ROPOLIVY
ROCHE
polatuzumabe vedotina
Anticorpo monoclonal.
Apresentações.
Pó liofilizado para solução injetável.
Cada cartucho contém 1 frasco-ampola de dose única de 30 mg ou 140 mg de polatuzumabe vedotina.
VIA INTRAVENOSA
USO ADULTO
COMPOSIÇÃO
Composição.
RoPolivy® 30 mg
Cada frasco-ampola de dose única contém 30 mg de polatuzumabe vedotina. Após a reconstituição, cada mL conterá 20 mg de polatuzumabe vedotina.
Princípio ativo: polatuzumabe vedotina 30 mg. Excipientes: ácido succínico, hidróxido de sódio, sacarose e polissorbato 20
RoPolivy® 140 mg
Cada frasco-ampola de dose única contém 140 mg de polatuzumabe vedotina. Após a reconstituição, cada mL conterá 20 mg de polatuzumabe vedotina.
Princípio ativo: polatuzumabe vedotina 140 mg. Excipientes: ácido succínico, hidróxido de sódio, sacarose e polissorbato 20.
Informações técnicas.
1. INDICAÇÕES
RoPolivy®, em combinação com rituximabe, ciclofosfamida, doxorrubicina e prednisona (R-CHP), é indicado para o tratamento de pacientes adultos com linfoma difuso de grandes células B (LDGCB) não tratados previamente.
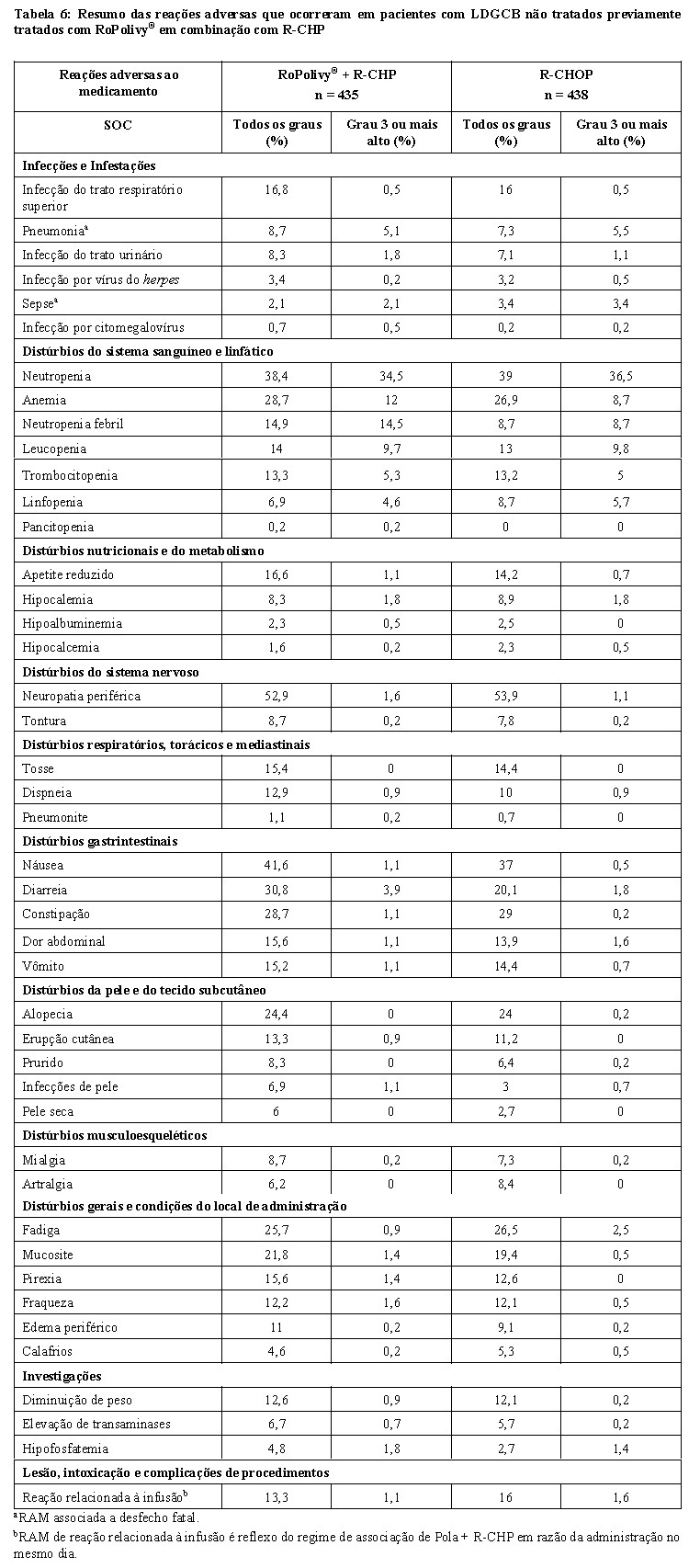
2. RESULTADOS DE EFICÁCIA
Linfoma difuso de grandes células B (LDGCB) não tratado previamente¹
A eficácia do RoPolivy® foi avaliada em um estudo internacional, multicêntrico, randomizado duplo-cego e controlado por placebo (POLARIX, GO39942) em 879 pacientes com LDGCB não tratado previamente.
Os pacientes elegíveis tinham 18-80 anos de idade e apresentavam uma pontuação do International Prognostic Index (IPI) de 2-5 e um Status de Desempenho do Eastern Cooperative Oncology Group (ECOG) de 0-2. As histologias incluíam LDGCB (sem outra especificação [SOE], célula B ativada [ABC], célula B do centro germinativo [GCB]), linfoma de células B de alto grau (LCBAG; SOE, com translocações MYC e BCL-2 e/ou BCL-6 [double hit ou triple hit]) e outros subtipos de linfoma de grandes células B (positivo para vírus Epstein-Barr [VEB], rico em células T/rico em histiócitos). Os pacientes não apresentavam linfoma do sistema nervoso central (SNC) ou neuropatia periférica de > Grau 1 conhecidos.
