PHESGO
ROCHE
pertuzumabe + trastuzumabe
Antineoplásico.
Apresentações.
Phesgo® 600 mg + 600 mg / 10 ml: solução injetável para administração subcutânea
Cada embalagem contém 1 (um) frasco-ampola de dose fixa com 600 mg de pertuzumabe e 600 mg de trastuzumabe em 10 ml de solução injetável (não reconstituir ou diluir).
Phesgo® 1200 mg + 600 mg / 15 ml: solução injetável para administração subcutânea
Cada embalagem contém 1 (um) frasco-ampola de dose fixa com 1200 mg de pertuzumabe e 600 mg de trastuzumabe em 15 ml de solução injetável (não reconstituir ou diluir).
VIA SUBCUTÂNEA
USO ADULTO
Composição.
Phesgo® 600 mg + 600 mg / 10 ml:
Princípio ativo: cada frasco-ampola de uso único com 10 ml contém 600 mg de pertuzumabe e 600 mg de trastuzumabe.
Phesgo® 1200 mg + 600 mg / 15 ml:
Princípio ativo: cada frasco-ampola de uso único com 15 ml contém 1200 mg de pertuzumabe e 600 mg de trastuzumabe.
Excipientes: hialuronidase humana recombinante (rHuPH20)*, histidina, cloridrato de histidina monoidratado, trealose di-hidratada, sacarose, polissorbato, levometionina e água para injetáveis.
*Hialuronidase humana recombinante (rHuPH20) é uma enzima usada para aumentar a dispersão e absorção do medicamento quando administrado por via subcutânea.
Informações técnicas.
As informações disponíveis nesta bula aplicam-se exclusivamente a Phesgo®.
1. INDICAÇÕES
Câncer de Mama Inicial
Phesgo® está indicado, em combinação com quimioterapia*, para:
- Tratamento neoadjuvante de pacientes com câncer de mama HER2-positivo localmente avançado, inflamatório ou em estágio inicial com elevado risco de recorrência (tanto para > 2 cm de diâmetro quanto para linfonodo positivo) como parte de um esquema terapêutico completo para o câncer de mama inicial.
- Tratamento adjuvante de pacientes com câncer de mama HER2-positivo em estágio inicial com elevado risco de recorrência.
*(Vide itens "2. RESULTADOS DE EFICÁCIA" e "8. POSOLOGIA E MODO DE USAR").
