REPEGOXA
DR. REDDY'S
doxorrubicina
Antineoplásico.
MEDICAMENTO SIMILAR EQUIVALENTE AO MEDICAMENTO DE REFERÊNCIA
Apresentações.
Suspensão injetável de liberação prolongada de 20 mg de REPEGOXA em embalagem com 1 frasco- ampola com 10 mL de suspensão injetável de liberação prolongada (2 mg/mL).
Suspensão injetável de liberação prolongada de 50 mg de REPEGOXA em embalagem com 1 frasco- ampola com 25 mL de suspensão injetável de liberação prolongada (2 mg/mL).
USO INTRAVENOSO
USO ADULTO
Composição.
REPEGOXA 10 mL:
Cada frasco-ampola contém 20 mg de REPEGOXA (equivalente a 18,74 mg de doxorrubicina) em 10 mL de suspensão injetável.
Excipientes: fosfatidilcolina hidrogenada de soja, diestearato de fosfatidiletanolamin carbonilmetoxipolietilenoglicol sódico, colesterol, álcool etílico, sulfato de amônio, sacarose, histidina, ácido clorídrico, hidróxido de sódio.
REPEGOXA 25 mL:
Cada frasco-ampola contém 20 mg de REPEGOXA (equivalente a 46,85 mg de doxorrubicina) em 25 mL de suspensão injetável.
Excipientes: fosfatidilcolina hidrogenada de soja, diestearato de fosfatidiletanolamin carbonilmetoxipolietilenoglicol sódico, colesterol, álcool etílico, sulfato de amônio, sacarose, histidina, ácido clorídrico, hidróxido de sódio.
Informações técnicas.
1. INDICAÇÕES
Câncer de Mama:
REPEGOXA é indicado para o tratamento de:
- Câncer de mama metastático em mulheres com indicação de uso de antraciclina.
- Câncer de mama metastático em mulheres que não responderam a um esquema contendo taxano.
Câncer de Ovário:
REPEGOXA é indicado para o tratamento de câncer de ovário avançado em mulheres com falha na terapia de primeira linha com quimioterapia à base de platina.
Mieloma Múltiplo:
REPEGOXA é indicado em combinação com bortezomibe para o tratamento do mieloma múltiplo em progressão em pacientes que receberam pelo menos uma terapia anterior e que já foram submetidos a transplante de medula óssea, ou seja, inadequados a ele.
Sarcoma de Kaposi relacionado à síndrome da imunodeficiência adquirida:
REPEGOXA também é indicado para o tratamento do sarcoma de Kaposi (SK) relacionado à síndrome da imunodeficiência adquirida em pacientes com baixa contagem de células CD4 ( < 200 linfócitos CD4/mm3) e doença mucocutânea ou visceral extensa.
REPEGOXA pode ser usado como quimioterápico sistêmico de primeira ou de segunda linha em pacientes com sarcoma de Kaposi relacionado à síndrome da imunodeficiência adquirida com doença que tenha evoluído durante quimioterapia sistêmica combinada prévia, incluindo pelo menos dois dos seguintes agentes: um alcaloide da vinca, bleomicina e doxorrubicina convencional (ou outras antraciclinas) ou em pacientes com intolerância a esses esquemas.
2. RESULTADOS DE EFICÁCIA
Câncer de Mama1-4: Um estudo randomizado fase III do REPEGOXA versus cloridrato de doxorrubicina em pacientes com câncer de mama metastático foi concluído em 509 pacientes. O objetivo especificado pelo protocolo de demonstrar a não inferioridade entre REPEGOXA e a doxorrubicina foi atingido: a razão de risco (RR) para a sobrevida livre de progressão (SLP) foi de 1,00 (IC de 95% para RR=0,82 a 1,22). A razão de risco do tratamento para a sobrevida livre de progressão quando ajustada para as variáveis prognósticas foi compatível com a SLP para a população com intenção de tratamento (IT).
Trezentas e uma pacientes com câncer de mama avançado que não responderam a um esquema contendo taxano foram randomizadas em um estudo comparativo fase III de REPEGOXA versus um esquema de resgate aprovado (vinorelbina ou mitomicina C + vimblastina). A SLP foi semelhante para o REPEGOXA e para o comparativo ativo, com uma forte tendência a favor do REPEGOXA (RR=1,26; IC de 95% de 0,98 a 1,62; p=0,11). Em todos os subgrupos analisados, inclusive daquelas pacientes ≥ 55 anos de idade (n=166), houve um efeito terapêutico compatível com a SLP a favor de REPEGOXA em relação ao comparativo ativo (todas as razões de risco foram > 1,00).
Câncer de Ovário5-7: Um estudo comparativo de fase III de REPEGOXA versus topotecana em pacientes com câncer de ovário epitelial após fracasso da quimioterapia de primeira linha à base de platina foi concluído em 474 pacientes. Houve benefício na sobrevida global (SG) em pacientes tratadas com REPEGOXA sobre pacientes tratadas com topotecana conforme indicado pela razão de risco (RR) de 1,216 (IC de 95% de 1,000 a 1,478), p=0,050. Os índices de sobrevida em 1, 2 e 3 anos foram 56,3%, 34,7% e 20,2%, respectivamente, para REPEGOXA em comparação com 54,0%, 23,6% e 13,2% para topotecana.
Para o subgrupo de pacientes com doença sensível à platina a diferença foi maior: RR de 1,432 (IC de 95% de 1,066 a 1,923), p=0,017. Os índices de sobrevida em 1, 2 e 3 anos foram 74,1%, 51,2% e 28,4%, respectivamente, para REPEGOXA, comparados a 66,2%, 31,0% e 17,5% para topotecana.
Os tratamentos foram similares nos subgrupos de pacientes com doença refratária à platina: RR de 1,069 (IC de 95% de 0,823 a 1,387), p=0,618. Os índices de sobrevida em 1, 2 e 3 anos foram 41,5%, 21,1% e 13,8%, respectivamente, para REPEGOXA em comparação com 43,2%, 17,2% e 9,5% para topotecana.
Mieloma múltiplo8: Um estudo fase III, randomizado, de grupo paralelo, aberto, multicêntrico comparando a segurança e a eficácia da terapia combinada de REPEGOXA mais bortezomibe com a monoterapia com bortezomibe em pacientes com mieloma múltiplo que receberam pelo menos uma terapia anterior foi conduzido em 646 pacientes. Houve uma melhora significativa no desfecho primário relativo ao tempo para progressão (TPP) nos pacientes tratados com terapia combinada de REPEGOXA mais bortezomibe quando comparada com pacientes tratados com a monoterapia com bortezomibe conforme indicado pela redução do risco (RR) de 35% (95% IC de 21 a 47%), p < 0,0001, baseado nos 407 eventos TPP. A mediana de TPP foi de 6,9 meses para os pacientes que receberam a monoterapia com bortezomibe em comparação com 8,9 meses para os pacientes que receberam a terapia combinada de REPEGOXA mais bortezomibe. Uma análise interina definida em protocolo (baseada nos 249 eventos/TPP) desencadeou o término precoce do estudo para eficácia. Essa análise interina mostrou a redução do risco no TPP de 45% (IC de 95% de 29 a 57%), p < 0,0001. A mediana de TPP foi de 6,5 meses para pacientes recebendo a monoterapia com bortezomibe em comparação com 9,3 meses para pacientes recebendo a terapia combinada de REPEGOXA mais bortezomibe. Esses resultados, embora não finalizados, constituíram a análise final definida em protocolo.
Referências
1. Clinical Study Report Protocol I97-328. SCH200746: A Randomized Phase 3 Trial of Caelyx/Doxil (SCH200746) VS Doxorubicin For The First-Line Treatment of Woman With Metastatic Breast Cancer. Schering-Plough Research Institute: January 2002.
2. Clinical Study Report Protocol C/I96-352. SCH200746: A Randomized Multicenter Trial Of Caelyx/Doxil (SCH200746) AS Monotherapy VS A Comparative Salvage Regimen For The Treatment Of Subjects With Advanced Breast Cancer Who Have Failed A Prior Taxane-Containing Regimen. Schering-Plough Research Institute; January 2002.
3. Clinical Overview, Caelyx (SCH200746) Breast Cancer Variation, January 2002.
4. Report CL 160-30-PK-98-01, Addendum 2, Breast Cancer subset.
5. Clinical Study Report Protocol No. 30-49, SCH 200746: A Phase III, Randomized, Open Label Comparative Study Of Doxil/Caelyx versus Topetecan HCL In Patients With Epithelial Ovarian Carcinoma Following Failure of First-Lone, Platinum Based Chemotherapy. Schering-Plough Research Institute; April 2000.
6. Clinical Protocol No. 30-49 A Phase III, Randomized, Open Label Comparative Study Of Doxil/Caelyx versus Topetecan HCL In Patients With Epithelial Ovarian Carcinoma Following Failure of First-Lone, Platinum Based Chemotherapy. Schering-Plough Research Institute.
