REMICADE
JANSSEN-CILAG
infliximabe
Anti-reumático. Imunomodulador.
USO ADULTO E PEDIÁTRICO (acima de 6 anos de idade).
Apresentações.
Pó liofilizado para solução concentrada para infusão em embalagem com 1 frasco-ampola com 100 mg de infliximabe.
USO INTRAVENOSO
USO ADULTO E PEDIÁTRICO ACIMA DE 6 ANOS
Composição.
REMICADE® 100 mg:
Cada frasco-ampola contém 100 mg de infliximabe para ser reconstituído com 10 mL de água para injetáveis e, posteriormente, diluído em cloreto de sódio 0,9% para infusão.
Excipientes: fosfato de sódio monobásico monoidratado, fosfato de sódio dibásico di-hidratado, polissorbato 80 e sacarose.
Indicações.
Artrite Reumatoide
REMICADE® é uma "Terapia Antirreumática Controladora da Doença" (DCART - Disease-Controlling Anti-Rheumatic Therapy) indicado para:
- redução de sinais e sintomas;
- prevenção de lesão articular estrutural (erosões e estreitamento do espaço articular);
- melhora na função física; em pacientes com doença ativa já tratados com metotrexato (artrite reumatoide estabelecida) e com doença ativa ainda não tratados com metotrexato (artrite reumatoide inicial).
Espondilite Anquilosante
REMICADE® é indicado para:
- redução dos sinais e sintomas;
- melhora na função física; em pacientes com doença ativa.
Artrite Psoriásica
REMICADE® é indicado para:
Tratamento da artrite psoriásica ativa e progressiva em adultos, que tiveram resposta inadequada às drogas modificadoras da doença (DMARDs). REMICADE® deve ser administrado:
- em associação com metotrexato;
- ou em monoterapia em pacientes que demonstraram intolerância ao metotrexato ou aqueles para os quais o metotrexato é contraindicado. REMICADE® demonstrou melhorar a função física e inibir a progressão da lesão estrutural da artrite ativa, de acordo com a avaliação de radiografias dos pacientes com artrite psoriásica.
Psoríase
REMICADE® é indicado para:
- redução dos sinais e sintomas da psoríase;
- melhora na qualidade de vida; no tratamento de pacientes adultos com psoríase em placa grave candidatos à terapia sistêmica, e para aqueles com psoríase moderada em que a fototerapia é inadequada ou imprópria.
Doença de Crohn adulto e pediátrico
REMICADE® é indicado para:
- redução de sinais e sintomas;
- indução e manutenção da remissão clínica;
- indução da cicatrização da mucosa em adultos;
- melhora na qualidade de vida; em pacientes com doença de Crohn de moderada a grave que tiveram uma resposta inadequada às terapias convencionais. A terapia com REMICADE® permite a redução ou suspensão do uso de corticosteroides pelos pacientes.
Doença de Crohn Fistulizante
REMICADE® é indicado para:
- redução no número de fístulas enterocutâneas com drenagem e fístulas retovaginais e manutenção da fístula cicatrizada;
- redução dos sinais e sintomas;
- melhora na qualidade de vida;
em pacientes com doença de Crohn fistulizante.
Colite ou Retocolite Ulcerativa
Em 95% dos casos de colite ulcerativa, a inflamação envolve o reto e estende-se proximalmente, de maneira contínua, por uma distância variável (retocolite ulcerativa).
REMICADE® é indicado para:
- redução dos sinais e sintomas;
- indução e manutenção da remissão clínica;
- indução da cicatrização da mucosa;
- melhora na qualidade de vida;
redução ou descontinuação do uso de corticosteroides;
- redução da hospitalização relacionada à colite ou retocolite ulcerativa; em pacientes com colite ou retocolite ulcerativa ativa com resposta inadequada aos tratamentos convencionais.
Resultados de eficácia.
Artrite Reumatoide
A segurança e a eficácia de REMICADE® foram avaliadas em dois estudos clínicos pilotos, duplo-cegos, randomizados, multicêntricos: ATTRACT (Estudo Clínico Anti-TNF em Artrite Reumatoide com Terapia Concomitante) e ASPIRE (Estudo com Controle Ativo de Pacientes que Receberam Infliximabe para o Tratamento de Artrite Reumatoide de Início Recente). Foi permitido o uso concomitante de doses estáveis de ácido fólico, corticosteroides orais (≤10 mg/dia) e/ou drogas anti-inflamatórias não esteroidais.
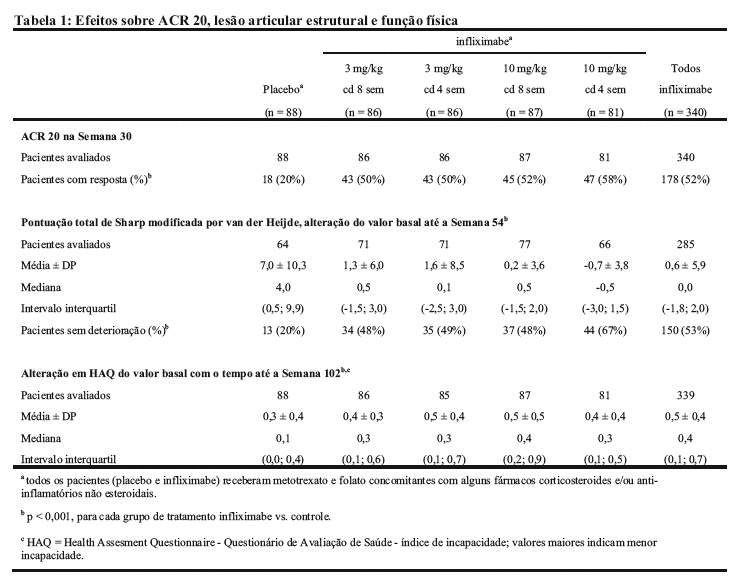
Os desfechos primários foram: redução de sinais e sintomas, conforme avaliação pelos critérios do American College of Rheumatology (ACR) (ACR 20 para ATTRACT, o marco ACR-N na Semana 54 para ASPIRE); prevenção de lesão estrutural; e a melhora na função física. A redução de sinais e sintomas foi definida como uma melhora de, pelo menos, 20% (ACR 20), em um número de articulações dolorosas e edemaciadas e em 3 dos 5 critérios seguintes: avaliação global do avaliador, avaliação global do paciente, medida funcional/incapacidade, escala analógica visual de dor e taxa da velocidade de hemossedimentação ou proteína C reativa. O ACR-N usa os mesmos critérios que o ACR 20, calculando-se a menor melhora percentual em número de articulações edemaciadas, número de articulações dolorosas e a mediana dos 5 componentes restantes da resposta ACR. A lesão estrutural articular (erosões e estreitamento articular) em mãos e pés foi medida pela alteração do valor basal na pontuação total Sharp modificada por van der Heijde (0 a 440). O Questionamento de Avaliação de Saúde (HAQ, escala de 0 a 3) foi usado para medir a alteração média na função física dos pacientes em relação à pontuação basal com o tempo até a Semana 102.
O estudo clínico ATTRACT avaliou respostas em 30 semanas (redução de sinais e sintomas), 54 semanas (a prevenção da lesão estrutural) e 102 semanas (a melhora na função física) em um estudo controlado com placebo de 428 pacientes com artrite reumatoide ativa, apesar do tratamento com metotrexato. Aproximadamente 50% dos pacientes estavam na Classe funcional III. Os pacientes receberam placebo, 3 mg/kg ou 10 mg/kg de REMICADE® nas Semanas 0, 2 e 6 e, depois, a cada 4 ou 8 semanas. Todos os pacientes estavam recebendo doses estáveis de metotrexato (mediana de 15 mg/semana) por 6 meses antes da inclusão e deveriam permanecer com doses estáveis durante todo o estudo.
Na Semana 30, uma porcentagem maior de pacientes, em todos os grupos tratados com REMICADE® , apresentou uma redução significativa de sinais e sintomas em comparação com metotrexato isolado (Tabela 1). Essa resposta foi observada a partir de 2 semanas e mantida durante as 102 semanas de tratamento (p < 0,001). Foi observada uma melhora no número de articulações edemaciadas e dolorosas, na avaliação de dor pelo paciente, na avaliação global do paciente e do avaliador sobre a doença, na rigidez matinal, fadiga e PCR em todos os grupos de REMICADE® (p < 0,05). Graus maiores de resposta clínica (ACR 50 e ACR 70) foram observados em todos os grupos de REMICADE® em 30, 54 e 102 semanas em comparação com o controle.
A prevenção da lesão estrutural articular (erosões e estreitamento de espaço articular) foi observada em todos os grupos de REMICADE® em 54 semanas (Tabela 1), que foi observada a partir de 30 semanas e mantida até 102 semanas (p < 0,001). Na população em estudo, 53% de todos os pacientes de REMICADE®, em comparação com 20% dos pacientes do grupo controle, não tinham apresentado nenhuma piora, definida como alteração 0 do valor basal na pontuação total de Sharp modificada por van der Heijde na Semana 54. Resultados semelhantes foram obtidos para as pontuações individuais (erosão e estreitamento de espaço articular). Além disso, foi também observada uma melhora maior no desempenho físico (HAQ) em 102 semanas, nos grupos tratados com REMICADE®, em comparação com os controles (Tabela 1) a partir de 54 semanas (p < 0,001).
O estudo clínico ASPIRE avaliou respostas em 54 semanas em 1.004 pacientes com artrite reumatoide ativa inicial (≤ 3 anos de duração da doença), sem tratamento prévio com metotrexato. Os pacientes randomizados tinham uma idade mediana de 51 anos com duração mediana de doença de 0,6 ano e um número mediano de articulações edemaciadas e dolorosas de 19 e 31, respectivamente. Todos os pacientes receberam metotrexato (otimizado para 20 mg/semana na Semana 8) e placebo, 3 mg/kg ou 6 mg/kg de infliximabe nas Semanas 0, 2 e 6 e a cada 8 semanas a partir daí. Nesse estudo, foram administradas infusões acima de 2 horas para as primeiras 3 infusões. A duração das infusões subsequentes pode ser reduzida para não menos que 40 minutos em pacientes que não apresentaram reações sérias à infusão.
Depois de 54 semanas de tratamento, as doses de infliximabe + metotrexato resultaram em uma melhora maior dos sinais e sintomas, de forma estatisticamente significativa, em comparação com metotrexato isolado, de acordo com a proporção de pacientes que atingiram respostas ACR 20, 50 e 70. Nos grupos infliximabe + metotrexato, 15% dos pacientes atingiram uma resposta clínica importante vs. 8% tratados apenas com metotrexato (p = 0,003).
No ASPIRE, mais de 90% dos pacientes apresentaram, pelo menos, duas radiografias para avaliação. A inibição da progressão do dano estrutural foi observada nas Semanas 30 e 54 nos grupos infliximabe + metotrexato, em comparação com metotrexato isolado. O infliximabe + metotrexato interromperam a progressão de doença articular em mais pacientes em comparação com metotrexato isolado, 97% vs. 86%, respectivamente. O infliximabe + metotrexato mantiveram um estado livre de erosão em uma proporção maior, de forma estatisticamente significativa, de pacientes do que o metotrexato isolado, 79% vs. 57%, respectivamente. Menos pacientes nos grupos infliximabe + metotrexato (48%) desenvolveram erosões em articulações não envolvidas em comparação com metotrexato isolado (59%).
Os dois grupos de tratamento com infliximabe apresentaram melhora maior, de forma estatisticamente significativa, em HAQ em relação ao valor basal ponderado ao longo do tempo até a Semana 54 em comparação com metotrexato isolado; 0,7 para infliximabe + metotrexato vs. 0,6 para metotrexato isolado (p 0,001). Não houve piora na pontuação resumida de componente mental SF-36.
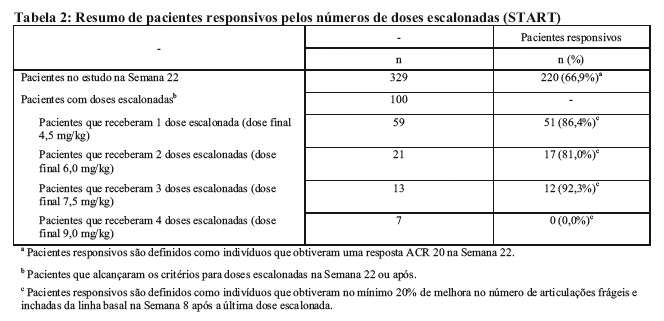
Dados que sustentam o ajuste de dose de REMICADE® em artrite reumatoide foram extraídos tanto dos estudos clínicos ATTRACT e ASPIRE, como do estudo START. O estudo clínico START foi um estudo randomizado, multicêntrico, duplo-cego, com 3 braços e estudos paralelos de segurança. Em um dos braços, o objetivo secundário foi avaliar a segurança e a eficácia da dose escalonada acima de 3 mg/kg de infliximabe em aumentos de 1,5 mg/kg para um máximo de 9 mg/kg, administrados a cada 8 semanas, em indivíduos com uma resposta inadequada para 3 mg/kg na Semana 22 e infusões subsequentes. Os resultados estão demonstrados na tabela a seguir.
- Artrite reumatoide associada à anemia
Existem evidências de que o TNF-alfa inibe a eritropoiese na doença inflamatória crônica. Em três estudos clínicos em pacientes com artrite reumatoide (ATTRACT, ASPIRE e START), 39,8% dos pacientes com hemoglobina basal < 12 g/dL tiveram aumento na hemoglobina ≥1 g/dL na Semana 22 do tratamento com infliximabe mais metotrexato vs. 19,3% dos pacientes que receberam metotrexato em monoterapia (p < 0,001). Adicionalmente, 12,1% dos pacientes tratados com infliximabe mais metotrexato tiveram aumento ≥ 2 g/dL na hemoglobina vs. 4,5% dos pacientes do braço do metotrexato em monoterapia (p < 0,001). Resultados significativos também foram encontrados nos pacientes com hemoglobina basal < 10 g/dL. A análise dos dados do estudo clínico ASPIRE mostrou que a terapia com infliximabe melhorou a anemia relacionada à artrite reumatoide, independentemente dos seus efeitos na resposta ACR 20. Além disso, demonstrou-se que, entre os respondedores ACR 20, a terapia infliximabe mais metotrexato melhorou de forma significativa a anemia em relação ao metotrexato em monoterapia. A melhora da hemoglobina está significativamente correlacionada com a melhora na função física e qualidade de vida na Semana 22.
