e prednisona) / alfainterferona também demonstraram melhorias significativas nas taxas de resposta, nos parâmetros dependentes do tempo e sobrevida global. Os principais resultados dos quatro estudos estão resumidos na Tabela 1 a seguir.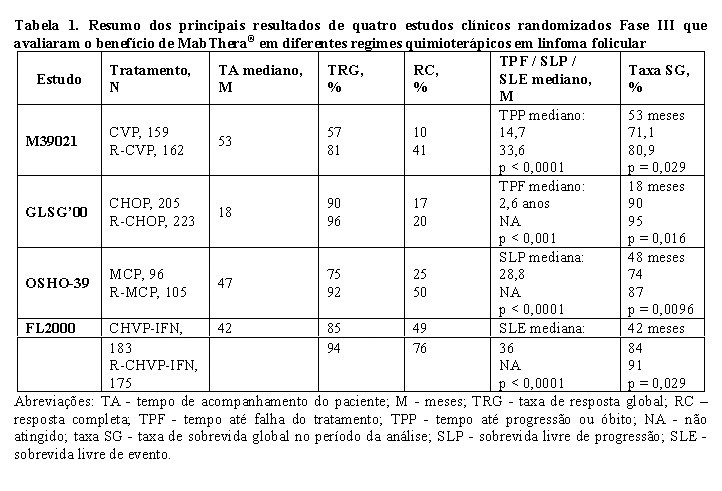
Terapia de manutenção
- Linfoma não Hodgkin folicular não tratado previamente
Em um estudo prospectivo, aberto, internacional, multicêntrico, fase III, 1.193 pacientes com linfoma folicular avançado não tratado previamente receberam terapia de indução com R-CHOP (n = 881), R-CVP (n = 268) ou R-FCM (ciclofosfamida, fludarabina e mitoxantrona) (n = 44), de acordo com a escolha do investigador. Um total de 1.078 pacientes respondeu à terapia de indução, dos quais 1.018 foram randomizados para terapia de manutenção com MabThera® (n = 505) ou observação (n = 513). Os dois grupos de tratamento foram bem equilibrados com relação às características basais e condição da doença. O tratamento de manutenção com MabThera® consistiu em infusão simples de MabThera® na dose de 375 mg/m2 de superfície corpórea a cada dois meses, até a progressão da doença, ou por período máximo de dois anos.
Após o tempo mediano de observação de 25 meses da randomização, a terapia de manutenção com MabThera® resultou em melhora clínica e estatisticamente significante no desfecho primário de sobrevida livre de progressão avaliada pelo investigador (SLP), quando comparada ao grupo de observação em pacientes com Linfoma não Hodgkin folicular não tratado previamente (Tabela 2). Essa melhora na SLP foi confirmada por um comitê de revisão independente (IRC) (Tabela 2).
O benefício significativo do tratamento de manutenção com MabThera® foi também observado para os desfechos secundários de sobrevida livre de eventos (SLE), tempo para o próximo tratamento anti-linfoma (TNLT), tempo para a próxima quimioterapia (TNCT) e taxa de resposta global (TRG) (Tabela 2).
A atualização da análise correspondente ao tempo mediano de observação de 73 meses a partir da randomização confirma os resultados da análise primária (Tabela 2).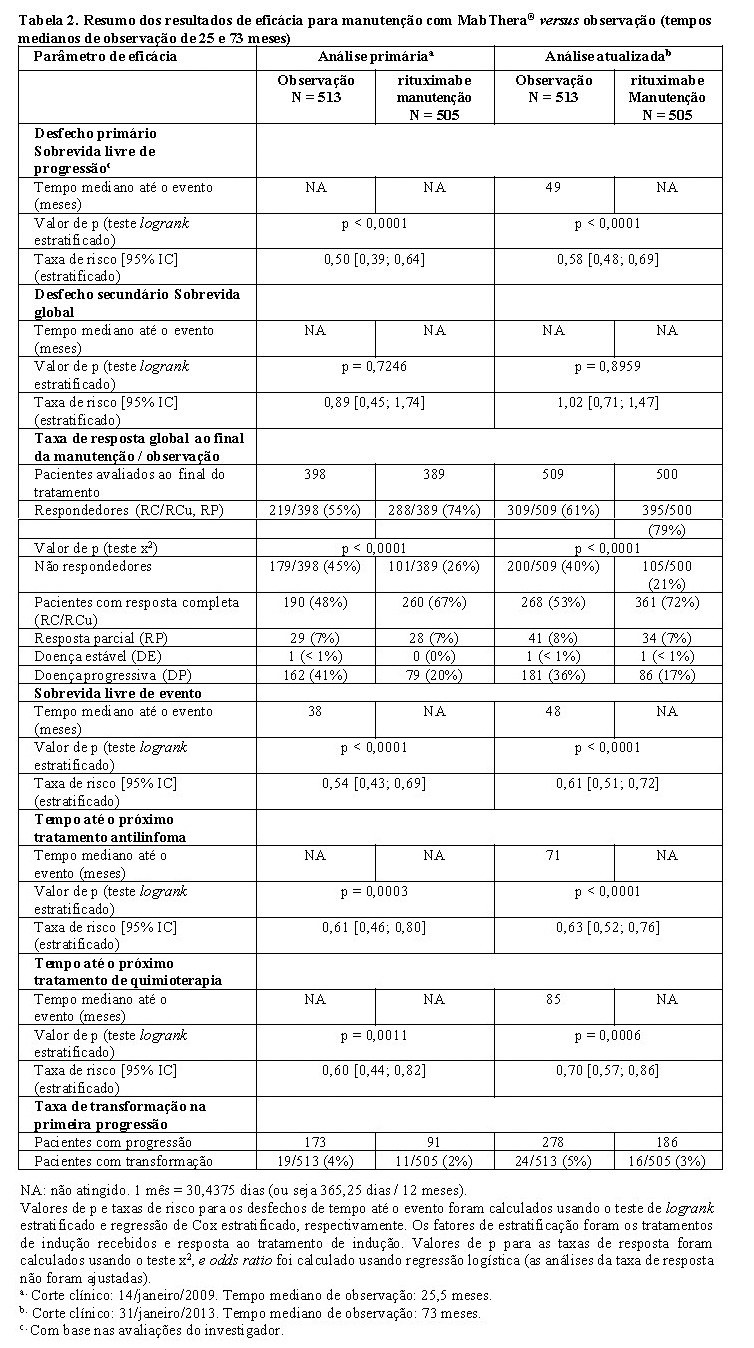
A terapia de manutenção com MabThera® proporcionou benefício consistente em todos os subgrupos avaliados: gênero (homens, mulheres), idade ( < 60 anos, ≥ 60 anos), índice de prognóstico internacional para linfoma folicular (FLIPI) (1, 2 ou 3), terapia de indução (R-CHOP, R-CVP ou R-FCM) e independente da qualidade de resposta ao tratamento de indução (RC ou RP).
