PIQRAY
NOVARTIS
alpelisibe
Antineoplásico.
Apresentações.
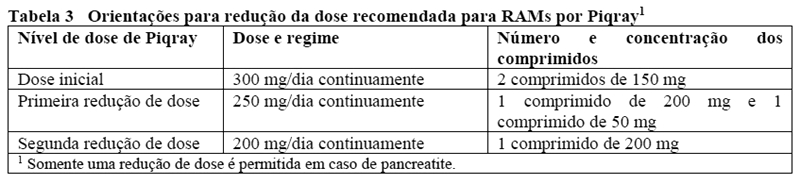
Piqray® 150 mg - embalagens contendo 28 ou 56 comprimidos revestidos de 150 mg.
Piqray® 200 mg + 50 mg - embalagens contendo 14 comprimidos revestidos de 200 mg e 14 comprimidos revestidos de 50 mg ou 28 comprimidos revestidos de 200 mg e 28 comprimidos revestidos de 50 mg.
Piqray® 200 mg - embalagens contendo 28 comprimidos revestidos de 200 mg.
VIA ORAL
USO ADULTO
Composição.
Cada comprimido revestido de Piqray® 50 mg, 150 mg e 200 mg contém, respectivamente, 50 mg, 150 mg e 200 mg de alpelisibe.
Excipientes: celulose microcristalina, manitol, amidoglicolato de sódio, hipromelose, estearato de magnésio, água purificada.
Excipientes do revestimento: hipromelose, macrogol, talco, óxido de ferro preto, óxido de ferro vermelho, dióxido de titânio.
Informações técnicas.
1. INDICAÇÕES
Piqray é indicado para o tratamento de mulheres na pós-menopausa e homens com câncer de mama avançado ou metastático com mutação PIK3CA, receptor hormonal (RH) positivo e receptor para o fator de crescimento epidérmico humano tipo 2 (HER2) negativo, em combinação com fulvestranto após progressão da doença que tenha ocorrido durante ou após o uso de terapia inicial de base endócrina.
2. RESULTADOS DE EFICÁCIA
Eficácia clínica e segurança
Estudo C2301 (SOLAR-1)
Piqray foi avaliado em um estudo pivotal de fase III, randomizado, duplo-cego, controlado por placebo de alpelisibe em combinação com fulvestranto em homens e mulheres na pós-menopausa com câncer de mama localmente avançado ou metastático positivo para HR e negativo para HER2, cuja doença havia progredido ou recidivado durante ou após um tratamento à base de inibidor da aromatase (com ou sem a combinação com inibidor de CDK4/6).
Um total de 572 pacientes foram incluídos em duas coortes, uma coorte com câncer de mama com a mutação PIK3CA e uma coorte com câncer de mama sem a mutação PIK3CA. O status da mutação PIK3CA foi determinado por ensaios do estudo clínico. Os pacientes foram randomizados para receber 300 mg de Piqray mais fulvestranto ou placebo mais fulvestranto na proporção de 1:1. A randomização foi estratificada com base na presença de metástases pulmonares e/ou hepáticas e tratamento anterior com inibidor(es) de CDK4/6.
Na coorte com a mutação PIK3CA, 169 pacientes foram randomizados para receber Piqray em combinação com fulvestranto e 172 pacientes foram randomizados para receber placebo em combinação com fulvestranto. Nesta coorte, 170 (49,9%) pacientes tinham metástases hepáticas/pulmonares e 20 (5,9%) pacientes haviam recebido tratamento anterior com inibidor de CDK4/6.
Na coorte sem mutação PIK3CA, 115 pacientes foram randomizados para receber Piqray em combinação com fulvestranto e 116 pacientes foram randomizados para receber placebo em combinação com fulvestranto. Nesta coorte, 112 (48,5%) pacientes tinham metástases hepáticas/pulmonares e 15 (6,5%) pacientes haviam recebido tratamento anterior com inibidor de CDK4/6.
Na coorte com mutação PIK3CA, 97,7% dos pacientes haviam recebido terapia hormonal anterior. 47,8% dos pacientes haviam progredido doença já no cenário metastático, enquanto que 51,9% progrediram após tratamento de adjuvancia. De modo geral, considerou-se que 85,6% dos pacientes tinham doença resistente a tratamento endócrino, resistência endócrina primária foi observada em 13,2% e resistência endócrina secundária em 72,4% dos pacientes.
Em ambas as coortes (com ou sem mutação PIK3CA), as características demográficas e avaliação inicial da doença, status de desempenho ECOG - Eastern Cooperative Oncology Group (eletrocorticografia), carga tumoral e terapia antineoplásica anterior foram bem equilibrados entre os braços do estudo.
Durante a fase de tratamento randomizado, uma dose de 300 mg de Piqray ou do placebo correspondente foi administrada oralmente, uma vez ao dia, em regime contínuo. Uma dose de 500 mg de fulvestranto foi administrada por via intramuscular nos dias 1 e 15 do ciclo 1 e depois no dia 1 de um ciclo de 28 dias durante a fase de tratamento (administração ± 3 dias).
Não foi permitida a troca de pacientes do braço do placebo para Piqray durante o estudo ou após a progressão da doença.
O desfecho primário do estudo foi a sobrevida livre de progressão (SLP) usando os Critérios de avaliação de resposta em tumores sólidos (RECIST v1.1), com base na avaliação do investigador em pacientes com câncer de mama avançado ou metastático com a mutação PIK3CA. O desfecho secundário principal foi a sobrevida global (SG) para pacientes com a mutação PIK3CA.
Outros desfechos secundários incluíram SLP para pacientes sem a mutação PIK3CA, SG para pacientes sem a mutação PIK3CA, bem como a taxa de resposta objetiva (TRO) e taxa de benefício clínico (TBC) na coorte de PIK3CA.
Coorte com a mutação PIK3CA
Os pacientes incluídos com uma mutação PIK3CA tinham idade mediana de 63 anos (intervalo: 25 a 92 anos). Desses pacientes, 44,9% tinham 65 anos ou mais e ≤ 85 anos. Os pacientes eram de etnia branca (66,3%), asiática (21,7%) e negra ou afro-americana (1,2%).
A duração mediana do acompanhamento na coorte com a mutação PIK3CA foi de 20 meses.