Espondilite Anquilosante
A eficácia e a segurança de infliximabe foram avaliadas em dois estudos multicêntricos, duplo-cegos e controlados com placebo em pacientes com espondilite anquilosante ativa (pontuação BASDAI ≥ 4 e dor espinhal ≥ 4 numa escala de 1 a 10). A melhora dos sinais e sintomas foi medida utilizando o critério de resposta ASAS 20 e/ou BASDAI 50. A melhora da função física foi avaliada usando o Índice Bath de Atividade de Doença Espondilite Anquilosante (BASDAI - Bath Ankylosing Spondylitis Disease Activity Index). A melhora na variação do movimento axial foi avaliada usando o índice metrológico Bath para espondilite anquilosante (BASMI - Bath Ankylosing Spondylitis Metrology Index) e/ou medidas clínicas de expansão peitoral. A qualidade de vida relacionada à saúde foi avaliada usando o SF-36 (função física, modelo físico, dor física, saúde geral, vitalidade, funcionamento social, modelo emocional e saúde mental).
No primeiro estudo (P01522), que teve uma fase duplo-cega de 3 meses, os pacientes receberam 5 mg/kg de infliximabe ou placebo nas Semanas 0, 2, 6 (35 pacientes em cada grupo). Começando na Semana 12, os pacientes do grupo placebo foram trocados para infliximabe e, todos os pacientes, subsequentemente, receberam 5 mg/kg de infliximabe a cada 6 semanas até a Semana 54. Após o primeiro ano do estudo, 53 pacientes continuaram na extensão do braço aberto até a Semana 102. Na Semana 12, o tratamento com infliximabe resultou em melhora de sinais e sintomas, conforme avaliado pelo BASDAI, em que 57% dos pacientes tratados com infliximabe atingiram uma redução de, pelo menos, 50% em relação ao valor basal na pontuação BASDAI (a pontuação basal mediana foi de 6,5 no grupo recebendo infliximabe e de 6,3 no grupo recebendo placebo) em comparação com 9% dos pacientes do grupo placebo (p < 0,01). A melhora foi observada a partir da Semana 2 e foi mantida até a Semana 102. Houve melhora semelhante na função física, variação do movimento e qualidade de vida (SF-36).
No segundo estudo clínico (ASSERT) 279 pacientes (78 pacientes no grupo recebendo placebo e 201 no grupo infliximabe) foram randomizados para receber placebo (Grupo 1) ou 5 mg/kg de infliximabe (Grupo 2) nas Semanas 0, 2 e 6 e depois a cada 6 semanas, até a Semana 96. Na Semana 24, os pacientes recebendo placebo (Grupo 1) receberam 5 mg/kg de infliximabe a cada 6 semanas até a Semana 96. Com infusões iniciadas na Semana 36 e mantidas até a Semana 96, um paciente no Grupo 2, que tinha um BASDAI ≥ 3 em duas visitas consecutivas recebeu uma infusão de infliximabe de 7,5 mg/kg, que foi mantida a cada 6 semanas até a Semana 96.
Nas 24 semanas, o tempo para a eficácia primária, melhora dos sinais e sintomas, conforme medido pela proporção de pacientes que atingiram uma resposta ASAS 20, foi de 61% no grupo tratado com infliximabe versus 19% no grupo recebendo placebo (p < 0,001). A melhora foi observada no início da Semana 2. Uma melhora significativa nos sinais e sintomas também foi avaliada pelo BASDAI, com 51% dos pacientes tratados com infliximabe que atingiram redução de, pelo menos, 50% do valor basal na pontuação BASDAI (a pontuação basal mediana foi de 6,5 no grupo recebendo infliximabe e de 6,2 no grupo placebo), comparado com 10,7% dos pacientes recebendo placebo (p < 0,001). A melhora mediana a partir do valor basal na variação do movimento axial, conforme avaliado pelo BASMI, foi 1,0 no grupo tratado com infliximabe vs. 0,0 no grupo recebendo placebo (p = 0,019). O percentual mediano de melhora a partir do valor basal na expansão peitoral foi de 17% para o grupo tratado com infliximabe e de 0% para o grupo recebendo placebo (p = 0,037). A função física e a qualidade de vida, conforme medido pelo BASFI e pelo SF-36, também melhoraram significativamente na Semana 24.
Todas as melhoras foram mantidas até a Semana 102 e os pacientes do grupo recebendo placebo que passaram para o grupo recebendo infliximabe na Semana 24 mostraram melhoras em todas as pontuações, que foram semelhantes ao grupo tratado com infliximabe na Semana 102.
Artrite Psoriásica
A eficácia e a segurança foram avaliadas em um estudo multicêntrico, duplo-cego e controlado com placebo em 104 pacientes com artrite psoriásica poliarticular ativa.
Durante a fase duplo-cega de 16 semanas, os pacientes receberam 5 mg/kg de infliximabe ou placebo nas Semanas 0, 2, 6 e 14 (52 pacientes em cada grupo). A partir da Semana 16, os pacientes do grupo placebo passaram para o grupo recebendo infliximabe e todos os pacientes, subsequentemente, receberam 5 mg/kg de infliximabe a cada 8 semanas até a Semana 46.
O tratamento com infliximabe resultou em melhora dos sinais e sintomas, avaliados pelo critério ACR, com 65% dos pacientes tratados com infliximabe atingindo ACR 20 na Semana 16, comparado com 10% dos pacientes tratados com placebo (p < 0,01). A resposta foi semelhante, independentemente do uso concomitante de metotrexato. Foi observada melhoria (ACR 20 e 50) já na Semana 2 e mantida até a Semana 50 (ACR 20, 50 e 70). Na Semana 16, o grupo tratado com infliximabe apresentava um Escore de Atividade da Doença (DAS 28) médio de 3,0 (um DAS 28 < 3,2 é considerado indicativo de baixa atividade da doença), enquanto nenhuma alteração foi observada no grupo de tratados com placebo (p < 0,01). No final da Semana 50, ambos os grupos de indivíduos com escore DAS 28 semelhantes, indicando que os indivíduos tratados com infliximabe mantiveram escore DAS 28 ao longo do estudo, enquanto os indivíduos tratados com placebo tiveram uma redução no DAS 28, apenas após o cruzamento de tratamento com infliximabe. O total de pacientes que tiveram resposta ao DAS 28 foi de 88,5% no grupo infliximabe na Semana 16 em comparação com 25% no grupo placebo (p < 0,01). O critério de Resposta para Artrite Psoriásica (PsARC) apresentou melhora rápida. Na Semana 2, 56% dos indivíduos do grupo infliximabe apresentaram melhora em comparação com 17% dos indivíduos no grupo placebo (p < 0,01). Na Semana 16, os resultados mostraram 75% de resposta nos indivíduos tratados com infliximabe comparados com 21% em indivíduos do grupo placebo (p < 0,01). Reduções nos parâmetros da característica de atividade periférica da artrite psoriásica (como número de articulações edemaciadas, número de articulações dolorosas, dactilites e presença de entesopatia) foram observadas nospacientes tratados com infliximabe. Em um subgrupo de pacientes com valor basal de Índice de Área e Gravidade de Psoríase (PASI) ≥ 2,5, a melhora acentuada no PASI foi alcançada na Semana 16, com 68% (15/22) dos pacientes tratados com infliximabe atingindo, pelo menos, 75% de melhoria do valor basal vs. 0% (0/16) dos pacientes tratados com placebo a melhoria foi mantida até a Semana 50. Os pacientes que receberam infliximabe apresentaram melhora na função física avaliada por HAQ (alteração média do início até a Semana 16 de 0,6 vs. 0 para os pacientes tratados com placebo). A melhora foi mantida até a Semana 50.
Psoríase
A eficácia de infliximabe foi avaliada em dois estudos multicêntricos, randomizados, duplo-cegos: SPIRIT eEXPRESS. Os pacientes dos dois estudos apresentavam psoríase em placas (Área de Superfície Corporal [BSA] ≥ 10% e Escore do Índice de Área e Gravidade de Psoríase [PASI] ≥ 12). O desfecho primário nos dois estudos foi a porcentagem de pacientes que atingiram melhora ≥ 75% de PASI na Semana 10 em relação ao valor basal. Os que apresentaram resposta acentuada foram identificados como pacientes que atingiram melhora de PASI ≥ 90% em relação ao valor basal.
O SPIRIT avaliou a eficácia da terapia de indução com infliximabe em 249 pacientes com psoríase em placa que tinham recebido anteriormente PUVA ou terapia sistêmica. Os pacientes receberam 3 ou 5 mg/kg de infliximabe ou infusões de placebo nas Semanas 0, 2 e 6. Os pacientes com pontuação de PGA ≥ 3 foram elegíveis a receber uma infusão adicional do mesmo tratamento na Semana 26.
A proporção de pacientes com melhora de PASI ≥ 75% em relação ao valor basal (PASI 75) na Semana 10 foi de 71,7% no grupo com 3 mg/kg de infliximabe, 87,9% no grupo com 5 mg/kg de infliximabe e 5,9% no grupo placebo (p < 0,001 para cada grupo infliximabe vs. placebo comparativo). Na Semana 10, uma proporção significativamente maior de pacientes tratados com infliximabe (3 mg/kg: 45,5%; 5 mg/kg: 57,6%) atingiu uma resposta acentuada (melhora ≥ 90% do PASI em relação ao valor basal), em comparação com os pacientes tratados com placebo (2,0%). No grupo 3 mg/kg, 60,6% dos pacientes mantiveram a resposta até a Semana 14 e 75,3% dos pacientes no grupo 5 mg/kg mantiveram a resposta até a Semana 18. Na Semana 26, vinte semanas depois da última dose de indução, 30% dos pacientes no grupo 5 mg/kg e 13,8% no grupo 3 mg/kg estavam no grupo de resposta PASI 75, sugerindo a necessidade de uma terapia de manutenção.
A qualidade de vida relacionada à saúde foi avaliada com o DLQI. O DLQI basal mediano foi de 12. A alteração mediana na Semana 10 em relação ao valor basal de DLQI foi de -8,0 e -10,0 para os grupos infliximabe 3 mg/kg e 5 mg/kg, respectivamente, em comparação com 0,0 no grupo placebo (p < 0,001 para todos com infliximabe vs. placebo comparativo), demonstrando uma melhora substancial na qualidade de vida para pacientes que receberam terapia com infliximabe.
O EXPRESS avaliou a eficácia da terapia de indução e manutenção com infliximabe em 378 pacientes com psoríase em placa que eram candidatos a fototerapia ou terapia sistêmica. Os pacientes receberam 5 mg/kg de infliximabe ou infusões de placebo nas Semanas 0, 2 e 6 seguidas por terapia de manutenção a cada 8 semanas até a Semana 22 no grupo placebo e até a Semana 46 no grupo infliximabe. Na Semana 24, o grupo placebo cruzou para terapia de indução com infliximabe (5 mg/kg) seguida por terapia de manutenção com infliximabe (5 mg/kg).
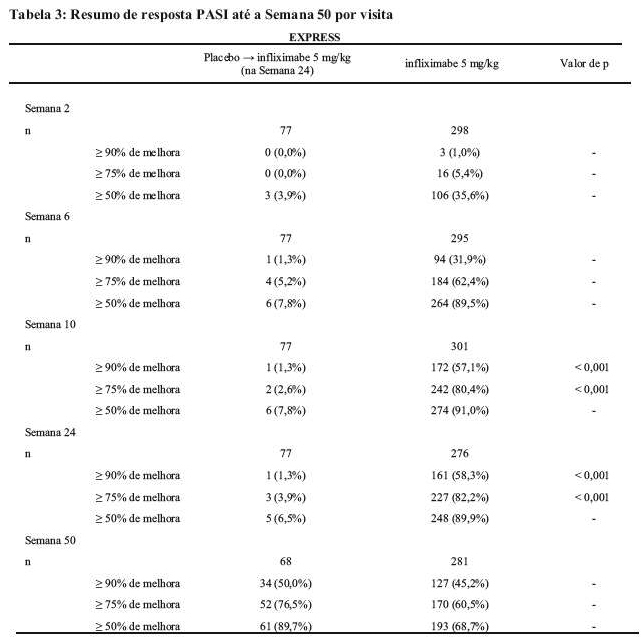
No EXPRESS, a BSA basal mediana foi de 29%, o escore PASI basal mediana foi de 21,1 e a maioria dos pacientes (89,9%) apresentava um escore de PGA moderada, acentuada ou grave. Um total de 71,4% dos pacientes tinha recebido terapia prévia com PUVA, metotrexato, ciclosporina ou acitretina. Na Semana 10, 80,4% do grupo infliximabe atingiram resposta PASI 75 em comparação com 2,6% no grupo placebo (p < 0,001). O tempo mediano para atingir PASI 75 ficou entre 2 e 6 semanas. A melhora de PASI foi constante entre os subgrupos, definidos por demografia basal, características clínicas da doença e histórico de medicação para psoríase. Respostas acentuadas (PASI 90) na Semana 10 foram atingidas por 57,1% do grupo infliximabe em comparação com 1,3% no grupo placebo (p < 0,001). A resposta foi mantida até a Semana 24, período controlado com placebo. As taxas de resposta PASI até a Semana 50 são apresentadas na Tabela 3.
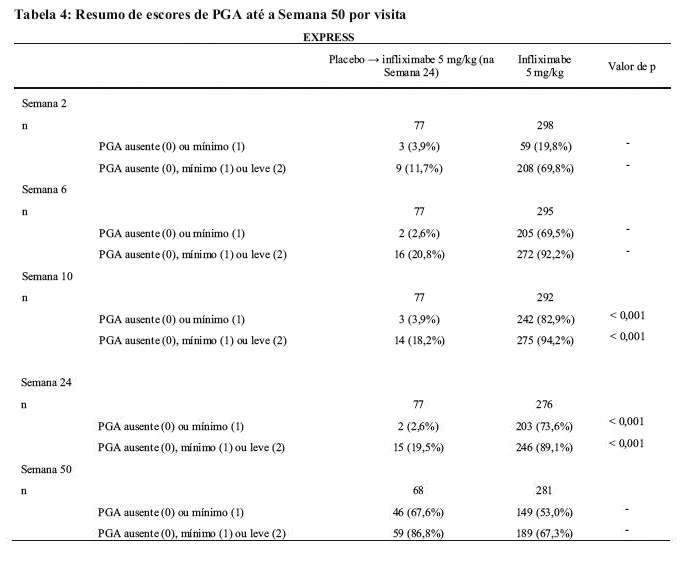
Na Semana 10, 82,9% dos pacientes do grupo infliximabe atingiram pontuação de PGA de mínimo ou ausente em comparação com 3,9% do grupo placebo (p < 0,001). Os escores de PGA nas Semanas 6, 10, 24 e 50 são apresentados na Tabela 4.
O valor basal mediano de DLQI foi de 12,5. Os valores basais médios foram de 45,6 para componente físico de SF-36 e 45,7 para o componente mental. A qualidade de vida melhorou significativamente em comparação com placebo nas Semanas 10 e 24, quando avaliada por DLQI e SF-36.