- Linfoma não Hodgkin folicular recidivado/refratário
Em um estudo prospectivo, aberto, internacional, multicêntrico, fase III, 465 pacientes com LNH folicular recidivado/refratário foram randomizados em uma primeira etapa para terapia de indução com CHOP (ciclofosfamida, doxorrubicina, vincristina, prednisolona, n = 231) ou com MabThera® mais CHOP (R-CHOP, n = 234). Os dois grupos de tratamento foram bem equilibrados em relação às características basais e condição da doença. Um total de 334 pacientes alcançou remissão completa ou parcial na fase de indução e foi randomizado em uma segunda etapa para o tratamento de manutenção com MabThera® (n = 167) ou observação (n = 167). O tratamento de manutenção com MabThera® consistiu em infusão simples de MabThera® na dose de 375 mg/m2 de superfície corpórea a cada três meses, até a progressão da doença, ou por período máximo de dois anos.
A análise final da eficácia incluiu todos os pacientes randomizados para ambas as fases do estudo.
Após o tempo mediano de observação de 31 meses para pacientes randomizados na fase de indução, R-CHOP melhorou significativamente o resultado em pacientes com LNH folicular recidivado / refratário, quando comparado com o CHOP (Tabela 3).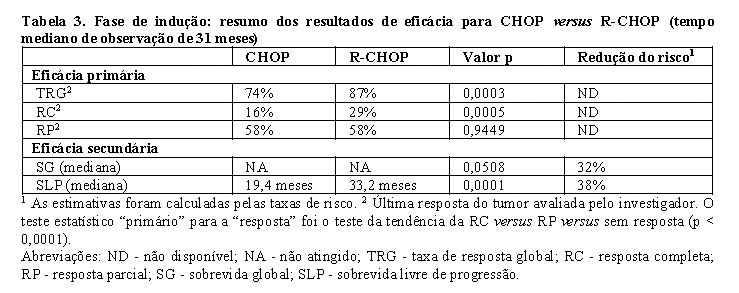
Em pacientes randomizados para a fase de manutenção do estudo, o tempo mediano de observação foi de 28 meses, a partir da randomização para manutenção. O tratamento de manutenção com MabThera® conduziu a melhora clinicamente relevante e estatisticamente significativa no desfecho primário, a SLP (tempo desde a randomização para manutenção até a recidiva, progressão da doença ou óbito), quando comparado somente com a observação (p < 0,0001 teste logrank). A mediana da SLP foi 42,2 meses no braço de manutenção com MabThera®, em comparação com 14,3 meses no braço de observação. Usando a análise de regressão de Cox, o risco de ocorrer progressão da doença ou óbito foi reduzido em 61% no grupo de tratamento de manutenção com MabThera®, quando comparado com a observação (IC95%; 45% - 72%). As taxas livres de progressão em 12 meses estimadas por Kaplan-Meier foram de 78% no grupo de manutenção com MabThera® versus 57% no grupo de observação. A análise da sobrevida global confirmou benefício significativo da manutenção com MabThera® sobre a observação (p = 0,0039 teste logrank). A manutenção com MabThera® reduziu o risco de morte em 56% (IC95%; 22% - 75%).
O tempo mediano para novo tratamento contra o linfoma foi significativamente mais longo no grupo que recebeu tratamento de manutenção com MabThera®, em comparação com a observação (38,8 meses versus 20,1 meses, p < 0,0001 teste logrank). A probabilidade de iniciar novo tratamento foi reduzida em 50% (IC95%; 30% - 64%). Em pacientes que atingiram RC/RCu (resposta completa / resposta completa não confirmada) como a melhor resposta durante o tratamento de indução, o tratamento de manutenção com MabThera® prolongou significativamente a mediana de sobrevida livre de doença (SLD), em comparação com o grupo de observação (53,7 versus 16,5 meses, p = 0,0003 teste logrank) (Tabela 4). O risco de recidiva em pacientes com respostas completas foi reduzido em 67% (IC95%; 39% - 82%).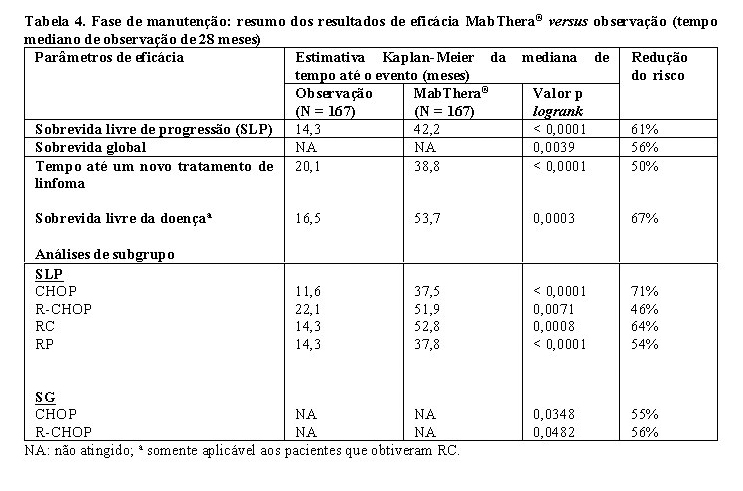
O benefício do tratamento de manutenção com MabThera® foi confirmado em todos os subgrupos analisados, independentemente do regime de indução (CHOP ou R-CHOP) ou da qualidade da resposta para o tratamento de indução (RC ou RP) (Tabela 4). Prolongou também significativamente a mediana da SLP em pacientes respondedores à terapia de indução com CHOP (SLP mediana 37,5 meses versus 11,6 meses, p < 0,0001), bem como em pacientes que responderam à indução com R-CHOP (mediana da SLP 51,9 meses versus 22,1 meses, p = 0,0071). O tratamento de manutenção com MabThera® promoveu benefícios clinicamente significativos em sobrevida global para ambos os pacientes que responderam à terapia CHOP e R-CHOP na fase de indução do estudo.