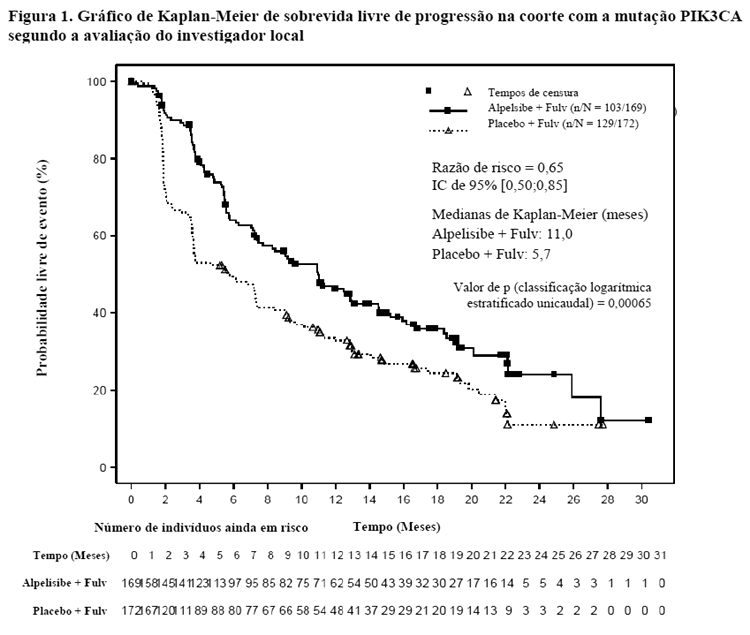
Os resultados de eficácia na coorte com a mutação PIK3CA demonstraram uma melhora estatisticamente significativa na SLP em pacientes que receberam Piqray mais fulvestranto em comparação com pacientes que receberam placebo mais fulvestranto (razão de risco [RR] de 0,65 com intervalo de confiança (IC) de 95%: 0,50, 0,85, teste de classificação logarítmica (log-rank) estratificado unicaudal p = 0,00065), com estimativa de redução do risco de progressão da doença ou óbito de 35%. Os resultados de eficácia do estudo estão resumidos na Tabela 1 e na Figura 1.
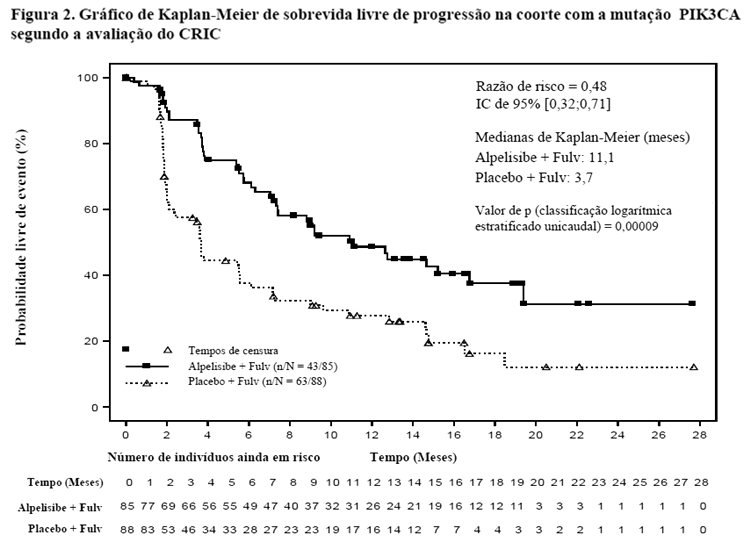
Os resultados primários de SLP para a coorte com a mutação PI3K3CA foram corroborados pelos resultados consistentes de uma avaliação do comitê de revisão independente em caráter cego (CRIC) nesta coorte, a qual incluiu um subconjunto selecionado aleatoriamente de 50% dos pacientes randomizados (RR: 0,48 com IC de 95%: 0,32, 0,71) (vide detalhes apresentados na Tabela 1 e Figura 2).
As análises dos subgrupos de SLP pelos fatores de estratificação da randomização demonstraram um efeito do tratamento homogêneo e consistente de modo geral, segundo a avaliação do investigador, independente de tratamento anterior com CDK4/6 e presença ou ausência de metástases pulmonares/hepáticas, embora o número de pacientes com tratamento anterior com CDK4/6 tenha sido limitado.
• Embora o número de pacientes seja limitado para a análise do subgrupo de tratamento anterior com inibidor de CDK4/6, a razão de risco (IC de 95%) foi 0,48 (0,17, 1,36).
• No subgrupo de pacientes com presença de metástases pulmonares/hepáticas, a RR(IC de 95%) foi de 0,62 (0,44, 0,89).
No momento da análise final de SLP, os dados de sobrevida global ainda não estavam maduros, com 92 de 178 óbitos para a análise final relatados, correspondendo a uma fração das informações de 51,7%. O limite de interrupção predefinido de O'Brien Fleming não foi ultrapassado na primeira análise preliminar da SG.
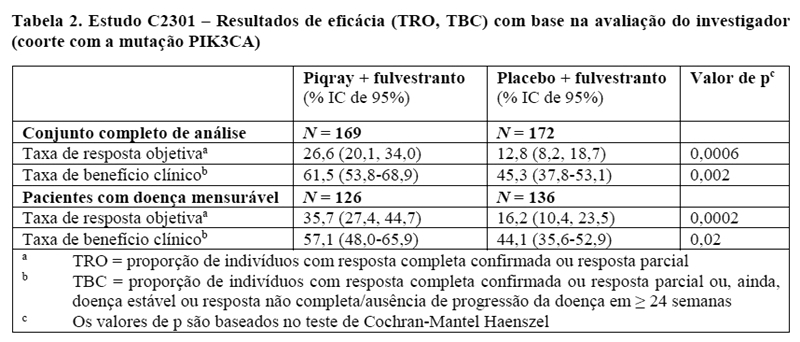
O tratamento com a combinação de Piqray mais fulvestranto foi associado com melhoras bem definidas na TRO e TBC em relação à combinação de placebo com fulvestranto. A TRO foi de 26,6% (IC de 95%: 20,1, 34,0) no braço de Piqray mais fulvestranto e 12,8% (IC de 95%: 8,2, 18,7) no braço do placebo mais fulvestranto (p = 0, 0006). A TBC foi de 61,5% (IC de 95%: 53,8, 68,9) no braço de Piqray mais fulvestranto e 45,3% (IC de 95%: 37,8, 53,1) no braço do placebo mais fulvestranto (p = 0,002) (vide detalhes apresentados na Tabela 2).
Em pacientes com doença mensurável na avaliação inicial, a TRO foi de 35,7% (IC de 95%: 27,4, 44,7) no braço de Piqray mais fulvestranto e 16,2% (IC de 95%: 10,4, 23,5) no braço do placebo mais fulvestranto (p = 0,0002). A TBC foi de 57,1% (IC de 95%: 48,0, 65,9) no braço de Piqray mais fulvestranto e 44,1% (IC de 95%: 35,6, 52,9) no braço do placebo mais fulvestranto (p = 0,02).
As análises de subgrupo de SLP também demonstraram um efeito de tratamento homogêneo e consistente de modo geral em todos os principais subgrupos demográficos e com outros prognósticos.
Os resultados de estado geral de saúde/qualidade de vida (QoL - quality of life) foram similares entre o braço de Piqray mais fulvestranto e o braço do placebo mais fulvestranto. O tempo até a deterioração (TTD) no estado geral de saúde, segundo o questionário EORTC QLQ-C30, foi definido como o tempo entre a avaliação inicial e a primeira ocorrência de agravamento ≥ 10 pontos do estado geral de saúde (pontuação na escala de saúde geral EORTC QLQ-C30) em relação à avaliação inicial, sem melhora posterior acima desse limite durante o período de tratamento ou óbito por qualquer causa. A combinação de Piqray com o fulvestranto não demonstrou diferença relevante no TTD na pontuação na escala de saúde geral EORTC QLQ C30 em comparação com o placebo mais fulvestranto (RR=1,03 [0,72, 1,48]).