Os pacientes foram randomizados 1:1 para receber RoPolivy® mais rituximabe, ciclofosfamida, doxorrubicina e prednisona (R-CHP) ou rituximabe, ciclofosfamida, doxorrubicina, vincristina e prednisona (R-CHOP) por seis ciclos de 21 dias, seguidos por dois ciclos adicionais de rituximabe em monoterapia em ambos os braços. Os pacientes foram estratificados por pontuação do IPI (2 vs. 3-5), presença ou ausência de doença volumosa (lesão de ≥ 7,5 cm) e região geográfica.
RoPolivy® foi administrado por via intravenosa com 1,8 mg/kg no Dia 1 dos ciclos 1-6. O regime de R-CHP ou R-CHOP foi administrado com início no Dia 1 dos Ciclos 1-6, seguido por rituximabe isolado no Dia 1 dos Ciclos 7-8. A administração em cada braço de tratamento foi feita de acordo com o seguinte:
• Braço de RoPolivy® + R-CHP: 1,8 mg/kg de RoPolivy®, 375 mg/m² de rituximabe, 750 mg/m² de ciclofosfamida, 50 mg/m² de doxorrubicina e 100 mg/dia de prednisona por via oral, nos dias 1-5 de cada ciclo.
• Braço de R-CHOP: 375 mg/m² de rituximabe, 750 mg/m² de ciclofosfamida, 50 mg/m² de doxorrubicina, 1,4 mg/m² de vincristina e 100 mg/dia de prednisona por via oral, nos dias 1-5 de cada ciclo.
Os dois grupos de tratamento apresentaram equilíbrio em relação aos dados demográficos e às características basais da doença. A idade mediana foi de 65 anos (faixa de 19 a 80 anos), 53,6% dos pacientes eram brancos e 53,8% eram homens. Um percentual de 43,8% apresentava doença volumosa, 38,0% apresentavam uma pontuação do IPI de 2, 62,0% apresentavam uma pontuação do IPI de 3-5 e 88,7% apresentavam doença em Estágio 3 ou 4. A maioria dos pacientes (84,2%) apresentava LDGCB (incluindo SOE, ABC e GCB). Por determinação dos perfis de expressão gênica, 25,1% dos pacientes apresentavam LDGCB do tipo células B ativadas (ABC) e 40,0% dos pacientes apresentavam LDGCB do tipo células B do centro germinativo (GCB).
O desfecho primário do estudo foi a sobrevida livre de progressão avaliada pelo investigador. A duração mediana do acompanhamento foi de 28,2 meses. Os resultados de eficácia estão resumidos na Tabela 1 e na Figura 1.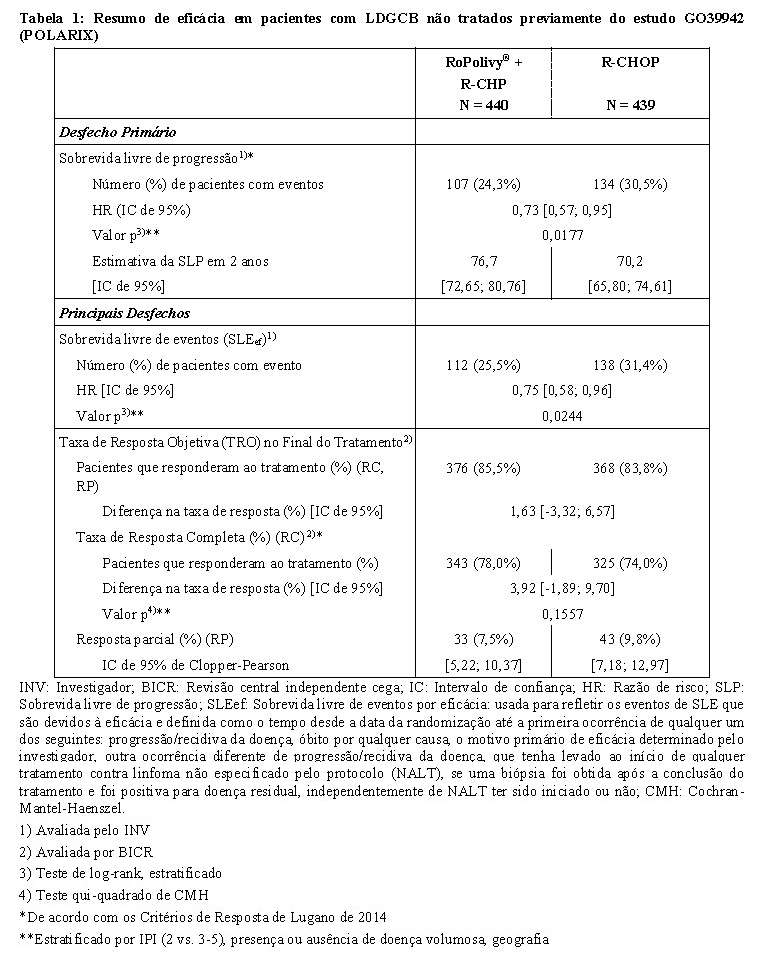
Na análise final, a sobrevida global (SG), um relevante desfecho secundário do estudo, apesar de mais maduro não apresentou resultados estatisticamente diferentes (razão de risco estratificada de 0,94 [IC de 95%: 0,67; 1,33]; p = 0,7326). Não houve diferença entre os braços de tratamento em relação a esse desfecho.
A taxa de RC no final do tratamento foi de 78% no braço de RoPolivy® mais R-CHP e de 74% no braço de R-CHOP. A durabilidade da RC foi avaliada com análise da sobrevida livre de doença (SLD) entre os braços de RoPolivy® mais R-CHP e R-CHOP (HR = 0,70, IC de 95% [0,50, 0,98]) em pacientes que obtiveram uma melhor resposta global de RC por avaliação do investigador enquanto estavam no estudo, com taxas de SLD de referência em 1 ano após a primeira RC documentada de 90% e 83%, respectivamente. A duração da resposta (DR) foi avaliada para pacientes com uma melhor resposta global de RC ou RP com base em avaliação do investigador (HR = 0,74, IC de 95% [0,56, 0,98]); 84% e 78% dos pacientes ainda estavam em remissão em 1 ano após a primeira resposta documentada.