O tratamento de manutenção com MabThera® promoveu benefícios consistentes em todos os subgrupos testados: gênero (masculino, feminino), idade (≤ 60 anos, > 60 anos), estágio (III, IV), condição de desempenho da Organização Mundial de Saúde (OMS) (0 versus > 0), sintomas B (ausentes, presentes), infiltração da medula óssea (não versus sim), IPI (0 - 2 versus 3 - 5), escore FLIPI (0 - 1, versus 2 versus 3 - 5), número de sítios extranodais (0 - 1 versus > 1), número de sítios nodais ( < 5 versus ≥ 5), número de regimes prévios (1 versus 2), melhor resposta à terapia prévia (RC/RP versus NC/DP), hemoglobina ( < 12 g/dL versus ≥ 12 g/dL), b2 microglobulina ( < 3 mg/L versus ≥ 3 mg/L), LDH (elevado, não elevado), exceto para um pequeno grupo de pacientes com doença volumosa.
2. Linfoma não Hodgkin (LNH) difuso de grandes células B
Em um estudo randomizado, aberto, 399 pacientes idosos (idade de 60 a 80 anos) com LNH difuso de grandes células, sem tratamento prévio, receberam a quimioterapia padrão CHOP (ciclofosfamida 750 mg/m2, doxorrubicina 50 mg/m2, vincristina 1,4 mg/m2 até o máximo de 2 mg no dia 1 e prednisolona 40 mg/m2/dia, nos dias 1 a 5), a cada três semanas, por oito ciclos, ou MabThera® 375 mg/m2 + CHOP (R-CHOP). MabThera® foi administrado no primeiro dia de cada ciclo.
A análise de eficácia incluiu todos os pacientes randomizados (197 CHOP, 202 R-CHOP), com acompanhamento mediano de 31 meses de duração. Os dois grupos de tratamento foram bem balanceados nas suas características basais e condição da doença. A análise final confirmou que o R-CHOP aumenta significativamente a duração de sobrevida livre de eventos (o parâmetro primário de eficácia, no qual eventos considerados foram: óbito, recidiva, progressão do linfoma ou instituição de novo tratamento contra o linfoma) (p = 0,0001). Pelo método de Kaplan-Meier, a estimativa mediana da duração da sobrevida livre de eventos foi de 35 meses no braço de R-CHOP, comparada a 13 meses no braço de CHOP (redução de risco de 41%). Aos 24 meses, a estimativa para sobrevida global foi de 68,2% no braço de R-CHOP, comparada a 57,4% no braço de CHOP. Uma análise da sobrevida global realizada com tempo de seguimento mediano de 60 meses de duração confirmou os benefícios do R-CHOP sobre o tratamento com CHOP (p = 0,0071), representando redução de risco de 32%.
A análise de todos os parâmetros secundários (taxa de resposta, sobrevida livre de progressão, sobrevida livre de doença, duração da resposta) comprovou o efeito do tratamento com R-CHOP, em comparação ao CHOP. A taxa de resposta completa após o ciclo 8 foi de 76,2% no grupo R-CHOP e 62,4% no grupo CHOP (p = 0,0028). O risco de progressão da doença foi reduzido em 46%, e o risco de recaída, em 51%.
Em todos os subgrupos de pacientes (gênero, idade, IPI ajustado à idade, estágio de Ann Arbor, ECOG, b2microglobulina, LDH, albumina, sintomas B, doença volumosa, sítios extranodais, comprometimento da medula óssea), as taxas de risco para sobrevida livre de eventos e sobrevida global (R-CHOP comparado ao CHOP) foram menores que 0,83 e 0,95, respectivamente. R-CHOP associou-se à melhora no resultado, para pacientes com alto ou baixo risco, de acordo com o IPI ajustado por idade.
3. Leucemia linfoide crônica (LLC) sem tratamento prévio e com recaída / refratária
Em dois estudos randomizados, abertos, um total de 817 pacientes com LLC sem tratamento prévio e 552 pacientes com LLC recaída / refratária foram escolhidos para receber quimioterapia FC (25 mg/m2 fludarabina, ciclofosfamida 250 mg/m2, nos dias 1 - 3), a cada quatro semanas, durante seis ciclos, ou MabThera® em combinação com FC (R-FC).
MabThera® foi administrado na dosagem de 375 mg/m2 durante o primeiro ciclo um dia antes da quimioterapia e na dosagem de 500 mg/m2 no primeiro dia de cada ciclo de tratamento subsequente. Um total de 810 pacientes (403 R-FC, 407 FC) no estudo de primeira linha (Tabela 5 e Tabela 6) e 552 pacientes (276 R-FC, 276 FC) para o estudo de recaída / refratária (Tabela 7) foi analisado para eficácia.