7. Clinical Overview, Caelyx (SCH200746) Ovarian Cancer HRD, November 1999.
8. 2.7.3 Summary of Clinical Efficacy - DOXIL-MMY-3001 study - section 2.1.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Descrição: REPEGOXA é o cloridrato de doxorrubicina encapsulado em lipossomas com metoxipolietilenoglicol (MPEG) conjugado na superfície. Esse processo é conhecido como peguilação e protege os lipossomas da detecção pelo sistema fagocítico mononuclear (SFM), o que prolonga o tempo de circulação sanguínea.
Cada frasco-ampola de REPEGOXA contém 2 mg/mL de cloridrato de doxorrubicina em uma formulação lipossomal peguilada contendo 10 mL (20 mg) em suspensão concentrada para infusão destinada a uma única aplicação intravenosa e é apresentada como uma suspensão vermelha, translúcida e estéril. O ingrediente ativo de REPEGOXA é o cloridrato de doxorrubicina peguilhado, uma antraciclina (antibiótico citotóxico) obtido a partir de Streptomyces peucetius, var. caesius
Informação Pré-Clínica
Em estudos com doses múltiplas realizados em animais, o perfil de toxicidade de REPEGOXA parece muito semelhante ao descrito em humanos quando recebe infusões prolongadas de cloridrato de doxorrubicina convencional. Com REPEGOXA, a encapsulação do cloridrato de doxorrubicina em lipossomas peguilados resulta em uma potência diferente desses efeitos, conforme apresentado abaixo:
- Cardiotoxicidade: Estudos em coelhos mostraram que a cardiotoxicidade de REPEGOXA é reduzida em comparação com a das preparações convencionais de cloridrato de doxorrubicina.
- Toxicidade dérmica: Em estudos realizados após a administração múltipla de REPEGOXA em ratos e cães, foram observadas inflamações dérmicas graves e formações de úlceras em doses clinicamente relevantes. No estudo em cães, a ocorrência e a gravidade dessas lesões foram reduzidas com a diminuição da dose ou com o aumento dos intervalos entre as doses. Lesões dérmicas semelhantes, que são descritas como eritrodisestesia palmo-plantar, também foram observadas em alguns pacientes depois de administrações múltiplas (vide "Reações Adversas").
- Resposta anafilactoide: Durante estudos de toxicologia com doses múltiplas em cães, uma resposta aguda caracterizada por hipotensão, palidez de membranas mucosas, salivação, êmese e períodos de hiperatividade seguidos por hipoatividade e letargia foram observados depois da administração de lipossomas peguilados (placebo). Também foi observada uma resposta semelhante, mas menos grave, em cães tratados com REPEGOXA ou doxorrubicina convencional. A resposta de hipotensão foi reduzida em magnitude pelo pré-tratamento com anti- histamínicos. No entanto, a resposta não levou ao risco de morte e os cães se recuperaram rapidamente ao interromper o tratamento.
- Toxicidade local: Estudos de tolerância subcutânea indicam que REPEGOXA, quando comparado ao cloridrato de doxorrubicina convencional, provoca relativamente menos irritação local ou lesão tecidual depois de um eventual extravasamento.
- Mutagenicidade e carcinogenicidade: Embora não tenha sido conduzido nenhum estudo com REPEGOXA, o cloridrato de doxorrubicina, ingrediente farmacologicamente ativo de REPEGOXA, é mutagênico e carcinogênico. Lipossomas peguilados placebo não são mutagênicos nem genotóxicos.
- Toxicidade reprodutiva: REPEGOXA resultou em atrofia leve a moderada de ovários e testículos em camundongos após dose única de 36 mg/kg. Pesos testiculares reduzidos e hipospermia estavam presentes em ratos depois de doses múltiplas ≥ 0,25 mg/kg/dia. Degeneração difusa dos túbulos seminíferos e redução acentuada na espermatogênese foram observadas em cães após doses múltiplas de 1 mg/kg/dia.
- Nefrotoxicidade: Um estudo mostrou que o REPEGOXA em uma dose intravenosa única duas vezes maior do que a dose clínica produz toxicidade renal em macacos. A toxicidade renal foi observada mesmo com doses únicas mais baixas de cloridrato de doxorrubicina em ratos e coelhos. Entretanto, esses achados em macacos podem não ter relevância para a avaliação de risco em pacientes, uma vez que a avaliação do banco de dados de segurança pós-comercialização referente a REPEGOXA não sugeriu risco significativo de nefrotoxicidade.
Farmacologia Clínica
REPEGOXA é uma fórmula lipossomal peguilada de ação prolongada do cloridrato de doxorrubicina que proporciona uma concentração maior de doxorrubicina em tumores de sarcoma de Kaposi do que na pele normal. Os lipossomas peguilados contêm segmentos do polímero hidrófilo metoxipolietilenoglicol (MPEG) inseridos em sua superfície. Esses grupos lineares de MPEG se estendem desde a superfície do lipossoma, criando uma camada protetora que reduz as interações entre a membrana de dupla camada lipídica e os componentes do plasma, o que permite que os lipossomas de REPEGOXA circulem por períodos prolongados na corrente sanguínea. Os lipossomas peguilados são suficientemente pequenos (diâmetro médio de aproximadamente 100 nm) para passarem intactos (extravasar) através dos vasos sanguíneos defeituosos que alimentam os tumores. A evidência de penetração dos lipossomas peguilados a partir dos vasos sanguíneos e de sua entrada e acúmulo nos tumores foi observada em camundongos com tumores de carcinoma de cólon C-26 e em camundongos transgênicos com lesões semelhantes às do sarcoma de Kaposi. Os lipossomas peguilados também apresentam uma matriz lipídica de baixa permeabilidade e um sistema tampão aquoso interno que se combinam para manter o cloridrato de doxorrubicina encapsulado durante o tempo de permanência do lipossoma em circulação.
Propriedades Farmacocinéticas
Em doses equivalentes, os valores de concentração plasmática e ASC (área sob a curva) de REPEGOXA, que representam a maior parte do cloridrato de doxorrubicina encapsulado em lipossomas (contendo 90% a 95% da doxorrubicina medida), são significativamente maiores do que os obtidos com as preparações de cloridrato de doxorrubicina convencional.
A farmacocinética plasmática de REPEGOXA em humanos é significativamente diferente daquela relatada na literatura para as preparações de cloridrato de doxorrubicina convencional. Em baixas doses (10 mg/m2 a 20 mg/m2), REPEGOXA mostrou farmacocinética linear. No intervalo de doses de 10 mg/m2 a 60 mg/m2, REPEGOXA apresentou farmacocinética não-linear. O cloridrato de doxorrubicina convencional apresenta uma distribuição tecidual extensa (volume de distribuição de 700 a 1.100 L/m2) e depuração de eliminação rápida (24 a 73 L/h/m2). Diferentemente, o perfil farmacocinético de REPEGOXA indica que o agente fica restrito em grande parte ao volume de líquido vascular e que a depuração da doxorrubicina do sangue depende do carregador lipossomal. A doxorrubicina fica disponível depois que os lipossomas extravasam e entram no compartimento tecidual.
Farmacocinética da população: A farmacocinética de REPEGOXA foi avaliada em 120 pacientes de 10 diferentes estudos clínicos usando a abordagem de farmacocinética populacional. A farmacocinética de REPEGOXA em doses no intervalo de 10 mg/m2 a 60 mg/m2 foi melhor descrita por um modelo não-linear de dois compartimentos, com entrada de ordem zero e eliminação de Michaelis-Menten. A depuração intrínseca média de REPEGOXA foi de 0,030 L/h/m2 (intervalo de 0,008 a 0,152 L/h/m2) e o volume de distribuição central médio foi de 1,93 L/m2 (intervalo de 0,96 a 3,85 L/m2), aproximando-se do volume plasmático. A meia-vida aparente variou de 24 a 231 horas, com uma média de 73,9 horas.
Pacientes com câncer de mama: A farmacocinética de REPEGOXA determinada em 18 pacientes com câncer de mama foi semelhante à farmacocinética determinada na população de 120 pacientes com diversos tipos de câncer. A depuração intrínseca média foi de 0,016 L/h/m2 (intervalo de 0,009 a 0,027 L/h/m2) e o volume de distribuição central médio foi de 1,46 L/m2 (intervalo de 1,10 a 1,64 L/m2). A meia-vida aparente média foi de 71,5 horas (intervalo de 45,2 a 98,5 horas).
Pacientes com câncer de ovário: A farmacocinética de REPEGOXA determinada em 11 pacientes com câncer de ovário foi semelhante à farmacocinética determinada na população de 120 pacientes com diversos tipos de câncer. A depuração intrínseca média foi de 0,021 L/h/m2 (intervalo de 0,009 a 0,041 L/h/m2) e o volume de distribuição central médio foi de 1,95 L/m2 (intervalo de 1,67 a 2,40 L/m2). A meia-vida aparente média foi de 75,0 horas (intervalo de 36,1 a 125 horas).