Coorte sem a mutação PIK3CA
Os critérios da prova de conceito para concluir que há um benefício no tratamento com Piqray e fulvestranto com relação à SLP em indivíduos na coorte sem a mutação PIK3CA não foram atendidos (RR = 0,85; IC de 95%: 0,58, 1,25) (vide seção 8. Posologia e Modo de Usar).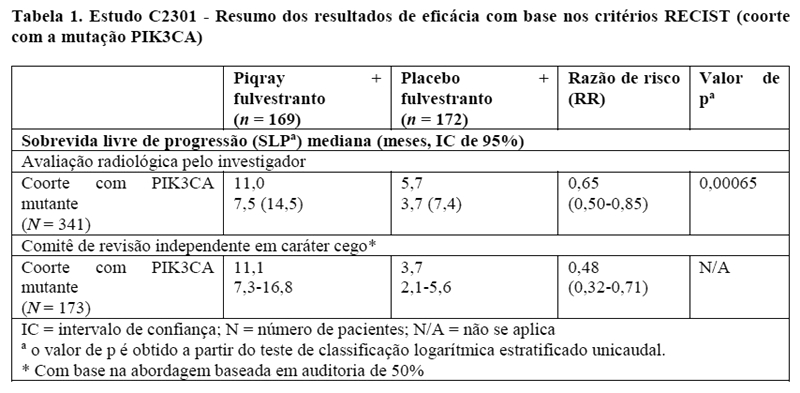
Referências bibliográficas
Summary of Clinical Efficacy - BYL719 - 2.7.3 Summary of Clinical Efficacy. Novartis. 2018.
Andre F, Ciruelos Gil EM, Rubovsky G, et al. Alpelisib (ALP) + fulvestrant (FUL) for advanced breast cancer (ABC): results of the Phase III SOLAR-1 trial. Presented at European Society of Medical Oncology (ESMO) 2018 Congress;October 19-23, 2018.Abstract:LBA3_PR.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Grupo farmacoterapêutico: Agentes antineoplásicos, outros agentes antineoplásicos. Código ATC: L01XX47.
Mecanismo de ação
O alpelisibe é um inibidor a específico de classe I da fosfatidilinositol 3-quinase (PI3Kalfa). As quinases lipídicas PI3K de classe I são componentes chave da via de sinalização PI3K/AKT/mTOR (alvo de mamíferos da rapamicina). Mutações que promovem ganho de função no gene que codifica a subunidade alfa catalítica da PI3K (PIK3CA) levam à ativação da PI3Kalfa manifestada pelo aumento da atividade da quinase lipídica, da ativação independente de fator de crescimento da sinalização da AKT, da transformação celular e da geração de tumores em diversos tipos de modelos pré-clínicos.
In vitro, o tratamento com o alpelisibe inibiu de forma potente a fosforilação do alvo AKT no downstream da PI3K, bem como de seus vários efetores downstream em células de câncer de mama e demonstrou seletividade em relação à linhagens celulares que abrigam uma mutação de PIK3CA.
In vivo, o alpelisibe mostrou tolerabilidade, bem como inibição dependente de dose e tempo da via da PI3K/AKT e inibição do crescimento tumoral dependente da dose em modelos de xenoenxertos tumorais relevantes, inclusive modelos de câncer de mama.
Demonstrou-se que a inibição da PI3K pelo tratamento com alpelisibe induz um aumento na transcrição do receptor estrogênico (RE) em células de câncer de mama e, portanto, sensibiliza tais células à inibição do RE pelo tratamento com fulvestranto. A combinação de alpelisibe e fulvestranto demonstrou maior atividade antitumoral que qualquer um dos tratamentos isolado em modelos de xenoenxerto derivados de linhagens celulares de câncer de mama RE positivas e com mutação PIK3CA (MCF-7 e KPL1).
Efeitos farmacodinâmicos
Em ensaios bioquímicos, o alpelisibe inibiu o tipo selvagem de PI3Kalfa (CI50=4,6 nmol/L) e suas duas mutações somáticas mais comuns (H1047R, E545K) (CI50 aprox. 4 nmol/L) de maneira mais potente do que as isoformas PI3Kdelta (CI50=290 nmol/L) e PI3Kgamma (CI50=250 nmol/L) e demonstrou atividade significativamente reduzida contra PI3Kbeta (CI50=1156 nmol/L).
A potência e seletividade do alpelisibe foram confirmadas à nível celular em linhagens celulares tumorais mecanísticas e relevantes.
Eletrofisiologia cardíaca
Foram coletados ECGs (eletrocardiogramas) seriados, em triplicata, após uma dose única e no estado estacionário para avaliar o efeito do alpelisibe sobre o intervalo QTcF em pacientes com câncer avançado. A análise farmacocinética-farmacodinâmica incluiu um total de 134 pacientes tratados com doses de alpelisibe variando entre 30 mg e 450 mg.
As análises demonstraram a ausência de um prolongamento de QTcF clinicamente significativo na dose recomendada de 300 mg com ou sem fulvestranto. A média estimada da alteração do intervalo QTcF em relação à avaliação inicial foi < 10 ms (7,2 ms; IC de 90%: 5,62, 8,83) na Cmáx geométrica média observada no estado estacionário (2900 ng/mL) após a administração de agente único na dose recomendada de 300 mg.
Propriedades farmacocinéticas
A farmacocinética do alpelisibe foi investigada em pacientes sob um regime de administração oral de doses variando de 30 a 450 mg diariamente. Indivíduos saudáveis receberam doses orais únicas variando de 300 a 400 mg. A farmacocinética foi comparável em pacientes oncológicos e indivíduos saudáveis.
Absorção
Após a administração oral do alpelisibe, o tempo mediano para atingir o pico de concentração plasmática (Tmáx) variou entre 2,0 a 4,0 horas, independentemente da dose, tempo ou regime. Com base em modelos de absorção, a biodisponibilidade foi estimada como muito alta ( > 99%) em condições após refeições, mas mais baixa em condições de jejum (aprox. 68,7% com uma dose de 300 mg). Pode-se esperar que níveis plasmáticos de alpelisibe no estado estacionário, após a administração diária, sejam atingidos no terceiro dia após o início do tratamento na maioria dos pacientes.