Câncer de Mama Metastático
Phesgo® está indicado, em combinação docetaxel, para pacientes com câncer de mama HER2-positivo metastático ou localmente recorrente não ressecável, que não tenham recebido tratamento prévio com medicamentos anti-HER2 ou quimioterapia para doença metastática.
2. RESULTADOS DE EFICÁCIA
ESTUDOS CLÍNICOS
Tratamento Neoadjuvante e Adjuvante de Câncer de Mama
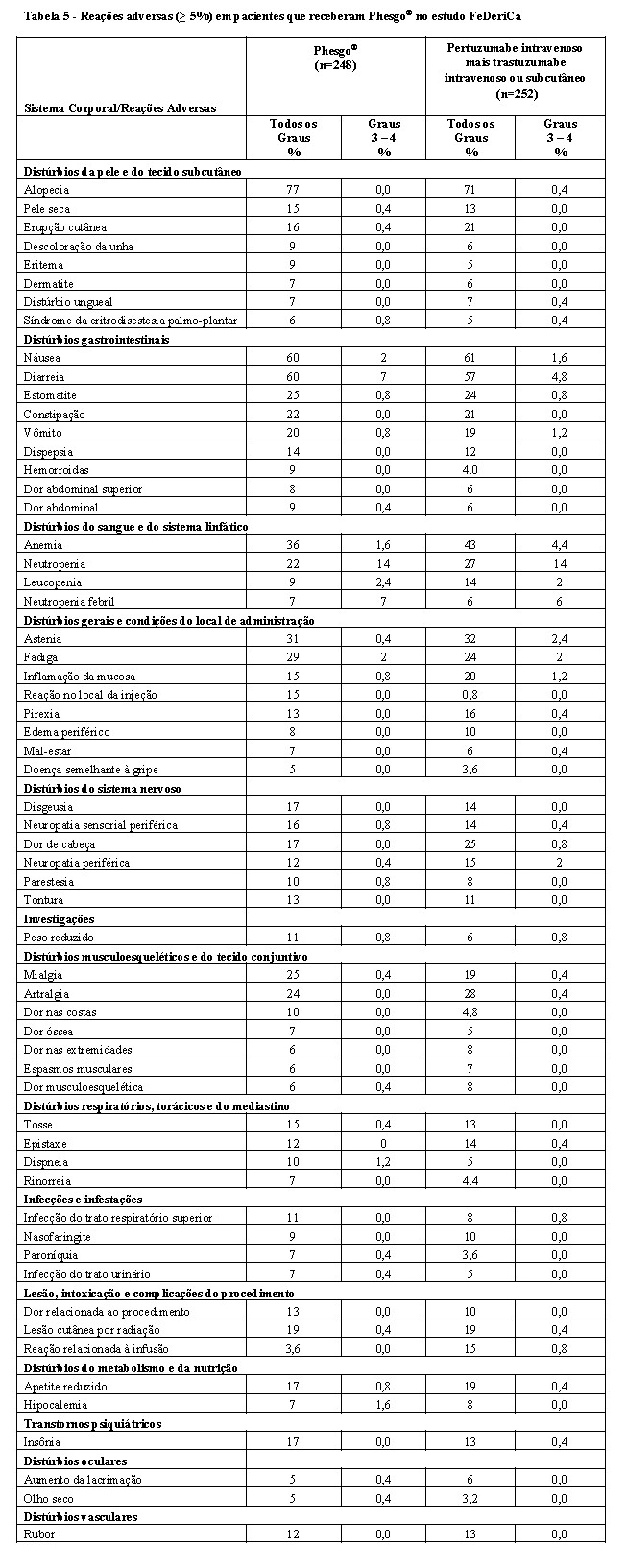
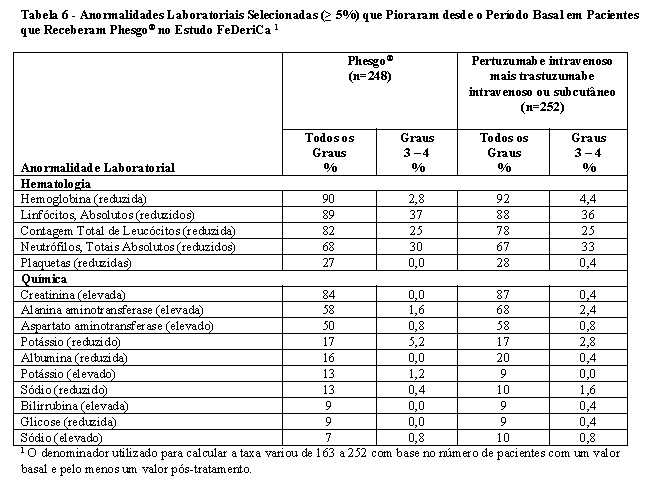
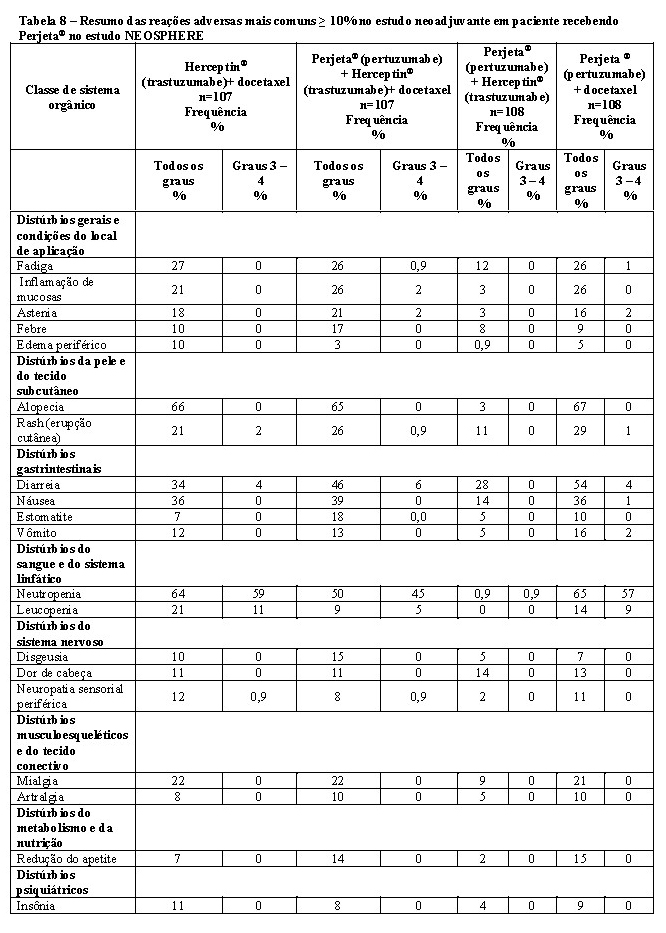
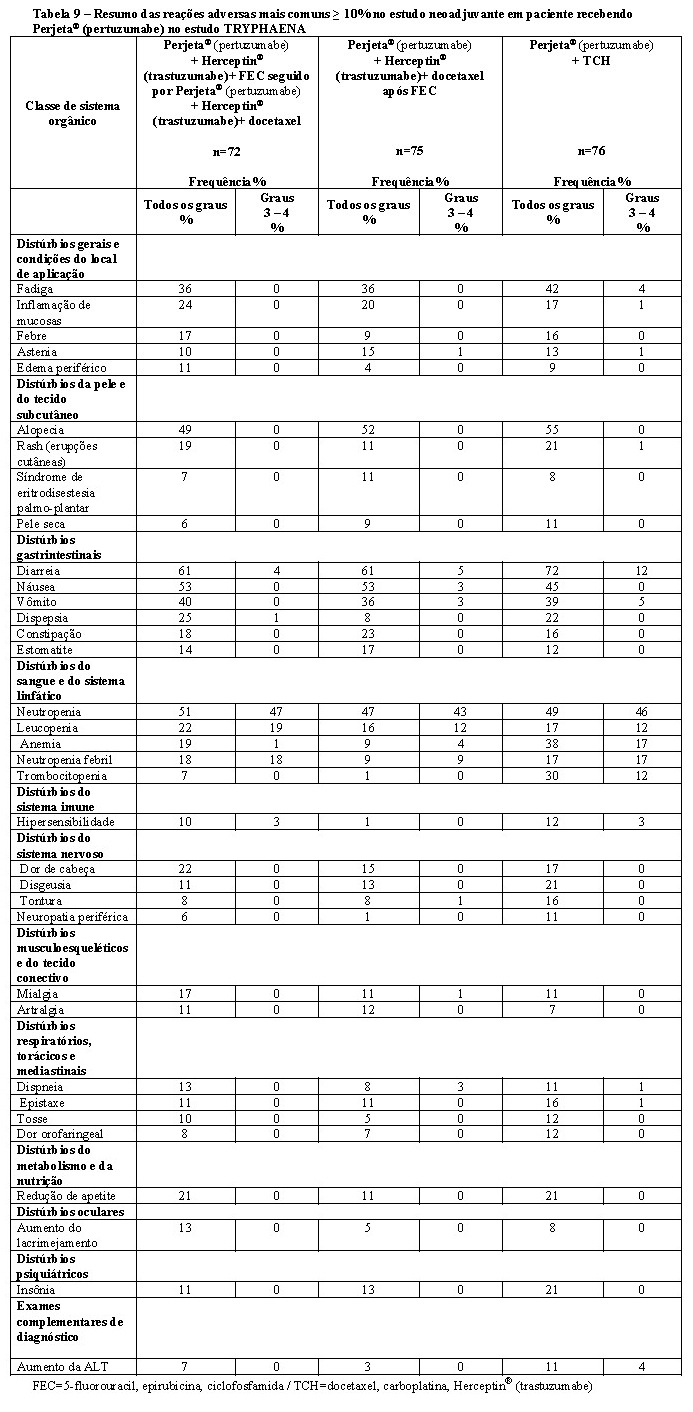
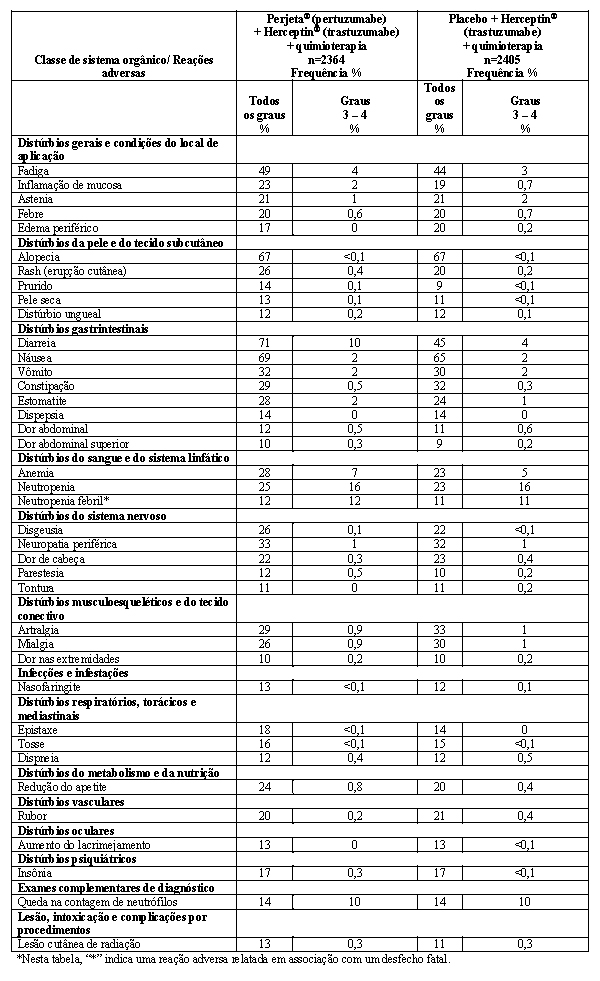
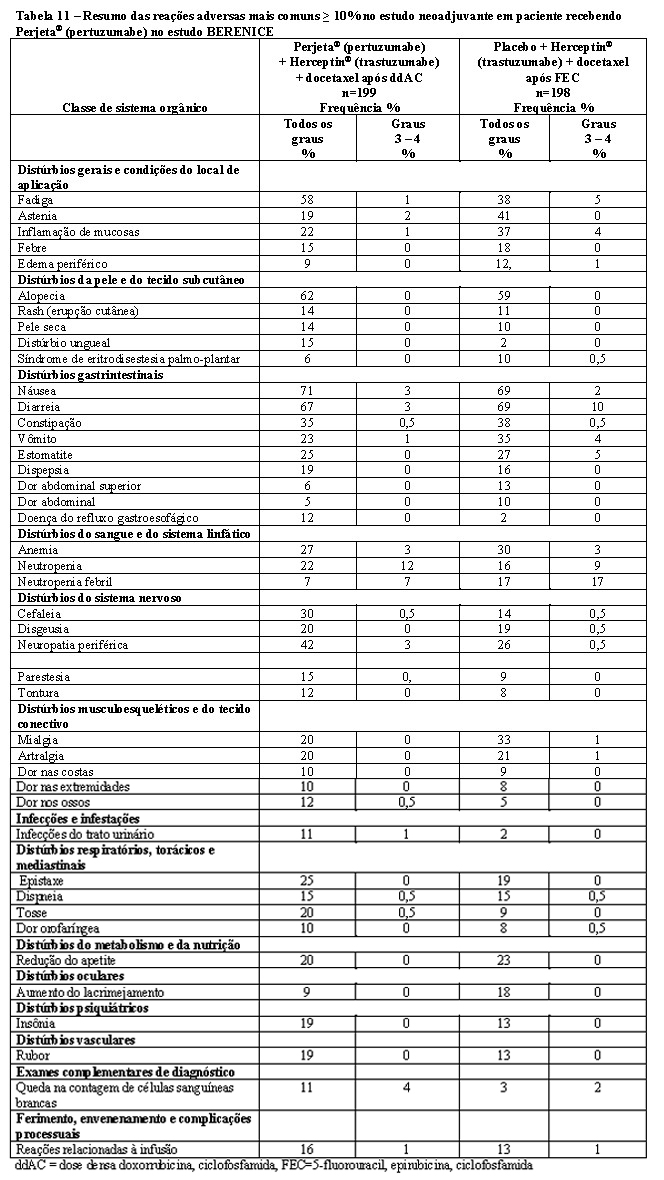
A eficácia do Phesgo® para uso em combinação com quimioterapia foi estabelecida para o tratamento de pacientes com câncer de mama em estágio inicial HER2-positivo. O uso de Phesgo® para essa indicação é corroborado por evidências de estudos adequados e bem controlados conduzidos com pertuzumabe e trastuzumabe intravenosos administrados em combinação com quimioterapia em adultos com câncer de mama em estágio inicial com superexpressão de HER2 (NEOSPHERE2, TRYPHAENA3, BERENICE4, APHINITY5) e dados de segurança e farmacocinéticos adicionais que demonstraram perfis de segurança e farmacocinética semelhantes entre Phesgo® e pertuzumabe e trastuzumabe intravenosos no estudo FeDeriCa1 (Vide itens "9. REAÇÕES ADVERSAS" e "3. CARACTERÍSTICAS FARMACOLÓGICAS").
FeDeriCa1
O Estudo FeDeriCa foi um estudo aberto, multicêntrico e randomizado, conduzido em 500 pacientes com câncer de mama HER2-positivo, operável ou localmente avançado (incluindo inflamatório), com tamanho de tumor > 2 cm ou nódulo-positivo. A superexpressão de HER2 foi definida como IHQ 3+ em > 10% das células imunorreativas ou amplificação do gene HER2 por ISH (taxa entre sinais do gene HER2 e sinais do centrômero 17 > 2,0) utilizando um teste aprovado. Os pacientes foram randomizados para receber oito ciclos de quimioterapia neoadjuvante com administração concomitante de quatro ciclos de Phesgo® ou pertuzumabe e trastuzumabe por via intravenosa durante os ciclos 5 a 8, seguidos por cirurgia. Os investigadores selecionaram um dos dois regimes de quimioterapias neoadjuvantes a seguir para pacientes individuais:
- Quatro ciclos de doxorrubicina (60 mg/m2) e ciclofosfamida (600 mg/m2) a cada duas semanas, seguidos de paclitaxel (80 mg/m2) semanalmente por 12 semanas.
- Quatro ciclos de doxorrubicina (60 mg/m2) e ciclofosfamida (600 mg/m2) a cada três semanas, seguidos por quatro ciclos de docetaxel (75 mg/m2 no primeiro ciclo e, em seguida, 100 mg/m2 nos ciclos subsequentes, a critério do investigador) a cada três semanas
Após a cirurgia, os pacientes continuaram a terapia com Phesgo® ou pertuzumabe e trastuzumabe por via intravenosa, conforme foram tratados antes da cirurgia, por mais 14 ciclos, a fim de concluir 18 ciclos de terapia anti HER2. Os pacientes também receberam radioterapia adjuvante e terapia endócrina, a critério do investigador. No período adjuvante, a substituição de trastuzumabe por via intravenosa por trastuzumabe por via subcutânea foi permitida a critério do investigador. Os pacientes receberam terapia direcionada ao HER2 a cada três semanas, de acordo com a Tabela 1, conforme segue:
FeDeriCa1 foi desenhado para demonstrar a não inferioridade da Cvale sérica de pertuzumabe do ciclo 7 (ou seja, ciclo 8 pré-dose) de pertuzumabe de Phesgo® em comparação ao pertuzumabe intravenoso (desfecho primário). Os desfechos secundários incluíram a Cvale sérica de trastuzumabe no ciclo 7, a eficácia (resposta patológica completa [pCR], definida como a ausência de células neoplásicas invasivas na mama e nos linfonodos axilares) e segurança. A idade mediana era de 51 anos (faixa: 25-81) e a maioria dos pacientes era caucasiana (66%). A maioria dos pacientes tinham doença receptor hormonal-positiva (61%) ou doença nódulo-positiva (58%).