No estudo de primeira linha, após o tempo mediano de observação de 20,7 meses, a sobrevida livre de progressão (desfecho primário) mediana foi de 40 meses no grupo de R-FC e 32 meses no grupo de FC (p < 0,0001, teste logrank) (Tabela 5). As análises da sobrevida global mostraram melhora da sobrevida em favor do braço de R-FC (p = 0,0427 teste logrank). Esses resultados foram confirmados no acompanhamento estendido: após o tempo mediano de observação de 48,1 meses, a mediana de SLP foi 55 meses no grupo de R-FC e 33 meses no grupo de FC (p < 0,0001, teste logrank) e a análise da sobrevida global continuou a mostrar um benefício significante do tratamento R-FC sobre a quimioterapia FC (p = 0,0319, teste logrank). O benefício em termos de SLP foi observado consistentemente na maioria dos subgrupos de pacientes analisados de acordo com o risco da doença no período basal (isto é, classificação de Binet A-C) e foi confirmado no acompanhamento estendido (Tabela 6).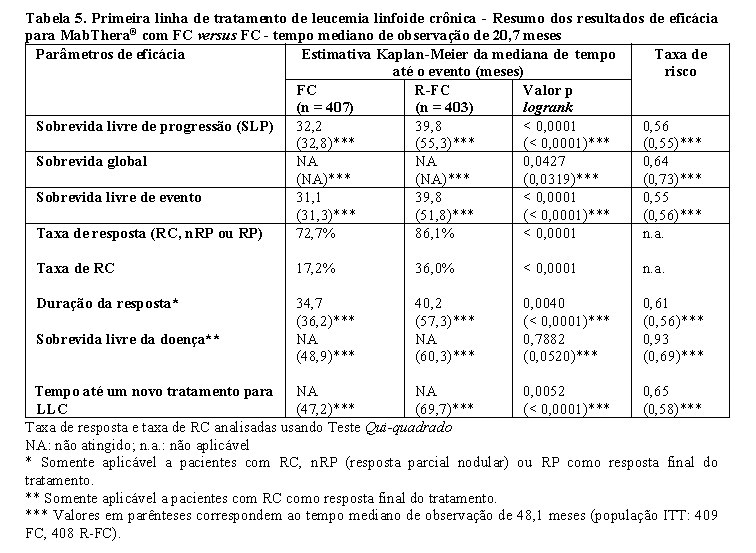
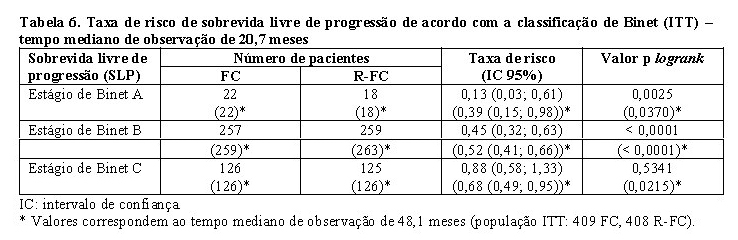
No estudo de pacientes com LLC recaída / refratária, a mediana da sobrevida livre de progressão (desfecho primário) foi de 30,6 meses no grupo de R-FC e 20,6 meses no grupo do FC (p = 0,0002, teste logrank). O benefício em termos de SLP foi observado na maioria dos subgrupos de pacientes analisados de acordo com o risco da doença no período basal. Melhora discreta, mas não significativa, na sobrevida global foi relatada no R-FC, em comparação com o braço de FC.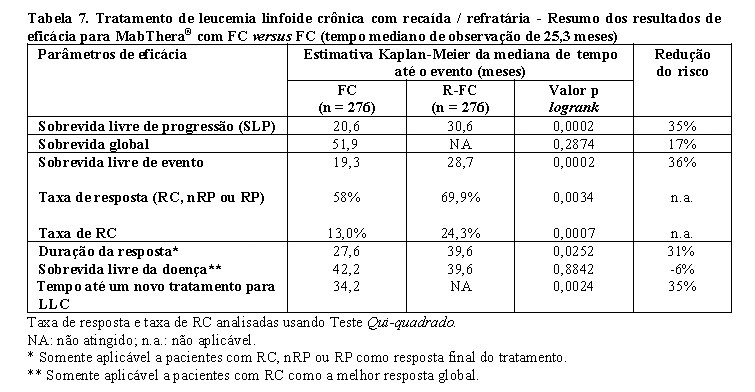
Resultados de outros estudos suporte que utilizaram MabThera® em combinação com outras quimioterapias (incluindo CHOP, FCM, PC, PCM, bendamustina e cladribina) para o tratamento de pacientes com LLC têm também demonstrado elevada taxa de resposta global com taxas de SLP promissoras, sem acrescentar toxicidade para o tratamento.