Efeito da ingestão alimentar
A absorção de alpelisibe é afetada pela alimentação. Em voluntários sadios, após uma dose única oral de 300 mg de alpelisibe, comparado à condição em jejum, uma refeição com alto teor de gordura e caloria (985 calorias com 58,1 g de gordura) aumentou a ASCinf em 73% e a Cmáx em 84%, e uma refeição com baixo teor de gordura e caloria (334 calorias com 8,7 g de gordura) aumentou a ASCinf em 77% e a Cmáx em 145%. Não foi identificada diferença significativa na ASCinf entre os dois tipos de refeição, com uma proporção geométrica média de 0,978 (IC: 0,876, 1,09), demonstrando que nem o teor de gordura e nem a ingestão calórica de modo geral têm um impacto considerável na absorção. O aumento na solubilidade gastrointestinal pela bile, secretada em resposta à ingestão de alimentos, é a causa potencial do efeito da ingestão de alimentos. Portanto, Piqray deve ser tomado imediatamente após refeições, aproximadamente no mesmo horário todos os dias.
Agentes redutores de pH
O Piqray pode ser coadministrado com medicamentos que sejam agente redutores da acidez, se o Piqray for tomado imediatamente após alimentação. A coadministração do antagonista do receptor de H2 ranitidina em combinaç com uma dose oral única de 300 mg de alpelisibe reduziu ligeiramente a biodisponibilidade do alpelisibe e diminuiu a exposição ao alpelisibe de forma geral. Na presença de uma refeição de baixo teor de gordura e caloria, a ASCinf foi diminuída em média em 21% e a Cmáx em 36% com a ranitidina. Na ausência de alimentos, o efeito foi mais proeminente com uma redução de 30% na ASCinf e uma diminuição de 51% na Cmáx com a ranitidina em comparação com a condição em jejum sem a coadministração de ranitidina. A análise da farmacocinética populacional não revelou efeito significativo da coadministração de agentes redutores da acidez, inclusive inibidores da bomba de prótons, antagonistas do receptor de H2 e antiácidos, na farmacocinética de Piqray.
Distribuição
O alpelisibe se liga moderadamente a proteínas com uma fração livre de 10,8% independentemente da concentração. O alpelisibe foi distribuído igualmente entre os glóbulos vermelhos do sangue e o plasma, com uma razão média sangue-plasma in vivo de 1,03. Não houve evidência de distribuição nos glóbulos vermelhos causada por metabólitos. O alpelisibe não penetrou na barreira hematoencefálica em ratos. Como o Piqray é um substrato de transportadores de efluxo humano, não se espera que haja penetração da barreira hematoencefálica em humanos. O volume de distribuição do alpelisibe no estado estacionário (Vss/F) é estimado em 114 litros (CV% entre indivídos 46%).
Biotransformação
Estudos in vitro demonstraram que a formação por hidrólise do metabólito BZG791 por hidrólise química e enzimática de amida foi uma via metabólica importante, seguida de uma contribuição menos significativa da CYP3A4. A hidrólise do alpelisibe ocorreu sistematicamente tanto por decomposição química como por hidrólise enzimática através de enzimas de alta capacidade ubiquamente expressas (esterases, amidases, colina esterase), não limitadas ao fígado. Os metabólitos mediados pela CYP3A4 e as glicuronidas somaram aproximadamente 15% da dose; o BZG791 representou cerca de 40% a 45% da dose. O restante da fração absorvida da dose foi excretada como alpelisibe.
Eliminação
O alpelisibe exibe taxa de eliminação baixa, com 9,2l/h (CV% 21%), com base na análise de farmacocinética populacional em condições após refeição. A meia-vida derivada da população, independente de dose e tempo, foi de 8 a 9 horas no estado estacionário com uma dose de 300 mg uma vez ao dia.
Em um estudo de equilíbrio de massa em humanos, após a administração oral, o alpelisibe e seus metabólitos foram excretados nas fezes (81,0%), principalmente através de exportação hepatobiliar e/ou secreção intestinal do alpelisibe, ou metabolizado em BZG791. A excreção na urina é pouco significativa (13,5%), com alpelisibe inalterado (2%). Após uma dose oral única de alpelisibe marcado com [14C], 94,5% da dose radioativa total administrada foi recuperada dentro de 8 dias.
Linearidade/não-linearidade
Verificou-se que a farmacocinética é linear com relação à dose e tempo em condições após refeição entre 30 mg e 450 mg. Após múltiplas doses, a exposição ao alpelisibe (ASC) em estado estacionário é apenas ligeiramente maior do que a exposição com uma dose única, com um acúmulo médio de 1,3 a 1,5 em um regime de dose diária.
Interação metabólica
Com base nos resultados de estudos metabólicos in vitro de indução e inibição, o alpelisibe pode induzir à depuração metabólica de medicamentos coadministrados metabolizados por CYP2B6, CYP2C9 e CYP3A e pode inibir a eliminação metabólica de medicamentos coadministrados metabolizados pelo CYP3A4 (inibição dependente do tempo) se concentrações suficientemente altas forem atingidas in vivo.
Em um estudo de interação medicamentosa, a coadministração de alpelisibe com everolimo, um substrato sensível de CYP3A4, confirmou a ausência de interações farmacocinéticas clinicamente significativas (aumento na ASC em 11,2%) entre o alpelisibe e substratos do CYP3A4. Não foi observada alteração na exposição ao everolimo com doses de alpelisibe variando de 250 mg a 300 mg, o que também foi confirmado por modelagem PBPK com everolimo e midazolam (aumento ≤ 15% na ASC). Devido à indução concorrente e à inibição dependente do tempo pelo alpelisibe, simulações PBPK com substratos de CYP3A4, que também possuem inibição adicional dependente do tempo e potencial de indução da CYP3A4, que afeta o seu próprio metabolismo, preveem alterações na exposição (diminuição ou aumento) inferiores a 2 vezes, dependendo do substrato.
Substratos do CYP2C9
Avaliações in vitro indicaram que a atividade farmacológica pode ser reduzida pelos efeitos de indução da CYP2C9 pelo alpelisibe.
Interação baseada em transportador
O alpelisibe demonstrou apenas inibição fraca in vitro de transportadores de efluxo expressos ubiquamente (P-gp, BCRP, MRP2, BSEP), transportadores carreadores de solutos na entrada do fígado (OATP1B1, OATP1B3, OCT1) e transportadores carreadores de solutos no rim (OAT1, OAT3, OCT2, MATE1, MATE2K). Como as concentrações sistêmicas de alpelisibe não ligadas no estado estacionário (ou concentrações na entrada do fígado), tanto na dose terapêutica e quanto na dose máxima tolerada, são significativamente menores do que as constantes de inibição não ligada ou CI50 determinadas experimentalmente, a inibição não se traduzirá em relevância clínica. Pode-se excluir um efeito clinicamente relevante sobre os substratos de P-gp.
Fulvestranto
Dados de um estudo clínico em pacientes com câncer de mama não indicaram nenhum efeito do fulvestranto sobre a exposição ao alpelisibe (e vice-versa) após a coadministração desses medicamentos.
Populações especiais
Efeitos da idade, peso e gênero
A análise farmacocinética populacional mostrou que não existem efeitos clinicamente relevantes da idade, do peso corporal ou do gênero sobre a exposição sistêmica do alpelisibe que possam requerer o ajuste de dose de Piqray.
Pacientes pediátricos (abaxio de 18 anos)
A farmacocinética de Piqray em pacientes pediátricos não foi determinada.
