REKOVELLE
FERRING
deltafolitropina
Indicado para o desenvolvimento de múltiplos folículos maduros.
Apresentações.
Solução injetável 33,3 mg/mL disponível nas seguintes apresentações:
Rekovelle 12 microgramas: 0.36 mL de solução injetável em caneta aplicadora pré-envasada. Embalagem com 1 caneta aplicadora com Rekovelle e 3 agulhas para serem utilizadas com a caneta.
Rekovelle 36 microgramas: 1.08 mL de solução injetável em caneta aplicadora pré-envasada. Embalagem com 1 caneta aplicadora com Rekovelle e 6 agulhas para serem utilizadas com a caneta.
Rekovelle 72 microgramas: 2.16 mL de solução injetável em caneta aplicadora pré-envasada. Embalagem com 1 caneta aplicadora com Rekovelle e 9 agulhas para serem utilizadas com a caneta.
VIA SUBCUTÂNEA
USO ADULTO
Composição.
Cada mL de Rekovelle contém: deltafolitropina 33,3 mg. Excipientes: fenol, polissorbato 20, L-metionina, sulfato de sódio decahidratado, hidrogeno fosfato dissódico dodecahidratado, ácido fosfórico, hidróxido de sódio e água para injeção
Informações técnicas.
1. INDICAÇÕES
Este medicamento é indicado para o desenvolvimento de folículos múltiplos na estimulação ovariana controlada em mulheres sob tratamento de reprodução assistida (TRA), como na fertilização in vitro (FIV) ou no ciclo de injeção intracitoplasmática de espermatozoides (ICSI).
2. RESULTADOS DE EFICÁCIA
Eficácia e Segurança Clínica
O estudo ESTHER-1 foi um estudo randomizado, assessor-cego, controlado em 1.326 pacientes sob FIV/ICSI, comparando o esquema de dose individual de Rekovelle (com dose fixa) com um esquema de dose padrão de alfafolitropina padronizada por massa (dose inicial de 11 microgramas (150 UI) nos primeiros 5 dias seguido de ajustes de dose a partir do dia 6 da estimulação com base no desenvolvimento folicular). A faixa etária das pacientes era até 40 anos de idade (idade média de 33,3 ± 3,90) e possuíam ciclos menstruais regulares considerados ovulatórios. Da população estudada 59% era < 35 anos e 41% era ≥35 anos, dos quais 25% entre 35-37 anos e 16% era 38-40 anos. A transferência única de blastócito no dia 5 foi compulsória com exceção de pacientes entre 38-40 anos, nas quais foi realizada transferência dupla de blastócito, se não houvesse blastócito de boa qualidade disponível. Os dois desfechos co-primários foram índice de gravidez em andamento e índice de implantação em andamento, definido como pelo menos um feto intrauterino viável 10 - 11 semanas após a transferência e número de fetos intrauterinos viáveis 10-11 semanas após transferência dividido pelo número de blastócitos transferidos, respectivamente.
O estudo demonstrou que Rekovelle é, no mínimo, tão efetivo quanto a alfafolitropina em termos de índice de gravidez em andamento e índice de implantação em andamento, como mostra a Tabela 1.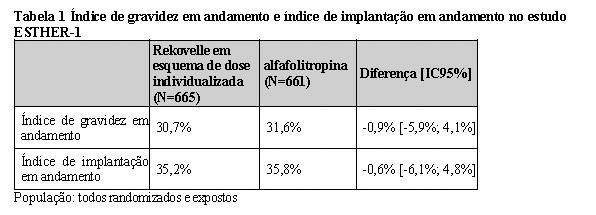
O valor clínico do esquema de dose de Rekovelle baseado no hormônio anti-Mülleriano (AMH - anti-Müllerian hormone) também foi avaliado no desfecho secundário, assim como a resposta ovariana, o gerenciamento do risco de síndrome de hiperestimulação ovariana (SHO) e consumo de gonadotropina.
Resposta ovariana e dose total de FSH
Resposta ovariana excessiva levando à indução com agonista de GnRH ocorreu em menor número em pacientes que receberam o esquema de dose individualizada de Rekovelle quando comparado ao esquema de dose de alfafolitropina (p < 0,05). Baixa resposta ovariana com consequente cancelamento do ciclo ocorreu em índices comparáveis em Rekovelle e alfafolitropina.