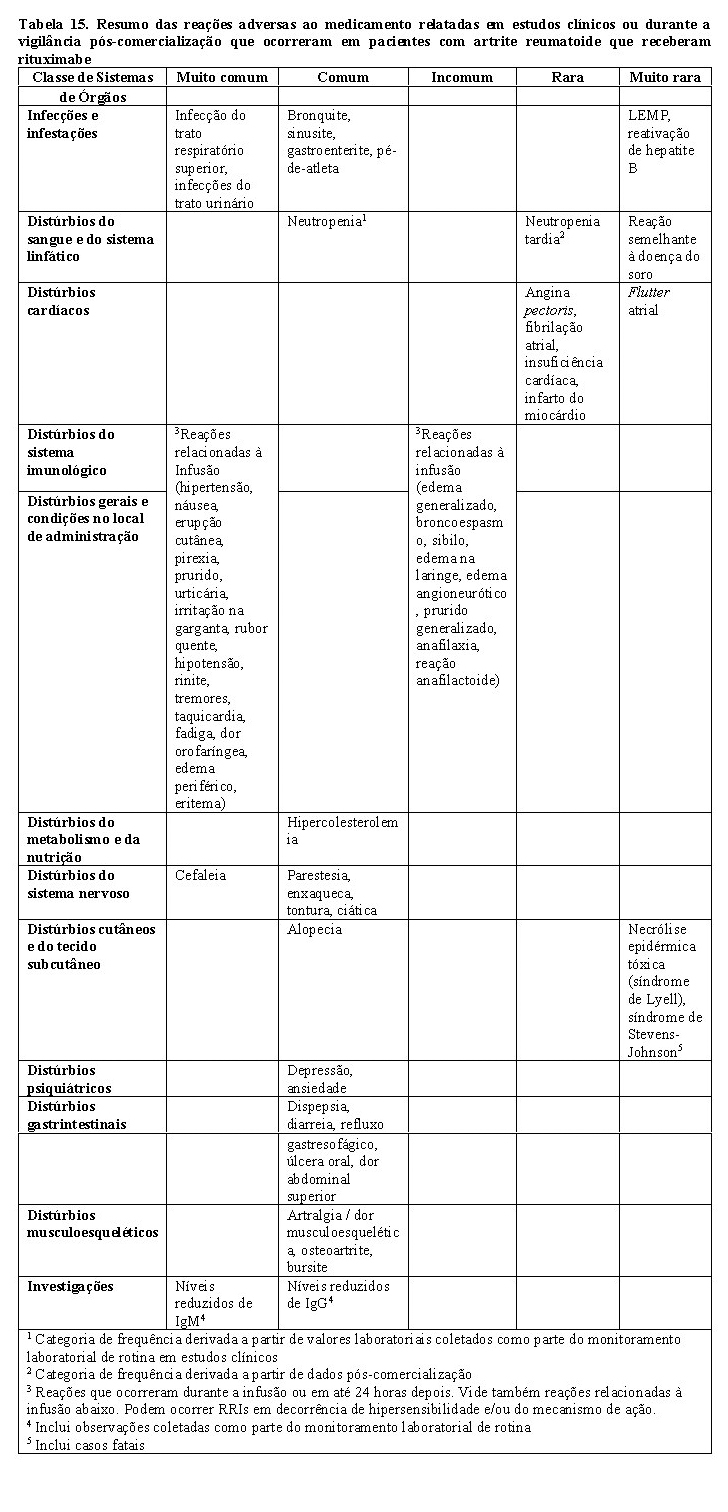
4. Artrite reumatoide
A eficácia e a segurança de MabThera® em aliviar os sinais e sintomas da artrite reumatoide foram demonstradas em três estudos randomizados, controlados, duplos-cegos e multicêntricos.
O Estudo 1 foi um estudo duplo-cego, comparativo, que incluiu 517 pacientes com resposta inadequada ou intolerância a um ou mais inibidores de TNF. Os pacientes elegíveis tinham artrite reumatoide ativa grave diagnosticada de acordo com os critérios do American College of Reumatology (ACR). O desfecho primário foi a proporção de pacientes que alcançou resposta ACR 20 na semana 24. Os pacientes receberam 2 x 1.000 mg de MabThera®, cada uma precedida por 100 mg de metilprednisolona IV, separadas por um intervalo de 15 dias. Todos os pacientes receberam concomitantemente metotrexato (MTX) oral (10 - 25 mg/semana) e 60 mg de prednisolona oral nos dias 2 - 7 e 30 mg nos dias 8 - 14 após a primeira infusão. Os pacientes foram acompanhados além da semana 24 em relação aos desfechos tardios, incluindo avaliação radiográfica em 56 semanas. Nesse período, os pacientes poderiam ter recebido outros cursos de rituximabe durante um prolongamento aberto do protocolo de estudo.
O Estudo 2 foi randomizado, duplo-cego, duplo-mascarado, multifatorial, que comparou duas doses diferentes de rituximabe administradas com ou sem um dos dois regimes de corticosteroide pré-infusional em combinação com metotrexato semanal em pacientes com artrite reumatoide ativa que não responderam ao tratamento com um a cinco outros DMARDs.
O Estudo 3 foi um estudo duplo-cego, duplo-mascarado, controlado e avaliou a monoterapia com rituximabe e rituximabe em combinação com ciclofosfamida ou metotrexato em pacientes com artrite reumatoide ativa que não responderam a um ou mais DMARDs anteriores.
O grupo de comparação, em todos os três estudos, recebeu metotrexato semanal (10 - 25 mg/semana).
Resultados de atividade da doença
Nos três estudos, a proporção de pacientes que alcançou melhora de, pelo menos, 20% no escore ACR foi significativamente maior após rituximabe (2 x 1.000 mg), em comparação com pacientes tratados com metotrexato apenas (Tabela 8). Em todos os estudos de desenvolvimento, o benefício do tratamento foi similar em todos os pacientes, independentemente de idade, gênero, superfície corporal, raça, número de tratamentos anteriores ou status do fator reumatoide.
Melhora clínica e estatisticamente significativa também foi notada em todos os componentes individuais da resposta ACR [número de articulações dolorosas e edemaciadas, avaliação global do paciente e do médico, escore do índice de incapacidade (HAQ), avaliação da dor e PCR (mg/dL)].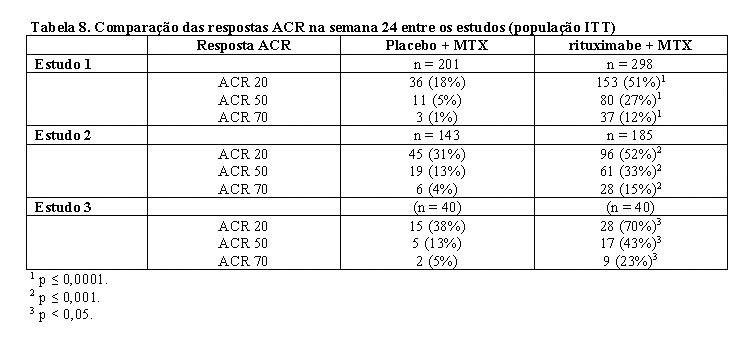
No Estudo 3, a resposta ACR 20, em pacientes tratados apenas com rituximabe, foi de 65%, em comparação com 38% em pacientes tratados apenas com metotrexato (p = 0,025).