O escore basal mediano de NAPSI para psoríase ungueal foi de 4 e o número mediano de unhas envolvidas com psoríase foi de 10. Os pacientes tratados com infliximabe apresentaram uma melhora evidente na psoríase ungueal, em relação ao período basal, em comparação com pacientes tratados com placebo, conforme aferição pelo escore de NAPSI e pela redução do número de unhas envolvidas.
Doença de Crohn em pacientes adultos
A segurança e a eficácia de doses únicas e múltiplas de REMICADE® foram avaliadas em dois estudos duplocegos, randomizados e controlados com placebo em pacientes com doença de Crohn ativa moderada a grave[Índice de Atividade de Doença de Crohn (CDAI) ≥ 220 ≤ 400], com uma resposta inadequada às terapias convencionais prévias. O uso concomitante de doses estáveis de terapias convencionais foi permitido e 92% dos pacientes continuaram a receber essas medicações.
No estudo clínico de dose única com 108 pacientes, 22/27 (81%) dos pacientes tratados com REMICADE® 5 mg/kg atingiram uma resposta clínica (redução do CDAI ≥ 70 pontos) vs. 4/25 (16%) dos tratados com placebo (p < 0,001). Também na Semana 4, 13/27 (48%) dos pacientes tratados com REMICADE® atingiram uma remissão clínica (CDAI < 150) vs. 1/25 (4%) dos tratados com placebo.
No estudo clínico com doses múltiplas, 573 pacientes receberam 5 mg/kg na Semana 0 e, então, foram randomizados para um dos três grupos de tratamento: o grupo de manutenção com placebo recebeu placebo nas Semanas 2 e 6 e, depois, a cada 8 semanas; o grupo de manutenção 5 mg/kg recebeu 5 mg/kg nas Semanas 2 e 6 e, depois, a cada 8 semanas e o grupo de manutenção 10 mg/kg recebeu 5 mg/kg nas Semanas 2 e 6 e, depois, 10 mg/kg a cada 8 semanas. Os pacientes em resposta na Semana 2 foram randomizados e analisados separadamente daqueles que não apresentaram resposta.
Na Semana 2, 58% (335/573) dos pacientes apresentavam resposta clínica (redução em CDAI ≥ 25% e ≥ 70 pontos). Uma proporção significativamente maior de pacientes nos grupos de manutenção 5 mg/kg e 10 mg/kg atingiu remissão clínica na Semana 30, em comparação com os pacientes no grupo de manutenção com placebo. Os pacientes nos grupos de manutenção com infliximabe apresentaram um tempo significativamente maior até a perda de resposta do que os pacientes no grupo de manutenção com placebo (p < 0,001). O tempo mediano até a perda de resposta foi de 46 semanas, no grupo de tratamento de manutenção com infliximabe combinado vs. 19 semanas no grupo de manutenção com placebo. Os pacientes que atingiram uma resposta e, subsequentemente, a perderam, foram candidatos a receber infliximabe em uma dose 5 mg/kg maior do que aquela para a qual tinham sido randomizados. Oitenta e nove por cento (50/56) dos pacientes que deixaram de apresentar a resposta clínica com a dose de manutenção de 5 mg/kg de infliximabe a cada oito semanas responderam a uma infusão de 10 mg/kg de infliximabe.
Na Semana 30, foi observada uma melhora significativa das medidas de qualidade de vida no IBDQ (Inflammatory Bowel Disease Questionnaire - Questionário de Doença Inflamatória Intestinal) e pontuação SF36 (p < 0,001) nos pacientes tratados com REMICADE® .
Para pacientes que receberam corticosteroides no período basal, a proporção desses pacientes em remissão clínica e que não estavam recebendo corticosteroides na Semana 30 foi de 31% para o grupo de manutenção 5 mg/kg e 37% para o grupo de manutenção 10 mg/kg, em comparação com 11% dos pacientes no grupo de manutenção com placebo (p = 0,001 para os grupos de manutenção 5 e 10 mg/kg). A dose mediana de corticosteroides no período basal (20 mg/dia) foi reduzida para 10 mg/dia no grupo de manutenção com placebo e 0 mg/dia nos grupos de manutenção com infliximabe combinado na Semana 30, indicando que, pelo menos 50% dos pacientes do grupo de manutenção com infliximabe, conseguiram descontinuar o uso de esteroides.
Na Semana 10, uma proporção significativamente maior de pacientes nos grupos de manutenção com infliximabe combinados (31%) teve cicatrização da mucosa em comparação com os do grupo placebo (0%, p = 0,010). Os resultados foram semelhantes na Semana 54.
A segurança e eficácia também foram avaliadas em um estudo randomizado, duplo-cego, controlado com placebo, incluindo 94 pacientes com doença de Crohn fistulizante, com fístulas de pelo menos 3 meses de duração. Trinta e um desses pacientes foram tratados com REMICADE® 5 mg/kg. Aproximadamente 93% dos pacientes tinham recebido antibiótico ou terapia imunossupressora anteriormente.
Foi permitido o uso concomitante de doses estáveis de terapias convencionais e 83% dos pacientes continuaram a receber pelo menos uma dessas medicações. Os pacientes receberam três doses de placebo ou REMICADE® nas Semanas 0, 2 e 6 e foram acompanhados durante 26 semanas. O desfecho primário foi a proporção de pacientes que apresentou uma resposta clínica definida como uma redução ≥ 50% em relação ao valor basal em número de fístulas drenadas com uma suave compressão realizada em pelo menos duas visitas consecutivas (com intervalo de 4 semanas) sem aumento da medicação ou cirurgia para doença de Crohn.
Sessenta e oito por cento (21/31) dos pacientes tratados com REMICADE® que receberam um esquema de administração de 5 mg/kg, atingiram uma resposta clínica vs. 26% (8/31) dos tratados com placebo (p = 0,002). O tempo mediano para o início da resposta no grupo tratado com REMICADE® foi de 2 semanas. A duração mediana de resposta foi de 12 semanas. Além disso, o fechamento de todas as fístulas foi atingido em 55% dos pacientes tratados com REMICADE® em comparação com 13% dos tratados com placebo (p = 0,001).
A segurança e eficácia de infusões repetidas de infliximabe em pacientes com doença de Crohn fistulizante foram avaliadas em um estudo clínico de um ano. Um total de 306 pacientes recebeu 3 doses de 5 mg/kg de infliximabe nas Semanas 0, 2 e 6. Entre os pacientes randomizados no período basal, 87% tinham fístulas perianais, 14% fístulas abdominais e 9% fístulas retovaginais. O escore CDAI mediano foi de 180. Cento e noventa e cinco pacientes que responderam às 3 doses (para definição de resposta, ver descrição de desfecho primário para o estudo apresentado) foram randomizados na Semana 14 para receber placebo ou 5 mg/kg de infliximabe a cada 8 semanas até a Semana 46. Na Semana 14, 65% (177/273) dos pacientes randomizados apresentaram respostas às fístulas. Os pacientes randomizados para manutenção com infliximabe apresentaram um tempo significativamente mais longo até a perda de resposta para fístulas em comparação com o grupo de manutenção com placebo (p < 0,001). O tempo mediano até a perda da resposta foi > 40 semanas no grupo infliximabe, em comparação com 14 semanas no grupo placebo. Na Semana 54, 38% (33/87) dos pacientes tratados com REMICADE® não apresentavam fístulas com drenagem, em comparação com 22% (20/90) dos pacientes tratados com placebo (p = 0,02). O grupo infliximabe apresentou maior melhora no escore CDAI em relação ao valor basal em comparação com placebo (p = 0,04). Os pacientes que atingiram uma resposta de fístula e, subsequentemente, perderam-na, foram candidatos a receber terapia de manutenção com REMICADE® em uma dose 5 mg/kg maior que aquela para a qual haviam sido randomizados. Dos pacientes em manutenção com placebo, 66% (25/38) responderam a 5 mg/kg de REMICADE® e 57% (12/21) daqueles em manutenção com REMICADE® responderam a 10 mg/kg. Em comparação à manutenção com placebo, pacientes em manutenção com REMICADE® tiveram uma tendência a um número menor de internações.
Doença de Crohn ativa em pacientes pediátricos
A segurança e a eficácia de doses únicas e múltiplas de REMICADE® foram avaliadas em um estudo de Fase II randomizado, de dose única e multicêntrico em 21 pacientes pediátricos com doença de Crohn ativa e em um estudo Fase III randomizado, de doses múltiplas, aberto e multicêntrico em 112 pacientes pediátricos com doença de Crohn (estudo REACH).
No estudo Fase II de dose única em 21 pacientes (11 a 17 anos, idade média de 15 anos), todos os pacientes atingiram resposta clínica (diminuição em CDAI ≥ 70 pontos ou diminuição em PCDAI ≥ 10) em algum ponto no período de 20 semanas após a dose única de infliximabe e remissão clínica (definida como uma redução na pontuação CDAI modificada para abaixo de 150 pontos ou uma redução no PCDAI para menos de 10) foi atingida por 10 (47,6%) pacientes. Entre as 3 doses administradas (1, 5 ou 10 mg/kg), os grupos de tratamento com 5 mg/kg e 10 mg/kg apresentaram uma maior proporção de pacientes atingindo remissão clínica (16,7% no grupo de tratamento com 1 mg/kg de infliximabe comparado a 57,1% e 62,5% nos grupos de tratamento com 5 mg/kg e 10 mg/kg de infliximabe, respectivamente). Todos os 7 pacientes que apresentavam doença fistulizante tiveram suas fístulas fechadas em pelo menos 1 visita de avaliação (8 semanas).
Em um estudo clínico Fase III de doses múltiplas (REACH), 112 pacientes (6 a 17 anos, idade média de 13 anos) foram tratados com 5 mg/kg de infliximabe nas Semanas 0, 2 e 6. Os pacientes que, de acordo com a avaliação do investigador, responderam clinicamente na Semana 10, foram randomizados e receberam 5 mg/kg de infliximabe a cada 8 ou 12 semanas, como um esquema de tratamento de manutenção. Quando havia perda de resposta durante o tratamento de manutenção, permitia-se o cruzamento para uma dose maior ou para um intervalo menor de dosagem.
No estudo clínico REACH, a resposta clínica na Semana 10 foi de 88,4% (99/112) em comparação com 66,7% (128/192) em adultos (ACCENT 1). Similarmente, a proporção de indivíduos que atingiram remissão clínica na Semana 10 foi de 58,9% (66/112) em comparação com 39,1% (75/192) em adultos (ACCENT 1).
Na Semana 30, a proporção de indivíduos em resposta clínica foi significativamente maior no grupo de tratamento de manutenção a cada 8 semanas (73,1%, 38/52) do que no grupo de tratamento de manutenção a cada 12 semanas (47,1%, 24/51; p = 0,007). Na Semana 54, a proporção de indivíduos em resposta clínica foi, também, significativamente maior nos indivíduos do grupo de tratamento de manutenção a cada 8 semanas (63,5%, 33/52) que no grupo de tratamento de manutenção a cada 12 semanas (33,3%, 17/51; p = 0,002).
Na Semana 30, a proporção de pacientes em remissão clínica foi significativamente maior no grupo de tratamento de manutenção a cada 8 semanas (59,6%, 31/52) que no grupo de tratamento de manutenção a cada 12 semanas (35,3%, 18/51; p = 0,013). Na Semana 54, a proporção de pacientes em remissão clínica foi, também, significativamente maior nos pacientes no grupo de tratamento de manutenção a cada 8 semanas (55,8%, 29/52) que no grupo de tratamento de manutenção a cada 12 semanas (23,5%, 12/51; p < 0,001).
No estudo clínico REACH, a alteração em relação aos valores basais no uso diário médio de corticosteroide foi significativa nas Semanas 10, 30 e 54 (p < 0,001). Nos pacientes em tratamento com corticosteroides no período basal no estudo clínico REACH, a remissão clínica atingida sem corticosteroides na Semana 30 foi de 45,8% no grupo de tratamento de manutenção a cada 8 semanas e 33,3% no grupo de tratamento de manutenção a cada 12 semanas. Na Semana 54, 45,8% dos pacientes do grupo de tratamento de manutenção a cada 8 semanas e 16,7% dos indivíduos no grupo de tratamento de manutenção a cada 12 semanas encontravam-se em remissão clínica e não estavam recebendo corticosteroides.
A qualidade de vida (QV) foi avaliada com o escore do IMPACT III (um questionário de QV desenvolvido e validado especificamente para pacientes pediátricos com doença inflamatória intestinal). Foi aplicado apenas em indivíduos da América do Norte. As alterações médias (uma alteração negativa indica melhora) em relação aos valores basais do escore do IMPACT III nas Semanas 10, 30 e 54 (-22,9, -21,1 e -24,3, respectivamente) foram todas significativas (p < 0,001).
A pontuação z de altura é uma quantificação do desvio da altura do paciente pediátrico em relação àquela esperada para a população da mesma idade e sexo. Na população estudada, a pontuação z mediana no período basal foi de -1,6. As alterações medianas em relação aos valores basais nas pontuações z foram de 0,3 e 0,4 para a Semana 30 e Semana 54, respectivamente. As pontuações z foram significativamente melhores em relação ao período basal, tanto na Semana 30 (p < 0,001) quanto na Semana 54 (p < 0,001).
Colite ou Retocolite Ulcerativa
A segurança e a eficácia do infliximabe foram avaliadas em dois estudos clínicos (ACT 1 e ACT 2) randomizados, duplo-cegos e controlados com placebo, que incluíram pacientes adultos com colite ou retocolite ulcerativa moderada a gravemente ativa (pontuação Mayo 6 a 12; subpontuação de endoscopia ≥ 2) com resposta inadequada às terapias convencionais [corticosteroides orais, aminossalicilatos e/ou imunomoduladores (6-MP, AZA)]. Foram permitidas doses estáveis concomitantes de aminossalicilatos orais, corticosteroides e/ou agentes imunomoduladores. Nos dois estudos, os pacientes foram randomizados para receber placebo, 5 mg/kg de infliximabe ou 10 mg/kg de infliximabe nas Semanas 0, 2, 6, 14 e 22. A redução de corticosteroides foi permitida depois da Semana 8.