Os resultados de farmacocinética (PK) para o desfecho primário de pertuzumabe ciclo 7 Cvale (ou seja, pré-dose ciclo 8), mostraram não inferioridade de pertuzumabe em Phesgo® (média geométrica 88,7 mcg / mL) em comparação com pertuzumabe intravenoso (média geométrica 72,4 mcg / mL) com uma razão média geométrica de 1,22 (IC de 90%: 1,14 1,31). O limite inferior do intervalo de confiança bilateral de 90% para a razão média geométrica de pertuzumabe em Phesgo® e pertuzumabe intravenoso foi de 1,14, ou seja, maior do que a margem predefinida de 0,8.
Os resultados de PK para o desfecho secundário, trastuzumabe no ciclo 7 Cvale (ou seja, pré-dose ciclo 8), mostraram não inferioridade de trastuzumabe em Phesgo® (média geométrica 57,5 mcg / mL) em comparação com trastuzumabe intravenoso (média geométrica 43,2 mcg / mL) com uma razão média geométrica de 1,33 (IC de 90%: 1,24 1,43).
Os parâmetros farmacocinéticos de pertuzumabe e trastuzumabe são descritos no item "3. CARACTERÍSTICAS FARMACOLÓGICAS".
O resultado do desfecho secundário de eficácia, taxa de pCR, foi de 59,7% (IC de 95%: 53,3, 65,8) no braço de Phesgo® e 59,5% (IC de 95%: 53,2, 65,6) no braço de pertuzumabe e trastuzumabe por via intravenosa.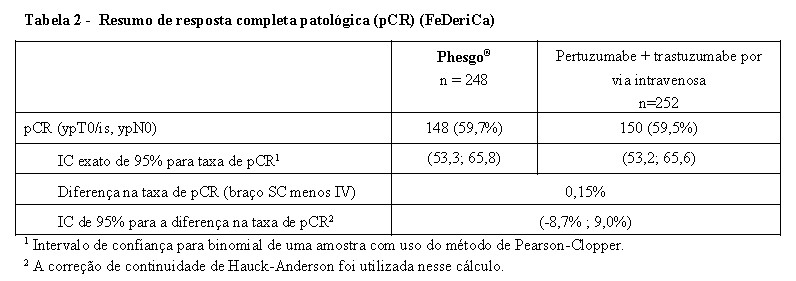
Câncer de Mama Metastático
A eficácia de Phesgo® para uso em combinação com docetaxel foi estabelecida para o tratamento de pacientes com câncer de mama metastático HER2-positivo que não receberam terapia anti-HER2 ou quimioterapia para doença metastática previamente. O uso de Phesgo® para essa indicação é corroborado por evidências de estudos adequados e bem controlados conduzidos com pertuzumabe e trastuzumabe intravenosos administrados em combinação com quimioterapia em adultos com câncer de mama metastático com superexpressão de HER2 (CLEOPATRA6) e dados de segurança e farmacocinéticos adicionais que demonstraram perfis de segurança e farmacocinética semelhantes entre Phesgo® e pertuzumabe e trastuzumabe intravenosos no estudo FeDeriCa1 (vide itens "9. REAÇÕES ADVERSAS" e "3. CARACTERÍSTICAS FARMACOLÓGICAS").
Referências bibliográficas
1. Primary Clinical Study report WO40324 (FEDERICA). A phase III, randomized, multicenter, open-label, two-arm study to evaluate the pharmacokinetics, efficacy, and safety of subcutaneous administration of the fixed-dose combination of pertuzumab and trastuzumab in combination with chemotherapy in patients with HER2-positive early breast cancer. Report No. 1096382. December 2019 (CDS 1.0).
2. Relatório de Estudo Clínico - WO20697 (NEOSPHERE) - A randomized, multicenter, multinational Phase II study on trastuzumab plus docetaxel versus trastuzumab plus docetaxel plus pertuzumab versus trastuzumab plus pertuzumab versus pertuzumab and docetaxel in patients with locally advanced, inflammatory or early stage HER2 positive breast cancer. Report No. 1032196. June 2011 (CDS vs. 1.0).
3. Relatório de Estudo Clínico - BO22280 (TRYPHAENA) - A randomized, multicentre, multinational Phase II study to evaluate pertuzumab in combination with trastuzumab, given either concomitantly or sequentially with standard anthracycline-based chemotherapy or concomitantly with a non-anthracycline-based chemotherapy regimen, as neoadjuvant therapy for patients with locally advanced, inflammatory or early stage HER2-positive breast cancer. June 2009 (CDS 1.0).
4. Relatório de Estudo Clínico - WO29217 (BERENICE) - A Multicenter, Multinational, Phase II Study to Evaluate Perjeta in Combination with Herceptin and Standard Neoadjuvant Anthracycline-Based Chemotherapy in Patients with HER2-Positive, Locally Advanced, Inflammatory, or Early-Stage Breast Cancer. December 2016. (CDS 1.0)
5. Relatório de Estudo Clínico - BO25126 (APHINITY): A randomized multicenter, double-blind, placebo-controlled comparison of chemotherapy plus trastuzumab plus placebo versus chemotherapy plus trastuzumab plus pertuzumab as adjuvant therapy in patients with operable HER2-positive primary breast cancer. Report No. 1075429. Updated December 2017 (CDS 1.0).
6. Relatório de Estudo Clínico - WO20698/TOC4129g (CLEOPATRA) - A Phase III, Randomized, Double-Blind, Placebo Controlled Clinical Trial to Evaluate the Efficacy and Safety of Pertuzumab + Trastuzumab + Docetaxel vs. Placebo + Trastuzumab + Docetaxel in Previously Untreated HER2-Positive Metastatic Breast Cancer: Report No: 1046288, October 2011 (CDS 1.0).