Os pacientes tratados com MabThera® tiveram redução significativamente maior no escore de atividade da doença (DAS 28) que os pacientes tratados apenas com metotrexato. Resposta EULAR boa e moderada foi alcançada por um número significativamente maior de pacientes tratados com rituximabe, em comparação com pacientes tratados com metotrexato apenas (Tabela 9).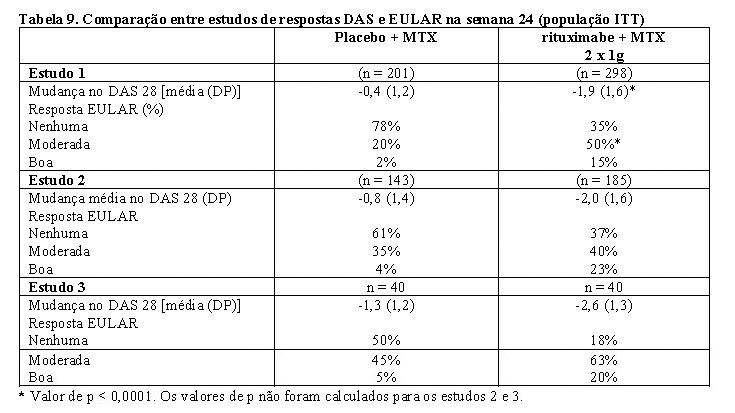
Inibição do dano articular
No Estudo 1, o dano estrutural articular foi avaliado radiograficamente e expresso como alteração no Escore Total de Sharp Modificado (mTSS) e seus componentes, escore de erosão e de estreitamento do espaço articular. Esse estudo conduzido em pacientes TNF-IR que receberam MabThera® em combinação com metotrexato demonstrou redução significativa da progressão radiográfica em 56 semanas, em comparação com pacientes que receberam apenas metotrexato. Maior proporção de pacientes que receberam MabThera® também não apresentou progressão da erosão após 56 semanas.
Também foi observada inibição da taxa de progressão do dano articular em longo prazo. A análise radiográfica em dois anos, no estudo 1, demonstrou progressão significativamente menor no dano estrutural articular em pacientes que receberam MabThera® (2 x 1.000 mg) + MTX, em comparação com pacientes que receberam MTX apenas, bem como uma proporção significativamente maior de pacientes sem nenhuma progressão de dano articular em um período de dois anos.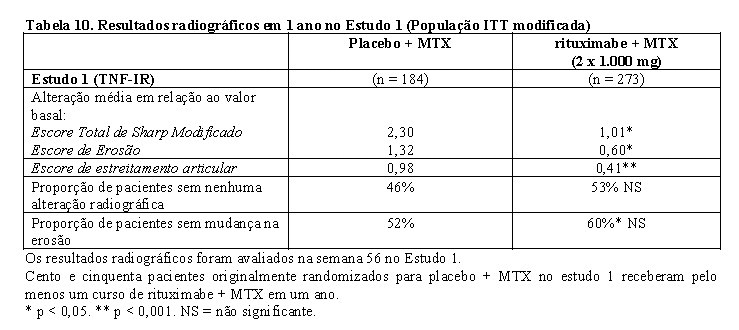
Resultados de qualidade de vida
Os pacientes tratados com MabThera® apresentaram melhora em todos os resultados (Questionários HAQ-DI, FACIT-Fadiga e SF-36). Reduções significativas no índice de invalidez (HAQ-DI), fadiga (FACIT-Fadiga) e melhoras nos domínios físico e mental do SF-36 foram observadas em pacientes tratados com MabThera®, em comparação aos pacientes tratados apenas com metotrexato.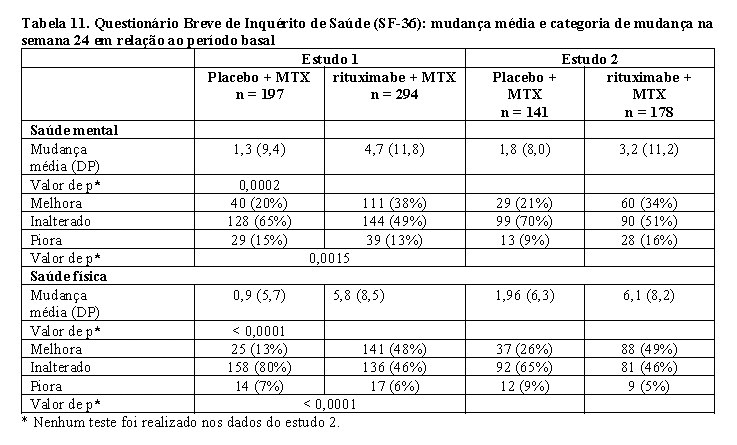
Categoria de mudança da saúde mental: mudança > 6,33 = melhora, -6,33 ≤ mudança < 6,33 = inalterado, mudança < -6,33 = piora.
Categoria de mudança da saúde física: mudança > 5,42 = melhora, -5,42 ≤ mudança < 5,42 = inalterado, mudança < -5,42 = piora.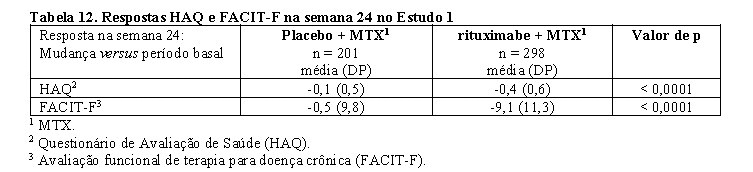
Na semana 24, em todos os três estudos, a proporção de pacientes que apresentaram melhora clinicamente relevante no HAQ-DI (definido como uma diminuição no escore total > 0,25) foi maior nos pacientes tratados com rituximabe que entre os pacientes que receberam metotrexato em monoterapia.