Pola: polatuzumabe vedotina
NE: Não pode ser avaliado
Em uma análise de subgrupos exploratória da SLP, os resultados foram geralmente corroborativos do benefício de RoPolivy® + R-CHP (HR de < 1), embora o estudo não tenha sido desenhado para demonstrar diferenças em subgrupos.
Resultados relatados pelo paciente
A taxa de neuropatia periférica relatada pelo paciente foi avaliada usando o questionário Functional Assessment of Cancer Therapy/Gynecologic Oncology Group - Neurotoxicity (FACT/GOG-Ntx). As pontuações variam entre 0-44, com pontuações mais altas refletindo baixos sintomas de neuropatia periférica e alta qualidade de vida relacionada com saúde (health-related quality of life - HRQoL). Os pacientes em ambos os braços relataram baixos níveis de neuropatia periférica no período basal. Durante a administração do tratamento, a maioria dos aumentos na neuropatia periférica (isto é, diminuições na pontuação) foi menor no braço de RoPolivy® mais R-CHP (faixa média ajustada em relação ao valor basal: 0,22 a -2,71) do que no braço de R-CHOP (faixa média ajustada em relação ao valor basal: 0,01 a -3,51). Os pacientes no braço de R-CHOP apresentaram aumentos na neuropatia periférica mais cedo (Ciclo 4) do que os pacientes no braço de RoPolivy® mais R-CHP (Ciclo 6) (vide item "9. Reações Adversas"). Após a conclusão do tratamento, os níveis de neuropatia periférica em ambos os braços voltaram a níveis próximos dos basais.
Imunogenicidade
Assim como observado em todas as proteínas terapêuticas, existe o potencial de uma resposta imunológica em pacientes tratados com polatuzumabe vedotina. No Estudo GO39442 (POLARIX), 1,4% (6/427) dos pacientes apresentaram resultados de exames positivos para anticorpos contra polatuzumabe vedotina, dos quais nenhum foi positivo para anticorpos neutralizantes. Em razão do número limitado de pacientes positivos para o anticorpo antipolatuzumabe vedotina, não é possível chegar a nenhuma conclusão com relação a um possível efeito da imunogenicidade sobre a eficácia ou segurança.
Os resultados de ensaios de imunogenicidade são altamente dependentes de diversos fatores, incluindo a sensibilidade e a especificidade dos ensaios, a metodologia dos ensaios, o manuseio de amostras, o momento da coleta de amostras, as medicações concomitantes e a doença subjacente. Por esses motivos, a comparação da incidência de anticorpos contra o polatuzumabe vedotina à incidência de anticorpos contra outros produtos pode ser enganosa.
Referências bibliográficas
¹ Tilly H, et al. Polatuzumab Vedotin in Previously Untreated Diffuse Large B-Cell Lymphoma. N Engl J Med. 2022; 386:351-363
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades farmacodinâmicas
Mecanismo de ação
Polatuzumabe vedotina é um anticorpo droga-conjugado direcionado a CD79b que, preferencialmente, entrega um potente agente antimitótico (monometil auristatina E, ou MMAE) às células B, resultando na destruição de células B malignas. A molécula de polatuzumabe vedotina consiste em MMAE ligado de forma covalente a um anticorpo monoclonal humanizado imunoglobulina G1, por meio de um agente de ligação clivável. O anticorpo monoclonal liga-se, com alta afinidade e seletividade, ao CD79b, um componente da superfície celular do receptor da célula B. A expressão de CD79b é restrita às células normais na linhagem das células B (com exceção das células plasmáticas) e células B malignas. Ele é expresso em > 95% do linfoma difuso de grandes células B. Com a ligação ao CD79b, polatuzumabe vedotina é rapidamente internalizado, e o agente de ligação é submetido à clivagem pelas proteases lisossômicas para permitir a entrega de MMAE intracelular. MMAE liga-se aos microtúbulos e destrói as células em divisão, o que inibe a divisão celular e induz a apoptose.
Propriedades farmacocinéticas
A exposição plasmática do anticorpo conjugado MMAE (acMMAE) aumentou de forma proporcional à dose com a faixa de dose de 0,1 a 2,4 mg/kg de polatuzumabe vedotina. Após a primeira dose de 1,8 mg/kg de polatuzumabe vedotina, a concentração máxima média (Cmáx) de acMMAE foi de 803 (± 233) ng/mL, e a área sob a curva de concentração-tempo do tempo zero até o infinito (ASCinf) foi de 1.860 (± 966) dia•ng/mL. Com base na análise da farmacocinética da população, a ASC do acMMAE no ciclo 3 aumentou, aproximadamente, 30% em relação à ASC do ciclo 1 e alcançou mais de 90% da ASC do ciclo 6. A meia-vida terminal no ciclo 6 foi de, aproximadamente, 12 dias (IC 95% de 8,1 - 19,5 dias) para acMMAE.
As exposições de MMAE não conjugado, o componente citotóxico de polatuzumabe vedotina, aumentaram de forma proporcional à dose no intervalo de dose de 0,1 a 2,4 mg/kg de polatuzumabe vedotina. As concentrações plasmáticas de MMAE seguiram a cinética limitada da taxa de formação. Após a primeira dose de 1,8 mg/kg de polatuzumabe vedotina, a Cmáx foi de 6,82 (± 4,73) ng/mL, o tempo até a concentração plasmática máxima é de, aproximadamente, 2,5 dias, e a meia-vida terminal é de, aproximadamente, 4 dias. As exposições plasmáticas de MMAE não conjugado são < 3% das exposições de acMMAE. Com base na análise da farmacocinética da população, há diminuição da exposição plasmática de MMAE não conjugado (ASC) após a administração repetida a cada três semanas.