Pacientes idosos (65 anos ou mais)
Dos 284 pacientes que receberam Piqray no estudo de fase III (no braço de Piqray mais fulvestranto), 117 pacientes tinham ≥ 65 anos de idade e 34 pacientes tinham ≥ 75 anos de idade. Em pacientes tratados com Piqray em combinação com fulvestranto, houve uma maior incidência de hiperglicemia de Grau 3 e 4 em pacientes com ≥ 65 anos de idade (44%) em comparação com pacientes com < 65 anos de idade (32%). Não foi observada nenhuma diferença de modo geral na segurança e eficácia de Piqray entre esses pacientes e pacientes mais jovens (vide seção 8. Posologia e modo de usar).
Raça/Etnia
Análises farmacocinéticas populacionais e análises farmacocinéticas de um estudo de agente único em pacientes oncológicos japoneses mostraram que não há efeitos clinicamente relevantes da etnia sobre a exposição sistêmica de Piqray.
Os parâmetros farmacocinéticos não compartimentais após doses diárias únicas e múltiplas de Piqray em pacientes japoneses foram bastante semelhantes aos parâmetros relatados na população caucasiana.
Insuficiência renal
Não é necessário nenhum ajuste de dose em pacientes com insuficiência renal leve ou moderada (CLcr 30 a < 90 mL/min). Pacientes com insuficiência renal severa (CLcr < 30 mL/min) não foram estudados e deve-se ter cautela. Com base em uma análise farmacocinética populacional que incluiu 117 pacientes com função renal normal (eGFR [taxa estimada de filtração glomerular] ≥ 90 mL/min/1,73 m2) / (CLcr ≥ 90 mL/min), 108 pacientes com insuficiência renal leve (eGFR 60 a < 90 mL/min/1,73 m2) / (CLcr 60 a < 90 mL/min) e 45 pacientes com insuficiência renal moderada (eGFR 30 a < 60 mL/min/1.73 m2), a insuficiência renal leve e moderada não teve efeito sobre a exposição ao alpelisibe (vide seção 8. Posologia e modo de usar).
Insuficiência hepática
Não é necessário nenhum ajuste de dose em pacientes com insuficiência hepática leve, moderada ou severa (Child-Pugh A, B e C).
Com base em um estudo farmacocinético em pacientes com insuficiência hepática, a insuficiência hepática moderada e severa teve efeito negligenciável sobre a exposição ao alpelisibe (vide seção 8. Posologia e modo de usar). A exposição média ao alpelisibe aumentou em 1,26 vezes em pacientes com insuficiência hepática severa (IMG[razão média geométrica]: 1,00 para Cmáx; 1,26 para ASCúltima/ASCinf).
Com base em uma análise farmacocinética populacional que incluiu 230 pacientes com função hepática normal, 45 pacientes com insuficiência hepática leve e nenhum paciente com insuficiência hepática moderada, o que corrobora com os achados do estudo específico de insuficiência hepática, a insuficiência hepática leve e moderada não teve efeito sobre a exposição ao alpelisibe (vide seção 8. Posologia e modo de usar).
Dados de segurança pré-clínica
O alpelisibe foi avaliado em estudos de farmacologia de segurança, toxicidade de dose única e doses repetidas, genotoxicidade e fototoxicidade.
Farmacologia de segurança e toxicidade de doses repetidas
A maioria dos efeitos observados do alpelisibe estavam relacionados à atividade farmacológica do alpelisibe como um inibidor específico de p110alfa da via da PI3K, como a influência sobre a homeostase da glicose, resultando em hiperglicemia e risco de aumento da pressão arterial.
A medula óssea e o tecido linfoide, o pâncreas e alguns órgãos reprodutivos em ambos os sexos foram os principais órgãos alvo de efeitos adversos que, em geral, foram reversíveis com a interrupção do tratamento. O alpelisibe não demonstrou nenhum efeito sobre as funções neuronal e pulmonar. Não foi observada evidência de irritação ou corrosão cutânea com alpelisibe.
Farmacologia de segurança cardiovascular
Em um teste in vitro de hERG (no qual avalia-se a funcionalidade do canal de hERG (gene humano relacionado ao ether-a-go-go) cardíaco humano expresso de forma heteróloga em células HEK293 in vitro), uma CI50 de 9,4 microM (4,2 micrograma/mL) foi encontrada. Não foi observado efeito eletrofisiológico relevante em cães em diversos estudos, com doses únicas de até 180 mg/kg in vivo. Um estudo de telemetria in vivo em cães revelou pressão arterial elevada, começando com uma exposição menor que a exposição em humanos, na dose máxima recomendada de 300 mg/dia.
Carcinogenicidade e mutagenicidade
Não foram realizados estudos de carcinogenicidade.
O alpelisibe não foi mutagênico em um teste de mutação reversa de Salmonella em cinco cepas, nem aneugênico ou clastogênico em testes in vitro de micronúcleos de células humanas e de aberração cromossômica. Além disso, um teste de micronúcleo in vivo em reticulócitos de sangue periférico, obtidos na semana 4 de um estudo de 13 semanas de toxicidade de doses repetidas em ratos, com doses de até 20 mg/kg/dia, com níveis de exposição plasmática de cerca de 1,7 vezes a exposição em humanos com a dose máxima recomendada de 300 mg/dia com base na ASC, foi negativo.
Toxicidade reprodutiva
Estudos de desenvolvimento embriofetal em ratos e coelhos demonstraram que a administração oral do alpelisibe durante a organogênese induziu à embriotoxicidade, fetotoxicidade e teratogenicidade. Em ratos e coelhos, após a exposição pré-natal ao alpelisibe, foram observadas incidências elevadas de perda pós-implantação e redução do peso fetal, bem como incidências aumentadas de anormalidades fetais, começando em exposições de doses abaixo da exposição em humanos com a dose máxima recomendada de 300 mg.
Em estudos de desenvolvimento embriofetal em ratos e coelhos, animais fêmeas grávidas receberam doses orais de alpelisibe de até 30 mg/kg/dia durante o período de organogênese.
Em ratos, a administração oral do alpelisibe foi associada à perda ou estagnação do peso corporal materno, baixa ingestão alimentar e morte embrionária com 30 mg/kg/dia, cerca de 3,2 vezes (com base na ASC) a exposição em humanos na dose máxima recomendada de 300 mg. Baixo ganho de peso corporal materno, incidências aumentadas de ventrículo cerebral aumentado em fetos, peso fetal reduzido, diminuição da ossificação e malformações esqueléticas foram observados com 10 mg/kg/dia, o que é igual a aproximadamente 0,9 vezes abaixo da exposição em humanos com a dose máxima recomendada.
Em coelhos, com doses ≥ 25 mg/kg/dia, observou-se perda de peso corporal materno com redução da ingestão alimentar. Com 15 mg/kg/dia, observou-se uma pequena variação no peso corporal. Com ≥ 15 mg/kg/dia, houve aumento das mortes e malformações embriofetais, em sua maioria relacionadas à cauda e à cabeça, e foram associadas ao aumento dos níveis de glicose sérica nas gestantes. Com 25 mg/kg/dia, observou-se redução do peso fetal médio. A dose de 15 mg/kg/dia em coelhos é equivalente a aproximadamente cerca de 5,5 vezes (com base na ASC) a exposição atingida na dose máxima recomendada em humanos.