3. CARACTERÍSTICAS FARMACOLÓGICAS
Farmacodinâmica
Mecanismo de ação
Pertuzumabe tem como alvo o domínio de dimerização extracelular (subdomínio II) do HER2 e, assim, bloqueia a heterodimerização dependente de ligante de HER2 com outros membros da família HER, incluindo EGFR, HER3 e HER4. Como resultado, pertuzumabe inibe a sinalização intracelular iniciada por ligante através de duas principais vias de sinalização, a proteína quinase ativada por mitógenos (MAP) e a fosfoinositídeo 3-quinase (PI3K). A inibição dessas vias de sinalização pode resultar em parada no crescimento celular e apoptose, respectivamente
Trastuzumabe liga-se ao subdomínio IV do domínio extracelular da proteína HER2 para inibir a proliferação celular mediada por HER2, independentes do ligante, e a via de sinalização de PI3K em células tumorais humanas que superexpressam HER2.
Demonstrou-se que tanto a citotoxicidade celular dependente de anticorpos (CCDA) mediada por pertuzumabe quanto por trastuzumabe demonstraram ser exercidas preferencialmente em células cancerígenas que superexpressam HER2, em comparação com células cancerígenas que não superexpressam HER2.
Enquanto pertuzumabe isolado inibiu a proliferação de células tumorais humanas, a combinação de pertuzumabe e trastuzumabe aumentou a atividade antitumoral nos modelos de xenoenxerto com superexpressão de HER2
O hialuronano é um polissacarídeo encontrado na matriz extracelular do tecido subcutâneo. É despolimerizado pela enzima hialuronidase de ocorrência natural. Ao contrário dos componentes estruturais estáveis da matriz intersticial, o hialuronano tem meia-vida de, aproximadamente, 0,5 dia. A hialuronidase aumenta a permeabilidade do tecido subcutâneo ao despolimerizar o hialuronano. Nas doses administradas, a hialuronidase em Phesgo® atua de modo transitório e localmente.
Os efeitos da hialuronidase são reversíveis e a permeabilidade do tecido subcutâneo é restaurada em 24 a 48 horas.
Foi demonstrado que a hialuronidase aumenta a taxa de absorção de um produto de trastuzumabe na circulação sistêmica quando administrada por via subcutânea em Minisuínos Göttingen.
Eletrofisiologia Cardíaca
O efeito do pertuzumabe intravenoso com uma dose inicial de 840 mg seguida de uma dose de manutenção de 420 mg a cada três semanas no intervalo QTc foi avaliado em um subgrupo de 20 pacientes com câncer de mama positivo para HER2 (NCT00567190). Não foram detectadas grandes alterações no intervalo QT médio (ou seja, superior a 20 ms) em relação ao placebo com base no método de correção de Fridericia no estudo. Um pequeno aumento no intervalo QTc médio (ou seja, menos de 10 ms) não pode ser excluído devido às limitações do desenho do estudo.
Os efeitos do trastuzumabe nos desfechos eletrocardiográficos (ECG), incluindo duração do intervalo QTc, foram avaliados em pacientes com tumores sólidos HER2-positivos. O trastuzumabe não teve nenhum efeito clinicamente relevante na duração do intervalo QTc e não houve nenhuma relação evidente entre as concentrações séricas de trastuzumabe e a alteração na duração do intervalo QTcF em pacientes com tumores sólidos HER2-positivos.
Farmacocinética
Absorção
A concentração sérica máxima média (Cmáx) de pertuzumabe dentro de Phesgo® e o tempo para a concentração máxima (Tmáx) foram 157 mcg/ml e 3,82 dias, respectivamente. Com base na análise farmacocinética da população, a biodisponibilidade absoluta foi de 0,712 e a taxa de absorção de primeira ordem (Ka) é de 0,348 (1/dia).
A mediana Cmáx de trastuzumabe em Phesgo® e Tmáx foi de 114 mcg/mL e 3,84 dias, respectivamente. Com base na análise farmacocinética da população, a biodisponibilidade absoluta foi de 0,771 e o Ka é de 0,404 (1/dia).
Distribuição
Com base na análise farmacocinética da população, o volume de distribuição do compartimento central (Vc) de pertuzumabe em Phesgo® no paciente típico foi de 2,77 litros.
Com base na análise farmacocinética da população, o Vc do trastuzumabe subcutâneo no paciente típico foi de 2,91 litros.
Biotransformação
O metabolismo de Phesgo® não foi estudado diretamente. Os anticorpos são eliminados principalmente por catabolismo.
Eliminação
Com base na análise farmacocinética da população, a depuração de pertuzumabe em Phesgo® foi de 0,163 L/dia e a meia-vida de eliminação (t1/2) foi de aproximadamente 24,3 dias.
Com base na análise farmacocinética da população, a depuração de trastuzumabe em Phesgo® foi de 0,111 L/dia. Estima-se que o trastuzumabe alcance concentrações < 1 mcg/mL (aproximadamente 3% da Cmin, ss da população ou cerca de 97% de washout) em pelo menos 95% dos pacientes 7 meses após a última dose.
Pacientes idosos
Não foram realizados estudos para investigar a farmacocinética de Phesgo® em pacientes idosos.
Em análises de farmacocinética populacional de pertuzumabe em Phesgo® e pertuzumabe intravenoso, a idade não afetou significativamente a farmacocinética de pertuzumabe.
Em análises farmacocinéticas populacionais de trastuzumabe subcutâneo ou intravenoso, a idade demonstrou não ter efeito sobre a distribuição de trastuzumabe.
Insuficiência renal
Não foram realizados estudos para investigar a farmacocinética de Phesgo® em pacientes com insuficiência renal.
Com base nas análises farmacocinéticas da população de pertuzumabe em Phesgo® e pertuzumabe intravenoso, o comprometimento renal demonstrou não afetar a exposição ao pertuzumabe; no entanto, apenas dados limitados de pacientes com insuficiência renal grave foram incluídos nas análises farmacocinéticas da população.
Numa análise farmacocinética populacional de trastuzumabe subcutâneo e intravenoso, a insuficiência renal demonstrou não afetar a disposição de trastuzumabe.
Insuficiência hepática
Nenhum estudo de PK formal foi conduzido em pacientes com insuficiência hepática. Com base nas análises farmacocinéticas da população de pertuzumabe em Phesgo®, a insuficiência hepática leve demonstrou não afetar a exposição ao pertuzumabe. No entanto, apenas dados limitados de pacientes com insuficiência hepática leve foram incluídos nas análises farmacocinéticas da população. As moléculas de IgG1, como pertuzumabe e trastuzumabe, são catabolizadas por enzimas proteolíticas amplamente distribuídas, não restritas ao tecido hepático. Portanto, é improvável que as alterações na função hepática tenham efeito sobre a eliminação de pertuzumabe e trastuzumabe.