Absorção
RoPolivy® é administrado como uma infusão intravenosa (IV). Não foram realizados estudos com outras vias de administração.
Distribuição
A estimativa do volume de distribuição central na população para acMMAE foi de 3,15 L, que se aproximou do volume plasmático.
In vitro, MMAE liga-se moderadamente (71% - 77%) às proteínas plasmáticas humanas. MMAE não se segmenta de forma significativa em células vermelhas do sangue humano in vitro; a razão entre a quantidade no sangue e a quantidade no plasma é de 0,79 a 0,98.
Dados in vitro indicam que MMAE é um substrato da glicoproteína P (P-gp), mas não inibe a P-gp em concentrações clinicamente relevantes.
Metabolismo
Estima-se que polatuzumabe vedotina passe por catabolismo nos pacientes, o que resulta na produção de pequenos peptídeos, aminoácidos, MMAE não conjugado e catabólitos relacionados ao MMAE não conjugado.
Estudos in vitro indicam que MMAE é um substrato da CYP3A4/5, mas não induz as principais enzimas CYP. MMAE é um fraco inibidor dependente do tempo da CYP3A4/5, mas não inibe competitivamente a CYP3A4/5 em concentrações clinicamente relevantes.
MMAE não inibe CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 ou CYP2D6.
Eliminação
Com base na análise da farmacocinética populacional, o conjugado (acMMAE) é eliminado principalmente pela via da depuração linear não específica, com valor de 0,9 L/dia.
Estudos in vivo em ratos que receberam polatuzumabe vedotina (radiomarcado no MMAE) demonstraram que a maior parte da radioatividade é excretada nas fezes e a menor parte da radioatividade é excretada na urina.
Farmacocinética em populações especiais
População pediátrica
Não foram conduzidos estudos para investigar a farmacocinética de polatuzumabe vedotina na população pediátrica ( < 18 anos de idade).
População geriátrica
A idade não teve efeito na farmacocinética de acMMAE e de MMAE não conjugado, com base em uma análise da farmacocinética da população em pacientes com idade entre 19 - 80 anos. Não foi observada nenhuma diferença significativa na farmacocinética de acMMAE e MMAE não conjugado entre pacientes < 65 anos de idade (n = 207) e pacientes ≥ 65 anos de idade (n = 222).
Comprometimento renal
Em pacientes com comprometimento renal leve (depuração de creatinina 60 - 89 mL/min, n = 200) ou moderado (depuração de creatinina 30 - 59 mL/min, n = 54), as exposições de acMMAE e MMAE não conjugado são semelhantes às de pacientes com função renal normal (depuração de creatinina ≥ 90 mL/min, n = 171), com base em uma análise de farmacocinética da população. Não há dados suficientes para avaliar o impacto do comprometimento renal grave (depuração de creatinina 15 - 29 mL/min, n = 1) na farmacocinética. Não há dados disponíveis de pacientes com doença renal em estágio terminal e / ou que estão em diálise (vide item "8. Posologia e Modo de Usar").,
Comprometimento hepático
Em pacientes com comprometimento hepático leve [TGO (aspartato aminotransferase) de > 1,0 - 2,5 × LSN (limite superior da normalidade) ou TGP (alanina aminotransferase) de > 1,0 - 2,5 × LSN ou bilirrubina total de > 1,0 - 1,5 × LSN, n = 79], as exposições do acMMAE são semelhantes, enquanto as ASC de MMAE não conjugado são no máximo de 40% mais altas, em comparação com pacientes com função hepática normal (n = 338), com base na análise da farmacocinética da população.
Existem dados insuficientes para avaliar o impacto do comprometimento hepático moderado (bilirrubina total > 1,5 - 3 × LSN, n = 9) na farmacocinética. Há dados limitados de pacientes com comprometimento hepático grave ou transplante de fígado (vide item "8. Posologia e Modo de Usar").
Segurança não-clínica
Carcinogenicidade
Não foram realizados estudos específicos de carcinogenicidade em animais com RoPolivy® e / ou MMAE.
Genotoxicidade
Não foram realizados estudos específicos de mutagenicidade em animais com RoPolivy®.
O MMAE foi genotóxico no estudo do micronúcleo da medula óssea de ratos, provavelmente por meio de um mecanismo aneugênico. Esse mecanismo é consistente com o efeito farmacológico de MMAE como um agente de ruptura de microtúbulos. MMAE não foi mutagênico no teste de mutação reversa bacteriana (teste de Ames) ou no teste de mutação direta do linfoma de camundongo L5178Y.
Comprometimento da fertilidade
Não foram realizados estudos dedicados de fertilidade em animais com RoPolivy®. No entanto, os resultados do estudo de toxicidade em ratos indicam o potencial de polatuzumabe vedotina de comprometer a função reprodutiva e a fertilidade masculina. No estudo de toxicidade de doses repetidas de 4 semanas em ratos com a administração semanal de 2, 6 e 10 mg/kg, foi observada degeneração dos túbulos seminíferos testiculares dose-dependente com conteúdo anormal do lúmen no epidídimo. Os achados nos testículos e no epidídimo não foram revertidos e foram correlacionados com diminuição de peso dos testículos e achados macroscópicos na recuperação da necropsia de testículos pequenos e/ou moles nos machos que receberam ≥ 2 mg/kg.
Toxicidade reprodutiva
Não foram realizados estudos dedicados de teratogenicidade em animais com RoPolivy®. No entanto, MMAE foi avaliado em ratos em um estudo de desenvolvimento embriofetal e toxicocinético de acordo com as BPL, no qual ratos fêmeas prenhes receberam 2 doses intravenosas de 0,2 mg/kg de MMAE durante o período de organogênese nos dias de gestação 6 e 13. O tratamento com MMAE com 0,2 mg/kg causou malformações externas fetais, incluindo protrusão da língua, má rotação de membros, gastrosquise e agnatia. A exposição sistêmica (ASC) em ratos a uma dose de 0,2 mg/kg de MMAE é de, aproximadamente, 50% da ASC em pacientes que receberam a dose recomendada de 1,8 mg/kg de RoPolivy® a cada 21 dias.