Pacientes com sarcoma de Kaposi relacionado à síndrome da imunodeficiência adquirida: A farmacocinética plasmática de REPEGOXA foi avaliada em 23 pacientes com sarcoma de Kaposi (SK) que receberam doses únicas de 20 mg/m2 administradas em infusão de 30 minutos. Os parâmetros farmacocinéticos de REPEGOXA (em sua maior parte constituído por cloridrato de doxorrubicina encapsulado em lipossomas e por baixas concentrações de cloridrato de doxorrubicina não encapsulado) observados após a administração de dose de 20 mg/m2 são apresentados na Tabela 1.
Propriedades Farmacodinâmicas
Ação: O exato mecanismo de atividade antitumoral da doxorrubicina é desconhecido. Acredita-se que a inibição do DNA, do RNA e da síntese proteica seja responsável pela maior parte do efeito citotóxico, provavelmente como resultado da interposição da antraciclina entre os pares de bases adjacentes da dupla hélice do DNA, impedindo que essas se desenrolem para a sua replicação.
4. CONTRAINDICAÇÕES
Este medicamento é contraindicado para uso por:
- pacientes com histórico de reação de hipersensibilidade ao cloridrato de doxorrubicina ou a qualquer um de seus componentes. REPEGOXA não deve ser administrado durante a amamentação.
- pacientes com sarcoma de Kaposi relacionado à síndrome da imunodeficiência adquirida que podem ser tratados de forma eficaz com terapia local ou com alfainterferona sistêmica.
5. ADVERTÊNCIAS E PRECAUÇÕES
Dada a diferença entre os perfis farmacocinéticos e os esquemas de administração, REPEGOXA não pode ser usado de forma intercambiável com outras formulações de cloridrato de doxorrubicina.
A associação de quimioterapias com REPEGOXA foi extensamente estudada em populações com tumores sólidos. REPEGOXA foi administrado com segurança concomitantemente às doses convencionais dos agentes quimioterápicos frequentemente empregados no tratamento do câncer de mama ou de ovário avançado; no entanto, a eficácia desses esquemas de associação não foi estabelecida.
Risco cardíaco: todos os pacientes em terapia com REPEGOXA devem ser monitorados rotineiramente com ECGs frequentes. As alterações transitórias do ECG, como nivelamento da onda T, depressão do segmento S-T e arritmias benignas, não são consideradas alterações que obrigatoriamente levam à suspensão do tratamento. No entanto, uma redução do complexo QRS é considerada uma indicação de toxicidade cardíaca. Se essa alteração ocorrer, deverá ser considerado um exame mais definitivo para lesão miocárdica por antraciclina, ou seja, uma biópsia do endomiocárdio (vide "Reações Adversas").
Os métodos mais específicos para avaliação e monitoração de funções cardíacas, em comparação com o ECG, são uma medição da fração de ejeção ventricular esquerda (FEVE) por ecocardiografia ou, preferivelmente, por angiografia de múltiplas entradas (MUGA - prova para aferir função cardíaca). Esses métodos devem ser utilizados rotineiramente antes de se iniciar o tratamento com REPEGOXA e devem ser repetidos periodicamente durante o mesmo.
Em um estudo clínico fase III que comparou REPEGOXA (50 mg/m2 a cada 4 semanas) versus doxorrubicina (60 mg/m2 a cada 3 semanas), o risco de desenvolvimento de um evento cardíaco como uma consequência da dose cumulativa da antraciclina foi significativamente menor com REPEGOXA quando comparado a doxorrubicina (RR[doxorrubicina/ REPEGOXA]=3,16; p < 0,001). Nas doses cumulativas entre 450 mg/m2 e 600 mg/m2, não houve risco aumentado de toxicidade cardíaca com REPEGOXA. A avaliação da função do ventrículo esquerdo é considerada obrigatória antes de cada administração adicional de REPEGOXA que exceda uma dose cumulativa de antraciclina de 600 mg/m2 durante toda a vida, em pacientes sem exposição anterior à antraciclina. Para pacientes que receberam antraciclinas adjuvantes anteriores (epirrubicina ou doxorrubicina) as avaliações de fração de ejeção do ventrículo esquerdo (FEVE) devem ser realizadas antes de cada administração adicional de REPEGOXA que exceda uma dose cumulativa de 450 mg/m2 de antraciclina, equivalente à doxorrubicina, durante toda a vida.
Os testes de avaliação e os métodos mencionados acima referentes à monitoração do desempenho cardíaco durante o tratamento com antraciclina devem ser empregados na seguinte ordem: monitoração do ECG, medições da fração de ejeção ventricular esquerda e biópsia do endomiocárdio. Se um resultado de exame indicar possibilidade de uma lesão cardíaca associada ao tratamento com REPEGOXA, deverá ser considerado cuidadosamente o benefício de se continuar o tratamento em relação ao risco de provocar lesão do miocárdio.
Os pacientes com histórico de doença cardiovascular devem receber REPEGOXA apenas quando o benefício for maior do que o risco para o paciente.
Deve-se agir com cautela em pacientes com comprometimento da função cardíaca que recebem REPEGOXA.
Quando houver suspeita de uma cardiomiopatia, por exemplo, a fração de ejeção ventricular esquerda estiver substancialmente diminuída em relação aos valores do pré-tratamento e/ou a fração de ejeção ventricular esquerda for menor que os valores relevantes no prognóstico (por exemplo, < 45%), uma biópsia do endomiocárdio deve ser considerada e o benefício de se continuar a terapia deve ser cuidadosamente avaliado em relação ao risco de desenvolvimento irreversível de dano cardíaco.
Insuficiência cardíaca congestiva devido a cardiomiopatia pode ocorrer repentinamente sem mudanças anteriores no ECG e também pode ocorrer várias semanas após a descontinuação da terapia.
Deve-se agir com cautela em pacientes que tenham recebido outras antraciclinas. A dose total de cloridrato de doxorrubicina também deve levar em conta qualquer tratamento anterior (ou concomitante) com compostos cardiotóxicos como outras antraciclinas/antraquinonas ou, por exemplo, o 5-fluoruracila. A toxicidade cardíaca também pode ocorrer em doses cumulativas de antraciclina menores do que 450 mg/m2 em pacientes com irradiação prévia do mediastino ou nos que receberam terapia concomitante com ciclofosfamida.
O perfil de segurança cardíaca para o esquema de administração recomendado tanto para câncer de mama quanto para câncer de ovário (50 mg/m2) é semelhante ao perfil em pacientes com sarcoma de Kaposi relacionado à síndrome da imunodeficiência adquirida com 20 mg/m2.
Mielossupressão: muitos pacientes tratados com REPEGOXA apresentam mielossupressão basal decorrente de fatores como doença provocada pelo HIV preexistente, ou administração prévia ou concomitante de vários medicamentos ou tumores comprometendo a medula óssea. No estudo pivotal em pacientes com câncer de ovário tratadas com dose de 50 mg/m2, a mielossupressão foi geralmente de leve a moderada, reversível e não foi associada a episódios de infecção neutropênica ou sepse. Além disso, em estudo clínico controlado de REPEGOXA vs. topotecana, a incidência de sepse relacionada ao tratamento foi substancialmente menor em pacientes com câncer de ovário tratadas com REPEGOXA em comparação com o grupo de tratamento que recebeu topotecana. Uma incidência baixa semelhante de mielossupressão foi observada em pacientes com câncer de mama metastático que receberam REPEGOXA em um estudo clínico de primeira linha. Diferentemente da experiência em pacientes com câncer de mama ou câncer de ovário, a mielossupressão parece ser o efeito adverso limitante da dose em pacientes com sarcoma de Kaposi relacionado à síndrome da imunodeficiência adquirida. Devido ao potencial de supressão da medula óssea, devem ser realizados hemogramas periódicos no decorrer do tratamento com REPEGOXA e, no mínimo, antes da administração de cada dose de REPEGOXA.
A mielossupressão grave e persistente, embora não observada em pacientes com câncer de mama ou de ovário, pode resultar em infecções graves ou hemorragias.
Em um estudo clínico pivotal de mieloma múltiplo, foi observada uma alta incidência de sangramento em pacientes recebendo DLP + bortezomibe comparado com os pacientes recebendo bortezomibe isolado. Nesses casos, o médico deve avaliar criteriosamente a relação risco-benefício nas populações com histórico de sangramento espontâneo, distúrbios idiossincrásicos e pacientes com potencial para sangramento ou que irão se submeter a algum procedimento cirúrgico.
Pacientes diabéticos: deve-se notar que cada frasco de REPEGOXA contém sacarose e que é administrado em soro glicosado a 5% para infusão intravenosa.