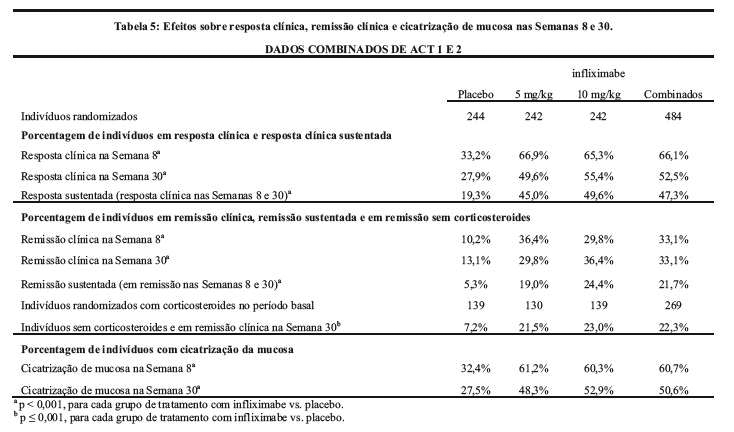
Nos dois estudos, uma porcentagem significativamente maior de pacientes nos grupos infliximabe apresentava resposta clínica e remissão clínica na Semana 8 comparada com placebo. Além disso, tanto no ACT 1 quanto no ACT 2, uma proporção significativamente maior de pacientes tratados com 5 mg/kg ou 10 mg/kg de infliximabe apresentou resposta clínica e remissão clínica na Semana 30 em comparação ao tratamento com placebo. Além disso, a proporção de pacientes com resposta sustentada (isto é, com resposta clínica na Semana 8 e também na Semana 30) nos grupos infliximabe foi de pelo menos o dobro daquela do grupo placebo. Os resultados das Semanas 8 e 30 são mostrados na Tabela 5.
Dos pacientes tratados com corticosteroides no período basal, uma proporção significativamente maior de pacientes nos grupos tratados com infliximabe estava em remissão clínica na Semana 30 e com capacidade de descontinuar os corticosteroides comparada com aqueles tratados com placebo (22,3% vs. 7,2%, respectivamente, ver Tabela 5).
Além disso, nas Semanas 8 e 30, uma proporção significativamente maior de pacientes nos grupos com 5 mg/kg e 10 mg/kg nos estudos ACT 1 e ACT 2 atingiu cicatrização da mucosa em comparação com os pacientes que receberam placebo. A proporção de indivíduos com cicatrização de mucosa foi semelhante entre os 2 grupos de dose com infliximabe nos dois estudos (ver Tabela 5).
O infliximabe melhorou a qualidade de vida, o que foi confirmado por melhora estatística e clinicamente significativa nas medidas específicas de doença, IBDQ e também no formulário abreviado de 36 itens SF-36. Entre o período basal e a Semana 30, nos dados agrupados dos estudos ACT 1 e ACT 2, o número médio de internações foi 50% menor no grupo de tratamento combinado com infliximabe do que no grupo placebo (9 vs.
18 internações por 100 indivíduos, p = 0,005). Nenhuma diferença relevante foi observada entre os grupos de tratamento com 5 mg/kg e 10 mg/kg de infliximabe.
Referências bibliográficas:
1. Reich K, Nestle FO, Papp K, Ortonne JP, Evans R, Guzzo C, Li S, Dooley LT, Griffiths CEM, for the EXPRESS study investigators. Infliximab induction and maintenance therapy for moderate-to-severe psoriasis: a phase III, multicentre, double-blind trial. Lancet 2005;
366: 1367-74.
2. Gottlieb AB, Evans R, Li S, Dooley LT, Guzzo CA, Baker D, Bala M, Marano CW, and Alan Menter A. Infliximab induction therapy for patients with severe plaque-type psoriasis: A randomized, double-blind, placebo-controlled trial. J Am Acad Dermatol 2004;51(4):534-42.
3. Rahman MU, Strusberg I, Geusens P, Berman A, Yocum D, Baker D, Wagner C, Han J. and Westhovens R. Double-blinded infliximab dose escalation in patients with rheumatoid arthritis Ann Rheum Dis 2007; published online 1 Jun 2007. doi: 10.1136/ard.2006.065995.
4. Antoni CE, Kavanaugh A, Kirkham B, Tutuncu Z, Burmester GR, Schneider U, Furst DE, Molitor J, Keystone E, Gladman D, Manger B, Wassenberg S, Weier R, Wallace DJ,Weisman MH, Kalden JR, and Smolen J. Sustained Benefits of Infliximab Therapy for Dermatologic and Articular Manifestations of Psoriatic Arthritis. Results From the Infliximab Multinational Psoriatic Arthritis Controlled Trial (IMPACT). Arthritis & Rheumatism, 2005;52(4):1227-1236. DOI 10.1002/art.20967
5. Antoni C, Krueger GG, de Vlam K, Birbara C, Beutler A, Guzzo C, Zhou B, Dooley LT. and Kavanaugh A. Infliximab improves signs and symptoms of psoriatic arthritis: results of the IMPACT 2 trial. Ann. Rheum. Dis published online 24 Feb 2005; doi:10.1136/ard.2004.032268
6. Smolen JS, van der Heijde DMFM, St.Clair EW, Emery P, Bathon JM, Keystone E, Maini RN, Kalden JR, Schiff M, Baker D, Han C, Han J, and Bala M, for the Active-Controlled Study of Patients Receiving Infliximab for the Treatment of Rheumatoid Arthritis of Early Onset (ASPIRE) Study Group. Predictors of Joint Damage in Patients With Early Rheumatoid Arthritis Treated With High-Dose Methotrexate With or Without Concomitant Infliximab. Results From the ASPIRE Trial. Arthritis & Rheumatism, 2006;54(3): 702-710. DOI 10.1002/art.21678
7. Breedveld FC, Han C, Bala M, van der Heijde D, Baker D, Kavanaugh AF, Maini RN, Lipsky PE. Association between baseline radiographic damage and improvement in physical function after treatment of patients with rheumatoid arthritis. Ann Rheum Dis 2005;64:52-55. doi: 10.1136/ard.2003.017160.
8. Hanauer SB, Feagan BG, Lichtenstein GR, Mayer LF, Schreiber S, Colombel JF, Rachmilewitz D, Wolf DC, Olson A, Bao W, Rutgeerts P, and The ACCENT I Study Group. Maintenace infliximab for Chron's disease: The ACCENT I randomized trial. Lancet, 2002;359:1541-1549.
9. Sands BE, Anderson FH, Bernstein CN, Chey WY, Feagan BG, Fedorak BN, Kamm MA, Korzenik JR, Lashner BA, Onken JE, Rachmilewitz D, RutgeertsP, Wild G, Wolf DC, Marsters PA, Travers SB, MBlank MA, and van Deventer SJ. Infliximab Maintenance Therapy for Fistulizing Crohn's Disease. N Engl J Med 2004;350:876-85.
10. Rutgeerts P; Sandborn WJ.; Feagan BG.; Reinisch W; Olson A; Johanns J; Travers S; Rachmilewitz D; Hanauer SB.; Lichtenstein GR.; de Villiers W.S.; Present D; Sands BE.; Colombel JF. Infliximab for Induction and Maintenance Therapy for Ulcerative Colitis. N Engl J Med 2005;353: 2462-76.
11. Braun, J.; Brandt, J.; Listing, J.; Zink, A.; Alten, R.; Golder, W.; Gromnica-Ihle, E.; Kellner, H.; Krause, A.; Schneider, M; Sörensen, H.; Zeiddler, H.; Thriene, W.; Sieper, J. Treatment ofactive ankylosing spondylitis with infliximab: a randomised controlled multicentre trail. The Lancet. n.359, p.1187-93, 2002.
12. Braun, J.; Brandt, J.; Listing, J.; Zink, A.; Alten, R.; Burmester, G.; Golder, W.; Gromnica-Ihle, E.; Kellner, H.; Schneider, M; Sörensen, H.; Zeiddler, H.; Reddig, J.; Sieper, J. Long-term efficacy and safety of infliximab in the treatment of ankylosing spondylitis. Arthritis & Rheumatism. v.48,n.8, p.2224-2233, 2003.
13. Braun, J.; Brandt, J.; Listing, J.; Zink, A.; Alten, R.; Burmester, G.; Gromnica-Ihle, E.; Kellner, H.; Schneider, M; Sörensen, H.; Zeiddler, H.; Sieper, J. Two year maintenance of efficacy and safety of infliximab in thte treatent of ankylosing spondylitis. Annals of the Rheumatic Diseases. v.64, p.229-234, 2005.
14. Heijde, D.; Dijkmans, B.; Geusens, P.; Sieper, J.; DeWoody, K.; Williamson, P.; Braun, J. and the Ankylosing Spondylitis Study for the Evaluation of Recombinant Infliximab Therapy Study Group. Efficacy and safety of infliximab in patients with ankylosing spondylitis. Arthritis & Rheumatism. v.52, n.2, p.582-591, 2005.
15. Braun, J.; Deodhar, A.; Dijkmans, B.; Geusens, P.; Sieper, J.; Williamson, P.; Xu, W.; Visvanathan,S.; Baker, D.; Goldstein, N.; Heijde and the Ankylosing Spondylitis Study for the Evaluation of Recombinant Infliximab Therapy Study Group. Efficacy and Safety of Infliximab in patientes with ankylosing spondylitis over a two-year period. Arthritis & Rheumatism. v.59, n.9, p.1270-1278, 2008.
Caract. farmacológicas.
Ação: O infliximabe é um anticorpo monoclonal quimérico humano-murino, que se liga com alta afinidade a formas solúveis e de membranas do Fator de Necrose Tumoral alfa (TNF-alfa), mas não à linfotoxina alfa (TNF-beta). O infliximabe inibe a atividade funcional do TNF-alfa em vários tipos de bioensaios in vitro. O infliximabe previne o surgimento de doenças em camundongos transgênicos que desenvolvem poliartrite, resultante da expressão constitutiva do TNF-alfa humano e, quando administrado após o início da doença, promove a cura de articulações com erosão. In vivo, o infliximabe forma rapidamente complexos estáveis com o TNF-alfa humano, um processo paralelo à perda de bioatividade do TNF-alfa.
Foram encontradas concentrações elevadas de TNF-alfa nas articulações de pacientes com artrite reumatoide, que se correlacionam com atividade elevada da doença, bem como aumentos nas concentrações de TNF-alfa nos tecidos/fluidos articulares e em lesões psoriásicas de pele nos pacientes com artrite psoriásica. Na artrite reumatoide, o tratamento com REMICADE® reduz a infiltração de células inflamatórias em áreas inflamadas da articulação, bem como a expressão de moléculas mediadoras da adesão celular, quimiotaxia e degradação tecidual. Depois do tratamento com REMICADE®, os pacientes apresentaram níveis reduzidos de interleucina 6 (IL-6) sérica e proteína C reativa, em comparação ao período basal. Os linfócitos do sangue periférico não apresentaram redução significativa em número ou respostas proliferativas à estimulação mitogênica in vitro, quando comparados a células de pacientes não tratados. Nos pacientes com psoríase, o tratamento com infliximabe resultou na diminuição da inflamação epidérmica e normalização da diferenciação queratinocítica na placa psoriásica. Na artrite psoriásica, o tratamento com REMICADE® resultou na redução do número de células-T e vasos sanguíneos na sinóvia e pele psoriásica, assim como redução de macrófagos na sinóvia.
Propriedades Farmacodinâmicas: A avaliação histológica de biópsias de cólon obtida antes e 4 semanas após a administração de REMICADE® revelou uma redução substancial do TNF-alfa detectável. O tratamento de pacientes com a doença de Crohn com REMICADE® também foi associado com redução substancial do marcador inflamatório sérico comumente elevado, a proteína C reativa (PCR). O número total de leucócitos no sangue periférico foi muito pouco afetado em pacientes tratados com REMICADE®, embora alterações em linfócitos, monócitos e neutrófilos refletissem desvios dentro de intervalos normais. Células mononucleares de sangue periférico (PBMCs - peripheral blood mononuclear cells) de pacientes tratados com REMICADE®, não apresentaram diminuição das respostas proliferativas a estímulos em comparação com pacientes não tratados. Não foram observadas alterações substanciais na produção de citocinas pelas PBMCs estimuladas, depois do tratamento com REMICADE®. A análise de células mononucleares da lâmina própria obtida por biópsia da mucosa intestinal mostrou que o tratamento com REMICADE® provoca redução do número de células capazes de expressar o TNF-alfa e a gamainterferona. Estudos histológicos adicionais forneceram evidência de que o tratamento com REMICADE® reduz a infiltração de células inflamatórias em áreas afetadas do intestino e a presença de marcadores inflamatórios nesses sítios.
Propriedades Farmacocinéticas: Infusões intravenosas únicas de 1, 3, 5, 10 ou 20 mg/kg de REMICADE® promoveram aumentos proporcionais à dose na concentração sérica máxima (Cmáx) e na área sob a curva de concentração vs. tempo (AUC). O volume de distribuição em estado de equilíbrio (Vd médio de 3 a 4,1 litros) não foi dependente da dose administrada e indica que REMICADE® é distribuído predominantemente no compartimento vascular. Não foi observada dependência da farmacocinética em relação ao tempo. As vias de eliminação para REMICADE® não foram caracterizadas. Não foram observadas diferenças importantes na depuração ou volume de distribuição em subgrupos de pacientes distribuídos por idade, peso ou função hepática ou renal. Não foram observadas diferenças significativas nos parâmetros farmacocinéticos de dose única entre pacientes pediátricos e adultos com doença de Crohn.
Em doses únicas de 3, 5 e 10 mg/kg, os valores farmacocinéticos médios para a Cmáx foram de 77, 118 e 277 mcg/mL, respectivamente. A mediana da meia-vida nessas doses variou de 8 a 9,5 dias. Na maioria dos pacientes, o infliximabe foi detectado no soro durante, pelo menos, 8 semanas após uma única infusão. Um pequeno acúmulo de infliximabe foi observado no soro após a segunda dose do esquema habitual de 3 doses e não houve acúmulo clinicamente relevante posteriormente. Na maioria dos pacientes com doença de Crohn fistulizante, o infliximabe foi detectado no soro por 12 semanas (variação de 4 a 28 semanas) após a administração do esquema de dose.
Toxicologia pré-clínica: O infliximabe não produziu anticorpos com TNF-alfa em espécies que não humanos e chimpanzés. Contudo, dados de segurança pré-clínicos convencionais com infliximabe são limitados. Em estudos de toxicidade reprodutiva conduzidos em camundongos, utilizando anticorpos análogos que, seletivamente, inibem a atividade funcional do TNF-alfa, não houve indicação de prejuízo da função reprodutiva, toxicidade materna, embriotoxicidade ou teratogenicidade. Estudos de longa duração não foram efetuados para avaliar o potencial carcinogênico do infliximabe. Estudos de tumorigenicidade em camundongos deficientes de TNF-alfa, não demonstraram aumento nos tumores quando avaliados com indicadores de tumor e/ou promotores.
Contraindicações.
Este medicamento é contraindicado para uso por pacientes com:
- hipersensibilidade conhecida a qualquer componente do produto ou a proteínas murinas;
- infecções graves como tuberculose, sepse, abscessos e infecções oportunistas; e
- insuficiência cardíaca moderada ou grave (NYHA - New York Heart Association - de classe funcional III/IV). Nestes pacientes, o tratamento com REMICADE® na dose de 10 mg/kg foi associado a uma incidência aumentada de morte e hospitalização devido à piora da insuficiência cardíaca. Portanto, REMICADE® é contraindicado em pacientes com insuficiência cardíaca moderada a grave em doses maiores que 5 mg/kg.