A média geral de número de oócitos recuperados foi similar em pacientes tratados com Rekovelle e alfafolitropina, com mais pacientes tratadas com Rekovelle alcançando 8-14 oócitos em comparação com alfafolitropina, com dose inicial de 11 microgramas (150 UI) e ajustes durante a estimulação (p < 0,05). A média da dose diária de Rekovelle foi de 0,16 mg/kg. A resposta ovariana e dose total de FSH e de acordo com a concentração de AMH estão dispostas na Tabela 2.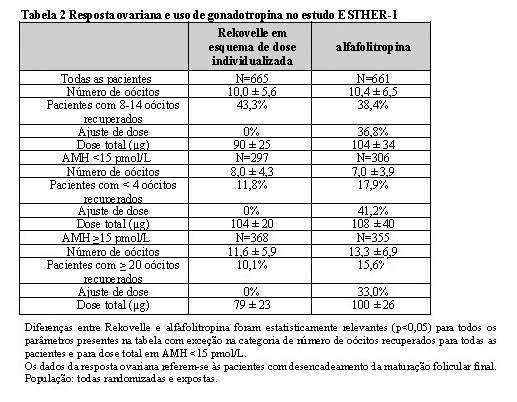
Segurança - gerenciamento de risco da SHO
A incidência de pacientes que necessitaram de intervenções preventivas para SHEO precoce, como para a indução com agonista GnRH ou administração de agonista de dopamina, foi reduzido em 50% nas pacientes tratadas com Rekovelle quando comparado com as pacientes tratadas com alfafolitropina (p < 0,05). A SHO precoce e/ou intervenções preventivas, assim como SHO precoce e tardia e/ou intervenções preventivas ocorreram em menor frequência com o esquema de dose individualizada de Rekovelle quando comparado ao esquema de dose padrão de alfafolitropina (p < 0,05). Os parâmetros para o gerenciamento de risco de SHO encontram-se resumidos na Tabela 3.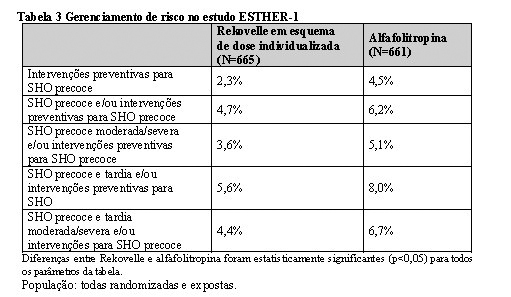
Segurança - Imunogenicidade
Anticorpos anti-FSH foram mensurados na pré-dose e pós-dose em pacientes submetidas a até três ciclos de tratamento repetidos com Rekovelle (665 pacientes no ciclo 1 no estudo ESTHER-1 assim como 252 pacientes no ciclo 2 e 95 pacientes no ciclo 3 no estudo ESTHER-2). A incidência de anticorpos anti-FSH após o tratamento com Rekovelle foi de 1,1% no ciclo 1, 0,8% no ciclo 2 e 1,1% no ciclo 3. Esses índices foram similares ao índice de anticorpos anti-FSH pré-existente anteriormente à exposição ao Rekovelle no ciclo 1, o qual foi de 1,4%, e comparável aos índices de anticorpos anti-FSH após o tratamento com alfafolitropina. Em todas as pacientes com anticorpos anti-FSH, os níveis eram indetectáveis ou muito baixos e sem capacidade neutralizante. O tratamento repetido com Rekovelle em pacientes com anticorpos anti-FSH pré-existente ou induzido por tratamento não ocasionou aumento do nível do anticorpo, não foi associado à diminuição da resposta ovariana e não induziu eventos adversos imuno relacionados.
Dados de segurança pré-clínica
Os dados não-clínicos não revelaram nenhum risco especial para humanos com base nos estudos convencionais de segurança farmacológica, toxicidade de dose repetida e de tolerância local.
A superdose de Rekovelle resultou em ação farmacológica ou ação farmacológica exagerada. Rekovelle demonstrou efeito negativo na fertilidade e no desenvolvimento precoce embrionário em ratos quando administrado em doses ≥ 0,8 mg/kg/dia, acima da dose máxima recomendada em humanos.
Considerando que Rekovelle é contraindicado durante a gravidez, estas observações possuem significância clínica limitada.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades farmacodinâmicas
Grupo farmacoterapêutico: Hormônio sexual, gonadotropina.
Mecanismo de ação
O efeito mais importante, resultado da administração parenteral do FSH, é o desenvolvimento de múltiplos folículos maduros.
O Rekovelle é um FSH humano recombinante produzido em uma linha celular humana por tecnologia de DNA recombinante. As sequências de aminoácido das duas subunidades de FSH do Rekovelle são idênticas às sequências do FSH endógeno recombinante. A linha celular expressante pode influenciar as características do FSH recombinante, e diferenças no perfil de glicosilação, padrão de ácido siálico e perfil de isoformas foram documentadas entre Rekovelle e produtos de FSH recombinante como alfafolitropina e betafolitropina em linhas celulares de ovário de hamster Chinês (CHO). A glicosilação de FSH no Rekovelle contém tanto o ácido siálico ligado à a2,3 como o ligado à a2,6 (o 2,6-ácido siálico está ausente no FSH recombinante derivado de CHO), diferentes açúcares como N-acetilgalactosamina, carregam ligações adicionais entre carboidratos como N-acetilglicosamina bissectante e a fucose ramificada, e tem uma maior proporção de estruturas tetra-antenárias e maior conteúdo geral de ácido siálico do que no FSH recombinante derivado de CHO.
Efeitos farmacodinâmicos
As comparações realizadas entre Rekovelle e alfafolitropina indicam que as diferenças na glicosilação influenciam tanto na farmacocinética quanto no perfil farmacodinâmico.
Após administração diária de doses iguais (UI) de Rekovelle e alfafolitropina como determinado em bioensaio in vivo realizado em ratos (ensaio de Steelman-Pohley), maior exposição FSH e maior resposta ovariana (por exemplo, estradiol, inibina B e volume folicular) foram observadas em pacientes após administração de Rekovelle quando comparado com alfafolitropina. Como o bioensaio em ratos pode não refletir totalmente a potência de FSH em Rekovelle em humanos, Rekovelle é dosado em microgramas e não em UI. Além do mais, a dose em microgramas de Rekovelle suficiente para obter a mesma resposta farmacodinâmica é 11 a 27% menor, considerando 11 microgramas (150 UI) de alfafolitropina em termos de desenvolvimento folicular e hormônios relacionados. Consequentemente, a dose recomendada de Rekovelle em microgramas não é aplicável para outras preparações de FSH recombinante.
O número de oócitos recuperados aumenta com a dose de Rekovelle e a concentração de AMH sérico. Inversamente, o aumento de peso corpóreo leva a uma diminuição dos oócitos recuperados (clinicamente relevante apenas para doses de Rekovelle abaixo de 12 microgramas).
Consequentemente, o esquema de dose de Rekovelle baseia-se na concentração de AMH sérico e, além disso, no peso corpóreo para doses abaixo de 12 microgramas.