Reações associadas com a infusão: reações de infusão grave e, às vezes com risco de morte, caracterizadas como reações alérgicas ou anafilactoides, com sintomas que incluem asma, rubor, urticária, dor torácica, febre, hipertensão, taquicardia, prurido, sudorese, falta de ar, edema facial, calafrios, dor lombar, aperto no peito e garganta e/ou hipotensão podem ocorrer dentro de minutos do início da infusão de REPEGOXA. Muito raramente, foram observadas convulsões em relação às reações de infusão. Uma suspensão temporária da infusão geralmente resolve esses sintomas sem a necessidade de uma terapia adicional. No entanto, medicamentos para tratar esses sintomas (por exemplo, anti-histamínicos, corticosteroides e adrenalina), assim como equipamento de emergência, devem estar disponíveis para uso imediato. Na maioria dos pacientes, o tratamento pode ser recomeçado após a resolução dos sintomas, sem recorrência. Reações de infusão raramente se repetem após o primeiro ciclo de tratamento. Para minimizar o risco das reações de infusão, a dose inicial deve ser administrada a uma velocidade não superior a 1 mg/minuto (vide "Reações Adversas").
Para eventos adversos comuns que requerem modificação da dose ou descontinuidade do tratamento em pacientes com mieloma múltiplo vide "Reações Adversas".
Neoplasias orais secundárias: foram relatados casos muito raros de câncer oral secundário em pacientes com longo período de exposição (mais de um ano) a REPEGOXA ou em pacientes recebendo dose cumulativa de REPEGOXA superior a 720 mg/m2. Foram diagnosticados casos de câncer oral secundário durante o tratamento com REPEGOXA e até 6 anos após a última dose. Os pacientes devem ser examinados em intervalos regulares para a presença de ulceração oral ou qualquer desconforto oral que possa ser um indicativo de câncer oral secundário.
Efeitos sobre a capacidade de dirigir veículos ou operar máquinas: embora REPEGOXA não deva afetar o desempenho para dirigir veículos automotores, em estudos clínicos realizados até hoje, raramente ( < 5%) a administração de REPEGOXA esteve associada a tonturas e sonolência. Os pacientes que apresentarem esses efeitos devem evitar dirigir veículos e operar máquinas.
Gravidez (Categoria D): REPEGOXA é embriotóxico em ratos e embriotóxico e abortivo em coelhos. Não se pode descartar a possibilidade de teratogenicidade. Não existe experiência com REPEGOXA em gestantes e, por isso, a administração de REPEGOXA em gestantes não é recomendada. Mulheres com potencial para engravidar devem ser orientadas a evitar a gravidez enquanto elas ou seus parceiros estiverem recebendo REPEGOXA e por seis meses depois de sua interrupção.
Lactação
Não se sabe se esse medicamento é excretado no leite humano e devido ao potencial de efeitos adversos graves nos lactentes por causa de REPEGOXA, as mães devem suspender o aleitamento antes de receber esse agente. Especialistas em saúde recomendam que mulheres infectadas pelo HIV não amamentem seus filhos em nenhuma hipótese para evitar a transmissão do HIV.
Este medicamento contém álcool em traços.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
Este medicamento não deve ser usado por pessoas com síndrome de má-absorção de glicose-galactose.
Este medicamento não deve ser usado por pessoas com insuficiência de sacarose-isomaltase.
Atenção: Deve ser usado com cautela por portadores de Diabetes.
Atenção: Contém 94,00 mg de sacarose.
6. INTERAÇÕES MEDICAMENTOSAS
Não foram realizados estudos formais sobre interações farmacológicas de REPEGOXA, embora estudos de fase II de associações com agentes quimioterápicos convencionais tenham sido conduzidos em pacientes com doenças ginecológicas malignas. Deve-se proceder com cautela quando do uso concomitante de medicamentos que tenham interações conhecidas com o cloridrato de doxorrubicina convencional. REPEGOXA, assim como outras preparações contendo cloridrato de doxorrubicina, pode potencializar a toxicidade de outros tratamentos contra o câncer.
REPEGOXA já foi administrado como parte de um esquema terapêutico (associado com ciclofosfamida, taxanos ou vinorelbina) em 230 pacientes com tumores sólidos (incluindo câncer de ovário ou de mama). As doses de REPEGOXA e do agente associado usadas nesses estudos foram as seguintes: ciclofosfamida 600 mg/m2 + REPEGOXA 30 mg/m2 a cada 3 semanas, paclitaxel 175 mg/m2 + REPEGOXA 30 mg/m2 a cada 3 semanas, docetaxel 60 mg/m2 + REPEGOXA 30 mg/m2 a cada 3 semanas e vinorelbina 30 mg/m2 a cada 2 semanas + v 40 mg/m2 a cada 4 semanas. Não foram observadas novas toxicidades aditivas. Em pacientes com sarcoma de Kaposi relacionado à síndrome da imunodeficiência adquirida, a exacerbação da cistite hemorrágica induzida por ciclofosfamida e o aumento da hepatotoxicidade da 6-mercaptopurina foram descritos com o cloridrato de doxorrubicina convencional. Deve-se agir com cautela ao se administrar concomitantemente outros agentes citotóxicos, especialmente os mielotóxicos.
Incompatibilidade: NÃO MISTURE COM OUTROS FÁRMACOS.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Armazenar em geladeira. Não congelar.
O prazo de validade do medicamento é de 18 meses a partir da data de fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Depois de diluir em soro glicosado a 5%, a solução diluída de REPEGOXA deve ser usada imediatamente.
Após preparo, manter sob refrigeração (entre 2 e 8° C) por no máximo 24 horas.
Aspecto físico: é uma suspensão de cor vermelha e translúcida.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
REPEGOXA apresenta propriedades farmacocinéticas exclusivas e não deve ser usado de forma intercambiável com outras formulações de cloridrato de doxorrubicina.
REPEGOXA deve ser administrado apenas sob supervisão de um profissional qualificado, especializado na administração de agentes citotóxicos.
É recomendado o uso da agulha de calibre 21 para reconstituição do frasco para diluição.
Câncer de ovário/mama: REPEGOXA é administrado por via intravenosa em dose de 50 mg/m2 uma vez a cada 4 semanas enquanto a doença não progredir e a paciente continuar a tolerar o tratamento.
Para doses < 90 mg: diluir REPEGOXA em 250 mL de soro glicosado a 5%. Para doses ≥ 90 mg: diluir REPEGOXA em 500 mL de soro glicosado a 5%.
Para reduzir o risco de reações à infusão, a dose inicial é administrada em velocidade igual ou inferior a 1 mg/minuto. Se não for observada nenhuma reação à infusão, as infusões subsequentes de REPEGOXA podem ser administradas ao longo de um período de 60 minutos.
No programa de estudos clínicos do câncer de mama, foi permitida uma modificação da infusão para aquelas pacientes que apresentam uma reação à infusão, da seguinte maneira:
Cinco por cento (5%) da dose total foi infundida lentamente ao longo dos primeiros 15 minutos. Se tolerada sem reação, a velocidade de infusão foi dobrada nos 15 minutos seguintes. Novamente tolerada, a infusão foi completada ao longo da hora seguinte, para um tempo total de infusão de 90 minutos.
Infusões subsequentes de REPEGOXA podem ser administradas ao longo de um período de 60 minutos.
Mieloma Múltiplo: REPEGOXA é administrado na dose de 30 mg/m2 no dia 4 da terapia de 3 semanas com bortezomibe a uma infusão de 1 hora administrada imediatamente após a infusão de bortezomibe. A terapia com bortezomibe consiste na administração de 1,3 mg/m2 nos dias 1, 4, 8 e 11 a cada 3 semanas. A dose deve ser repetida enquanto os pacientes responderem satisfatoriamente e tolerarem o tratamento.
Para doses < 90 mg: dilua REPEGOXA em 250 mL de soro glicosado a 5% (50 mg/mL) para infusão. Para doses ≥ 90 mg: dilua REPEGOXA em 500 mL de soro glicosado a 5% (50 mg/mL) para infusão.
O cateter e o tubo intravenosos devem ser lavados com soro glicosado a 5% para infusão entre a administração dos 2 medicamentos. A dose do Dia 4 de ambos os medicamentos pode ser adiada por até 48 horas, se clinicamente necessário. As doses de bortezomibe devem ser administradas em intervalos de, no mínimo, 72 horas. A primeira infusão de REPEGOXA deve ser administrada durante 90 minutos, conforme segue:
• 10 mL nos primeiros 10 minutos;
• 20 mL nos próximos 10 minutos;
• 40 mL nos próximos 10 minutos;
• então, conclua a infusão durante um total de 90 minutos.
As doses subsequentes de REPEGOXA serão administradas durante 1 hora, conforme toleradas. Se ocorrer uma reação à infusão ao REPEGOXA, interrompa a infusão. Após os sintomas terem sido resolvidos, tentar administrar o conteúdo remanescente de REPEGOXA durante 90 minutos, conforme segue:
• 10 mL nos primeiros 10 minutos;
• 20 mL nos próximos 10 minutos;
• 40 mL nos próximos 10 minutos;
• então, concluir a infusão remanescente durante um total de 90 minutos.
A infusão pode ser administrada através de uma veia periférica ou um acesso central.