Advertências e precauções.
A fim de melhorar a rastreabilidade dos medicamentos biológicos, o nome comercial e o número de lote do produto administrado devem ser devidamente registrados (ou descritos) na ficha do paciente.
Infecções
Infecções bacterianas (incluindo sepse e pneumonia), micobacterianas [incluindo tuberculose, (frequentemente disseminada ou extrapulmonar na apresentação clínica)], fúngicas invasivas, virais e outras infecções oportunistas foram relatadas em pacientes tratados com REMICADE®. Algumas destas infecções foram fatais.
Os pacientes devem ser avaliados quanto aos fatores de risco para tuberculose, incluindo contato próximo com uma pessoa com tuberculose ativa, e testados para a presença de tuberculose latente, antes de iniciar o tratamento com REMICADE®. Essa avaliação deve incluir uma anamnese detalhada com o histórico pessoal de tuberculose ou um possível contato prévio com tuberculose e de terapia imunossupressora prévia e/ou atual. Testes de rastreamento apropriados, por exemplo, teste cutâneo de tuberculina e raio-x do tórax, devem ser realizados em todos os pacientes (conforme recomendações locais). Os prescritores devem ser lembrados quanto ao risco de falso-negativo. O teste cutâneo de tuberculina pode produzir resultado falso-negativo, especialmente em pacientes gravemente doentes ou imunocomprometidos. Pacientes que manifestaram clinicamente infecções e/ou abscessos devem ser tratados para essas condições antes do tratamento com REMICADE® .
O tratamento da tuberculose latente deverá ser iniciado antes do tratamento com REMICADE® .
A administração de terapia antituberculose deverá ser considerada antes do início do tratamento com REMICADE® em pacientes com histórico de tuberculose ativa ou latente e naqueles em que o curso adequado do tratamento não pode ser confirmado.
A administração de terapia antituberculose antes do início do tratamento com REMICADE® também deve ser considerada em pacientes com vários fatores de risco ou fatores de risco altamente significativos para tuberculose e que tenham teste negativo para tuberculose latente.
A decisão de iniciar a terapia antituberculose nesses pacientes deverá ser tomada somente após consulta com um médico especializado no tratamento de tuberculose, levando em consideração tanto o risco de infecção por tuberculose latente quanto os riscos da terapia antituberculose.
Os pacientes em tratamento com REMICADE® devem ser cuidadosamente monitorados para sinais e sintomas de tuberculose ativa durante e após o tratamento, incluindo pacientes com resultado negativo para tuberculose latente.
Para pacientes que residiram ou viajaram para regiões onde as infecções fúngicas invasivas, tais como a histoplasmose, a coccidioidomicose ou a blastomicose são endêmicas, os benefícios e riscos do tratamento com REMICADE® devem ser considerados cuidadosamente antes de iniciar o tratamento com REMICADE® .
Em pacientes tratados com REMICADE®, deve-se suspeitar de uma infecção fúngica invasiva, tal como aspergilose, candidíase, pneumocistose, histoplasmose, coccidioidomicose ou blastomicose, se eles desenvolverem doença sistêmica grave. As infecções fúngicas invasivas podem ocorrer mais como doença sistêmica do que como doença localizada, e os testes de antígenos e anticorpos podem ser negativos em alguns pacientes com infecção ativa. Enquanto os procedimentos para diagnóstico estão em andamento, deve-se considerar a introdução de tratamento antifúngico empírico apropriado. A decisão deve ser tomada, se possível, consultando um especialista no diagnóstico e tratamento de infecções fúngicas, levando em consideração o risco de infecção fúngica grave e os riscos do tratamento antifúngico.
REMICADE® não deve ser administrado em pacientes com infecção ativa clinicamente importante. Recomenda-se cautela ao considerar o uso de REMICADE® em pacientes com infecção crônica ou com histórico de infecção recorrente. Os pacientes devem ser informados sobre os fatores de risco potenciais de infecção e orientados para evitar a exposição a estes fatores.
Se for diagnosticada tuberculose ativa, a terapia com REMICADE® não deverá ser iniciada.
A supressão do TNF-alfa também pode mascarar sintomas de infecção como febre. O tratamento com REMICADE® deverá ser interrompido se o paciente desenvolver infecção grave ou sepse. Como a eliminação de REMICADE® pode levar até 6 meses, nesse período é importante o cuidadoso acompanhamento do paciente.
Pacientes com doença de Crohn fistulizante com fístulas supurativas agudas não devem iniciar a terapia com REMICADE® até que a fonte de uma possível infecção, especificamente abscesso, tenha sido excluída.
A experiência sobre a segurança de procedimentos cirúrgicos em pacientes tratados com REMICADE® é limitada. Pacientes que forem submetidos a cirurgias durante o tratamento com REMICADE® devem ser cuidadosamente monitorados quanto a infecções e ações apropriadas devem ser tomadas. Todos os pacientes deverão ser orientados a procurar o médico, se sinais/sintomas sugestivos de tuberculose (por exemplo, tosse persistente, perda de peso/debilidade, febre baixa) aparecerem durante ou após o tratamento com REMICADE® .
Insuficiência cardíaca congestiva
Em pacientes com insuficiência cardíaca moderada a grave (NYHA de classe funcional III/IV) não foi observado aumento da incidência de morte e hospitalização, devido à piora da insuficiência cardíaca, com a dose de 5 mg/kg. No entanto, a ocorrência de um evento adverso com esta dose ou com doses menores, ou na insuficiência cardíaca leve (NYHA de classe funcional I/II), particularmente durante o tratamento a longo prazo, não pode ser excluída. Portanto, REMICADE® deve ser usado apenas com extrema cautela em pacientes com insuficiência cardíaca e após considerar outras opções de tratamento para as condições indicadas. A dose de REMICADE® não deve exceder 5 mg/kg. Se a decisão for administrar REMICADE® em pacientes com insuficiência cardíaca, eles devem ser monitorados de perto durante o tratamento e REMICADE® não deve ser continuado se surgirem novos sintomas de insuficiência cardíaca ou se houver piora dos sintomas existentes.
Reações à infusão/Reações de hipersensibilidade
Para minimizar a incidência de reações de hipersensibilidade, incluindo reações à infusão e do tipo doença do soro, REMICADE® deve ser administrado como tratamento normal de manutenção após esquema de indução nas Semanas 0, 2 e 6.
REMICADE® tem sido associado a reações de hipersensibilidade que variam no seu tempo de início. As reações de hipersensibilidade, que incluem urticária, dispneia e/ou raramente broncoespasmo, edema de laringe, edema de faringe e hipotensão, ocorreram durante ou dentro de 2 horas após a infusão de REMICADE®. No entanto, em alguns casos foram observadas reações do tipo doença do soro em pacientes com doença de Crohn no período entre 1 a 14 dias após o tratamento com REMICADE®. Os sintomas associados a estas reações incluem febre, erupção cutânea, cefaleia, dor de garganta, mialgias, poliartralgias, edema facial e palmar e/ou disfagia. REMICADE® deve ser descontinuado na presença de reações graves. Medicamentos para tratamento de reações de hipersensibilidade devem estar disponíveis para uso imediato caso ocorra alguma reação.
Os dados do estudo ATTRACT indicam que o tratamento profilático (paracetamol e/ou anti-histamínicos) prévio dos pacientes para as reações à infusão reduziu a ocorrência de reações à infusão subsequentes. A velocidade da infusão deve ser reduzida a fim de diminuir as reações à infusão, especialmente se estas reações ocorreram anteriormente.
Reações agudas à infusão podem ser desenvolvidas imediatamente ou poucas horas após a infusão. Se ocorrerem reações agudas à infusão, a mesma deverá ser imediatamente interrompida. Alguns desses efeitos foram descritos como anafilaxia. Medicamentos como anti-histamínicos, corticosteroides, adrenalina e/ou paracetamol, equipamentos para respiração artificial e outros materiais apropriados para o tratamento desses efeitos, devem estar disponíveis para uso imediato. Os pacientes podem ser previamente tratados com, por exemplo, anti-histamínicos, hidrocortisona e/ou paracetamol para prevenir efeitos leves e transitórios.
Uma reação de hipersensibilidade tardia foi observada em um número significativo de pacientes (25% em um único estudo clínico) com doença de Crohn retratados com REMICADE® depois de um período de 2 a 4 anos sem tratamento com REMICADE®. Sinais e sintomas incluíram mialgia e/ou artralgia com febre e/ou erupção cutânea no período de 12 dias após o retratamento. Alguns pacientes também apresentaram prurido, edema facial, edema de mãos ou lábios, disfagia, urticária, dor de garganta e/ou cefaleia. Algumas vezes, esses efeitos são descritos como uma reação do tipo doença do soro. Aconselha-se que os pacientes procurem imediatamente atendimento médico, caso apresentem qualquer evento adverso tardio.
Se os pacientes forem retratados após um período prolongado, deverão ser cuidadosamente acompanhados em relação aos sinais e sintomas de hipersensibilidade tardia.
Reações à infusão após readministração de REMICADE®
Em um estudo clínico em psoríase, a indução com 3 doses de REMICADE® após um período sem tratamento resultou em incidência mais elevada de reações graves à infusão durante o esquema de repetição da indução do que a observada nos estudos de artrite reumatoide, psoríase e doença de Crohn, nos quais o período sem tratamento medicamentoso foi seguido por tratamento de manutenção normal sem tratamento de repetição da indução.
Quando o tratamento de manutenção com REMICADE® para psoríase for interrompido, REMICADE® deve ser reiniciado como dose única seguida por tratamento de manutenção.
Em geral, a relação risco-benefício da readministração de REMICADE® após um período sem tratamento, especialmente como esquema de repetição da indução administrada nas Semanas 0, 2 e 6, deve ser cuidadosamente considerada.
Processo autoimune
O tratamento com REMICADE® pode resultar na formação de autoanticorpos e, raramente, no desenvolvimento de uma síndrome semelhante ao lúpus. Se o paciente desenvolver sintomas sugestivos de síndrome semelhante ao lúpus após o tratamento com REMICADE®, o tratamento deverá ser descontinuado.
Eventos neurológicos
REMICADE® e outros agentes que inibem o TNF-alfa têm sido associados, em casos raros, a convulsões e novo início ou exacerbação dos sintomas clínicos e/ou evidência radiográfica de doenças desmielinizantes do sistema nervoso central, incluindo esclerose múltipla e neurite óptica, e doenças desmielinizantes periféricas, como a síndrome de Guillan-Barré. Os prescritores devem ter cuidado ao considerar o uso de REMICADE® em pacientes com esses distúrbios neurológicos e devem considerar a descontinuação de REMICADE® se esses distúrbios se desenvolverem.
Eventos hepatobiliares
Casos muito raros de icterícia e hepatite não infecciosa, alguns com características de hepatite autoimune, têm sido observados na experiência pós-comercialização de REMICADE® . Ocorreram casos isolados de insuficiência hepática que resultaram em transplante hepático ou morte. Não foi estabelecida uma relação causal entre REMICADE® e esses eventos. Pacientes com sinais ou sintomas de disfunção hepática devem ser avaliados para evidência de dano hepático. Se houver desenvolvimento de icterícia e/ou aumento da ALT ≥ 5 vezes o limite superior dos valores normais, REMICADE® deve ser descontinuado e uma investigação completa da anormalidade deve ser realizada. Como observado também com outros imunossupressores, o uso de bloqueadores de TNF-alfa, incluindo REMICADE®, tem sido associado à reativação do vírus da hepatite B em pacientes portadores crônicos desse vírus (por exemplo, antígeno de superfície positivo). Os pacientes devem ser testados quanto à presença de infecção pelo vírus da hepatite B (HBV) antes de iniciarem o tratamento com imunossupressores, incluindo REMICADE®. Para pacientes que apresentarem teste positivo para o antígeno de superfície da hepatite B, recomenda-se consultar um especialista com experiência no tratamento da hepatite B. Portadores crônicos da hepatite B devem ser apropriadamente avaliados e monitorados antes e durante o tratamento com REMICADE® e por vários meses após a descontinuação do tratamento.
Doenças malignas
- Linfomas
Na porção controlada dos estudos clínicos de todos os agentes bloqueadores de TNF, a maioria dos casos de linfoma foi observada nos pacientes que receberam bloqueadores de TNF em comparação aos pacientes do grupo controle. Durante os estudos clínicos de REMICADE® em pacientes com artrite reumatoide, doença de Crohn, artrite psoriásica, espondilite anquilosante e colite ou retocolite ulcerativa, a incidência de linfoma em pacientes tratados com REMICADE® foi maior que a esperada na população geral, porém, a ocorrência de linfoma foi rara. Pacientes com doença de Crohn ou artrite reumatoide, particularmente aqueles com doença altamente ativa e/ou exposição crônica à terapia imunossupressora, podem ter maior risco (até várias vezes maior) de desenvolvimento de linfoma do que a população geral, mesmo na ausência de terapia com bloqueador de TNF.
- Malignidade pediátrica
Após o início da comercialização foram relatados casos de tumores malignos, sendo alguns fatais, em crianças, adolescentes e adultos jovens (até 22 anos de idade) que receberam agentes bloqueadores do TNF (início do tratamento em idade ≤ 18 anos), incluindo REMICADE® para tratar a artrite juvenil idiopática, doença de Crohn ou outras condições. Aproximadamente a metade dos relatos foi de linfomas. Os outros casos representaram uma variedade de diferentes doenças malignas e incluíram malignidades que normalmente não são observadas em crianças e adolescentes. A maioria dos pacientes estava recebendo concomitantemente imunossupressores, tais como metotrexato, azatioprina ou 6-mercaptopurina. O papel dos bloqueadores de TNF no desenvolvimento de doenças malignas em crianças e adolescentes continua incerto.
- Linfoma de célula-T hepatoesplênica
Após o início da comercialização, casos raros de linfoma de célula-T hepatoesplênica foram relatados em pacientes tratados com agentes bloqueadores de TNF, incluindo REMICADE®. Esse tipo raro de linfoma de célula-T apresenta uma evolução muito agressiva da doença e, geralmente, é fatal. Todos os casos tratados com REMICADE® ocorreram em pacientes com doença de Crohn ou colite ulcerativa e a maioria deles foi relatada em adolescentes ou adultos jovens do sexo masculino. Todos esses pacientes haviam recebido tratamento com azatioprina ou 6-mercaptopurina concomitantemente ou imediatamente antes de REMICADE® . Casos de linfoma de célula-T hepatoesplênica também ocorreram em pacientes com doença de Crohn e colite ulcerativa recebendo azatioprina ou 6-mercaptopurina que não foram tratados com REMICADE®. Antes de iniciar ou continuar o tratamento com REMICADE® em um paciente que apresenta doença inflamatória intestinal crônica e que esteja recebendo um imunossupressor, tal como a azatioprina ou 6-mercaptopurina, deve-se avaliar cuidadosamente a necessidade de continuar o tratamento com o imunossupressor, considerando os potenciais riscos do tratamento concomitante. A relação causal entre a ocorrência de linfoma de célula-T hepatoesplênica e a terapia com REMICADE® permanece desconhecida.