4. CONTRAINDICAÇÕES
RoPolivy® é contraindicado a pacientes com hipersensibilidade conhecida a polatuzumabe vedotina ou quaisquer um dos excipientes.
5. ADVERTÊNCIAS E PRECAUÇÕES
Geral
Para aumentar a rastreabilidade dos medicamentos biológicos, o nome comercial e o número do lote do produto administrado devem ser claramente registrados (ou declarados) no prontuário médico do paciente.
Mielossupressão
Neutropenia e neutropenia febril graves e severas foram relatadas em pacientes tratados com RoPolivy® logo no primeiro ciclo de tratamento (vide item "9. Reações Adversas"). A administração profilática de fator estimulante de crescimento de colônias de granulócitos (G-CSF) deve ser considerada. Trombocitopenia ou anemia graus 3 ou 4 também pode ocorrer em pacientes que utilizam RoPolivy® (vide item "9. Reações Adversas"). Os hemogramas completos devem ser monitorados antes de cada dose de RoPolivy®. Monitoramento laboratorial mais frequente e / ou atrasos ou descontinuação de RoPolivy® devem ser considerados para pacientes com neutropenia e trombocitopenia grau 3 ou grau 4 (vide item "8. Posologia e modo de usar").
Neuropatia periférica (NP)
Neuropatia periférica foi relatada em pacientes tratados com RoPolivy® logo no primeiro ciclo de tratamento, e o risco aumenta com as doses sequenciais (vide item "9. Reações Adversas"). Pacientes com neuropatia periférica preexistente podem apresentar agravamento dessa condição. A neuropatia periférica relatada com o tratamento com RoPolivy® é predominantemente neuropatia periférica sensorial. No entanto, neuropatia periférica motora e sensitivo motora também foram relatadas. Os pacientes devem ser monitorados em relação aos sintomas de neuropatia periférica, como hipoestesia, hiperestesia, parestesia, disestesia, dor neuropática, sensação de queimação, fraqueza muscular ou distúrbio de marcha. Os pacientes que apresentam neuropatia periférica nova ou agravada podem precisar de atraso da dose, redução da dose ou descontinuação de RoPolivy® (vide item "8. Posologia e modo de usar").
Infecções
Infecções graves, de ameaça à vida ou fatais, que incluem infecções oportunistas, como pneumonia (e isso inclui pneumonia por pneumocystis jirovecii e outras pneumonias fúngicas), bacteremia, sepse, infecção por herpes e infecção por citomegalovírus, foram relatadas em pacientes tratados com RoPolivy® (vide item "9. Reações adversas"). Os pacientes devem ser rigorosamente monitorados durante o tratamento quanto aos sinais de infecções bacterianas, fúngicas ou virais. A profilaxia anti-infecciosa deve ser considerada. RoPolivy® e qualquer quimioterapia concomitante devem ser descontinuados em pacientes que desenvolverem infecções graves.
Vírus da Imunodeficiência Humana (HIV)
RoPolivy® não foi avaliado em pacientes com HIV.
Leucoencefalopatia multifocal progressiva (LEMP)
LEMP tem sido relatada em pacientes tratados com RoPolivy® (vide item "9. Reações adversas"). Os pacientes devem ser monitorados rigorosamente quanto a alterações neurológicas, cognitivas ou comportamentais novas ou agravadas sugestivas de LEMP. RoPolivy® e qualquer quimioterapia concomitante devem ser suspensos se houver suspeita de LEMP e permanentemente descontinuados se o diagnóstico for confirmado.
Síndrome da lise tumoral
Pacientes com alta carga tumoral e tumor de proliferação rápida podem apresentar aumento do risco de síndrome da lise tumoral. Medidas apropriadas, de acordo com as diretrizes locais, devem ser tomadas antes do tratamento com RoPolivy®. Os pacientes devem ser rigorosamente monitorados quanto à síndrome da lise tumoral durante o tratamento com RoPolivy®.
Toxicidade embrionária fetal
Com base no mecanismo de ação e nos estudos não clínicos, RoPolivy® pode ser prejudicial ao feto quando administrado a uma mulher grávida (vide item "3. Características farmacológicas"). As mulheres grávidas devem ser aconselhadas em relação ao risco para o feto.
As mulheres com potencial para engravidar devem ser aconselhadas a utilizar métodos contraceptivos eficazes durante o tratamento com RoPolivy® e durante pelo menos 9 meses após a última dose. Os pacientes do sexo masculino com parceiras do sexo feminino com potencial para engravidar devem ser aconselhados a utilizar métodos contraceptivos eficazes durante o tratamento com RoPolivy® e durante pelo menos 6 meses após a última dose (vide item "3. Características farmacológicas").
Toxicidade hepática
Casos graves de toxicidade hepática que foram consistentes com lesão hepatocelular, que incluem elevações das transaminases e / ou bilirrubina, ocorreram em pacientes tratados com RoPolivy®. Doença hepática preexistente, enzimas hepáticas basais elevadas e medicamentos concomitantes podem aumentar o risco. As enzimas hepáticas e o nível de bilirrubina devem ser monitorados (vide item "8. Posologia e Modo de Usar").
Uso em populações especiais
Mulheres e homens com potencial reprodutivo
Fertilidade
Com base em estudos em animais, RoPolivy® pode comprometer a função reprodutiva e a fertilidade masculina (vide item "3. Características farmacológicas", subitem "Segurança não-clínica").
Contracepção
Mulheres
Mulheres com potencial reprodutivo devem ser aconselhadas a utilizar método contraceptivo eficaz durante o tratamento com RoPolivy® e por, pelo menos, 9 meses após a última dose.