Propriedades Farmacocinéticas
O perfil farmacocinético de Rekovelle foi investigado em pacientes sadios do sexo feminino e em pacientes em FIV/ICSI submetidas a COS. Após repetidas administrações subcutâneas, Rekovelle alcançou nível estacionário (steady state) dentro de 6 a 7 dias com uma concentração três vezes maior comparado com a concentração após a primeira dose. Os níveis séricos de Rekovelle são inversamente relacionados ao peso corpóreo, o qual suporta a individualização da dose com base no peso corpóreo.
Absorção
Após a administração subcutânea diária de Rekovelle, o tempo para concentração sérica máxima é de 10 horas. A biodisponibilidade absoluta está em torno de 64%.
Distribuição
O volume de distribuição no nível de equilíbrio estacionário está em torno de 9 L. Dentro da faixa de dose terapêutica, a exposição à Rekovelle aumenta proporcionalmente com a dose.
Eliminação
Após administração intravenosa, o clearance de Rekovelle é de 0,3 L/h. A meia-vida de eliminação final após administração subcutânea única é de 40 horas e após administrações subcutâneas múltiplas é de 28 horas. A comparação da farmacocinética de Rekovelle com a da alfafolitropina após administração subcutânea única da mesma dosagem em UIs por 7 dias, revelou que o clearance aparente é 1,6 vezes mais lento e a ASC e Cmax são 1,7 e 1,6 vezes maior para Rekovelle do que para alfafolitropina.
Espera-se que Rekovelle seja eliminado de forma similar às outras folitropinas, isto é, principalmente pela via renal. A fração de Rekovelle excretada de forma inalterada na urina foi estimada em 9%.
4. CONTRAINDICAÇÕES
Hipersensibilidade à substância ativa ou a qualquer outro ingrediente descrito na composição do produto.
• Tumores no hipotálamo ou na hipófise.
• Ovários aumentados ou presença de cisto ovariano não causado pela síndrome do ovário policístico.
• Hemorragias ginecológicas de etiologia desconhecida.
• Carcinoma ovariano, uterino ou mamário.
• Gravidez e lactação.
O Rekovelle não deve ser utilizado nos seguintes casos:
O Rekovelle não deve ser usado quando uma resposta efetiva não pode ser obtida, como nos seguintes casos:
• Insuficiência ovariana primária.
• Malformação dos órgãos sexuais tornando-os incompatíveis com gravidez.
• Tumores fibrosos no útero tornando-o incompatível com gravidez.
Este medicamento é contraindicado para uso por mulheres grávidas e/ou que estejam amamentando.
Categoria C para gravidez - Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
5. ADVERTÊNCIAS E PRECAUÇÕES
O Rekovelle contém uma potente substância gonadotrópica capaz de causar reações adversas moderadas a severas, e deve ser prescrito apenas por médicos completamente familiarizados com problema de infertilidade e seu gerenciamento.
A terapia com gonadotropina requer compromisso de tempo pelo médico e professional da saúde de suporte, assim como disponibilidade de instalações apropriadas para o devido monitoramento. A segurança e eficácia do uso de Rekovelle requer monitoramento da resposta ovariana com ultrassonografia isolada, ou em combinação com a medição dos níveis de estradiol sérico regularmente. A dose de Rekovelle deve ser individualizada para cada paciente a fim de obter uma resposta ovariana com perfil de segurança/eficácia favorável. Pode haver certo grau de variabilidade entre pacientes em resposta à administração de FSH, variando desde uma resposta baixa quanto uma resposta exagerada.
Não é recomendada a utilização de resultados obtidos com outros ensaios para a determinação da dose de Rekovelle, com exceção do imunoensaio ELECSYS® AMH Plus da Roche, visto que não existe atualmente uma uniformização dos ensaios do AMH disponíveis.
Antes do início do tratamento, a infertilidade do casal deve ser avaliada apropriadamente e as contraindicações putativas para gravidez avaliadas. Em particular, os pacientes devem ser avaliados quanto ao hipotireoidismo e hiperprolactnemia, caso necessário, o devido tratamento deve ser realizado.
Pacientes submetidas a estimulação de crescimento folicular podem apresentar aumento ovariano e correm o risco de desenvolver a síndrome de hiperestimulação ovariana. A aderência à dose de Rekovelle e ao regime de administração, assim como o monitoramento cuidadoso da terapia, minimizam a incidência de tais eventos.
Síndrome de Hiperestimulação Ovariana (SHO)
É esperado certo grau de aumento ovariano durante a estimulação ovariana controlada, sendo mais comum em pacientes com síndrome do ovário policístico e normalmente este efeito regride sem nenhum tratamento específico. Ao contrário do aumento ovariano descomplicado, a SHO é uma condição que pode se manifestar em crescentes níveis de severidade. Compreende o aumento ovariano agudo, altos níveis séricos de esteroides sexuais e aumento da permeabilidade vascular que pode resultar no acúmulo de fluídos no peritônio, pleura e, raramente, nas cavidades pericárdicas.
É importante enfatizar que cautela e o monitoramento frequente do desenvolvimento folicular são valores significantes para reduzir o risco de SHO. Os seguintes sintomas podem ser observados nos casos de SHO severa: dor, desconforto e distensão abdominal, aumento ovariano severo, ganho de peso, dispneia, oligúria e sintomas gastrointestinais incluindo náusea, vômito e diarreia. A avaliação clínica pode revelar hipovolemia, hemoconcentração, desequilíbrio eletrolítico, ascite, hemoperitôneo, efusão pleural, hidrotórax ou insuficiência pulmonar aguda.