- Leucemia
Após o início da comercialização do bloqueador de TNF para artrite reumatoide e outras indicações, foram relatados casos de leucemia aguda e crônica. Mesmo na ausência de tratamento com bloqueadores do TNF, pacientes com artrite reumatoide podem apresentar risco maior para desenvolver leucemia (aproximadamente 2 vezes) em relação à população geral.
- Malignidades que não sejam linfomas
Nas porções controladas de alguns estudos clínicos com agentes bloqueadores de TNF, mais casos de malignidades que não sejam linfomas foram observados entre os pacientes recebendo bloqueadores de TNF comparado com os pacientes do grupo controle. A taxa de malignidades que não sejam linfomas entre os pacientes tratados com REMICADE® foi semelhante à esperada na população em geral, enquanto que a taxa entre os pacientes controle foi menor que a esperada.
Em um estudo clínico exploratório avaliando o uso de REMICADE® em pacientes com Doença Pulmonar Obstrutiva Crônica (DPOC) de moderada a grave, foram relatados mais casos de malignidades em pacientes tratados com REMICADE® comparado com os pacientes do grupo controle. Todos os pacientes tinham histórico de tabagismo importante.
- Cânceres de pele
Foram relatados casos de melanoma e carcinoma de células de Merkel em pacientes tratados com bloqueadores de TNF, incluindo REMICADE®. Para todos os pacientes, recomenda-se que sejam realizados exames periódicos da pele, particularmente, naqueles pacientes com fatores de risco para câncer de pele.
O potencial papel da terapia com bloqueador de TNF, no desenvolvimento de malignidades, não é conhecido. Deve-se ter precaução adicional ao considerar a terapia com bloqueador de TNF em pacientes com histórico de malignidade ou ao continuar o tratamento em pacientes que desenvolveram malignidade.
Administração concomitante de inibidor de TNF-alfa e anacinra
Infecções graves e neutropenia foram observadas em estudos clínicos com uso concomitante de anacinra e outro agente bloqueador de TNF-alfa, o etanercepte, sem benefício clínico adicional quando comparado ao etanercepte em monoterapia. Devido à natureza dos eventos adversos observados com a terapia de combinação de etanercepte e anacinra, toxicidade semelhante pode resultar também da combinação de anacinra e outros agentes que bloqueiam o TNF-alfa. Portanto, a associação de REMICADE® e anacinra não é recomendada.
Administração concomitante de REMICADE® e abatacepte
Nos estudos clínicos, a administração concomitante de agentes bloqueadores de TNF e abatacepte foi associada com um risco aumentado de infecções, incluindo infecções graves em comparação com os agentes bloqueadores de TNF isoladamente, sem aumento do benefício clínico. Considerando a natureza dos eventos adversos observados com o tratamento de combinação de agentes bloqueadores de TNF com abatacepte, a combinação de REMICADE® e abatacepte não é recomendada.
Administração concomitante com outros biológicos
Não há informação suficiente sobre o uso concomitante de REMICADE® com outros biológicos usados para tratar as mesmas condições que REMICADE®. O uso concomitante de REMICADE® com outros biológicos não é recomendado, devido à possibilidade de um risco aumentado para infecções.
Substituição por outro biológico
Ao substituir um biológico por outro, os pacientes devem continuar a ser monitorados, uma vez que a sobreposição das atividades biológicas pode aumentar o risco de infecção.
Reações hematológicas
Foram relatados casos de pancitopenia, leucopenia, neutropenia e trombocitopenia em pacientes recebendo bloqueadores de TNF, incluindo REMICADE® . Recomenda-se cautela em pacientes tratados com REMICADE® que apresentam atualmente ou apresentaram previamente citopenias significativas.
Vacinas de vírus vivos/Agentes terapêuticos infecciosos
Os dados disponíveis sobre a resposta a vacinas de vírus vivos ou a transmissão secundária de infecção por vacinas com vírus vivos, são limitados em pacientes recebendo terapia anti-TNF. O uso de vacinas de vírus vivos pode resultar em infecções clínicas, incluindo infecções generalizadas. Não é recomendado o uso concomitante de vacinas de vírus vivos e REMICADE®.
O uso de outros agentes terapêuticos infecciosos, tais como bactérias vivas atenuadas (por exemplo, instilação da bexiga com BCG para o tratamento de câncer) podem resultar em infecções clínicas, incluindo infecções generalizadas. Não é recomendado que agentes terapêuticos infecciosos sejam administrados com REMICADE® .
Vacinas de vírus inativados
Em um subgrupo de pacientes do estudo ASPIRE, uma proporção semelhante de pacientes em cada grupo de tratamento apresentou uma duplicação efetiva dos títulos da vacina pneumocócica polivalente, indicando que REMICADE® não interferiu nas respostas imunes humorais de células T independentes. Recomenda-se que, se possível, os pacientes pediátricos estejam com todas as vacinas atualizadas, de acordo com as diretrizes atuais para vacinação antes de iniciar o tratamento com REMICADE® .
Efeitos sobre a capacidade de dirigir veículos e operar máquinas REMICADE® tem pouca probabilidade de afetar a capacidade de dirigir veículos ou operar máquinas. No entanto, pacientes com fadiga devem ser alertados para evitar tais atividades.
Uso em idosos, crianças e outros grupos de risco
Pacientes pediátricos
REMICADE® é indicado para redução dos sinais e sintomas, bem como para indução e manutenção da remissão clínica em pacientes pediátricos que tenham doença de Crohn de moderada a altamente ativa. Todos os pacientes pediátricos do estudo clínico de Fase III (REACH) foram mantidos em doses estáveis de 6mercaptopurina, azatioprina ou metotrexato.
REMICADE® não foi estudado em crianças com doença de Crohn com menos de 6 anos de idade.
A segurança e a eficácia de REMICADE® em pacientes com artrite reumatoide juvenil não foram estabelecidas.
A farmacocinética de REMICADE® foi avaliada em pacientes pediátricos com doença de Crohn.
Uso em idosos
Não foram conduzidos estudos específicos de REMICADE® em pacientes idosos. Em estudos clínicos não foram observadas diferenças importantes na depuração ou volume de distribuição relacionadas à idade.
A incidência de infecções graves nos pacientes com idade ≥ 65 anos tratados com REMICADE® foi maior do que nos pacientes com idade inferior a 65 anos. Além disso, existe maior incidência de infecções na população de idosos em geral, e por isso, recomenda-se cautela ao se tratar pacientes idosos.
Uso durante a gravidez (Categoria B) e lactação
Pelo fato do REMICADE® não apresentar reação cruzada com o TNF-alfa em espécies que não o homem e os chimpanzés, não foram realizados estudos de reprodução em animais. Em um estudo de toxicidade de desenvolvimento conduzido em camundongos, usando um anticorpo análogo ao que inibe seletivamente a atividade funcional do TNF-alfa de camundongos, não foram observadas evidências de toxicidade materna, embriotoxicidade ou teratogenicidade. Doses de 10 a 15 mg/kg em modelos animais de farmacodinâmica com o anticorpo análogo anti-TNF produziram eficácia farmacológica máxima. Doses até 40 mg/kg não produziram efeitos adversos em estudos de reprodução em animais. Não se sabe se REMICADE® pode causar dano fetal quando administrado a gestantes ou afetar a capacidade reprodutiva. REMICADE® deverá ser administrado a gestantes somente se for realmente necessário. No entanto, como são necessários 6 meses para garantir que REMICADE® não esteja mais presente no sangue, recomenda-se que métodos contraceptivos sejam mantidos durante pelo menos 6 meses após a última infusão.
Assim como para outros anticorpos IgG, REMICADE® atravessa a placenta e foi detectado no soro de recém-nascidos de mães tratadas com REMICADE® durante a gestação, até 6 meses depois do nascimento. Consequentemente, essas crianças podem apresentar aumento do risco de infecções e adverte-se que é preciso ter cautela na administração de vacinas de vírus vivos nesses lactentes.
Não se sabe se REMICADE® é excretado no leite humano ou absorvido sistemicamente após a ingestão. Como muitos fármacos e imunoglobulinas são excretados no leite humano e tendo em vista o potencial de REMICADE® para reações adversas em lactentes em aleitamento materno, uma decisão deve ser tomada a respeito da interrupção da amamentação ou descontinuação do tratamento, considerando-se a importância do medicamento para a mãe.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação do médico ou do cirurgião-dentista.
Atenção: Este medicamento contém Açúcar, portanto, deve ser usado com cautela em portadores de Diabetes.
Medicamentos imunossupressores podem ativar focos primários de tuberculose. Os médicos que acompanham pacientes sob imunossupressão devem estar alertas quanto à possibilidade de surgimento de doença ativa, tomando, assim, todos os cuidados para o diagnóstico precoce e tratamento.
Interações medicamentosas.
Não foram conduzidos estudos de interação medicamentosa específicos com REMICADE® .
Uso concomitante de REMICADE® com outro biológico
A combinação de REMICADE® com outro biológico utilizado para tratar as mesmas condições que REMICADE®, incluindo anacinra e abatacepte, não é recomendada.
Foi demonstrado que a formação de anticorpos contra infliximabe é reduzida em pacientes com doença de Crohn, artrite psoriásica e artrite reumatoide quando administrado concomitantemente ao metotrexato e outros imunomoduladores. Não há informações disponíveis em relação a possíveis efeitos de outros imunossupressores sobre a farmacocinética do infliximabe.
Vacinas de vírus vivos/Agentes terapêuticos infecciosos
Não é recomendada a administração concomitante de vacinas de vírus vivos e REMICADE® .
Não é recomendada a administração concomitante de agentes terapêuticos infecciosos e REMICADE® .
Cuidados de armazenamento.
Conservar sob refrigeração (entre 2°C e 8°C). Não congelar.
O prazo de validade do medicamento é de 36 meses a partir da data de fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Aspecto físico
REMICADE® é um pó liofilizado branco, sem nenhuma evidência de liquefação e livre de partículas estranhas. Após a reconstituição, a solução é de incolor a amarelada e opalescente. A solução pode desenvolver algumas partículas translúcidas finas, porque o infliximabe é uma proteína. Não use se houver partículas opacas, alteração de cor ou presença de outras partículas estranhas.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
Posologia e modo de usar.
Preparação e administração:
1. Calcule a dose e o número de frascos-ampola necessários de REMICADE®. Cada frasco-ampola contém 100 mg de infliximabe. Calcule o volume total necessário de solução de REMICADE®
a ser reconstituída.
2. Reconstitua cada frasco-ampola de REMICADE® com 10 mL de água para injeção, usando uma seringa equipada com uma agulha de calibre 21 (0,8 mm) ou menor. Cada mL de solução reconstituída contém 10 mg de infliximabe. Retire o revestimento da tampa do frasco e limpe com álcool a 70%. Introduza a agulha da seringa no frasco-ampola através do centro da rolha de borracha e direcione o jato de água para a injeção para a parede de vidro do frasco-ampola. Não use o frasco-ampola se não houver vácuo. Mexa suavemente a solução, rodando o frasco para dissolver o pó liofilizado. Evite agitação forte ou prolongada. NÃO AGITE. É comum a formação de espuma na solução reconstituída. Deixe que a solução reconstituída permaneça em repouso por 5 minutos. Verifique se a solução é de incolor a amarelada e opalescente. A solução pode desenvolver algumas partículas translúcidas finas, porque o infliximabe é uma proteína. Não administre se houver partículas opacas, alteração de cor ou presença de outras partículas estranhas.
3. Dilua o volume total da dose da solução reconstituída de REMICADE® para 250 mL com solução de cloreto de sódio a 0,9% p/v para infusão. Isso pode ser realizado se retirado da bolsa ou frasco um volume de cloreto de sódio a 0,9% p/v igual ao volume de REMICADE® reconstituído a ser introduzido. Introduza lentamente o volume total da solução de REMICADE® reconstituída no frasco ou na bolsa de 250 mL para a infusão. Misture suavemente.
4. Administre a solução para infusão em período não inferior ao tempo de infusão recomendado para a indicação específica. Use um equipo para infusão com filtro interno, estéril, não pirogênico, com baixa ligação a proteínas (poro de tamanho 1,2 micrômetro ou menor). Como não há presença de conservantes, recomenda-se que a administração da solução para infusão seja iniciada assim que possível, em até 3 horas após a reconstituição e diluição. Se a reconstituição e a diluição forem realizadas em condições assépticas, a solução de infusão de REMICADE® poderá ser utilizada dentro de 24 horas, se armazenada entre 2 e 8°C. Não estoque a sobra da solução de infusão não utilizada para uso posterior.
5. Não foram conduzidos estudos de compatibilidade física ou bioquímica para avaliar a coadministração de REMICADE® com outros agentes. Não administre REMICADE® concomitantemente no mesmo equipo com outros agentes.
6. Verifique visualmente os produtos parenterais, procurando partículas ou alteração de cor antes da administração. Não use se houver partículas opacas, alteração de cor ou partículas estranhas visíveis.
7. Descarte a parte não utilizada da solução.
ATENÇÃO: O FRASCO-AMPOLA E OS MATERIAIS PARA INJEÇÃO DEVEM SER DESCARTADOS APÓS O USO. COLOQUE AS SERINGAS E AS AGULHAS DE MODO SEGURO EM UM RECIPIENTE ADEQUADO.
Posologia
REMICADE® destina-se ao uso intravenoso em adultos (com idade acima ou igual a 18 anos) para todas as indicações presentes nesta bula. REMICADE® destina-se ao uso intravenoso em crianças e adolescentes (com idade entre 6 a 17 anos) somente para a Doença de Crohn.
O tratamento com REMICADE® deve ser iniciado e supervisionado por médicos especializados no diagnóstico e tratamento da artrite reumatoide, espondilite anquilosante, artrite psoriásica, psoríase ou doenças inflamatórias intestinais. A infusão de REMICADE® só pode ser delegada a profissionais de saúde qualificados, devidamente treinados para detectar qualquer complicação relacionada à infusão.
O tempo de duração recomendado para a infusão no paciente está relacionado a cada indicação terapêutica e encontra-se descrito em cada uma delas. Todos os pacientes que receberem REMICADE® deverão ser observados por pelo menos 1 hora após a infusão para verificação de efeitos colaterais. Medicamentos, respirador artificial e outros equipamentos apropriados devem estar disponíveis para o tratamento desses efeitos colaterais. A velocidade de infusão deve ser lenta para diminuir o risco de reações relacionadas à infusão, especialmente se a reação já ocorreu anteriormente.