Avaliações laboratoriais
Cerca de 10% dos pacientes com AR apresentaram anticorpo antidroga (ADA) positivo nos estudos clínicos. A emergência de ADA não se associou à piora clínica ou a maior risco de reações infusionais na maioria dos pacientes.
A presença de ADA pode estar associada à piora das reações à infusão ou alérgicas após a segunda infusão de cursos subsequentes. Raramente observou-se falha na depleção de células B após cursos adicionais do tratamento.
Em pacientes com fator reumatoide positivo (FR+), observou-se diminuições acentuadas nas concentrações do FR, após tratamento com rituximabe, nos três estudos (intervalo de 45% - 64%, figura 1).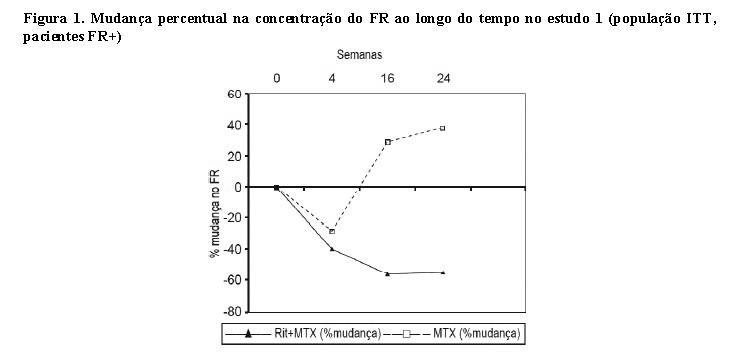
No geral, as concentrações plasmáticas totais de imunoglobulina (Ig) e as contagens totais de linfócitos e leucócitos permaneceram dentro dos limites de normalidade após tratamento com MabThera®, com exceção de queda transitória de leucócitos nas primeiras quatro semanas após o tratamento. Títulos IgG específicos para caxumba, rubéola, varicela, toxoide tetânico, influenza e Streptococcus pneumoniae permaneceram estáveis nas 24 semanas após exposição a MabThera® em pacientes com AR.
Os efeitos do rituximabe nos diversos biomarcadores foram avaliados em um subestudo que avaliou o impacto de um único curso de rituximabe nos níveis dos marcadores bioquímicos, incluindo marcadores de inflamação (interleucina 6, proteína C reativa, amiloide sérico A, proteína S100 isotipos A8 e A9), autoanticorpos (FR e anti-CCP) e marcadores de remodelação óssea [osteocalcina e peptídeo terminal procolágeno 1 N (P1NP)]. O tratamento com rituximabe, em monoterapia ou em combinação com MTX ou ciclofosfamida, reduziu significativamente os níveis dos marcadores inflamatórios versus MTX em monoterapia nas primeiras 24 semanas. Os níveis dos marcadores de renovação óssea, osteocalcina e P1NP aumentaram significativamente nos grupos rituximabe, em comparação aos grupos de MTX.
Retratamento
Após a conclusão do período do estudo duplo-cego, comparativo, de 24 semanas, os pacientes receberam permissão para se inscreverem em um estudo aberto, de longo prazo, de acompanhamento. Os pacientes receberam séries subsequentes de MabThera®, de acordo com a avaliação da atividade da doença pelo médico, independentemente da contagem de linfócitos B periféricos. O tempo de intervalo entre os cursos de tratamento foi variável, com a maioria dos pacientes recebendo terapia adicional de 6 - 12 meses após o curso inicial. Alguns pacientes necessitaram de retratamento com menor frequência. A resposta ao curso adicional foi de magnitude similar à do curso de tratamento inicial, conforme evidencia a mudança do DAS 28, em relação ao valor basal (Figura 2).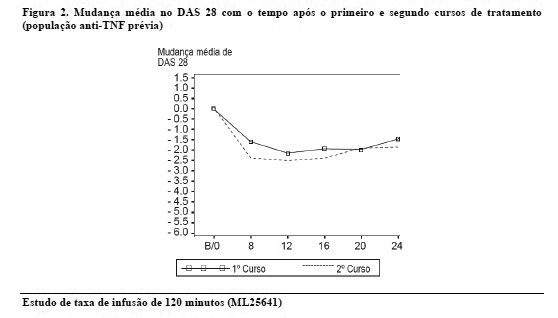
Em um estudo clínico multicêntrico, aberto, de um único braço, 351 pacientes com artrite reumatoide ativa, moderada a grave, que tiveram uma resposta inadequada a pelo menos um inibidor de TNF e estavam recebendo metotrexato, iriam receber dois cursos de tratamento de MabThera®. Pacientes virgens de tratamento com MabThera® (n = 306) e aqueles que tinham recebido 1 ou 2 cursos anteriores de MabThera® (n = 45), 6 - 9 meses antes do basal, eram elegíveis a participarem do estudo.
Os pacientes receberam dois cursos de tratamento com MabThera® (2 x 1000 mg) + metotrexato, sendo o primeiro curso administrado no Dia 1 e Dia 15 e o segundo curso, 6 meses depois, nos dias 168 e 182. A primeira infusão do primeiro curso (Dia 1) foi administrada ao longo de um período de 4,25h (255 minutos). A segunda infusão do primeiro curso (Dia 15) e ambas as infusões do segundo curso (Dia 168 e Dia 182) foram administrados ao longo de um período de 2 horas (120 minutos). Qualquer paciente que apresentasse uma reação grave relacionada à infusão em qualquer infusão era retirado do estudo.