Farmacocinética em situações clínicas especiais
Populações Específicas
O peso corporal magro e o nível de albumina sérica basal foram incluídos como covariáveis significativas no modelo PK populacional do pertuzumabe. No entanto, nenhum ajuste da dose com base no peso corporal ou no nível de albumina basal é necessário, pois as mudanças na exposição não são consideradas clinicamente relevantes.
O peso corporal demonstrou uma influência estatisticamente significativa na PK do trastuzumabe. Em pacientes com peso corporal < 58 kg, a AUC0-21 dias média de trastuzumabe no Ciclo 7 foi aproximadamente 34% maior após a administração de Phesgo® do que após o tratamento com trastuzumabe intravenoso, já no grupo com peso corporal mais alto ( > 77 kg), a AUC0-21 dia no Ciclo 7 foi 24% menor após a administração de Phesgo® do que após o tratamento com trastuzumabe intravenoso. No entanto, nenhum ajuste da dose com base no peso corporal é necessário, pois as mudanças na exposição não são consideradas clinicamente relevantes.
Nenhuma diferença clinicamente significativa na farmacocinética de pertuzumabe e trastuzumabe foi observada com base na idade (25 a 80 anos), na raça (Asiática e Não Asiática) e no comprometimento renal (depuração de creatinina determinada por Cockcroft-Gault de 30 mL/min ou mais). Os efeitos do comprometimento hepático na farmacocinética de pertuzumabe e trastuzumabe são desconhecidos.
Dados de segurança pré-clínica
Phesgo® contém pertuzumabe, trastuzumabe e hialuronidase.
Carcinogenicidade e mutagenicidade
- Pertuzumabe
Não foram realizados estudos para avaliar o potencial carcinogênico ou mutagênico do pertuzumabe.
- Trastuzumabe
Trastuzumabe não foi testado quanto ao potencial de carcinogenicidade.
Não foi observada evidência de atividade mutagênica quando o trastuzumabe foi testado nos ensaios padrão bacteriano de Ames e de mutagenicidade de linfócitos do sangue periférico humano em concentrações de até 5.000 mcg/ml. Em um ensaio in vivo de micronúcleos, não foram observadas evidências de danos cromossômicos às células da medula óssea de camundongos após doses intravenosas em bolus de até 118 mg/kg de trastuzumabe.
- Hialuronidase
As hialuronidases são encontradas na maioria dos tecidos do corpo. Não foram realizados estudos de longo prazo em animais para avaliar o potencial carcinogênico ou mutagênico da hialuronidase
Fertilidade
- Pertuzumabe
Nenhum estudo de fertilidade em animais específico foi realizado para avaliar o efeito de pertuzumabe.
Não foram observados efeitos adversos nos órgãos reprodutores masculinos e femininos em estudos de toxicidade de dose repetida com duração de até seis meses em macacos cynomolgus.
- Trastuzumabe
Um estudo de fertilidade foi realizado em macacos cynomolgus fêmeas em doses até 25 vezes a dose humana semanal recomendada de 2 mg/kg de trastuzumabe por via intravenosa, não revelando evidência de fertilidade comprometida, conforme medido pela duração do ciclo menstrual e pelos níveis de hormônios sexuais das fêmeas.
- Hialuronidase
Quando a hialuronidase (humana recombinante) subcutânea foi administrada a macacos cynomolgus por 39 semanas em níveis de dose de até 220.000 U/kg, ou seja > 223 e 335 vezes mais altas que a dose humana, com base nas doses de ataque e manutenção, respectivamente, nenhuma evidência de toxicidade para o sistema reprodutivo masculino ou feminino foi encontrada através do monitoramento periódico dos parâmetros em vida, por exemplo, análises de sêmen, níveis hormonais, ciclos menstruais e também de patologia macroscópica, histopatologia e dados de peso de órgãos.
Teratogenicidade
- Pertuzumabe
Macacas cynomolgus prenhes foram tratadas no Dia Gestacional (GD) 19, com doses de ataque de 30 a 150 mg/kg de pertuzumabe, seguidas de doses quinzenais de 10 a 100 mg/kg. Esses níveis de dose resultaram em exposições clinicamente relevantes 2,5 a 20 vezes maiores que as exposições em humanos que receberam a dose recomendada, com base na Cmáx. A administração intravenosa de pertuzumabe do GD19 ao GD50 (período de organogênese) foi embriotóxica, com aumentos dependentes da dose na morte embriofetal entre o GD25 e o GD70. As incidências de perda embriofetal foram de 33%, 50% e 85%. Na cesariana no GD100, foram identificados oligoidrâmnio, diminuição dos pesos relativos do pulmão e do rim e evidência microscópica de hipoplasia renal consistente com atraso no desenvolvimento renal em todos os grupos de dose de pertuzumabe. A exposição a pertuzumabe foi relatada na prole de todos os grupos tratados, em níveis de 29% a 40% dos níveis séricos maternos no GD100.
- Trastuzumabe
Em estudos em que trastuzumabe por via intravenosa foi administrado a macacas cynomolgus prenhes durante o período de organogênese em doses de até 25 mg/kg administradas duas vezes por semana (até 25 vezes a dose semanal recomendada em humanos de 2 mg/kg), trastuzumabe atravessou a barreira placentária durante a fase inicial (Dias de Gestação 20 a 50) e tardia (Dias de Gestação 120 a 150). As concentrações resultantes de trastuzumabe no soro fetal e no líquido amniótico foram de aproximadamente 33% e 25%, respectivamente, daquelas presentes no soro materno, mas não foram associadas a efeitos adversos no desenvolvimento.
- Hialuronidase
Em um estudo embriofetal, camundongos receberam administração diariamente por injeção subcutânea durante o período de organogênese com hialuronidase (humana recombinante) em níveis de dose de até 2.200.000 U/kg, ou seja, > 2.400 e 3.600 vezes superiores à dose humana, com base nas doses de ataque e manutenção, respectivamente. O estudo não encontrou evidências de teratogenicidade. Foram observados peso fetal reduzido e aumento do número de reabsorções fetais, sem efeitos encontrados na dose diária de 360.000 U/kg, que é > 400 e 600 vezes superior à dose humana, com base nas doses de ataque e manutenção, respectivamente.
Em um estudo de reprodução perinatal e pós-natal, camundongos receberam administração diariamente por injeção subcutânea com hialuronidase (humana recombinante) desde a implantação até a lactação e o desmame em níveis de dose de até 1.100.000 U/kg vezes superiores à dose humana, ou seja, > 1.200 e 1.800, com base nas doses de ataque e manutenção, respectivamente. O estudo não encontrou efeitos adversos em maturação sexual, aprendizado e memória ou fertilidade da prole.