Muito raramente, a SHO severa pode ser complicada pela torção ovariana ou eventos tromboembólicos como embolia pulmonar, acidente vascular cerebral isquêmico ou infarto do miocárdio. A resposta ovariana excessiva em resposta ao tratamento com gonadotropina raramente origina a SHO, a menos que seja administrado hCG para a indução final da maturação folicular. Além do mais, a síndrome pode ser mais severa e ser prolongada caso ocorra gravidez. Portanto, no caso de hiperestimulação ovariana, seria prudente suspender o hCG e aconselhar a paciente abster-se de coito ou fazer uso de um contraceptivo de barreira por pelo menos 4 dias. Outras medidas consideradas para reduzir o risco de SHO incluem a administração de agonista de GnRH ao invés de hCG para a indução final da maturação folicular. A administração de agonista de GnRH pode reduzir, mas não eliminar, o risco de SHO, sendo aplicável apenas para os ciclos de antagonista de GnRH.
A SHO pode progredir rapidamente (de 24 horas a vários dias) tornando-se um evento medico sério. Frequentemente ocorre após a descontinuação do tratamento hormonal. E também, o desenvolvimento tardio de SHO pode ocorrer como consequência das alterações hormonais durante a gravidez. Devido ao risco de desenvolver SHO, as pacientes devem ser acompanhadas por pelo menos duas semanas após a indução final da maturação folicular.
Eventos tromboembólicos
Mulheres com doença tromboembólica ou doença tromboembólica recente ou mulheres com risco conhecido para evento tromboembólico, como histórico familiar, obesidade severa (índice de massa corporal > 30kg/m2) ou trombofilia, podem ter risco aumentado para eventos tromboembólico arteriais ou venosos, durante ou após o tratamento com gonadotropina. O tratamento com gonadotropina pode aumentar adicionalmente o risco de ocorrer ou agravar tais eventos. Nessas mulheres, devem-se avaliar os benefícios da administração de gonadotropina contra seus respectivos riscos. Contudo, deve-se observar que a gravidez propriamente dita, assim como a SHO, também leva a um aumento no risco de eventos tromboembólicos.
Torção ovariana
A ocorrência de torção ovariana tem sido relatada durante os ciclos de tratamento de reprodução assistida. Pode estar associada com outros fatores de risco como a SHO, gravidez, cirurgia abdominal prévia, histórico passado de torção ovariana, cisto ovariano presente ou prévio e ovário policístico. Dano ovariano devido ao fluxo sanguíneo reduzido pode ser limitado pelo diagnóstico precoce e destorção imediata.
Gravidez múltipla
Gravidez múltipla traz um risco aumentado para desfechos adversos maternos e perinatais. Em pacientes submetidas a procedimentos para o tratamento de reprodução assistida o risco de gravidez múltipla relaciona-se tanto com o número de embriões substituídos, sua qualidade e idade do paciente, contudo a gravidez gemelar pode em raras ocasiões ocorrer a partir da transferência de um único embrião. As pacientes devem ser avisadas sobre o risco potencial de gravidez múltipla antes do início do tratamento.
Perda da gravidez
A incidência de perda de gravidez por aborto espontâneo é maior em pacientes submetidas a estimulação ovariana controlada durante o tratamento de reprodução assistida quando comparado com a concepção natural.
Gravidez ectópica
Mulheres com histórico de doença tubária correm risco de gravidez ectópica, independente de a gravidez ter sido obtida por concepção natural ou por tratamentos de fertilidade. A prevalência de gravidez ectópica após tratamento de reprodução assistida tem sido reportada como sendo maior do que na população em geral.
Neoplasmas no Sistema Reprodutivo
Existem relatos de neoplasmas ovarianos e em outros órgãos do Sistema reprodutivo, tanto benignos quanto malignos, em mulheres que se submeteram à múltiplos regimes de tratamento de infertilidade. Não foi estabelecido se o tratamento com gonadotropina aumenta o risco desses tumores em mulheres inférteis.
Malformação congênita
A prevalência de malformação congênita após tratamento de reprodução assistida pode ser ligeiramente maior quando comparado com a concepção espontânea. Isso pode ser devido às diferentes características paternais (por exemplo, idade maternal, características do esperma) e à gravidez múltipla.
Outras condições médicas
As condições médicas que contraindicam a gravidez também devem ser avaliadas antes do início do tratamento com Rekovelle.
Conteúdo de sódio
Rekovelle contém menos que 1 mmol de sódio (23 mg) por dose, isto é, essencialmente "livre de sódio".
Efeitos na capacidade de dirigir e operar máquinas
Espera-se que o Rekovelle tenha nenhuma influência ou influência negligenciável sobre a capacidade de dirigir e operar máquinas.
6. INTERAÇÕES MEDICAMENTOSAS
Não foram realizados estudos de interação medicamentosa com Rekovelle.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
O Rekovelle deve ser armazenado sob refrigeração (2°C - 8°C) e não deve ser congelado.
O Rekovelle possui prazo de validade de 36 meses quando seus cuidados de conservação são respeitados.
Antes do uso, manter o produto em sua embalagem original a fim de protegê-lo da luz.
Quando o carpule estiver em uso, mantê-lo dentro da caneta aplicadora de Rekovelle.
Após a primeira injeção o Rekovelle é válido por 28 dias quando armazenado abaixo de 30°C.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Após aberto (primeira aplicação), válido por até 28 dias.
Características físicas e organolépticas do produto
O Rekovelle é uma solução límpida e incolor, com pH entre 6,0 - 7,5.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
O tratamento com Rekovelle deve ser iniciado sob a supervisão de um médico experiente no tratamento de problemas de fertilidade. As pacientes devem ser devidamente orientadas sobre como usar a caneta de injeção de Rekovelle e em como realizar a administração.