Artrite Reumatoide
Para pacientes não tratados com REMICADE® previamente: Inicialmente, uma infusão intravenosa de 3 mg/kg, administrada por um período mínimo de 2 horas, seguida por doses de infusões adicionais de 3 mg/kg nas Semanas 2 e 6 após a primeira infusão e, depois, a cada 8 semanas. Para uma melhor resposta clínica, deve-se levar em consideração um ajuste de dose de até 10 mg/kg ou a administração de doses de 3 mg/kg a cada 4 semanas. Em pacientes com artrite reumatoide cuidadosamente selecionados, que toleraram 3 infusões iniciais de REMICADE® com 2 horas de duração, deve-se levar em consideração que as administrações subsequentes devem ocorrer por um período mínimo de 1 hora.
REMICADE® deve ser administrado em combinação com o metotrexato.
Dados avaliados sugerem que a resposta clinica é normalmente obtida dentro de 12 semanas de tratamento. Se o paciente apresentar uma resposta inadequada ou perda da resposta após esse período, a dose poderá ser ajustada conforme mencionado acima. Caso seja obtida uma resposta adequada, os pacientes deverão ser mantidos na dose selecionada ou na frequência de doses.
A continuação da terapia deverá ser reconsiderada com cautela em pacientes que não demonstraram benefício evidente com a terapêutica dentro das primeiras 12 semanas de tratamento ou após o ajuste da dose.
Espondilite Anquilosante
Infusão intravenosa de 5 mg/kg, administrada por um período mínimo de 2 horas, seguida por doses de infusões adicionais de 5 mg/kg nas Semanas 2 e 6 após a primeira infusão e, depois, a cada 6 a 8 semanas.
Artrite Psoriásica
Infusão intravenosa de 5 mg/kg, administrada por um período mínimo de 2 horas, seguida por doses de infusões adicionais de 5 mg/kg nas Semanas 2 e 6 após a primeira infusão e, depois, a cada 8 semanas. A eficácia e a segurança foram demonstradas em monoterapia ou em combinação com metotrexato.
Psoríase
Infusão intravenosa de 5 mg/kg, administrada por um período mínimo de 2 horas, seguida por doses de infusões adicionais de 5 mg/kg nas Semanas 2 e 6 após a primeira infusão e, depois a cada 8 semanas.
Doença de Crohn moderada a grave em adultos
Para a otimização do controle dos sintomas a longo prazo, infusões intravenosas no regime de indução de 5 mg/kg, administrado em dose única por um período mínimo de 2 horas, nas Semanas 0, 2 e 6 e, depois, em regime de manutenção de 5 mg/kg a cada 8 semanas. Para pacientes que apresentarem resposta incompleta durante o regime de manutenção, deve ser considerado o ajuste da dose para até 10 mg/kg. Alternativamente, infusão intravenosa inicial de 5 mg/kg, administrada por um período mínimo de 2 horas, pode ser seguida por infusões repetidas de 5 mg/kg, quando houver recorrência dos sinais e sintomas; entretanto, existem dados limitados em relação a intervalos de dose superiores a 16 semanas.
Doença de Crohn em pediatria
Infusão intravenosa de 5 mg/kg seguida por doses de infusões adicionais de 5 mg/kg nas Semanas 2 e 6 após a primeira infusão e, depois, a cada 8 semanas. Para os pacientes que tiveram resposta incompleta, deve-se levar em consideração a possibilidade de ajuste da dose para até 10 mg/kg. REMICADE® deve ser administrado concomitantemente com imunomoduladores, incluindo 6-mercaptopurina (6-MP), azatioprina (AZA) ou metotrexato (MTX). Dados disponíveis não sustentam o tratamento com infliximabe em pacientes pediátricos que não responderam dentro das 10 semanas da infusão inicial.
Doença de Crohn fistulizante em adultos
Infusão intravenosa de 5 mg/kg, administrada por um período mínimo de 2 horas, seguida por doses de infusões adicionais de 5 mg/kg administradas nas Semanas 2 e 6 após a primeira infusão para tratamento de fístula(s) na doença de Crohn. Se o paciente não responder após essas três doses, não se deve instituir tratamento adicional com infliximabe.
As estratégias para o tratamento continuado são:
-infusões adicionais de 5 mg/kg a cada 8 semanas ou
- readministração, se reaparecerem sinais e sintomas da doença, seguidas de infusões de 5 mg/kg a cada 8 semanas.
Na doença de Crohn, a experiência com readministração em caso de reaparecimento de sinais e sintomas da doença é limitada e não há dados comparativos a respeito do benefício/risco das estratégias alternativas para o tratamento continuado.
Colite ou Retocolite Ulcerativa
Infusão intravenosa de 5 mg/kg, administrada por um período mínimo de 2 horas, seguida por doses de infusões adicionais de 5 mg/kg nas Semanas 2 e 6 após a primeira infusão e, depois, a cada 8 semanas. Em alguns pacientes, deve-se considerar o ajuste da dose para até 10 mg/kg para sustentar a resposta clínica e a remissão.
Readministração para doença de Crohn e artrite reumatoide
Se os sinais e sintomas da doença reaparecerem, REMICADE® pode ser readministrado dentro de 16 semanas após a última infusão. Dez pacientes com doença de Crohn manifestaram reações de hipersensibilidade tardia com a readministração de uma formulação alternativa de infliximabe depois de uma prévia infusão, após intervalo sem droga de 2 a 4 anos. É desconhecido o risco de hipersensibilidade tardia depois da readministração, em um intervalo sem droga de 16 semanas a 2 anos. Por isso, depois de um intervalo de 16 semanas, não se pode recomendar a readministração.
Readministração para colite ou retocolite ulcerativa
No momento, não há dados disponíveis que suportem a readministração além de intervalos de 8 semanas.
Readministração para espondilite anquilosante
No momento, não há dados disponíveis que suportem a readministração além de intervalos de 6 a 8 semanas.
Readministração para artrite psoriásica
No momento, não há dados disponíveis que suportem a readministração além de intervalos de até 8 semanas.
Readministração para psoríase
A experiência do tratamento intermitente com REMICADE® na psoríase após um período sem tratamento sugere eficácia reduzida e incidência mais elevada de reações à infusão em comparação com a recomendação posológica aprovada.
Fica a critério do(a) médico(a) assistente a orientação em relação à perda de uma dose prescrita.
Reações adversas.
Nos estudos clínicos com REMICADE®, foram observadas reações adversas atribuíveis ao tratamento em 40% dos pacientes tratados com placebo e 60% dos tratados com REMICADE®.Os dados de segurança de estudos clínicos estão disponíveis para 5561 pacientes tratados com REMICADE® incluindo 1304 com artrite reumatoide, 117 com artrite reumatoide juvenil, 1566 com doença de Crohn (1427 adultos e 139 crianças), 347 com espondilite anquilosante, 293 com artrite psoriásica, 1373 com psoríase em placa, 544 com colite ulcerativa e 17 com outras condições. As reações adversas em estudos clínicos estão resumidas na Tabela 6. As reações relacionadas à infusão (por exemplo, dispneia, rubor, cefaleia e erupção cutânea) estão entre as causas mais comuns de descontinuação, exceto na colite ulcerativa, doença de Crohn pediátrica e artrite psoriásica.
Reações relacionadas à infusão: Nos estudos clínicos de Fase III, 18% dos pacientes tratados com REMICADE® apresentaram reação relacionada à infusão durante a infusão ou dentro de 1 hora após a infusão, comparado com 5% dos pacientes tratados com placebo. Dos pacientes tratados com infliximabe que apresentaram uma reação à infusão durante o período de indução, 27% apresentaram uma reação à infusão durante o período de manutenção. Dos pacientes que não apresentaram uma reação à infusão durante o período de indução, 9% apresentaram uma reação à infusão durante o período de manutenção.
Aproximadamente 3% das infusões de REMICADE® foram acompanhadas por sintomas não específicos como febre ou calafrios, < 1% por prurido ou urticária, 1% por reações cardiopulmonares (principalmente dor no tórax, hipotensão, hipertensão ou dispneia) ou por sintomas combinados de prurido/urticária e reações cardiopulmonares. Reações graves à infusão ocorreram em < 1% dos pacientes e incluíram anafilaxia, convulsões, erupção cutânea eritematosa e hipotensão. Aproximadamente 3% dos pacientes descontinuaram o tratamento devido a reações relacionadas à infusão e todos os pacientes se recuperaram com ou sem tratamento médico.
Em um estudo clínico em pacientes com artrite reumatoide (ASPIRE), 66% dos pacientes (686 de 1040) receberam pelo menos uma infusão abreviada de 90 minutos ou menos e 44% dos pacientes (454 de 1040) receberam pelo menos uma infusão com duração inferior a 60 minutos ou menos. Entre os pacientes tratados com REMICADE® que receberam pelo menos uma infusão abreviada, reações relacionadas à infusão ocorreram em 15% (74/494) dos pacientes e reações graves à infusão ocorreram em 0,4% (2/494) dos pacientes. Infusões abreviadas em doses > 6 mg/kg não foram estudadas.
Em um estudo clínico em pacientes com doença de Crohn (SONIC), reações relacionadas à infusão ocorreram em 16,6% (27/163) dos pacientes recebendo REMICADE® em monoterapia, 5,0% (9/179) recebendo REMICADE® em combinação com azatioprina e 5,6% (9/161) recebendo azatioprina em monoterapia. Um paciente apresentou uma reação grave à infusão de REMICADE® em monoterapia.
A vigilância pós-comercialização notou relatos de reações do tipo anafiláticas, incluindo edema de laringe, edema de faringe e broncoespasmo grave. Casos raros de convulsões foram associados à administração de REMICADE®. Também foram relatados casos extremamente raros de perda visual transitória e isquemia do miorcárdio/infarto do miocárdio, que ocorreram durante ou dentro de 2 horas após a infusão de REMICADE® .
Reações relacionadas à infusão após a readministração de REMICADE®: Nos estudos clínicos em artrite reumatoide, doença de Crohn e psoríase, a readministração de REMICADE® após um período sem tratamento resultou em incidência mais elevada de reações relacionadas à infusão do que no tratamento normal de manutenção.
Em um estudo clínico de pacientes com psoríase moderada a grave planejado para avaliar a eficácia do tratamento de manutenção a longo prazo em comparação ao retratamento com um ciclo de indução de REMICADE®, 4% (8/219) dos pacientes no grupo de tratamento intermitente apresentaram reações graves à infusão versus < 1% (1/222) no grupo de tratamento de manutenção. Os pacientes incluídos nesse estudo não receberam qualquer tratamento imunossupressor concomitante. O tratamento intermitente nesse estudo foi definido como a readministração de um ciclo de indução (máximo de 4 infusões nas Semanas 0, 2, 6 e 14) de REMICADE® na reativação da doença após o período sem tratamento. Nesse estudo, a maioria das reações graves à infusão ocorreu durante a segunda infusão na Semana 2. Os sintomas incluíram, mas não foram limitados a, dispneia, urticária, edema facial e hipotensão. Em todos os casos, o tratamento com REMICADE® foi descontinuado e/ou foi instituído outro tratamento com remissão completa dos sintomas e sinais.
Hipersensibilidade tardia/Reações após a readministração: Em um estudo clínico no qual 37 de 41 pacientes com doença de Crohn foram retratados com REMICADE®, após um período de 2 a 4 anos sem tratamento com REMICADE®, 10 pacientes apresentaram eventos adversos que se manifestaram 3 a 12 dias após a infusão. Em 6 desses, os eventos foram considerados graves. Os sinais e sintomas incluíram mialgia e/ou artralgia com febre e/ou erupção cutânea, com alguns pacientes apresentando, também, prurido, edema facial, edema palmar ou lábios, disfagia, urticária, dor de garganta e cefaleia. Os pacientes que apresentaram estes eventos adversos não haviam apresentado eventos adversos relacionados à infusão associados ao tratamento inicial com REMICADE®. Estes eventos adversos ocorreram em 39% (9/23) dos pacientes que haviam recebido a formulação líquida (usada em um pequeno número de estudos clínicos), e 7% (1/14) dos pacientes que receberam a formulação liofilizada. Os dados clínicos não foram suficientes para determinar se a ocorrência dessas reações é devida às diferentes formulações administradas nesses pacientes. Em todos os casos, os sinais e sintomas dos pacientes melhoraram significativamente ou foram resolvidos com o tratamento. Os dados são insuficientes em relação à incidência desses eventos após intervalos de 1 a 2 anos sem o uso do medicamento. Esses eventos foram observados com pouca frequência em estudos clínicos e em relatos pós-comercialização com intervalos de retratamento de até 1 ano. Nos 3 estudos de psoríase, 1% (15/1373) dos pacientes tiveram sintomas de artralgia, doença do soro, mialgia, febre e erupções cutâneas, frequentemente no início do tratamento após as infusões de REMICADE®. O tratamento com REMICADE® foi descontinuado e/ou outro tratamento foi instituído na maioria dos casos, com melhora ou resolução dos sinais e sintomas.
Imunogenicidade: Pacientes que produziram anticorpos contra REMICADE® apresentaram maior predisposição (aproximadamente 2 a 3 vezes) ao desenvolvimento de reações relacionadas à infusão. Anticorpos contra REMICADE® apareceram em 10% dos pacientes tratados com esquema de indução com 3 doses, seguido por terapia de manutenção. O uso concomitante de imunossupressores pareceu reduzir a frequência de anticorpos contra REMICADE® e de reações relacionadas à infusão. Em um estudo fase III de doença de Crohn em pacientes virgens de imunomoduladores, os anticorpos apareceram na Semana 30 em 14% dos pacientes tratados com REMICADE® em monoterapia e 1% dos pacientes tratados com REMICADE® em combinação com azatioprina. Uma maior incidência de anticorpos contra REMICADE® foi observada em pacientes com doença de Crohn tratados com REMICADE® após intervalos sem tratamento > 16 semanas. No estudo fase III de artrite psoriásica, onde os pacientes receberam 5 mg/kg com ou sem metotrexato concomitante, os anticorpos ocorreram em 15% dos pacientes. Nos dois estudos fase III de psoríase, REMICADE® foi administrado com indução seguida de terapia de manutenção sem tratamento concomitante com imunossupressores. Nestes estudos, anticorpos foram detectados em aproximadamente 25% a 30% dos pacientes que receberam tratamento de manutenção com 5 mg/kg a cada 8 semanas, durante 1 ano e em taxas maiores (até 1,6 vezes) com outros esquemas posológicos (3 mg/kg a cada 8 semanas, 3 mg/kg administrados conforme necessário e 5 mg/kg administrados conforme necessário). Apesar do aumento na formação de anticorpos, as taxas de reação à infusão nos dois estudos fase III de psoríase em pacientes tratados com 5 mg/kg na indução seguido por manutenção a cada 8 semanas, durante um ano (14,1%-23,0%), e as taxas de reações graves à infusão ( < 1%) foram semelhantes àquelas observadas em outras populações do estudo.