Pacientes com sarcoma de Kaposi relacionado à síndrome da imunodeficiência adquirida: REPEGOXA deve ser administrado por via intravenosa na dose de 20 mg/m2 a cada duas ou três semanas. Os intervalos menores que 10 dias devem ser evitados já que não se pode descartar a possibilidade de acúmulo do fármaco e de toxicidade crescente. Os pacientes devem ser tratados durante dois a três meses para obter uma resposta terapêutica. O tratamento deve continuar de acordo com a necessidade para manter a resposta terapêutica.
REPEGOXA, diluído em 250 mL de soro glicosado a 5%, é administrado por infusão intravenosa ao longo de 30 minutos.
Todos os pacientes: se o paciente apresentar sintomas ou sinais iniciais de reação à infusão, interrompa imediatamente a infusão, administre os medicamentos apropriados (anti-histamínicos e/ou corticosteroide de ação imediata) e reinicie em velocidade baixa.
Não se deve administrar REPEGOXA na forma de injeção em bolus, nem como dispersão não diluída. Recomenda-se que o equipo de infusão de REPEGOXA seja conectado à solução de glicose 5% através de um equipo com injetor lateral a fim de se obter diluição maior e reduzir ao mínimo o risco de trombose e extravasamento. A infusão pode ser administrada através de uma veia periférica.
REPEGOXA não deve ser administrado por via intramuscular ou subcutânea. Não use filtros em linha.
Orientações para Modificação da Dose de REPEGOXA
Para tratar eventos adversos, como eritrodisestesia palmo-plantar (EPP), estomatite ou toxicidade hematológica, a dose pode ser reduzida ou retardada. Orientações para modificação da dose de REPEGOXA secundária a esses efeitos adversos são fornecidas nas tabelas seguintes. A classificação de toxicidade nessas tabelas se baseia no National Cancer Institute Common Toxicity Criteria (NCI-CTC).
As tabelas para eritrodisestesia palmo-plantar (Tabela 2) e estomatite (Tabela 3) oferecem o esquema seguido para modificação de dose em estudos clínicos de tratamento de câncer de mama ou de ovário (modificação do ciclo de tratamento recomendado de 4 semanas). Se essas toxicidades ocorrerem em pacientes com sarcoma de Kaposi relacionado à síndrome da imunodeficiência adquirida, o ciclo de tratamento recomendado de 2 a 3 semanas pode ser modificado de forma semelhante.
A tabela para toxicidade hematológica (Tabela 4) oferece o esquema seguido para modificação da dose em estudos clínicos de tratamento de pacientes com câncer de mama ou de ovário. A modificação da dose em pacientes com sarcoma de Kaposi relacionado à síndrome da imunodeficiência adquirida é abordada em "REAÇÕES ADVERSAS".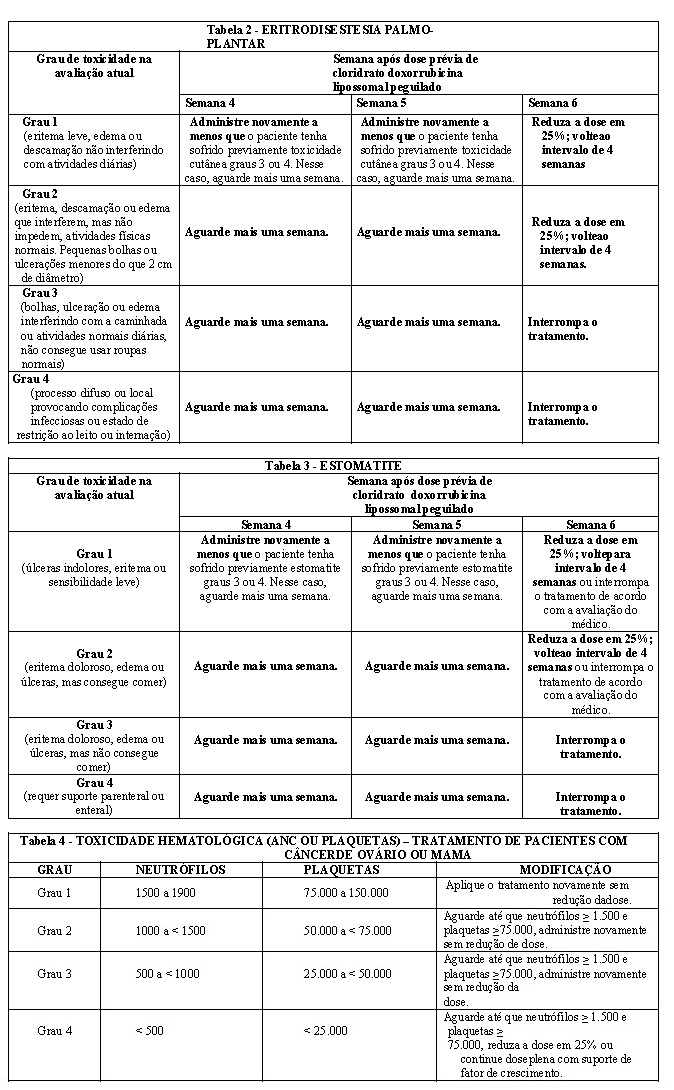
Para pacientes com mieloma múltiplo tratados com REPEGOXA em combinação com bortezomibe que apresentarem EPP ou estomatite, a dose de REPEGOXA deve ser modificada conforme descrito na Tabela 2 e na Tabela 3 acima, respectivamente. Para informações mais detalhadas sobre a dosagem e os ajustes da dosagem de bortezomibe, consulte a bula do bortezomibe.
Pacientes com função hepática comprometida: a farmacocinética de REPEGOXA determinada em pequeno número de pacientes com níveis elevados de bilirrubinas totais não difere daquela de pacientes com bilirrubinas totais normais; no entanto, até que se tenha mais experiência, a dose de REPEGOXA em pacientes com função hepática comprometida deve ser reduzida com base na experiência do programa de estudo clínico ovariano e de mama, como se segue: no início da terapia, se a bilirrubina estiver entre 1,2 e 3,0 mg/dL, a primeira dose é reduzida em 25%. Se a bilirrubina for > 3,0 mg/dL, a primeira dose é reduzida em 50%. Se o paciente tolerar a primeira dose sem aumento de bilirrubina sérica ou enzimas hepáticas, a dose para o ciclo 2 pode ser aumentada até o nível de dose seguinte, isto é, se reduzida em 25% para a primeira dose, aumente para dose completa para o ciclo 2; se reduzida em 50% para a primeira dose, aumente para 75% da dose completa para o ciclo 2. A dose pode ser aumentada até a dose completa para ciclos subsequentes, se tolerada. REPEGOXA pode ser administrado a pacientes com metástases hepáticas e elevação concomitante de bilirrubinas e enzimas hepáticas até 4 vezes o limite superior do intervalo normal. Antes da administração de REPEGOXA, avalie a função hepática usando os exames laboratoriais clínicos convencionais, como ALT/AST, fosfatase alcalina e bilirrubinas.
Pacientes com função renal comprometida:
Pacientes com sarcoma de Kaposi relacionado à síndrome da imunodeficiência adquirida e esplenectomizados: como não se tem experiência com REPEGOXA em pacientes esplenectomizados, não se recomenda a sua administração.
Pacientes pediátricos: dados limitados de segurança de estudos fase I indicam que doses de até 60 mg/m2 a cada 4 semanas são bem toleradas em pacientes pediátricos; no entanto, a eficácia em pacientes abaixo dos 18 anos de idade não foi estabelecida.
Pacientes idosos: A análise com base populacional demonstra que a idade no intervalo testado (21 a 75 anos) não altera significativamente a farmacocinética de REPEGOXA.
Instruções para uso e manipulação
NÃO USAR MATERIAL COM EVIDÊNCIA DE PRECIPITAÇÃO OU PRESENÇA DE QUALQUER OUTRO TIPO DE PARTÍCULAS.
Deve-se proceder com cuidado quando se manipula a dispersão de REPEGOXA. É necessário usar luvas. Se REPEGOXA entrar em contato com a pele ou as mucosas, lave imediata e minuciosamente com água e sabonete. REPEGOXA deve ser manipulado e descartado de forma semelhante à utilizada para outros medicamentos contra o câncer.
O medicamento não utilizado deve ser manuseado e descartado de maneira consistente com outros medicamentos anticancerígenos, de acordo com os requisitos locais.
Determinar a dose de REPEGOXA que vai ser administrada (com base na dose recomendada e na superfície corporal do paciente). Colocar o volume apropriado de REPEGOXA em uma seringa estéril. Deve-se adotar técnica estritamente asséptica, já que REPEGOXA não contém conservantes nem agentes bacteriostáticos.
A dose apropriada de REPEGOXA deve ser diluída em soro glicosado a 5% antes da administração. Para doses < 90 mg, dilua REPEGOXA em 250 mL e para doses ≥ 90 mg, dilua REPEGOXA em 500 mL de soro glicosado a 5%.