Em ratos e coelhos, não foram observados efeitos fetais na dose de 3 mg/kg/dia e esta foi considerada como o nível sem efeitos adversos observáveis (NOAEL) para anormalidadeas fetais. As exposições maternas sistêmicas (ASC) no NOAEL foram 0,12 (ratos) ou 0,86 (coelhos) vezes a exposição em humanos na dose máxima recomendada de 300 mg.
Infertilidade
Não foi realizado um estudo de fertilidade em ratos. No entanto, em estudos de toxicidade de doses repetidas de até 13 semanas de duração, foram observados efeitos adversos nos órgãos reprodutivos de machos e fêmeas, como atrofia vaginal e variações no ciclo menstrual em ratas (em doses de 6 mg/kg/dia ou acima, uma dose que fornece níveis de exposição plasmática abaixo da exposição em humanos na dose máxima recomendada de 300 mg/dia com base na ASC), ou atrofia de próstata em cães (com 15 mg/kg/dia, em níveis de exposição plasmática 2,8 vezes a exposição em humanos na dose máxima recomendada de 300 mg/dia com base na ASC) (vide seção 5. Advertências e Precauções).
Fototoxicidade
Um teste de fototoxicidade in vitro na linhagem celular de fibroblastos de camundongos Balb/c 3T3 não identificou um potencial de fototoxicidade relevante para o alpelisibe.
Estudos em animais jovens
Estudos em animais jovens não estão disponíveis.
4. CONTRAINDICAÇÕES
Hipersensibilidade ao princípio ativo ou a qualquer um dos excipientes.
5. ADVERTÊNCIAS E PRECAUÇÕES
Hipersensibilidade (incluindo reação anafilática)
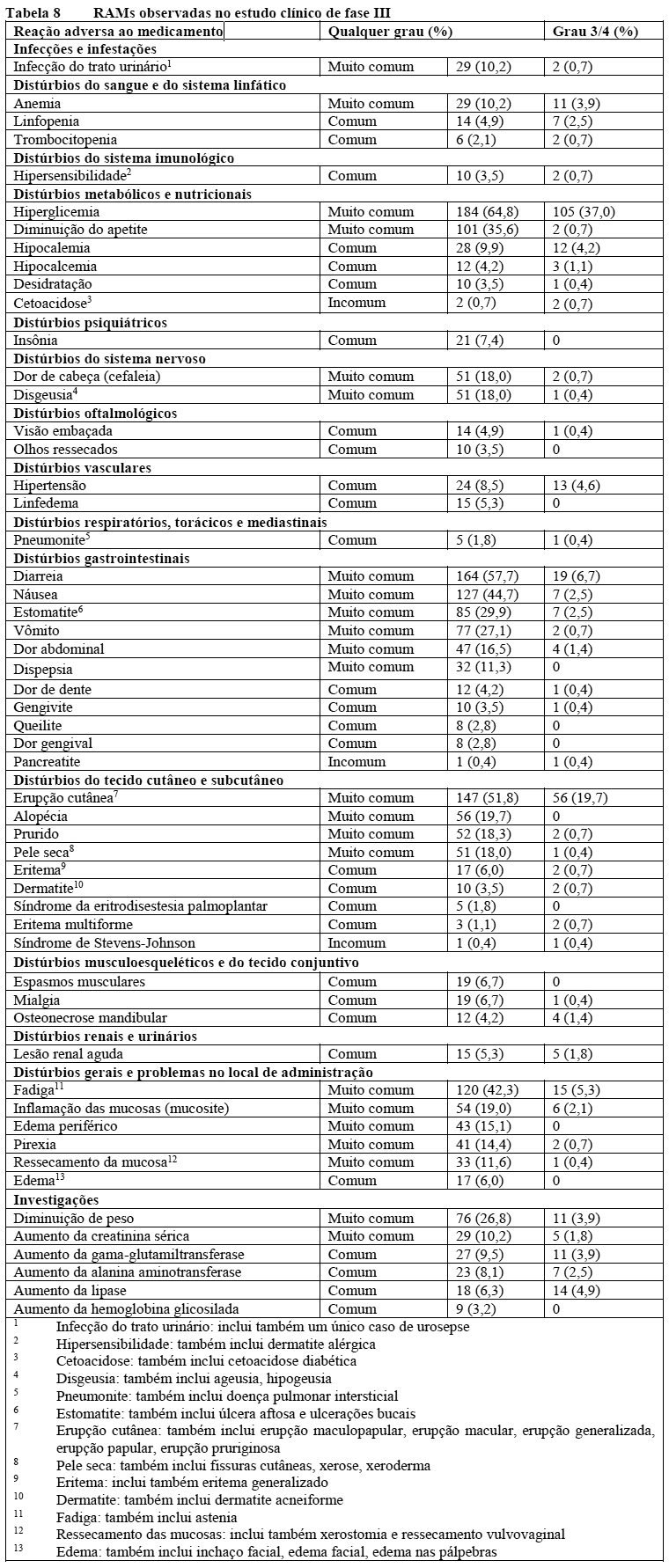
Reações de hipersensibilidade severa (incluindo reação anafilática e choque anafilático), manifestadas por sintomas que incluem, mas não limitados a dispneia, rubor, erupção cutânea, febre ou taquicardia, foram relatados em pacientes tratados com Piqray em estudos clínicos. A incidência de reações de hipersensibilidade de Grau 3 e 4 foi 0,7% (vide seção 9. Reações Adversas).
Os pacientes devem ser aconselhados sobre os sinais e sintomas de reações de hipersensibilidade severa. Piqray deve ser descontinuado permanentemente e não deve ser reiniciado em pacientes com reações de hipersensibilidade severa. Tratamento apropriado deve ser imediatamente iniciado.