Posologia
A posologia de Rekovelle é individualizada para cada paciente a fim de obter uma resposta ovariana com perfis de segurança e eficácia favoráveis.
O Rekovelle é dosado em microgramas (mg) e não em unidades internacionais (UI) de atividade biológica. O regime de dosagem é específico para Rekovelle e a dose em microgramas não pode ser aplicada à outras gonadotropinas.
Para o primeiro ciclo de tratamento, a dose individual diária será determinada com base no nível sérico do hormônio anti-Mülleriano (AMH - anti-Müllerian hormone) da mulher, o qual é utilizado como biomarcador da resposta ovariana à gonadotropina, em conjunto com o peso corpóreo da mulher. A dose deve ser baseada numa determinação recente de AMH (pelo menos dentro dos últimos 12 meses) mensurado através do teste de diagnóstico de imunoensaio ELECSYS® AMH Plus da Roche. A dose individual diária deve ser mantida durante o período de estimulação. Para mulheres com AMH < 15 pmol/L a dose diária é de 12 microgramas, independente do peso corpóreo. Para mulheres com AMH ≥15 pmol/L a dose diária diminui de 0,19 a 0,10 microgramas/kg corpóreo conforme a concentração de AMH aumenta (veja Tabela 1). A dose deve ser arredondada para o mais próximo de 0,33 microgramas para se equiparar à escala de dosagem da caneta de injeção. A dose máxima diária para o primeiro ciclo de tratamento é 12 microgramas.
A concentração de AMH deve ser expressa em pmol/L e deve ser arredondada para o número inteiro mais próximo (veja Tabela 1). Caso a concentração de AMH estiver em ng/mL, a concentração deve ser convertida para pmol/L multiplicando por 7,14 (ng/mL x 7,14 = pmol/L) antes do uso.
Para o cálculo da dosagem de Rekovelle, o peso corpóreo deve ser mensurado sem o uso de sapatos e casaco e logo antes o início da estimulação.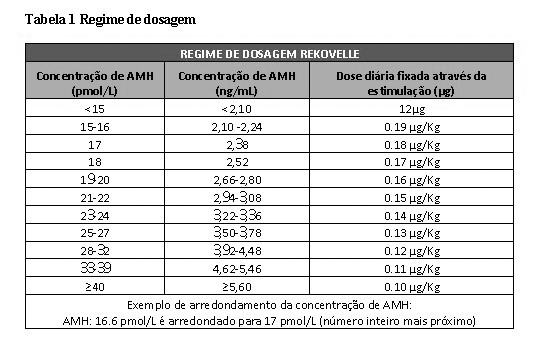
O tratamento com Rekovelle deve ser iniciado 2 ou 3 dias após o início do sangramento menstrual, e continuar até que o desenvolvimento folicular adequado seja alcançado, conforme avaliado pelo monitoramento de ultrassom isoladamente ou em combinação com a medida do nível sérico de estradiol. O desenvolvimento folicular adequado é alcançado em média no nono dia de tratamento (faixa de 5 a 20 dias). Assim que pelo menos 3 folículos ≥ 17 mm forem observados, uma injeção única de 250 microgramas de gonadotropina coriônica humana recombinante (hCG) ou 5.000 UI de hCG devem ser administrados para induzir a maturação folicular final. Em pacientes com resposta ovariana excessiva, com risco de desenvolver síndrome da hiperestimulação ovariana (SHO), deve-se considerar a administração de um agonista GnRH ao invés de hCG para induzir a maturação folicular final. A administração de agonista GnRH pode reduzir, mas não eliminar, o risco de SHO e é aplicável apenas para os ciclos de antagonistas de GnRH. No caso de administração de agonista de GnRH, os embriões não devem ser substituídos no ciclo fresco, mas criopreservado para uso posterior. Em pacientes com resposta ovariana excessiva com > 35 folículos de diâmetro ≥ 12 mm, não se deve realizar a maturação folicular final e o ciclo deve ser cancelado.
Para os ciclos de tratamento subsequentes, a dose diária de Rekovelle deve ser mantida ou modificada de acordo com a resposta ovariana da paciente no ciclo prévio. Se a paciente apresentou resposta ovariana adequada no ciclo prévio sem desenvolver SHO, a mesma dose diária de Rekovelle deve ser administrada. No caso de hiporresposta ovariana no ciclo de tratamento prévio, a dose diária de Rekovelle no ciclo subsequente deve ser aumentada em 25% ou 50%, dependendo da extensão da resposta observada. No caso de hiper-resposta ovariana no ciclo prévio, a dose diária de Rekovelle deve ser reduzida em 20% ou 33%, de acordo com a extensão da resposta observada. Em pacientes que desenvolveram SHO ou que apresentaram risco de desenvolver SHO no ciclo prévio, a dose de Rekovelle para o ciclo subsequente deve ser 33% menor do que a administrada no ciclo em que ocorreu a SHO ou o risco de desenvolvê-la. A dose máxima diária é 24 microgramas.
Não existe experiência em ensaios clínicos com Rekovelle no protocolo longo com agonistas da GnRH (veja item 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Pacientes com insuficiência renal ou hepática
A segurança, eficácia e farmacocinética de Rekovelle em pacientes com insuficiência renal ou hepática não foram estabelecidas.
Pacientes com Síndrome do Ovário Policístico com distúrbios anovulatórios
Pacientes com Síndrome do Ovário Policístico com desordem anovulatória não foram estudadas.
Uso em idosas (acima de 65 anos)
O uso de Rekovelle pela população idosa não é relevante. A segurança e eficácia de Rekovelle em pacientes idosas não foram estabelecidas.
Uso pediátrico
Considerando a indicação de Rekovelle, seu uso pela população pediátrica não é relevante.