Homens
Pacientes do sexo masculino com parceiras do sexo feminino com potencial reprodutivo devem ser aconselhados a usar método contraceptivo eficaz durante o tratamento com RoPolivy® e por, pelo menos, 6 meses após a última dose.
Gravidez
Categoria de risco na gravidez: D.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
RoPolivy® não é recomendado durante a gravidez, a menos que o potencial benefício para a mãe seja superior ao risco potencial para o feto. RoPolivy® pode causar dano ao feto, com base nos estudos em animais e no mecanismo de ação do medicamento (vide item "3. Características farmacológicas").
Dados em animais
Em estudos em animais, a monometil auristatina E (MMAE) causou genotoxicidade e toxicidade embrionária fetal (vide item "3. Características farmacológicas").
Parto e Trabalho de parto
O uso seguro de RoPolivy® durante o trabalho de parto e o parto não foi estabelecido.
Lactação
Não se sabe se polatuzumabe vedotina é excretado no leite materno humano. Não foi conduzido nenhum estudo para avaliar o impacto de RoPolivy® sobre a produção de leite ou a sua presença no leite materno. Uma vez que muitos medicamentos são excretados no leite materno e em razão do potencial para reações adversas graves em bebês amamentados devido ao RoPolivy®, mulheres devem descontinuar a amamentação durante o tratamento com RoPolivy® e por pelo menos 3 meses após a última dose.
Uso pediátrico
A segurança e a eficácia do RoPolivy® em pacientes pediátricos abaixo da idade de 18 anos não foram estabelecidas.
Uso geriátrico
No estudo GO39942, entre 435 pacientes com LDGCB não tratados previamente e tratados com RoPolivy® em combinação com R-CHP, 227 (52,2%) tinham ≥ 65 anos de idade. Pacientes com idade ≥ 65 anos tiveram uma incidência de reações adversas graves de 39,2% e 28,4% em pacientes com idade < 65 anos. Uma incidência semelhante de reações adversas graves foi observada em pacientes idosos no braço de tratamento com R-CHOP (vide itens "8. Posologia e Modo de Usar" e "3. Características farmacológicas").
Comprometimento renal
A segurança e a eficácia do RoPolivy® em pacientes com depuração de creatinina de < 30 mL/min não foram estudadas formalmente (vide itens "8. Posologia e Modo de Usar" e "3. Características farmacológicas").
Comprometimento hepático
A segurança e a eficácia do RoPolivy® em pacientes com (TGO de > 2,5 × LSN, TGP de > 2,5 × LSN ou bilirrubina total de > 1,5 × LSN) não foram estudadas formalmente e esses pacientes têm probabilidade de apresentar uma exposição aumentada à MMAE. A administração de RoPolivy® em pacientes com comprometimento hepático moderado ou severo (bilirrubina total maior do que 1,5 × [LSN]) deve ser evitada (vide itens "8. Posologia e Modo de Usar" e "3. Características farmacológicas").
Abuso e dependência do medicamento
RoPolivy® não tem o potencial para causar abuso e dependência.
Capacidade de dirigir e operar máquinas
RoPolivy® tem pequena influência na capacidade de dirigir veículos e operar máquinas.
Reações relacionadas à infusão, neuropatia periférica, fadiga e tontura podem ocorrer durante o tratamento com RoPolivy® (vide item "9. Reações adversas").
Até o momento, não há informações de que polatuzumabe vedotina possa causar doping.
Atenção: Este medicamento contém Açúcar, portanto, deve ser usado com cautela em portadores de Diabetes.
6. INTERAÇÕES MEDICAMENTOSAS
Não foi conduzido nenhum estudo clínico específico de interação medicamentosa em humanos com RoPolivy®.
Interações medicamentosas com medicamentos concomitantes que são inibidores, indutores ou substratos da CYP3A
Com base nas simulações do modelo de farmacocinética e de fisiologia de MMAE liberado de polatuzumabe vedotina, fortes inibidores da CYP3A (por exemplo, cetoconazol) podem aumentar a área sob a curva de concentração-tempo (ASC) do MMAE não conjugado em 48%. Os pacientes que receberem fortes inibidores da CYP3A devem ser rigorosamente monitorados quanto aos sinais de toxicidade. Fortes indutores da CYP3A (por exemplo, rifampicina) podem diminuir a ASC de MMAE não conjugado em 49%.
Não se espera que MMAE não conjugado altere a ASC de medicamentos concomitantes que são substratos da CYP3A (por exemplo, midazolam).
Interações medicamentosas de rituximabe, de ciclofosfamida e de doxorrubicina em combinação com polatuzumabe vedotina
A farmacocinética (PK) de rituximabe, de ciclofosfamida e de doxorrubicina não é afetada pela administração concomitante com RoPolivy®. Rituximabe administrado concomitantemente está associado ao aumento da ASC plasmática do anticorpo MMAE conjugado (acMMAE) em 24% e diminuição da ASC plasmática do MMAE não conjugado em 37%, com base na análise da PK da população. As ASCs plasmáticas de acMMAE e MMAE não conjugada para RoPolivy® mais R-CHP ficam de acordo com aquelas de outros estudos de RoPolivy®. Não é necessário ajuste da dose.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Armazenamento
Frasco-ampola
Conservar o frasco ampola fechado sob refrigeração (2 °C a 8 °C). Manter o frasco-ampola no cartucho para proteger da luz. Não congelar. Não agitar.
Prazo de validade
Prazo de validade do pó em frasco ampola fechado
Este medicamento possui prazo de validade de 30 meses a partir da data da fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Prazo de validade da solução reconstituída no frasco-ampola:
Do ponto de vista microbiológico, a solução reconstituída deve ser usada imediatamente. Se não for usada imediatamente, os tempos de armazenamento em uso e as condições antes do uso são de responsabilidade do usuário e normalmente seriam de, no máximo, 48 horas entre 2 °C e 8 °C e até 8 horas entre 9 °C e 30 °C antes da diluição. Descarte o frasco quando o tempo cumulativo de armazenamento antes da diluição exceder 48 horas.