Lactação
Em macacas cynomolgus em lactação, trastuzumabe estava presente no leite materno em cerca de 0,3% das concentrações séricas maternas após doses pré-parto (início no Dia de Gestação 120) e pós-parto (até o Dia 28 Pós-parto) de 25 mg/kg administradas duas vezes por semana (25 vezes a dose humana semanal recomendada de 2 mg/kg de trastuzumabe por via intravenosa). Macacos recém-nascidos com níveis séricos detectáveis de trastuzumabe não exibiram efeitos adversos no crescimento ou no desenvolvimento desde o nascimento até 1 mês de idade.
4. CONTRAINDICAÇÕES
Phesgo® é contraindicado a pacientes com hipersensibilidade conhecida a pertuzumabe, ou trastuzumabe ou a qualquer outro excipiente da fórmula.
5. ADVERTÊNCIAS E PRECAUÇÕES
Cardiomiopatia
Phesgo® pode causar hipertensão, arritmias, disfunção cardíaca do ventrículo esquerdo, insuficiência cardíaca incapacitante, cardiomiopatia e morte cardíaca. Phesgo® pode causar diminuição assintomática da FEVE (fração de ejeção ventricular esquerda).
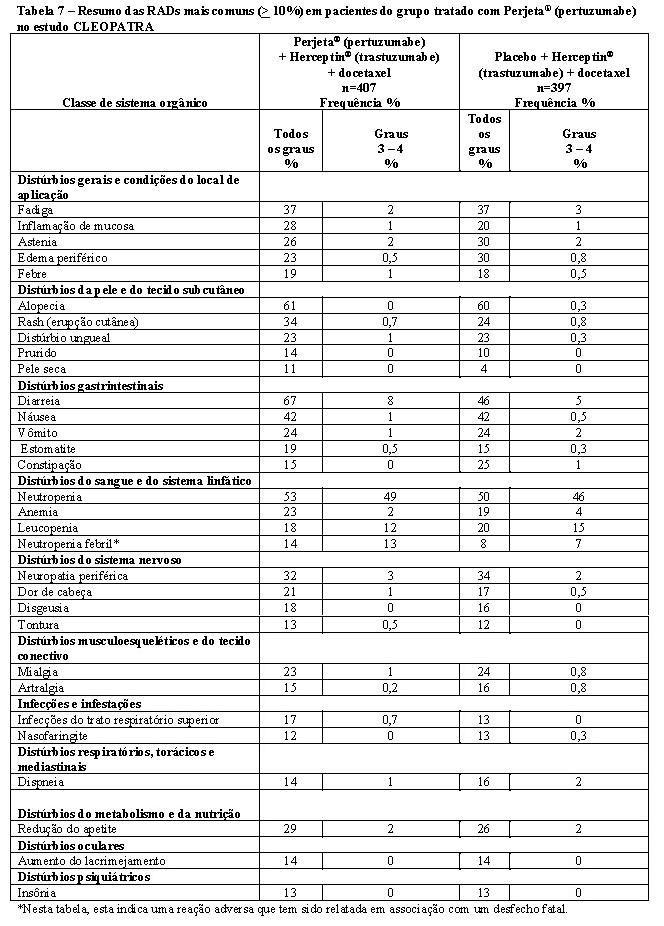
Uma maior incidência de diminuição da FEVE foi observada em pacientes tratados com pertuzumabe intravenoso, trastuzumabe intravenoso e docetaxel. Um aumento de 4 a 6 vezes na incidência de disfunção miocárdica sintomática foi relatado em pacientes que receberam trastuzumabe, com a incidência absoluta mais alta ocorrendo quando o trastuzumabe foi administrado com uma antraciclina.
Os pacientes que recebem antraciclina após a descontinuação de Phesgo® também podem estar em maior risco de desenvolver disfunção cardíaca (vide itens "6. INTERAÇÕES MEDICAMENTOSAS" e "3. CARACTERÍSTICAS FARMACOLÓGICAS").
Pacientes com histórico de doença cardíaca grave ou condições médicas cardíacas graves, histórico de disritmias ventriculares ou fatores de risco para disritmias ventriculares foram excluídos do estudo pivotal FeDeriCa de Phesgo® para câncer de mama em estágio inicial (neo) adjuvante.
O risco cardíaco deve ser cuidadosamente considerado e ponderado em relação à necessidade médica do paciente individual antes do uso de Phesgo® com uma antraciclina. Com base nas ações farmacológicas dos agentes direcionados ao HER2 e antraciclinas, pode-se esperar que o risco de toxicidade cardíaca seja maior com o uso concomitante de Phesgo® e antraciclinas do que com o uso sequencial.
O uso sequencial de Phesgo® (em combinação com um taxano) foi avaliado após o componente doxorrubicina de dois regimes baseados em antraciclina no estudo FeDeriCa, enquanto o uso sequencial de pertuzumabe intravenoso (em combinação com trastuzumabe e um taxano) foi avaliado após a epirrubicina ou o componente doxorrubicina de muitos regimes baseados em antraciclina nos estudos APHINITY e BERENICE. Apenas dados de segurança limitados estão disponíveis sobre o uso concomitante de pertuzumabe intravenoso em combinação com trastuzumabe e uma antraciclina. No estudo TRYPHAENA, pertuzumabe intravenoso em combinação com trastuzumabe foi administrado concomitantemente com epirrubicina, como parte do regime FEC (fluoruracila, epirrubicina, ciclofosfamida). Apenas pacientes que nunca tinham recebido quimioterapia foram tratados e receberam baixas doses cumulativas de epirrubicina (até 300 mg / m2). Neste estudo, a segurança cardíaca foi semelhante à observada em pacientes que receberam o mesmo regime, mas com pertuzumabe administrado sequencialmente (após quimioterapia FEC).
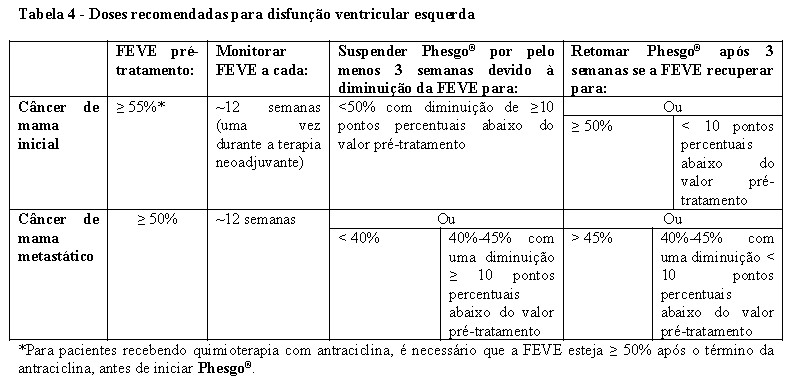
Monitoramento Cardíaco
Antes do início da administração de Phesgo®, realize uma avaliação cardíaca rigorosa, incluindo histórico, exame físico e determinação da FEVE por ecocardiograma ou exame MUGA (angiografia de múltipla entrada).