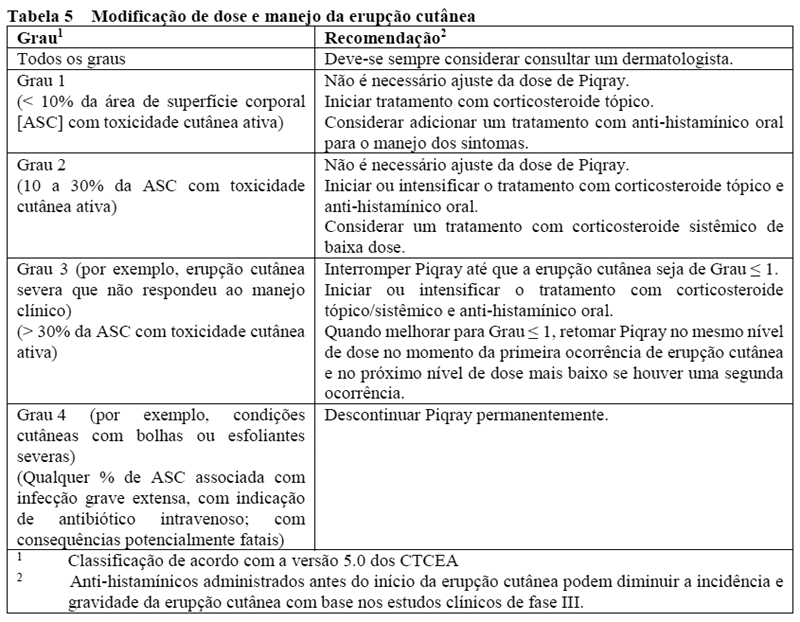
Reações cutâneas severas
Foram notificadas reações cutâneas graves com Piqray. Em estudo clínico de Fase III, a síndrome de Stevens-Johnson (SSJ) e o eritema multiforme (EM) foram relatados em 1 (0,4%) e 3 (1,1%) pacientes, respectivamente. A reação medicamentosa com eosinofilia e sintomas sistêmicos (DRESS) foi relatada no cenário pós-comercialização (vide seção 9. Reações Adversas).
O tratamento com Piqray não deve ser iniciado em pacientes com um histórico de reações cutâneas severas.
Os pacientes devem ser aconselhados quanto aos sinais e sintomas das reações cutâneas severas (por exemplo, sinais iniciais de febre, sintomas semelhantes à gripe, lesões nas mucosas ou erupção cutânea progressiva). Se sinais ou sintomas de reações cutâneas severas estiverem presentes, o Piqray deve ser interrompido até que a etiologia da reação tenha sido determinada. Recomenda-se consultar um dermatologista. Se reações cutâneas severas forem confirmadas, Piqray deve ser descontinuado permanentemente. O Piqray não deve ser reinicido em pacientes que tenham apresentado reações cutâneas severas anteriores. Se reações cutâneas severas não forem confirmadas, pode ser necessário interromper, reduzir a dose ou descontinuar o tratamento com Piqray, conforme descrito na Tabela 5 (vide seção 8. Posologia e modo de usar).
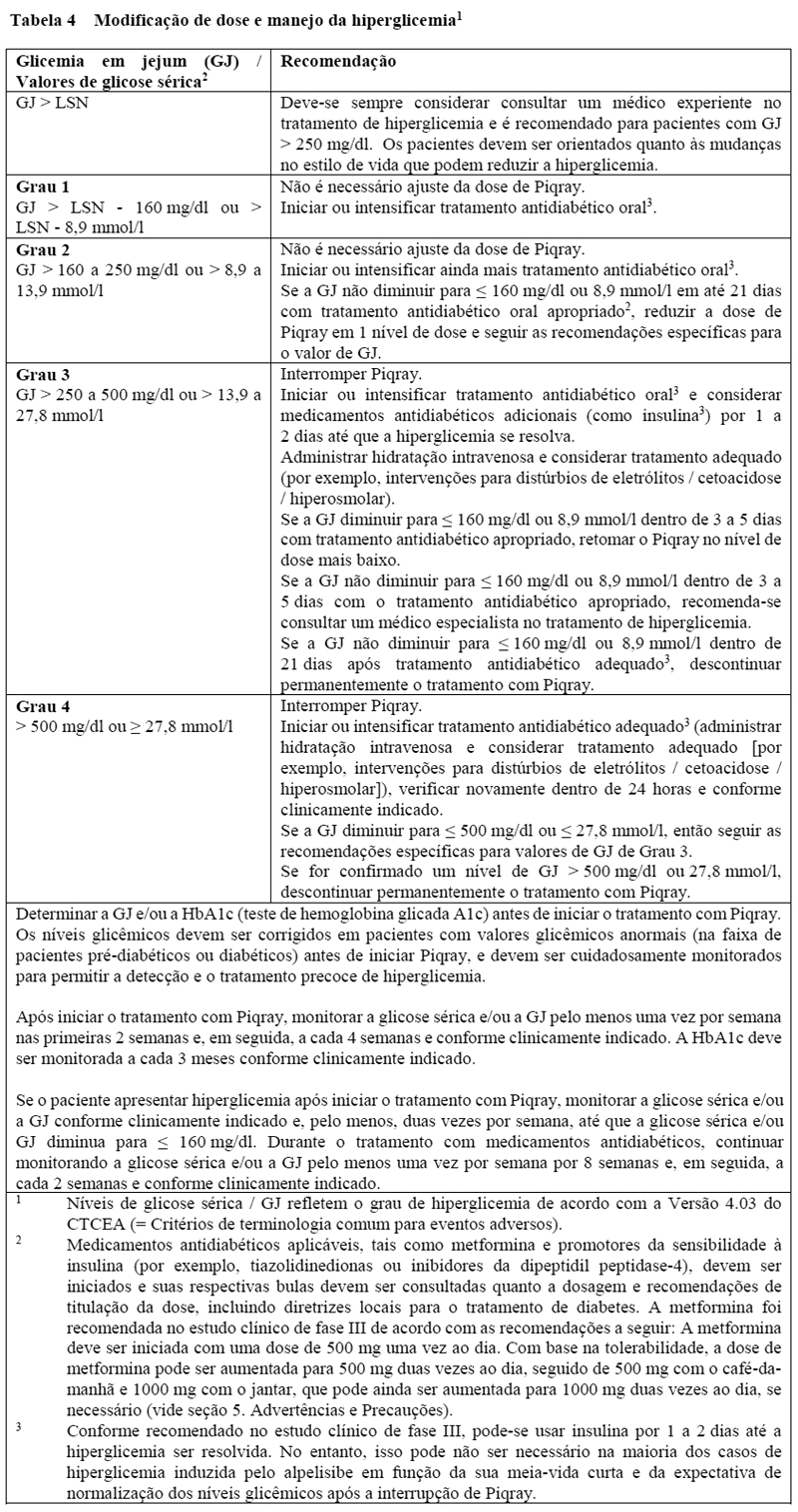
Hiperglicemia
A hiperglicemia foi reportada em 64,8% dos pacientes tratados com Piqray no estudo clínico de fase III. Hiperglicemia de grau 2 (GJ 160-250 mg/dl), 3 (GJ > 250-500 mg/dl) ou 4 (GJ > 500 mg/dl) foi relatada em 15,8%, 33,1% e 3,9% dos pacientes, respectivamente, no estudo clínico de fase III. Os pacientes devem ser aconselhados quanto aos sinais e sintomas de hiperglicemia (por exemplo, sede excessiva, micção mais frequente do que o normal ou maior quantidade de urina do que o normal, apetite aumentado com perda de peso).