O uso de qualquer diluente, exceto o soro glicosado 5% para infusão, ou a presença de qualquer agente bacteriostático, como álcool benzílico, por exemplo, pode provocar a precipitação de REPEGOXA.
Recomenda-se que o equipo de infusão de REPEGOXA seja conectado através de equipo em Y a um frasco com infusão intravenosa de soro glicosado a 5%. A infusão pode ser administrada através de veia periférica. Não use filtros em linha.
Incompatibilidades: NÃO MISTURE CLORIDRATO DE DOXORRUBICINA LIPOSSOAL PEGUILADO COM OUTROS MEDICAMENTOS.
9. REAÇÕES ADVERSAS
Ao longo desta seção, são apresentadas as reações adversas. Reações adversas são eventos adversos considerados razoavelmente associados ao uso de doxorrubicina lipossômica peguilada com base na avaliação abrangente da informação disponível sobre eventos adversos. Em casos individuais uma relação causal com doxorrubicina lipossomal peguilada não pode ser estabelecida de forma confiável. Além disso, como os ensaios clínicos são conduzidos sob condições muito variadas, as taxas de reações adversas observadas nos ensaios clínicos de um medicamento não podem ser diretamente comparadas às taxas nos estudos clínicos de outro medicamento e podem não refletir as taxas observadas na prática clínica.
Dados de ensaios clínicos
Pacientes com câncer de mama: 254 pacientes com câncer de mama avançado que não tinham recebido quimioterapia anterior para doença metastática foram tratadas com REPEGOXA, em uma dose de 50 mg/m2 a cada 4 semanas, em um estudo clínico fase III (I97-328). Os efeitos adversos relacionados ao tratamento mais frequentemente relatados incluíram eritrodisestesia palmo-plantar (EPP) (48,0%) e náuseas (37,0%) (ver Tabela 6). Esses efeitos foram, na maioria das vezes, leves e reversíveis, com casos graves (Grau III) relatados em 17,0% e 3,0%, respectivamente, e sem relatos de incidências de casos com risco de morte (Grau IV) para EPP ou náuseas. Esses efeitos raramente resultaram em descontinuidade permanente do tratamento (7,0% e 0%, respectivamente). Uma alopécia acentuada ou total foi observada em apenas 7,0% dos pacientes tratados com REPEGOXA, em comparação com 54,0% dos pacientes tratados com a doxorrubicina.
Os efeitos adversos hematológicos foram raramente relatados e foram, na maior parte, de gravidade leve ou moderada e controlável. Anemia, neutropenia, leucopenia e trombocitopenia foram raramente relatadas em incidências de 5,0%, 4,0%, 2,0% e 1,0%, respectivamente. Efeitos hematológicos com risco de morte (Grau IV) foram relatados em incidências < 1,0%. A necessidade de suporte de fator de crescimento ou de suporte transfusional foi mínima (5,1% e 5,5% dos pacientes, respectivamente).
As anormalidades laboratoriais clinicamente significativas (Graus III e IV) nesse grupo do câncer de mama incluíram aumentos nas bilirrubinas totais (2,4%) e em AST (1,6%). Os aumentos em ALT foram menos frequentes ( < 1%). Medições hematológicas clinicamente significativas foram raras, conforme medições de leucopenia (4,3%), anemia (3,9%), neutropenia (1,6%) e trombocitopenia (1,2%). Sepse foi relatada em uma incidência de 1%. Não houve relatos de aumentos clinicamente significativos na creatinina sérica.
Em 150 pacientes com câncer de mama avançado que não tinham respondido a uma quimioterapia anterior de primeira ou segunda linha contendo taxano e que foram subsequentemente tratadas com REPEGOXA em uma dose de 50 mg/m2 a cada 4 semanas em um estudo clínico fase III (C/I96-352), o perfil de segurança foi compatível com o relatado para o REPEGOXA em estudos anteriores que utilizaram o mesmo esquema de dosagem (ver Tabela 6). A proporção de pacientes que apresentaram anormalidades laboratoriais clinicamente significativas foi baixa e numericamente comparável à de 254 pacientes com câncer de mama que receberam REPEGOXA como terapia de primeira linha, com exceção da leucopenia (20%).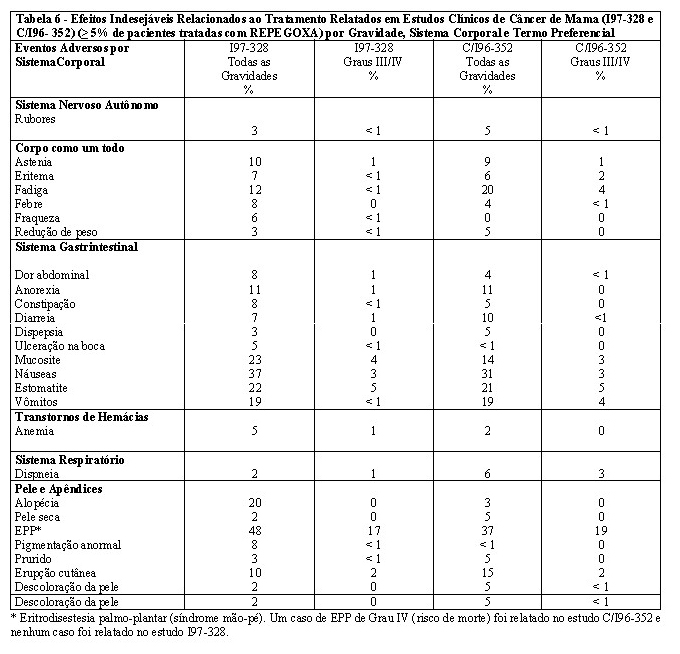
Outros dados de ensaios clínicos em câncer de mama
Os efeitos indesejáveis relatados entre 1% e < 5% dos 404 pacientes com câncer de mama tratados com REPEGOXA, não anteriormente relatados nos estudos clínicos de REPEGOXA, foram dor torácica, câimbras nas pernas, edema, edema na perna, neuropatia periférica, dor na boca, arritmia do ventrículo, foliculite, dor óssea, dor musculoesquelética, trombocitemia, ulcerações labiais (não herpéticas), infecção fúngica, epistaxe, infecção do trato respiratório superior, erupção bolhosa, dermatite, erupção cutânea eritematosa, transtorno ungueal, pele escamosa, lacrimejamento e visão turva.
Pacientes com câncer de ovário: um total de 512 pacientes com câncer de ovário (um subgrupo de 876 pacientes com tumores sólidos) foi tratado com REPEGOXA na dose de 50 mg/m2 a cada 4 semanas nos estudos clínicos. Os eventos adversos relacionados ao tratamento mais frequentemente relatados foram eritrodisestesia palmo-plantar (EPP) (46,1%) e estomatite (38,9%) (ver Tabela 7). Esses efeitos foram principalmente leves, com casos graves (Grau III) relatados em 19,5% e 8,0%, respectivamente, e casos com risco de morte (Grau IV) relatados em 0,6% e 0,8%, respectivamente. Esses resultados raramente levaram a uma descontinuidade permanente do tratamento ( < 5% e < 1%, respectivamente). 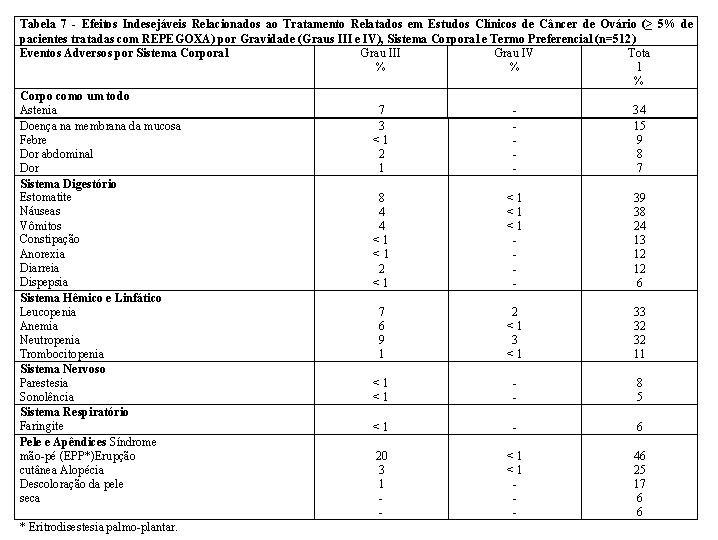
A mielossupressão foi principalmente de leve a moderada e passível de tratamento. A leucopenia foi o efeito adverso hematológico mais frequentemente relatado, seguido por anemia, neutropenia e trombocitopenia. Efeitos hematológicos com risco de morte (Grau IV) foram relatados em uma incidência de 1,6%, 0,4%, 2,9% e 0,2%, respectivamente. Raramente foi necessário suporte com fator de crescimento ( < 5%) e a transfusão foi necessária em aproximadamente 15% das pacientes (vide "Posologia e Modo de Usar").