Populações especiais
A experiência proveniente de estudo clínico de Rekovelle na população afrodescendente e hispânica é limitada. Contudo, durante a avaliação do medicamento e dos estudos clínicos foi considerado que a influência de fatores étnicos na segurança e eficácia de Rekovelle como pouco provável.
Modo de usar
Rekovelle deve ser administrado subcutaneamente, preferivelmente na parede abdominal. A primeira injeção de Rekovelle deve ser realizada sob supervisão médica direta. A autoadministração de Rekovelle deve ser realizada apenas por pacientes que estejam confiantes na aplicação, devidamente treinadas e com acesso a conselho de especialista.
Precauções especiais para descarte e informações adicionais
A solução não deve ser administrada caso contenha partículas ou caso não esteja límpida.
Qualquer solução remanescente no carpule deve ser descartada após 28 dias da primeira aplicação.
As agulhas utilizadas devem ser descartadas imediatamente após a aplicação.
Instruções de Uso da Caneta de Aplicação: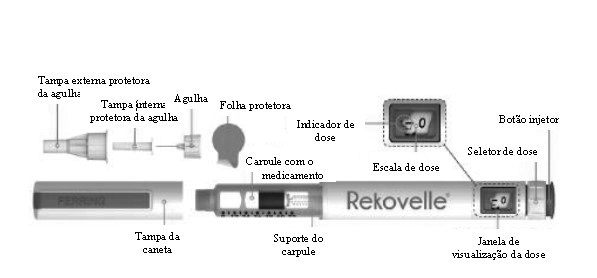
INSTRUÇÕES GERAIS:
• A caneta de Rekovelle deve ser utilizada apenas para fins de tratamento e apenas após o devido treinamento realizado por seu médico.
• A caneta de Rekovelle pré-envasada e respectivas agulhas devem ser utilizadas apenas por uma pessoa e não devem ser compartilhadas.
• Caso a paciente seja cega ou tenha problemas oftalmológicos que impossibilite o estabelecimento e a leitura da dose, garantir que a mesma tenha ajuda durante as aplicações e que este seja devidamente treinado pelo médico sobre como realizar o ajuste de dose e aplicação.
• A caneta de Rekovelle pode entregar doses de 0,33 microgramas a 20 microgramas de Rekovelle em acréscimos de 0,33 microgramas.
• A escala de dose da caneta é numerada de 0 a 20 microgramas.
• Cada número é separado por duas linhas, cada linha corresponde a um acréscimo de 0,33 microgramas.
• Quando girar o cursor para a dose correspondente, a paciente ouvirá um "clique" e sentirá uma resistência para girar para o próximo acréscimo, auxiliando que a dose correta seja estabelecida.
LIMPEZA:
• Caso necessário, a parte externa da caneta pode ser limpa com um pano umedecido com água.
• A caneta de Rekovelle não deve ser imersa em água e não deve estar em contato com nenhum outro líquido.
ARMAZENAGEM:
• A caneta deve ser armazenada sempre com tampa e sem agulha anexada.
• A caneta não deve ser utilizada após a data de validade impressa no rótulo.
• Não armazenar sob temperaturas extremas, sob luz solar ou em temperaturas muito baixas, como dentro de carro ou congelador.
• A caneta deve ser armazenada fora do alcance de crianças e de qualquer pessoas que não tenha sido treinada para o uso da caneta.
ANTES DE USAR:
• Armazene a caneta sob refrigeração, de 2°C a 8°C. Não congelar
APÓS O PRIMEIRO USO (CANETA EM USO):
• Após a primeira aplicação a caneta pode ser armazenada por até 28 dias sob temperatura de 2° a 30°C.
ANTES DO USO (ETAPA 1):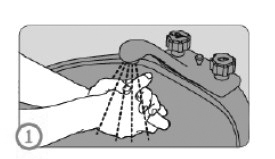
• Lavar as mãos.
• Checar se a caneta não está danificada. Não utilizar a caneta no caso de dano.
• Checar a caneta (carpule) para verificar se o medicamento está límpido e não contém partículas. Não utilizar a caneta caso o medicamento dentro do carpule não esteja límpido e livre de partículas.
• Garantir que a caneta correta com a dose correta está sendo manuseada.
• Checar a data de validade no rótulo da caneta.
ANEXANDO A AGULHA (ETAPAS 2 A 6)
Importante
• Sempre faça uso de uma agulha nova a cada injeção.
• Utilizar apenas a agulha que acompanha a caneta.
ETAPA 2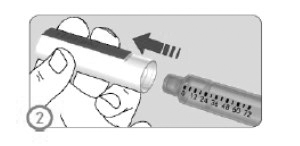
• Retirar a tampa da caneta.
ETAPA 3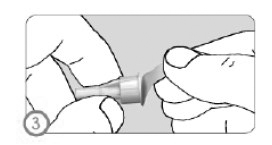
• Retirar a folha protetora da agulha.
ETAPA 4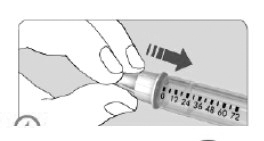
• Anexar a agulha à caneta.
• Será ouvido ou sentido um "clique" quando a agulha estiver seguramente anexada.
• A agulha também pode ser parafusada à caneta. Quando sentir uma certa resistência a agulha estará seguramente anexada.
ETAPA 5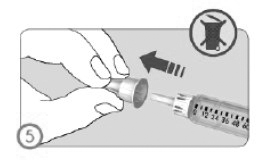
• Retirar a tampa externa protetora da agulha.
• Não descartar a tampa externa, ela será necessária para o descarte da agulha após a administração do medicamento.