Prazo de validade da solução para infusão após a diluição na bolsa de infusão intravenosa:
Do ponto de vista microbiológico, a solução para infusão preparada deve ser usada imediatamente. Se não for usada imediatamente, os tempos de armazenamento em uso e as condições antes do uso são de responsabilidade do usuário.
Se não for utilizada imediatamente, armazenar a solução diluída de RoPolivy®, conforme especificado na Tabela 2. Descartar a solução de RoPolivy® diluída se o tempo de armazenamento exceder os limites especificados na Tabela 2.
Características físicas e organolépticas
RoPolivy® apresenta-se sob forma de pó branco a branco acinzentado sem conservantes.
A solução reconstituída deve parecer incolor a ligeiramente marrom, clara a ligeiramente opalescente e sem partículas visíveis.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
Descarte de medicamentos não utilizados e / ou com data de validade vencida
RoPolivy® contém um componente citotóxico que é ligado covalentemente ao anticorpo monoclonal. Devem ser utilizados procedimentos para o manuseio e descarte adequados de medicamentos antineoplásicos e citotóxicos.
O descarte de medicamentos no meio ambiente deve ser minimizado. Os medicamentos não devem ser descartados no esgoto, e o descarte em lixo doméstico deve ser evitado. Utilize o sistema de coleta local estabelecido e apropriado, se disponível.
8. POSOLOGIA E MODO DE USAR
Para prevenir erros de medicação, é importante verificar o rótulo do frasco para garantir que o medicamento que está sendo preparado e administrado seja RoPolivy®.
RoPolivy® somente deve ser administrado sob a supervisão de um profissional da saúde com experiência no diagnóstico e tratamento de pacientes com câncer.
Para informações sobre rituximabe, ciclofosfamida, doxorrubicina ou prednisona, vide bula de tais medicamentos.
Posologia
A dose recomendada de RoPolivy® é de 1,8 mg/kg, administrada como infusão intravenosa a cada 21 dias, por 6 ciclos, em combinação com rituximabe, ciclofosfamida, doxorrubicina e prednisona (R-CHP). Administre RoPolivy®, rituximabe, ciclofosfamida e doxorrubicina em qualquer ordem no dia 1 de cada ciclo, após a administração de prednisona. Administre prednisona nos dias 1-5 de cada ciclo. Ciclos 7 e 8 consiste em administração de rituximabe como monoterapia.
Se o paciente ainda não estiver pré-medicado, administre um anti-histamínico e antipirético antes de RoPolivy®. Administre a dose inicial de RoPolivy® durante 90 minutos por infusão intravenosa. Monitore os pacientes quanto a reações relacionadas à infusão durante a infusão e por um período mínimo de 90 minutos após a conclusão da dose inicial. Se a infusão anterior foi bem tolerada, a dose subsequente de RoPolivy® pode ser administrada através de uma infusão de 30 minutos e os pacientes devem ser monitorados durante a infusão e por pelo menos 30 minutos após a conclusão da infusão.
Doses atrasadas ou perdidas
Se uma dose planejada de RoPolivy® for perdida, ela deve ser administrada assim que possível, e o cronograma de administração deve ser ajustado para manter um intervalo de 21 dias entre as doses.
Modificações na dose
A velocidade de infusão de RoPolivy® deve ser diminuída ou interrompida se o paciente desenvolver uma reação relacionada à infusão. RoPolivy® deve ser descontinuado imediatamente e permanentemente se o paciente apresentar uma reação que ameace a vida.
Há diferentes de modificação de dose possível para RoPolivy® em pacientes com LDGCB não tratados previamente (vide Tabela 3 e Tabela 4).
Para as modificações na dose a fim de manejar neuropatia periférica (vide item "5. Advertências e Precauções"), consulte a Tabela 3 a seguir.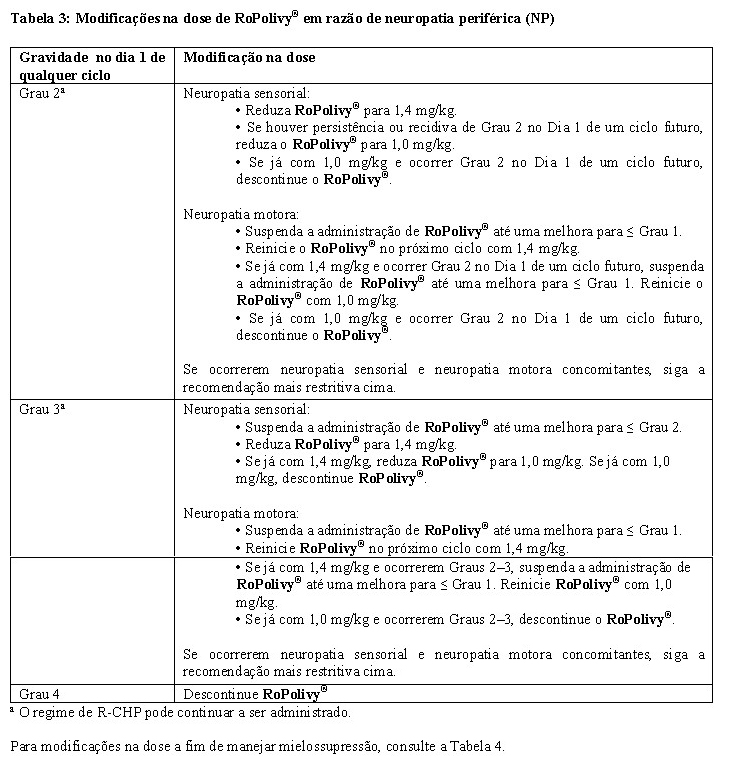
Para modificações na dose a fim de manejar mielossupressão, consulte a Tabela 4.