No estudo clínico de fase III, com base nos valores basais de GJ e HbA1c, 56% dos pacientes foram considerados pré-diabéticos (GJ > 100-126 mg/dL [5,6 a 6,9 mmol/L] e/ou HbA1c 5,7-6,4%) e 4,2% dos pacientes foram considerados diabéticos (GJ ≥ 126 mg/dl [≥ 7,0 mmol/L] e/ou HbA1c ≥ 6,5%). Não haviam
pacientes com diabetes mellitus tipo 1 com base no histórico médico relatado no estudo clínico de fase III. Dentre os pacientes que estavam pré-diabéticos na avaliação inicial, 74,2% apresentaram hiperglicemia (qualquer grau) quando tratados com Piqray. Dentre os pacientes com hiperglicemia de grau ≥ 2 (GJ ≥ 160 mg/dL), o tempo mediano até a primeira ocorrência foi de 15 dias (intervalo: 5 dias a 517 dias) (com base em achados laboratoriais). A duração mediana da hiperglicemia de grau 2 ou maior (com base em achados laboratoriais) foi de 10 dias (IC de 95%: 8 a 13 dias).
No estudo clínico de fase III, em pacientes com hiperglicemia, 163/187 (87,2%) foram tratados com medicamentos antidiabéticos e 142/187 (75,9%) relataram uso de metformina em monoterapia ou em combinação com outros medicamentos antidiabéticos. A dose máxima de metformina recomendada no estudo clínico de fase III foi de 2.000 mg/dia.
Em pacientes com hiperglicemia de, no mínimo, Grau 2, o tempo mediano para a melhora em pelo menos um grau do primeiro evento foi de 8 dias (IC de 95%: 8 a 10 dias). Em todos os pacientes com GJ elevada que continuaram o tratamento com fulvestranto após a descontinuação de Piqray, os níveis de GJ retornaram aos valores basais (normais).
No estudo clínico de fase III, pacientes com histórico de diabetes mellitus intensificaram o uso de medicamentos antidiabéticos durante o tratamento com Piqray, portanto, esses pacientes requerem monitoramento e, possivelmente, intensificação do tratamento antidiabético. Pacientes com baixo controle glicêmico podem apresentar maior risco de desenvolver hiperglicemia severa e complicações associadas.
Com base na gravidade da hiperglicemia, pode ser necessário interromper, reduzir ou descontinuar a dose de Piqray, conforme descrito na Tabela 4 (vide seção 8. Posologia e modo de usar).
Pneumonite
Pneumonite, incluindo casos graves de pneumonite/doença pulmonar intersticial aguda, foram relatados em pacientes tratados com Piqray em estudos clínicos. Deve-se aconselhar os pacientes a comunicar imediatamente o surgimento ou agravamento de quaisquer sintomas respiratórios. Em pacientes que apresentarem surgimento ou agravamento de sintomas respiratórios, ou que tiverem suspeita de pneumonite, o tratamento com Piqray deve ser imediatamente interrompido e o paciente deve ser avaliado quanto à pneumonite. Deve-se considerar um diagnóstico de pneumonite não infecciosa em pacientes que apresentarem sinais e sintomas respiratórios não específicos, tais como hipoxia, tosse, dispneia ou infiltrados intersticiais em exame radiológico e nos quais causas infecciosas, neoplásicas e outras tenham sido descartadas por métodos investigativos apropriados. O Piqray deve ser descontinuado permanentemente em todos os pacientes com pneumonite confirmada.
Diarreia
Diarreia grave, incluindo desidratação e lesão renal aguda, ocorreu em pacientes tratados com Piqray. A maioria dos pacientes (58%) apresentou diarreia durante o tratamento com Piqray. Diarreia de Grau 3 ocorreu em 7% (n=19) dos pacientes. Entre os pacientes com diarreia de Grau 2 ou 3 (n=71), o tempo mediano até o início foi de 46 dias (intervalo de variação: 1 a 442 dias).
Foram necessárias reduções da dose de Piqray em 6% dos pacientes e 2,8% dos pacientes descontinuaram permanetemente Piqray em função da diarreia. Dos 164 pacientes que apresentaram diarreia, o uso de medicamentos antidiarreicos (por exemplo, loperamida) foi necessário para manejar os sintomas em 63% (104/164) desses pacientes.
Com base na gravidade da diarreia, pode ser necessária a interrupção, redução ou descontinuação da dose de Piqray, conforme descrito na Tabela 6 (vide seção 8. Posologia e modo de usar).
Os pacientes devem ser aconselhados a iniciar o tratamento antidiarreico, aumentar os fluidos orais, e informar o seu médico se ocorrer diarreia enquanto estiver tomando Piqray.
Fertilidade, gravidez e lactação
Gravidez
Com base em dados animais e no seu mecanismo de ação, o Piqray pode causar danos fetais quando administrado a gestantes (vide seção 3. Características farmacológicas).
O estado gestacional de mulheres com potencial reprodutivo deve ser verificado antes de iniciar o tratamento com Piqray.
Não existem estudos adequados e bem controlados em gestantes. O Piqray não deve ser usado durante a gravidez, a menos que os benefícios para a mulher superem os riscos para o feto. Se o Piqray for usado durante a gravidez, a paciente deve ser aconselhada quanto ao risco potencial para o feto.
Lactação
Não se sabe se o alpelisibe é excretado no leite humano ou animal após a administração de Piqray. Não existem dados sobre os efeitos do alpelisibe em recém-nascidos/lactentes ou sobre os efeitos do alpelisibe na produção de leite.
Devido ao potencial para causar reações adversas graves em recém-nascidos/lactentes, recomenda-se que as mulheres não amamentem durante o tratamento e por 1 semana após a última dose de Piqray.
Fertilidade
Não há dados sobre os efeitos do alpelisibe sobre a fertilidade. Com base em estudos de toxicidade de doses repetidas em animais, o Piqray pode prejudicar a fertilidade em homens e mulheres com potencial reprodutivo com doses aproximadamente 2,8 vezes a exposição em humanos com a dose máxima recomendada de 300 mg/dia (com base na ASC) (vide seção 3. Características farmacológicas).
Mulheres com potencial reprodutivo devem ser aconselhadas de que estudos em animais e o mecanismo de ação demonstraram que o alpelisibe pode ser prejudicial ao feto em desenvolvimento. Mulheres sexualmente ativas com potencial reprodutivo devem usar contracepção eficaz (métodos que resultem em taxas de gravidez menores que 1%) enquanto estiverem tomando o Piqray e por 1 semana após encerrar o tratamento com Piqray. Não se sabe atualmente se o alpelisibe pode reduzir a eficácia de contraceptivos hormonais de ação sistêmica.
Homens com parceiras grávidas, possivelmente grávidas ou que podem engravidar devem usar preservativos nas relações sexuais enquanto estiverem tomando o Piqray e por 1 semana após interromperem o tratamento com Piqray.
Este medicamento pertence à categoria D de risco à gravidez e não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
Efeitos sobre a capacidade de conduzir e utilizar máquinas
O Piqray tem efeito pouco significativo sobre a capacidade de dirigir e operar máquinas. Os pacientes devem ser aconselhados para serem cautelosos ao dirigir ou operar máquinas no caso de apresentarem fadiga durante o tratamento com Piqray (vide seção 9. Reações Adversas).
6. INTERAÇÕES MEDICAMENTOSAS
Interação com