Outros efeitos indesejáveis descritos com menor frequência (1 a 5%) incluíram edema periférico, monilíase oral, vasodilatação, ulceração na boca, prurido, reação alérgica, desidratação, dispneia, erupção vésico-bolhosa, calafrios, infecção, perda de peso, esofagite, afecção de pele, dermatite esfoliativa, distúrbio cardiovascular, dor torácica, tontura, erupção maculopapular, gastrite, mialgia, dor nas costas, depressão, insônia, disfagia, aumento da tosse, sudorese, náuseas e vômitos, mal-estar, alteração do paladar, infecção do trato urinário, conjuntivite, acne, gengivite, herpes zoster, anemia hipocrômica, ansiedade, vaginite, dor de cabeça, flatulência, boca seca, caquexia, neuropatia, hipertonia, úlcera na pele e disúria.
No subgrupo de 410 pacientes com câncer de ovário, alterações laboratoriais clinicamente significativas em estudos clínicos com REPEGOXA incluíram aumentos da bilirrubina total (geralmente em pacientes com metástases hepáticas) (5%) e dos níveis séricos de creatinina (5%). Medidas clinicamente significativas, como neutropenia (11,4%), anemia (5,7%) e trombocitopenia (1,2%) Graus III e IV, foram baixas. Elevações de AST foram descritas com menor frequência ( < 1%). Sepse relacionada à leucopenia foi observada raramente ( < 1%).
Pacientes com tumores sólidos: Em um estudo maior de 929 pacientes com tumores sólidos (incluindo câncer de mama e de ovário), predominantemente tratados com uma dose de 50 mg/m2 a cada 4 semanas, o perfil de segurança e a incidência de efeitos adversos foram comparáveis àqueles de pacientes tratados em estudos clínicos pivotais de câncer de mama e de ovário.
Pacientes com mieloma múltiplo: 646 pacientes com mieloma múltiplo que receberam pelo menos uma terapia anterior, 318 pacientes foram tratados com a terapia combinada de REPEGOXA 30 mg/m2 administrado por infusão intravenosa por uma hora no dia 4 (D4) acompanhado da administração de bortezomibe na dose de 1,3 mg/m2 nos dias 1, 4, 8 e 11 a cada três semanas ou bortezomibe em monoterapia em um estudo clínico fase III. Ver Tabela 8 para os efeitos adversos relatados em ≥ 5% dos pacientes tratados com a terapia combinada de REPEGOXA mais bortezomibe.
Neutropenia, trombocitopenia e anemia foram os eventos hematológicos mais frequentemente relatados com a terapia combinada de REPEGOXA mais bortezomibe e com a monoterapia com bortezomibe. A incidência de neutropenia de grau 3 e 4 foi maior no grupo da terapia combinada do que no grupo da monoterapia (28% vs. 14%). A incidência de trombocitopenia de graus 3 e 4 foi maior no grupo da terapia combinada do que no grupo da monoterapia (22% vs. 14%). A incidência de anemia foi similar em ambos os grupos de tratamento (7% vs. 5%).
A estomatite foi mais frequentemente relatada no grupo da terapia combinada (16%) do que no grupo da monoterapia (3%), e a maioria dos casos foi de gravidade grau 2 ou menor. Estomatite de grau 3 foi reportado em 2% dos pacientes no grupo da terapia combinada. Nenhuma estomatite de grau 4 foi reportada.
Náuseas e vômitos foram mais frequentemente reportados no grupo da terapia combinada (40% e 28%) do que no grupo da monoterapia (32% e 15%) e foram na sua maioria de gravidade grau 1 e 2.
A descontinuidade do tratamento de um ou de ambos os fármacos devido a eventos adversos foi observada em 38% dos pacientes. Os eventos adversos comuns que levaram à descontinuidade do tratamento do bortezomibe e REPEGOXA incluíram EPP, neuralgia, neuropatia periférica, neuropatia sensorial periférica, trombocitopenia, diminuição da fração de ejeção e fadiga.
Pacientes com sarcoma de Kaposi relacionado à síndrome da imunodeficiência adquirida: os estudos clínicos abertos e controlados, realizados em pacientes com sarcoma de Kaposi relacionado à síndrome da imunodeficiência adquirida tratados com dose de 20 mg/m2 de REPEGOXA cada 2 a 3 semanas, demonstram que o efeito colateral mais frequente e considerado relacionado ao REPEGOXA foi a mielossupressão, que ocorreu em aproximadamente metade dos pacientes.
A leucopenia é o efeito indesejável mais frequente dessa população tratada com REPEGOXA, podendo-se também esperar neutropenia, anemia e trombocitopenia. Esses efeitos podem ocorrer no início do tratamento. A toxicidade hematológica talvez exija uma redução da dose ou que se suspenda ou adie o tratamento. A administração de REPEGOXA deverá ser suspensa temporariamente quando o número absoluto de neutrófilos for < 1.000/mm3 e/ou o número de plaquetas for < 50.000/mm3. O G-CSF (ou GM-CSF) pode ser administrado como tratamento concomitante para auxiliar a recuperação do hemograma quando o número absoluto de neutrófilos for < 1.000/mm3 nos ciclos seguintes. A toxicidade hematológica para pacientes com câncer de ovário é menos grave do que no grupo com sarcoma de Kaposi relacionado à síndrome da imunodeficiência adquirida (vide seção anterior "Pacientes com câncer de ovário").
Outros efeitos colaterais observados com frequência (≥ 5%) foram náuseas, astenia, alopécia, febre, diarreia, reações agudas associadas com a infusão e estomatite.
Nos estudos clínicos realizados com REPEGOXA, foram observados frequentemente (≥ 5%) efeitos adversos respiratórios, que talvez se relacionem com infecções oportunistas na população com síndrome da imunodeficiência adquirida. As infecções oportunistas são observadas em pacientes com sarcoma de Kaposi relacionado à síndrome da imunodeficiência adquirida depois da administração de REPEGOXA e com frequência em pacientes com imunodeficiência induzida pelo HIV. As infecções oportunistas observadas com maior frequência nos estudos clínicos foram candidíase, citomegalovirose, herpes simples, pneumonia por Pneumocystis carinii e complexo Mycobacterium avium.
Outros efeitos secundários observados com menor frequência ( < 5%) incluíram eritrodisestesia palmo-plantar, monilíase oral, náuseas e vômitos, perda de peso, erupção cutânea, ulcerações na boca, dispneia, dor abdominal, reação de hipersensibilidade incluindo reações anafiláticas, vasodilatação, tontura, anorexia, glossite, constipação, parestesias, retinite e confusão mental.
Alterações laboratoriais clinicamente significativas ocorreram frequentemente (≥ 5%) em estudos clínicos com REPEGOXA. Elas incluíram elevações de fosfatase alcalina e elevações em AST e bilirrubina que se acredita sejam relacionadas à doença e não ao REPEGOXA. A redução da hemoglobina e das plaquetas foi descrita menos frequentemente ( < 5%). Sepse relacionada à leucopenia foi observada raramente ( < 1%). Algumas dessas anormalidades podem ter sido relacionadas à infecção por HIV e não ao REPEGOXA.
Todos os pacientes: em 100 de 929 pacientes (10,8%) com tumores sólidos foi descrita reação associada à infusão durante o tratamento com REPEGOXA definida pelos seguintes termos COSTART: reação alérgica, reação anafilactoide, asma, edema de face, hipotensão, vasodilatação, urticária, dor nas costas, dor torácica, calafrios, febre, hipertensão, taquicardia, dispepsia, náuseas, tontura, dispneia, faringite, erupção cutânea, prurido, sudorese, reação no local da injeção e interação medicamentosa. Os índices de descontinuidade permanente do tratamento foram raramente relatados em 2%. Uma incidência semelhante de reações à infusão (12,4%) foi observada nos estudos clínicos pivotais de câncer de mama. O índice de descontinuidade permanente do tratamento também foi semelhante em 1,5%. Em pacientes com mieloma múltiplo em tratamento com REPEGOXA mais bortezomibe, reações relacionadas à infusão foram relatadas em 3%. Em pacientes com sarcoma de Kaposi relacionado à síndrome da imunodeficiência adquirida, as reações associadas com a infusão se caracterizaram por rubor, falta de ar, edema facial, cefaleia, calafrios, dor nas costas, aperto no peito ou na garganta e/ou hipotensão, e podem ser esperadas na incidência de 5% a 10%. Muito raramente, foram observadas convulsões em relação às reações de infusão. Em todos os casos, os efeitos colaterais ocorreram durante o primeiro ciclo de tratamento. A suspensão temporária da infusão resolve essas reações sem o uso de qualquer tratamento sintomático. Em praticamente todos os pacientes, o tratamento com REPEGOXA pode ser reiniciado depois da resolução de todos os sintomas, sem recorrência. As reações à infusão raramente ocorrem novamente depois do primeiro ciclo de tratamento com REPEGOXA.