Para modificações na dose a fim de manejar reações relacionadas à infusão, consulte a Tabela 5.
Instruções especiais de dose
Uso pediátrico
A segurança e a eficácia de RoPolivy® em crianças e adolescentes ( < 18 anos) não foram estabelecidas.
Uso geriátrico
Não é necessário nenhum ajuste na dose de RoPolivy® em pacientes ≥ 65 anos de idade (vide itens "3. Propriedades farmacocinéticas" e "5. Advertências e Precauções").
Comprometimento renal
Não é necessário nenhum ajuste na dose de RoPolivy® em pacientes com depuração da creatinina ≥ 30 mL/min. Uma dose recomendada não foi determinada para pacientes com depuração de creatinina < 30 mL/min (vide itens "3. Propriedades farmacocinéticas" e "5. Advertências e Precauções").
Comprometimento hepático
Evite a administração de RoPolivy® em pacientes com comprometimento hepático moderado ou grave (bilirrubina superior a 1,5 × LSN). Não é necessário ajuste na dose de RoPolivy® para pacientes com comprometimento hepático leve (bilirrubina total superior ao LSN e menor ou igual a 1,5 × LSN ou TGO superior ao LSN) (vide itens "3. Características Farmacológicas" e "5. Advertências e Precauções").
Método de administração
RoPolivy® deve ser reconstituído e diluído usando-se técnica asséptica sob a supervisão de um profissional da saúde. RoPolivy® deve ser administrado como uma infusão intravenosa por meio de uma linha de infusão dedicada equipada com um filtro em linha ou adicional estéril, não pirogênico, de baixa ligação à proteína (poro de tamanho 0,2 ou 0,22 micrômetros) e cateter. RoPolivy® não deve ser administrado por injeção intravenosa rápida ou em bolus.
Instruções especiais para uso e manipulação do produto
RoPolivy® contém um componente citotóxico que é ligado covalentemente ao anticorpo monoclonal. RoPolivy® deve ser administrado sob a supervisão de um médico com experiência no uso de agentes citotóxicos. Devem ser utilizados procedimentos para o manuseio e descarte adequados de medicamentos antineoplásicos e citotóxicos.
RoPolivy® deve ser reconstituído utilizando água estéril para injeção e diluído em uma bolsa de infusão intravenosa que contém cloreto de sódio a 0,9%, cloreto de sódio a 0,45% ou glicose a 5% por um profissional de saúde antes da administração.
Use técnica asséptica para a reconstituição e a diluição de RoPolivy®. Procedimentos apropriados para a preparação de produtos antineoplásicos devem ser usados.
O produto reconstituído não contém conservantes e é destinado somente para dose única. Descarte qualquer parte não usada.
A solução reconstituída e a solução para infusão não devem ser congeladas ou expostas à luz solar direta.
Deve ser usada uma linha de infusão específica equipada com um filtro em linha ou adicional estéril, não pirogênico e de baixa ligação à proteínas (tamanho de poro de 0,2 ou 0,22 mm) e um cateter para administrar RoPolivy® diluído.
Reconstituição
1. Usando uma seringa estéril, injete lentamente 1,8 mL de água estéril para injeção no frasco-ampola de 30 mg de RoPolivy® ou 7,2 mL de água estéril para injeção no frasco-ampola de 140 mg de RoPolivy®, para produzir uma solução de dose única que contém 20 mg/mL de polatuzumabe vedotina. Direcione o fluxo em direção à parede do frasco-ampola e não diretamente para o pó liofilizado.
2. Mexa o frasco-ampola suavemente até que o conteúdo esteja completamente dissolvido. Não agite.
4. Verifique a solução reconstituída quanto à descoloração e presença de material particulado. A solução reconstituída deve parecer incolor a ligeiramente marrom, clara a ligeiramente opalescente, e sem partículas visíveis. Não use se a solução reconstituída estiver descorada, turva ou contiver partículas visíveis.
Diluição
1. Polatuzumabe vedotina deve ser diluído até a concentração final de 0,72 - 2,7 mg/mL em uma bolsa de infusão IV, com um volume mínimo de 50 mL, que contém cloreto de sódio a 0,9% ou cloreto de sódio a 0,45% ou glicose a 5%.
2. Determine o volume de 20 mg/mL da solução reconstituída necessária com base na dose exigida:
3. Retire o volume exigido da solução reconstituída do frasco-ampola de RoPolivy®, utilizando uma seringa estéril, e dilua na bolsa de infusão IV. Descarte qualquer parte não utilizada restante no frasco-ampola.
4. Misture suavemente a bolsa IV invertendo-a lentamente. Não agite.
5. Verifique a bolsa IV quanto à presença de materiais particulados e descarte caso estejam presentes.
Evite o transporte da solução para infusão preparada, uma vez que o estresse da agitação pode resultar em agregação. Se a infusão preparada for transportada, remova o ar da bolsa de infusão e limite o transporte até 30 minutos, entre 9 °C e 25 °C, ou 24 horas, entre 2 °C e 8 °C. Se o ar for retirado, um conjunto de infusão com um perfurador ventilado é necessário para garantir a administração exata da dose durante a infusão.
Incompatibilidades
• Não misture RoPolivy® com outros medicamentos, nem administre pela mesma linha de infusão de outros medicamentos.
• Não foram observadas incompatibilidades entre RoPolivy® e bolsas de infusão intravenosa que contém os materiais policloreto de vinila (PVC) ou poliolefinas (PO), como polietileno (PE) e polipropileno (PP). Além disso, não foram observadas incompatibilidades com os conjuntos de infusão ou auxiliares de infusão com materiais que entram em contato com o medicamento e que são constituídos de PVC, PE, poliuretano (PU), polibutadieno (PBD), acrilonitrila butadieno estireno (ABS), policarbonato (PC), polieteruretano (PEU), ou etileno propileno fluorado (FEP), politetrafluoreti