Infecções: Em estudos clínicos, 36% dos pacientes tratados com REMICADE® infecções em comparação a 28% dos tratados com placebo. Não foi observado risco aumentado de infecções graves com REMICADE® comparado ao placebo, em estudos de doença de Crohn e no estudo fase III de artrite psoriásica. Em estudos de artrite reumatoide, a incidência de infecções graves, como pneumonia, foi maior em pacientes tratados com REMICADE® mais metotrexato comparado com metotrexato isolado, especialmente em doses de 6 mg/kg ou acima. Nos estudos de psoríase 1,5% dos pacientes (média de 41,9 semanas de acompanhamento) tratados com REMICADE® e 0,6% dos pacientes (média de 18,1 semanas de acompanhamento) que receberam placebo desenvolveram infecções graves.
Durante a experiência pós-comercialização foram observadas infecções por vários patógenos incluindo vírus, bactérias, fungos e protozoários. Infecções foram observadas em todos os sistemas de órgãos e relatadas em pacientes recebendo REMICADE® isolado ou em combinação com imunossupressores.
Eventos hepatobiliares: Na vigilância pós-comercialização, casos muito raros de icterícia e hepatite, alguns com características de hepatite autoimune, foram relatados em pacientes recebendo REMICADE® .
Em estudos clínicos, foram observadas elevações leves ou moderadas de ALT e AST em pacientes recebendo REMICADE®, sem progressão para lesão hepática grave. Foram observadas elevações de ALT ≥ 5 vezes o limite normal superior (ver Tabela 7). A elevação das aminotransferases (mais comum da ALT do que da AST) foi observada em maior proporção nos pacientes recebendo REMICADE® do que no grupo controle, quando administrado em monoterapia ou em combinação com outros agentes imunossupressores (ver Tabela 7). A maioria das anormalidades das aminotransferases foi transitória; entretanto, em um pequeno número de pacientes, as elevações foram mais prolongadas. Em geral, os pacientes que desenvolveram aumento de ALT e AST eram assintomáticos, e as anormalidades diminuíram ou foram resolvidas com a continuação ou descontinuação de REMICADE®, ou modificação da medicação concomitante.
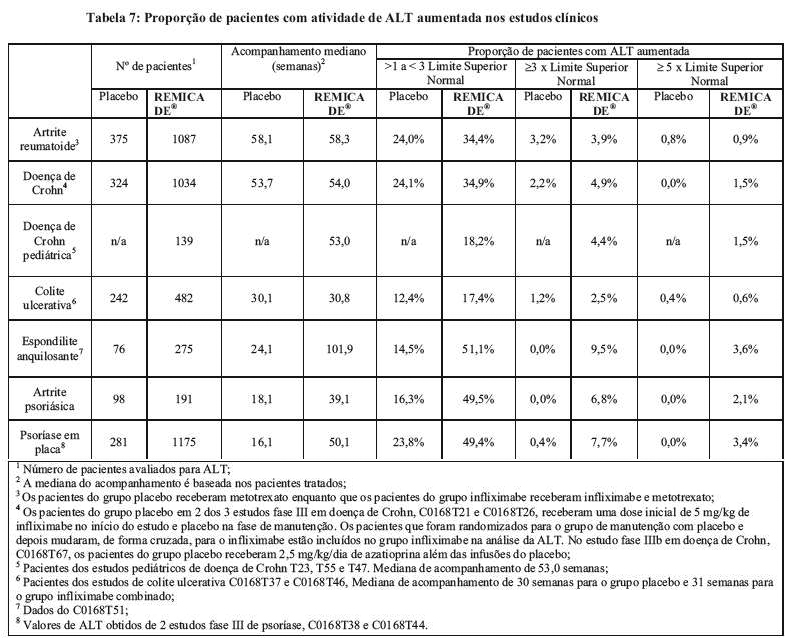
Malignidades: Durante os estudos clínicos de REMICADE® , foram relatadas malignidades novas ou recorrentes em pacientes tratados com REMICADE®. A incidência de linfoma nos pacientes tratados com REMICADE® foi maior do que a esperada na população geral. Pacientes com doença de Crohn ou artrite reumatoide, especialmente aqueles com a doença altamente ativa e/ou exposição crônica a terapias imunossupressoras, podem ter um risco maior do que a população em geral para o desenvolvimento de linfoma, mesmo na ausência de terapia bloqueadora de TNF. As incidências observadas de malignidades exceto linfoma foram semelhantes ao que se esperaria na população geral, enquanto que a taxa entre os pacientes controle foi menor que a esperado. Em um estudo clínico exploratório envolvendo pacientes com Doença Pulmonar Obstrutiva Crônica (DPOC) de moderada a grave, que eram fumantes ou ex-fumantes, foram relatados mais casos de malignidade em pacientes tratados com REMICADE® comparado com os controles. O papel potencial da terapia bloqueadora de TNF no desenvolvimento de malignidade não é conhecido.
Anticorpos antinucleares (ANA)/Anticorpos anti-DNA dupla-hélice (DNAds): Em estudos clínicos, aproximadamente metade dos pacientes tratados com REMICADE® que, inicialmente, eram negativos para ANA, desenvolveu ANA positivo durante o estudo, comparado com aproximadamente 20% dos pacientes tratados com placebo. Anticorpos anti-DNAds se desenvolveram em aproximadamente 17% dos pacientes tratados com REMICADE®, comparado com 0% dos pacientes tratados com placebo. No entanto, relatos de lúpus e síndrome semelhante ao lúpus permaneceram incomuns.
Insuficiência cardíaca congestiva: Em um estudo de Fase II para avaliar REMICADE® em insuficiência cardíaca congestiva (ICC) moderada a grave (NYHA classe III/IV com fração de ejeção do ventrículo esquerdo ≤ 35%), 150 pacientes foram randomizados para receber tratamento com 3 infusões de 10 mg/kg ou 5 mg/kg de REMICADE® ou placebo. Foi observada maior incidência de mortalidade e hospitalização devido ao agravamento da ICC em pacientes tratados com a dose de 10 mg/kg de REMICADE®. Em 28 semanas, três pacientes no grupo de 10 mg/kg vieram a óbito comparado com 1 óbito no grupo de 5 mg/kg de REMICADE® e nenhum óbito ocorreu no grupo placebo. Ao mesmo tempo nestas 28 semanas, 11 dos 51 pacientes no grupo 10 mg/kg de REMICADE® foram hospitalizados devido à piora da ICC comparado com 3 dos 50 pacientes no grupo 5 mg/kg de REMICADE® e 5 dos 49 pacientes no grupo placebo. Durante a fase de acompanhamento, no primeiro ano, 8 pacientes no grupo 10 mg/kg de REMICADE® vieram a óbito em comparação a 4 pacientes no grupo 5 mg/kg de REMICADE® e 4 pacientes no grupo placebo. REMICADE® não foi estudado em pacientes com insuficiência cardíaca leve (NYHA classe I/II). Houve relatos pós-comercialização de piora da insuficiência cardíaca com e sem fatores precipitantes identificáveis em pacientes usando REMICADE® .
Após o início da comercialização houve, também, raros relatos de novo início de insuficiência cardíaca, inclusive em pacientes sem doença cardiovascular pré-existente conhecida. Alguns desses pacientes tinham menos de 50 anos de idade.
Reações adversas na artrite reumatoide juvenil (ARJ): a segurança e a eficácia de REMICADE® foram avaliadas em um estudo duplo-cego, multicêntrico, randomizado, controlado por placebo durante 14 semanas, seguido por uma extensão de tratamento duplo-cego, todos ativos, por um máximo de 44 semanas. Um total de 122 pacientes com ARJ ativa, com idade entre 4 e 17 anos, que haviam sido tratados com metotrexato durante pelo menos 3 meses, foram admitidos. Cento e vinte (120) pacientes receberam o medicamento do estudo. O uso concomitante de ácido fólico, corticosteroides por via oral (≤ 10 mg/dia), anti-inflamatórios não esteroides e/ou metotrexato era permitido.
Doses de 3 mg/kg de REMICADE® ou placebo foram administradas por via intravenosa nas Semanas 0, 2, 6, 14 e 20; e depois a cada 8 semanas até a Semana 44. Para manter o tratamento cego, o grupo tratado com 3 mg/kg recebeu também uma única infusão de placebo na Semana 16, enquanto que os pacientes randomizados para placebo passaram a receber 6 mg/kg de REMICADE® nas semanas 14, 16 e 20 e depois a cada 8 semanas até a Semana 44.
A segurança e a eficácia de REMICADE® no tratamento de crianças com ARJ não foram estabelecidas. Quarenta e uma das 60 crianças (68,3%) com ARJ recebendo 3 mg/kg de REMICADE® com metotrexato apresentaram uma infecção durante 52 semanas de observação, comparada a 37 das 57 (64,9%) crianças com ARJ recebendo 6 mg/kg de REMICADE® concomitante ao metotrexato durante 38 semanas de observação e 28 das 60 (46,7%) crianças recebendo placebo concomitante ao metotrexato durante 14 semanas de observação. As infecções mais comumente relatadas foram as do trato respiratório superior e faringite, sendo a pneumonia a infecção grave mais comumente relatada. Outras infecções relevantes incluíram varicela primária em 1 paciente e herpes zoster em outro.
A incidência de reações à infusão em pacientes pediátricos com ARJ recebendo 3 mg/kg de REMICADE® foi de 35,0% comparado com 17,5% dos que receberam 6 mg/kg. As reações à infusão mais comuns foram: vômito, febre, cefaleia e hipotensão. No grupo recebendo 3 mg/kg de REMICADE®, 4 pacientes tiveram reações graves à infusão e três deles relataram uma possível reação anafilática (2 dos quais estavam entre as reações graves à infusão). No grupo recebendo 6 mg/kg de REMICADE®, 2 pacientes tiveram reações graves à infusão, um dos quais apresentou uma possível reação anafilática. Dois dos 6 pacientes que tiveram reações graves à infusão receberam REMICADE® por infusão rápida (menos de 2 horas de duração).
Um total de 37,7% dos pacientes com ARJ recebendo 3 mg/kg de REMICADE® desenvolveram anticorpos contra o infliximabe comparado aos 12,2% dos pacientes recebendo 6 mg/kg. Os títulos dos anticorpos foi significativamente maior para o grupo de 3 mg/kg comparado ao de 6 mg/kg.
Reações adversas em doença de Crohn pediátrica: Em geral, os eventos adversos ocorridos em pacientes pediátricos que receberam REMICADE® foram semelhantes em frequência e tipo àqueles observados em pacientes adultos com doença de Crohn. As diferenças dos adultos e outras considerações especiais são discutidas nos parágrafos a seguir.
Os seguintes eventos adversos foram mais frequentemente relatados em 103 pacientes pediátricos randomizados, com doença de Crohn, recebendo 5 mg/kg de REMICADE® durante 54 semanas comparado a 385 pacientes adultos com doença de Crohn recebendo um regime de tratamento similar: anemia (10,7%), sangue nas fezes (9,7%), leucopenia (8,7%), rubor (8,7%), infecção viral (7,8%), neutropenia (6,8%), fratura óssea (6,8%), infecção bacteriana (5,8%) e reação alérgica do trato respiratório (5,8%).
Infecções foram relatadas em 56,3% dos pacientes randomizados no estudo REACH e em 50,3% dos pacientes recebendo 5 mg/kg de REMICADE® no estudo ACCENT 1. No estudo clínico REACH, infecções foram relatadas mais frequentemente por pacientes que receberam infusão a cada 8 semanas em oposição aos que receberam infusão a cada 12 semanas (73,6% e 38,0%, respectivamente), enquanto que infecções graves foram relatadas por 3 pacientes no grupo de tratamento de manutenção a cada 8 semanas e 4 no grupo de tratamento de manutenção a cada 12 semanas. As infecções mais comumente relatadas foram as do trato respiratório superior e faringite e a infecção séria mais comumente relatada foi abscesso. A pneumonia foi relatada em 3 pacientes, 2 no grupo de tratamento de manutenção a cada 8 semanas e 1 no grupo de tratamento de manutenção a cada 12 semanas. Herpes zoster foi relatada em 2 pacientes no grupo de tratamento de manutenção a cada 8 semanas.
Em geral, no estudo clínico REACH, 17,5% dos pacientes randomizados apresentaram uma ou mais reações à infusão, com 17,0% e 18,0% dos pacientes no grupo de tratamento de manutenção a cada 8 e 12 semanas, respectivamente. Não houve reações graves à infusão e 2 pacientes no estudo clínico REACH apresentaram reação anafilática não séria.
Três pacientes pediátricos (2,9%) desenvolveram anticorpos para REMICADE.
Experiência pós-comercialização: Os eventos adversos adicionais, alguns com desfecho fatal, relatados na experiência pós-comercialização de REMICADE®, com base em dados mundiais, estão apresentados na Tabela 8. Devido ao fato desses eventos serem relatados voluntariamente por uma população de tamanho incerto, nem sempre é possível estimar com confiança sua frequência ou estabelecer uma relação causal com a exposição ao REMICADE® .
Os eventos adversos graves mais comumente relatados na experiência pós-comercialização em crianças foram infecções (algumas fatais), incluindo infecções oportunistas e tuberculose; reações à infusão e reações de hipersensibilidade. Os eventos adversos graves e espontâneos, ocorridos na experiência pós-comercialização com REMICADE® na população pediátrica incluíram malignidades, anormalidades transitórias nas enzimas hepáticas, síndrome semelhante ao lúpus e autoanticorpos positivos.
Após o início da comercialização, casos raros de linfoma em células-T hepatoesplênicas foram relatados em pacientes com doença de Crohn e colite ulcerativa tratados com REMICADE® , sendo a maioria deles adolescentes ou adultos jovens, do sexo masculino.
Em casos de eventos adversos, notifique ao Sistema de Notificações em Vigilância Sanitária - NOTIVISA, disponível em www.anvisa.gov.br/hotsite/notivisa/index.htm, ou para a Vigilância Sanitária Estadual ou Municipal.
Superdose.
Doses únicas de até 20 mg/kg foram administradas a pacientes sem efeitos tóxicos. Em caso de superdose, recomenda-se que os pacientes sejam acompanhados em relação a sinais e sintomas de reações ou efeitos adversos e que seja instituído tratamento sintomático apropriado imediatamente.
Em caso de intoxicação ligue para 0800 722 6001, se você precisar de mais orientações.
Dizeres legais.
MS- 1.1236.3403.
Venda sob prescrição médica.