Mielossupressão associada com anemia, trombocitopenia, leucopenia e raramente neutropenia febril foram relatadas em pacientes tratados com REPEGOXA.
Em pacientes que receberam infusões contínuas de cloridrato de doxorrubicina convencional, foi descrita estomatite, que também apareceu com frequência nos pacientes que receberam REPEGOXA. Esse distúrbio não impediu que os pacientes completassem o tratamento e, em geral, não exigiu ajustes de posologia, a não ser que a estomatite afetasse a capacidade de ingestão de alimentos pelo paciente. Nesse caso, os intervalos entre as doses podem ser prolongados em 1 a 2 semanas, ou deve-se reduzir a dose.
A eritrodisestesia palmo-plantar se caracteriza por erupções cutâneas dolorosas com eritema macular. Os pacientes que apresentam essa afecção geralmente passam a tê-la depois de dois ou três ciclos de tratamento. Na maioria dos casos, ela desaparece em uma ou duas semanas, com ou sem tratamento com corticosteroides. Piridoxina na dose de 50 a 150 mg por dia é usada para profilaxia e para tratamento de eritrodisestesia palmo-plantar. Outras estratégias para evitar e tratar essa afecção podem ser iniciadas de 4 a 7 dias depois do tratamento com REPEGOXA e incluem manter frios as mãos e os pés pela exposição à água fria (compressas, banhos ou natação), evitar calor e água quente em excesso e mantê-los descobertos (sem meias, luvas ou sapatos apertados). A eritrodisestesia palmo-plantar parece estar relacionada à dose e ao esquema posológico e pode ser reduzida se ampliando o intervalo de administração em 1 a 2 semanas ou se reduzindo a dose. No entanto, a reação pode ser grave e debilitante em alguns pacientes e pode ser necessário suspender o tratamento
Uma incidência aumentada de insuficiência cardíaca congestiva está associada à terapia com doxorrubicina em doses cumulativas > 450 mg/m2 ou em doses menores para pacientes com fatores de risco cardíaco. Biópsias endomiocárdicas em nove dentre dez pacientes com sarcoma de Kaposi relacionado à síndrome da imunodeficiência adquirida que receberam doses cumulativas dE REPEGOXA maiores que 460 mg/m2 indicam nenhuma evidência de miocardiopatia induzida por antraciclina. A dose de REPEGOXA recomendada para pacientes com sarcoma de Kaposi relacionado à síndrome da imunodeficiência adquirida é de 20 mg/m2 a cada duas a três semanas. A dose cumulativa em que a cardiotoxicidade se tornaria um motivo de preocupação para essas pacientes com sarcoma de Kaposi relacionado à síndrome da imunodeficiência adquirida ( > 400 mg/m2) exigiria mais de 20 ciclos de terapia com REPEGOXA durante 40 a 60 semanas.
Além disso, foram realizadas biópsias endomiocárdicas em 8 pacientes com tumor sólido com doses cumulativas de antraciclina de 509 mg/m2 a 1.680 mg/m2. A variação da pontuação de cardiotoxicidade de Billingham foi de graus 0 a 1,5. Esses critérios de classificação são compatíveis com toxicidade cardíaca ausente ou leve.
No estudo clínico pivotal fase III de câncer de mama comparando REPEGOXA (50 mg/m2 a cada 4 semanas) com doxorrubicina (60 mg/m2 a cada 3 semanas), 10 de 254 pacientes randomizados para receber REPEGOXA versus 48 de 255 pacientes randomizados para receber doxorrubicina satisfizeram os critérios definidos pelo protocolo para toxicidade cardíaca durante o tratamento e/ou acompanhamento. A toxicidade cardíaca foi definida como uma redução de 20 pontos percentuais ou mais em relação ao valor basal se a fração de ejeção do ventrículo esquerdo (FEVE) em repouso tiver permanecido na faixa normal, ou uma redução de 10 pontos percentuais ou mais, se a FEVE tiver se tornado anormal (abaixo do limite inferior da normalidade). O risco de desenvolvimento de um evento cardíaco em função do acúmulo da dose de antraciclina foi significativamente menor com REPEGOXA quando comparado à doxorrubicina (RR[doxorrubicina/ REPEGOXA]= 3,16, p < 0,001).
Os pacientes foram avaliados também quanto aos sinais e sintomas de insuficiência cardíaca congestiva (ICC). Nenhum dos 10 pacientes do grupo do REPEGOXA que apresentavam toxicidade cardíaca segundo os critérios de FEVE desenvolveu sinais e sintomas de ICC. Por outro lado, 10 dos 48 pacientes do grupo da doxorrubicina que apresentavam toxicidade cardíaca pelos critérios FEVE também desenvolveram sinais e sintomas de ICC.
Em pacientes com tumores sólidos, incluindo um subgrupo de pacientes com câncer de ovário e de mama tratados com dose de 50 mg/m2 por ciclo com doses cumulativas de até 1.532 mg/m2, a incidência de disfunção cardíaca clinicamente significativa foi baixa. Dos 929 pacientes tratados com 50 mg/m2 por ciclo de REPEGOXA, a medida inicial da fração de ejeção de ventrículo esquerdo (FEVE) e, pelo menos, uma medida de seguimento foram conduzidas em 418 pacientes e avaliadas pelo mapeamento MUGA. Desses 418 pacientes, 88 tinham uma dose cumulativa de antraciclina > 400 mg/m2, um nível de exposição associada a um risco aumentado de toxicidade cardiovascular com a formulação convencional de doxorrubicina. Apenas 13 desses 88 pacientes (15%) apresentaram pelo menos uma alteração clinicamente significativa de FEVE, definida como um valor de FEVE menor do que 45% ou uma redução de, pelo menos, 20% a partir do valor basal. Além disso, somente 1 paciente (que recebeu uma dose cumulativa de 944 mg/m2) interrompeu o tratamento em estudo por causa de sintomas clínicos de insuficiência cardíaca congestiva.
Embora necrose local tenha sido observada muito raramente após o extravasamento, deve-se considerar que REPEGOXA é um agente irritante. Os estudos em animais indicam que a administração de cloridrato de doxorrubicina como fórmula lipossomal reduz o potencial de ocorrência de uma lesão por extravasamento. Se ocorrerem sinais ou sintomas de extravasamento (como queimação e eritema), a infusão deve ser imediatamente suspensa e se deve prosseguir usando outra veia. A aplicação de gelo sobre o local de extravasamento durante aproximadamente 30 minutos talvez seja útil para aliviar a reação local. REPEGOXA não deve ser administrada por via intramuscular ou subcutânea.
Com a administração de REPEGOXA, raramente ocorreu um reaparecimento de reação cutânea provocada por radioterapia anterior.
Dados de Pós-comercialização
Foram identificadas reações adversas durante a experiência pós-comercialização com REPEGOXA, conforme descrito a seguir. As frequências são fornecidas de acordo com a seguinte convenção:
Muito Comum ≥ 1/10
Comum ≥ 1/100 e < 1/10
Incomum ≥ 1/1000 e < 1/100
Raro ≥ 1/10000 e < 1/1000
Muito Raro < 1/10000, incluindo casos isolados
Distúrbios vasculares
Pacientes com câncer possuem risco aumentado para doença tromboembólica. Em pacientes tratados com REPEGOXA, casos de tromboflebite e trombose venosa foram raramente relatados, assim como raros foram os casos de embolismo pulmonar.
Distúrbios na pele e no tecido subcutâneo
Condições graves na pele, incluindo eritema multiforme, síndrome de Stevens-Johnson, necrólise epidérmica tóxica e queratose liquenoide foram relatadas muito raramente.
Neoplasias orais secundárias
Foram relatados casos muito raros de câncer oral secundário em pacientes com longo período de exposição (mais de um ano) a REPEGOXA ou em pacientes recebendo dose cumulativa de REPEGOXA superior a 720 mg/m2.
Leucemia mieloide aguda secundária e síndrome mielodisplásica
Tal como ocorre com outros agentes antineoplásicos prejudiciais ao DNA, leucemia mieloide aguda secundária e síndrome mielodisplásica foram relatadas raramente em pacientes recebendo tratamento combinado com doxorrubicina.
Em casos de eventos adversos, notifique pelo Sistema VigiMed, disponível no Portal da Anvisa.
10. SUPERDOSE
Sinais e Sintomas
A superdose aguda com o cloridrato de doxorrubicina piora os efeitos tóxicos da mucosite, leucopenia e trombocitopenia.
Tratamento
O tratamento da superdose aguda do paciente gravemente mielossuprimido inclui internação, antibióticos, transfusões de plaquetas e granulócitos e tratamento sintomático da mucosite.
Em caso de intoxicação ligue para 0800 722 6001, se você precisar de mais orientações.
Dizeres legais.
Registro: 1.5143.0083
USO RESTRITO A ESTABELECIEMENTOS DE SAÚDE
VENDA SOB PRESCRIÇÃO
Esta bula foi atualizada conforme Bula Padrão aprovada pela Anvisa em 26/04/2021.