O objetivo primário do estudo foi avaliar a segurança de se administrar a segunda infusão do primeiro curso de MabThera®, ao longo de um período de 2 horas (120 minutos).
A incidência de reações relacionadas à infusão no Dia 15 foi 6,5 % (IC 95% [4,1% - 9,7%]), consistente com a taxa observada historicamente. Não foram observadas reações graves relacionadas à infusão. Os dados observados para as infusões no Dia 168 e Dia 182 (infusão de 120 minutos) demonstraram uma baixa incidência de reações relacionadas à infusão, similares a taxa observada historicamente, sem ocorrência de reações graves relacionadas à infusão (vide item 9. Reações Adversas - Experiência originada dos estudos clínicos em artrite reumatoide).
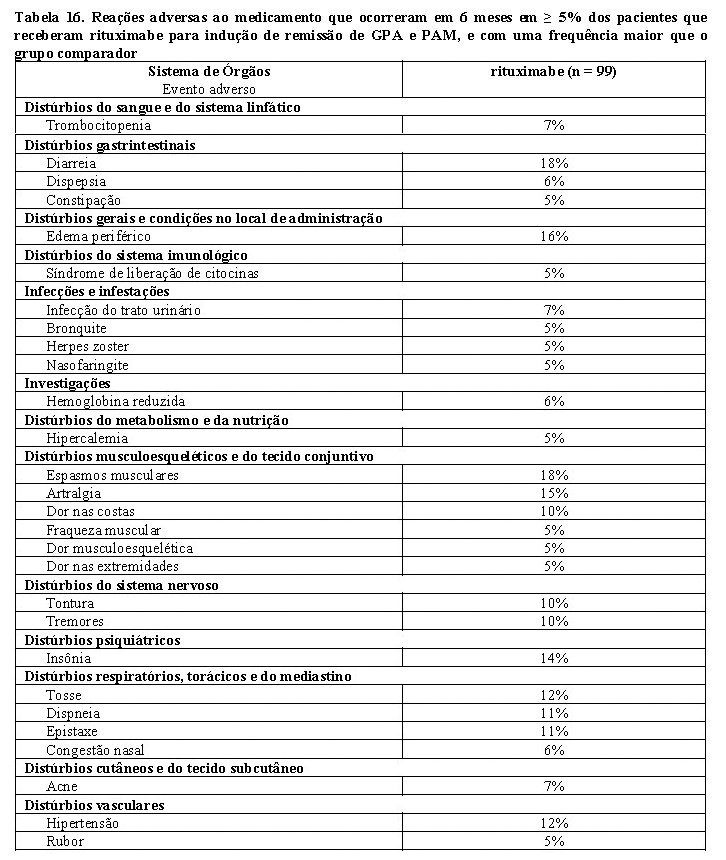
5. Granulomatose com poliangiite (Granulomatose de Wegener - GPA) e poliangiite microscópica (PAM)
Indução da remissão
Um total de 197 pacientes com granulomatose com poliangiite (Granulomatose de Wegener - GPA) e poliangiite microscópica (PAM) ativas graves foi incluído e tratado em um estudo de não inferioridade, multicêntrico, ativo controlado, randomizado e duplo-cego. Os pacientes tinham 15 anos ou mais e diagnóstico de Granulomatose com poliangiite (Granulomatose de Wegener) ativa grave (75% dos pacientes) ou Poliangiite Microscópica (PAM) ativa grave (24% dos pacientes), de acordo com o critério da Conferência do Consenso de Chapel Hill (1% dos pacientes tinha tipo de GPA e PAM desconhecido).
Os pacientes foram randomizados em uma taxa de 1:1 para receber ciclofosfamida oral diária (2 mg/kg/dia) por 3 - 6 meses, seguida de azatioprina ou MabThera® (375 mg/m2), uma vez por semana, por quatro semanas. Os pacientes de ambos os braços receberam 1.000 mg de metilprednisolona em pulsoterapia intravenosa (IV) (ou outro glicocorticoide dose equivalente) por dia, por um a três dias, seguida de prednisona oral (1 mg/kg/dia, não excedendo 80 mg/dia). A retirada da prednisona deveria estar completa em seis meses a partir do início do tratamento do estudo.
A medida do resultado primário foi a remissão completa em seis meses, definida como escore de Atividade de Vasculite de Birmingham para Granulomatose de Wegener (BVAS/WG) igual a zero, sem estar em uso de terapia com glicocorticoide. A margem de não inferioridade pré-especificada para a diferença de tratamento foi de 20%. O estudo demonstrou não inferioridade de MabThera® em relação à ciclofosfamida para a remissão completa em seis meses (Tabela 13). Adicionalmente, a taxa de remissão completa no braço de MabThera® foi significantemente maior que a taxa de remissão completa estimada em pacientes com GPA e PAM graves não tratados ou tratados apenas com glicocorticoides, baseado em dados de controle histórico.
A eficácia foi observada tanto nos pacientes com GPA e PAM recentemente diagnosticados como nos pacientes com doença recidivada.
Referências bibliográficas
1. Weaver R., Shen CD., Grillo-Lopez AJ.
Pivotal phase III multi-center study to evaluate the safety and efficacy of once weekly times four dosing of IDEC-C2B8 (IDEC-102) in patients with relapsed low-grade or follicular B-cell lymphoma. Protocol IDEC-102-05. IDEC Clinical Study Report 102-01-04, January 15, 1997.
2. McLaughlin P., Grillo-Lopez AJ., Link BK., et al. rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J Clin Oncol 1998; 16: 2825-2833.
3. Weaver R., Eldredge E., Alkuzweny B. Integrated summary of efficacy and safety of rituximab, September 27, 1999. Section 3.C.4. Claimed Effect, Response Rate/Time to Progression IDEC Pharmaceuticals Corporation, Rituxan® Biologic License Application Supplement, October 19, 1999.
4. Deardorff J. Clinical Study Report 102-01-06. A phase II multi-center stu