ETAPA 6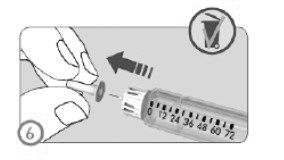
• Retirar a tampa interna protetora da agulha.
AJUSTANDO A CANETA (ETAPAS 7 A 9)
• Antes de usar a caneta pela primeira vez, será necessário remover as bolhas de ar do carpule para receber a dose correta do medicamento.
• O ajuste da caneta é necessário apenas no primeiro uso.
• As etapas 7 a 9 devem ser realizadas mesmo que a presença de bolhas de ar não sejam detectadas.
• Caso a caneta já tenha sido utilizada, proceda diretamente à etapa 10.
ETAPA 7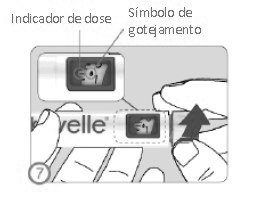
• Gire o seletor de dose até marcar o símbolo de gotejamento na janela de visualização da dose
ETAPA 8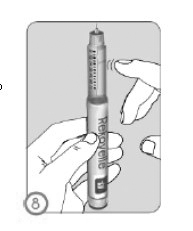
• Segure a caneta de Rekovelle com a agulha da caneta apontando para cima.
• Bata levemente no suporte do carpule para mover qualquer ar preso para o topo do carpule.
ETAPA 9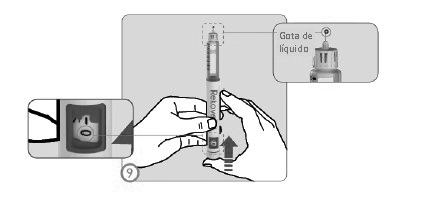
• Com a agulha apontada para cima (afastada do rosto) pressione o botão injetor até que o indicador de dose retorne ao número '0'.
• Checar que uma gota do líquido apareça na ponta da agulha.
• Caso nenhuma gota apareça, as etapas 7 a 9 devem ser refeitas até que uma gota apareça.
• Se após 5 tentativas a gota não aparecer, a agulha deve ser removida (vide Etapa 13), uma nova agulha deve ser anexada (vide Etapas 3 a 6) e um novo ajuste realizado (vide Etapas 7 a 9).
SELECIONANDO A DOSE (ETAPA 10)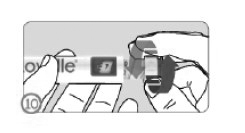
• Gire o seletor de dose no sentido horário até que a dose prescrita apareça no indicador de dose na janela de visualização da dose.
• A dose pode ser corrigida tanto para mais quanto para menos, girando o seletor de dose em uma das duas direções até que a dose esteja alinhada com a dose prescrita.
• Não pressione o botão injetor durante a seleção da dose para evitar perda de medicamento.
DOSE DIVIDIDA
• Pode ser necessária mais de uma caneta para completar a dose prescrita.
• Se não for possível selecionar a dose completa, isto significa que não há medicamento suficiente restante na caneta e, portanto, ou a dose será dividida ou a caneta em uso deverá ser descartada com o medicamento restante e uma nova caneta deve ser utilizada para a administração.
• Caso opte-se pela dose dividida, a paciente terá parte de sua dose administrada pela caneta em uso e o restante da dose administrada por uma caneta nova. Para tanto, siga as instruções abaixo, fazendo uso da tabela com exemplos para calcular a dose que será administrada por cada caneta:
- Na Coluna A encontram-se exemplos de doses prescritas pelo médico. Escreva qual a dose prescrita pelo seu médico.
- Na Coluna B encontram-se exemplos de doses restantes na canta em uso (que será igual a maior dose que você consegue selecionar na caneta).
- Escreva na Coluna B qual a dose restante na caneta em uso e realize a aplicação usando s dose remanescente da caneta em uso.
- Prepare e ajuste a nova caneta (Etapas 1 a 9)
- Calcule e escreva a dose faltante na Coluna C, subtraindo o número da Coluna B do número da Coluna A. Faça uso de uma calculadora para checar o resultado.
- A dose deve ser arredondada para o número mais próximo do acréscimo permitido pela caneta: X,00; X,33 ou X,66 microgramas. Por exemplo, se o número da Coluna C for 5,34 o arredondamento da dose faltante será 5,33. Se o número da Coluna C for 9,67 o arredondamento será para 9,66.
- Entre em contato com o médico no caso de qualquer dúvida sobre a divisão da dose.
- Proceda com a injeção da dose faltante (número da Coluna C) fazendo uso da caneta nova para completar a administração da dose prescrita.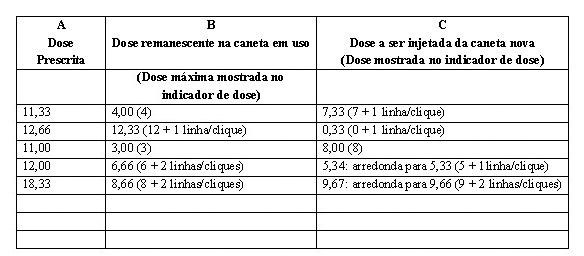
INJETANDO SUA DOSE (ETAPAS 11 A 12)
Importante
• O medicamento não deve ser utilizado caso apresentar partículas ou caso a solução não esteja límpida.
• Leia atentamente as Etapas 11 e 12 antes de realizar a injeção.
• Este medicamento deve ser administrado por injeção logo abaixo da pele (subcutaneamente) na região abdominal.
• Um novo local de injeção deve ser usado para cada injeção a fim de diminuir o risco de reações no local de injeção, como vermelhidão e irritação.
• A injeção não deve ser realizada em locais com feridas, sensíveis, com hematomas, vermelhidão, endurecidos, com cicatriz ou estrias.
ETAPAS 11 E 12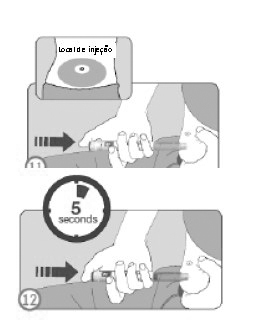
• O local da injeção deve ser limpo com algodão embebido em álcool e esta região não deve ser tocada até o momento da injeção.
• A caneta deve ser manuseada de modo que a janela de visualização da dose esteja visível.
• Deve realizar uma prega na pele da parede abdominal e a agulha da caneta deve ser inserida na pele da parede abdominal utilizando a técnica de injeção recomendada pelo seu médico. O botão injetor não deve ser pressionado ainda.
• Após a inserção da agulha na pele, o dedo polegar deve ser posicionado no botão injetor.
• O botão injetor deve ser pressionado completamente e mantido assim.
• Manter o botão injetor pressionado até o indicador de dose marcar '0', aguardar 5 segundos (contando lentamente) para garantir que a dose completa foi administrada.
• Após pressionar o botão por 5 segundos, soltar o botão injetor e lentamente remover a agulha do local de injeção, puxando-a para fora da pele.
• Caso aparecer sangue no local da injeção, o local deve ser levemente pressionado com gaze ou algodão.
Observações:
• A caneta não deve ser inclinada durante a injeção ou durante a remoção da agulha da pele.
• A inclinação da caneta pode fazer a agulha dobrar e até mesmo quebrar.
• Caso uma agulha quebrada permaneça presa no corpo ou embaixo da pele, deve-se procurar ajuda médica rapidamente.
DESCARTANDO A AGULHA (ETAPA 13)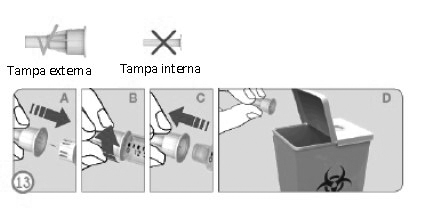
• Cuidadosamente recoloque a tampa protetora externa da agulha empurrando firmemente. (A)
• Desatarraxe a agulha da caneta na direção anti-horário para remover a agulha da caneta (B+C).
• Descarte cuidadosamente a agulha utilizada (D).
Observações:
Sempre remover a agulha logo após o uso.
A caneta não deve ser guardada com a agulha anexada.
RECOLOCANDO A TAMPA NA CANETA (ETAPA 14)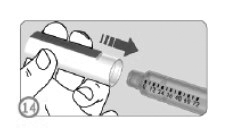
• Recoloque a tampa da caneta firmemente na caneta para protege-la entre as injeções.
Observações:
• A tampa da caneta não cabe na caneta com a agulha anexada.
• No caso de dose dividida, descarte a caneta apenas quando estiver vazia.
• Se uma nova caneta for utilizada para administrar a dose por completo ao invés da dose dividida, a caneta deve ser descartada quando não houver dose suficiente para administração completa.
• A caneta deve ser mantida tampada quando não estiver sendo utilizada.
Descarte
Agulhas:
As agulhas usadas e devidamente tampadas devem ser colocadas em um recipiente resistente a perfurações, como um recipiente para descarte de objetos cortantes imediatamente após o uso. O material cortante não deve ser descartado no lixo doméstico.
Caso a paciente não possuir recipiente para descarte de objetos cortantes, orientá-la a fazer uso de um recipiente doméstico que seja:
• Feito de plástico pesado/rígido
• Que possa ser fechado com tampa apertada e resistente a perfurações, sem que os objetos cortantes possam sair.
• Ereto e estável durante o uso.
• Resistente a vazamento.
• Devidamente rotulado/identificado para avisar de resíduos perigosos dentro do recipiente.
9. REAÇÕES ADVERSAS
As reações adversas mais comumente reportadas durante o tratamento com Rekovelle em estudos pivotais (1,012 ciclos) foram cefaleia (4,2%), desconforto pélvico (2,9%), síndrome da hiperestimulação ovariana (2,3%), dor pélvica (1,6%), náusea (1,4%), dor nos anexos uterinos (1,4%) e fadiga (1,2%).
A tabela abaixo (Tabela 2) apresenta as reações adversas em pacientes tratados com Rekovelle nos estudos clínicos pivotais de acordo com a Classe de Sistema de Órgãos e frequência; comum (≥1/100 a < 1/10) e incomum (≥1/1,000 a < 1/100). Dentro de cada grupo de frequência, as reações adversas estão dispostas em ordem decrescente de gravidade.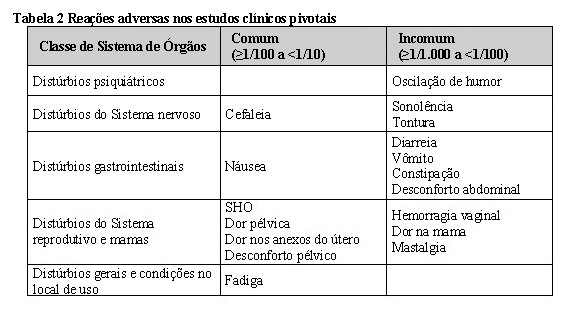
A SHO é um risco intrínseco da estimulação ovariana. Os sintomas gastrointestinais relacionados com a SHO conhecidos incluem dor, desconforto e distensão abdominal, náusea, vômito e diarreia.
A torção ovariana e eventos tromboembólicos são conhecidos como complicações raras no tratamento de estimulação ovariana.
A Imunogenicidade em termos de desenvolvimento de anticorpos anti-FSH é um r