Durante o tratamento com Phesgo®, avalie a FEVE em intervalos regulares (vide item "8. POSOLOGIA E MODO DE USAR").
Se, após uma avaliação repetida dentro de aproximadamente 3 semanas, a FEVE não tiver melhorado, tiver diminuído ainda mais e/ou se o paciente estiver sintomático, descontinue permanentemente a administração de Phesgo®.
Após a conclusão da administração de Phesgo®, continue realizando o monitoramento de cardiomiopatia e avalie as medições da FEVE a cada 6 meses durante pelo menos 2 anos como um componente da terapia adjuvante.
Phesgo®
No estudo FeDeriCa, a porcentagem de pacientes com pelo menos um distúrbio cardíaco foi de 22% no braço tratado com Phesgo®. A reação adversa cardíaca mais frequente no braço tratado com Phesgo® foi fração de ejeção reduzida.
A incidência de insuficiência cardíaca (Classe III/IV da NYHA) com diminuição de ≥ 10% da FEVE e uma diminuição para menos de 50% foi de 0,8% no braço tratado com Phesgo®. Diminuições assintomáticas ou levemente sintomáticas (Classe II da NYHA) confirmadas de ≥ 10% da FEVE e uma diminuição para menos de 50% foram de 1,2% no braço tratado com Phesgo® (Vide item "9. REAÇÕES ADVERSAS").
Phesgo® e/ou pertuzumabe e trastuzumabe por via intravenosa não foram estudados em pacientes com um valor de FEVE pré-tratamento < 55% (EBC) ou < 50% (MBC); histórico anterior de ICC, condições que poderiam prejudicar a função ventricular esquerda, como hipertensão não controlada, infarto do miocárdio recente, arritmia cardíaca séria que exige tratamento ou exposição cumulativa anterior a antraciclina > 360 mg/m2 de doxorrubicina ou equivalente.
Eventos pulmonares
Eventos pulmonares graves foram relatados com o uso de trastuzumabe no cenário pós-comercialização. Esses eventos foram ocasionalmente fatais. Além disso, foram relatados casos de doença pulmonar intersticial, incluindo infiltrados pulmonares, síndrome da angústia respiratória aguda, pneumonia, pneumonite, derrame pleural, dificuldade respiratória, edema pulmonar agudo e insuficiência respiratória. Os fatores de risco associados à doença pulmonar intersticial incluem terapia anterior ou concomitante com outras terapias antineoplásicas conhecidas por estarem associadas a ela, como taxanos, gencitabina, vinorelbina e radioterapia. Esses eventos podem ocorrer como parte de uma reação relacionada à infusão ou com início tardio. Pacientes que apresentam dispneia em repouso devido a complicações de malignidade avançada e comorbidades podem ter risco aumentado de eventos pulmonares. Portanto, esses pacientes não devem ser tratados com Phesgo®. Deve-se ter cuidado com a pneumonite, especialmente em pacientes tratados concomitantemente com taxanos.
Neutropenia febril
Pacientes tratados com Phesgo® em combinação com um taxano apresentam risco aumentado de neutropenia febril.
Os pacientes tratados com pertuzumabe intravenoso em combinação com trastuzumabe e docetaxel apresentam risco aumentado de neutropenia febril em comparação com pacientes tratados com placebo, trastuzumabe e docetaxel, especialmente durante os primeiros 3 ciclos de tratamento. No estudo CLEOPATRA em câncer de mama metastático, as contagens de neutrófilos nadir foram semelhantes em pacientes tratados com pertuzumabe e tratados com placebo. A maior incidência de neutropenia febril em pacientes tratados com pertuzumabe foi associada à maior incidência de mucosite e diarreia nesses pacientes. O tratamento sintomático para mucosite e diarreia deve ser considerado. Não foram relatados eventos de neutropenia febril após a interrupção do docetaxel.
Reações relacionadas à administração
Phesgo® foi associado a reações relacionadas com a injeção. As reações relacionadas à injeção foram definidas como qualquer reação sistêmica com sintomas como febre, calafrios, cefaléia, provavelmente devido à liberação de citocinas que ocorre 24 horas após a administração de Phesgo®.
Embora não tenham sido observados resultados fatais resultantes de reações relacionadas com a injeção com Phesgo®, deve ter-se cuidado, uma vez que reações fatais relacionadas com a infusão foram associadas a pertuzumabe intravenoso em combinação com trastuzumabe intravenoso e quimioterapia.
Reações relacionadas à administração ocorreram em 21% dos pacientes que receberam Phesgo®. No braço tratado com Phesgo®, as reações relacionadas à administração mais comuns foram reações no local da injeção (15%) e dor no local da injeção (2%).
Reações de hipersensibilidade/ anafilaxia
Os pacientes devem ser observados de perto quanto a reações de hipersensibilidade. Foram observadas reações de hipersensibilidade graves, incluindo anafilaxia e acontecimentos fatais, com pertuzumabe em combinação com trastuzumabe e quimioterapia. A maioria das reações anafiláticas ocorreram nos primeiros 6 - 8 ciclos de tratamento, quando pertuzumabe e trastuzumabe foram administrados em combinação com quimioterapia.
Os medicamentos para tratar essas reações, bem como o equipamento de emergência, devem estar disponíveis para uso imediato. Phesgo® deve ser descontinuado definitivamente em caso de reações de hipersensibilidade graves de grau 4 do NCI-CTCAE (anafilaxia), broncoespasmo ou síndrome da dificuldade respiratória aguda (vide item "8. POSOLOGIA E MODO DE USAR", subitem "Modificações de dose").
No estudo FeDeriCa, a incidência de hipersensibilidade foi de 1,2% no braço tratado com Phesgo®.
Diarreia
Phesgo® pode provocar diarreia grave. A diarreia é mais frequente durante a administração concomitante com a terapia com taxano. Pacientes idosos (≥ 65 anos) têm maior risco de diarreia em comparação com pacientes mais jovens ( < 65 anos). Trate a diarreia de acordo com a prática e as diretrizes padrão. A intervenção precoce com loperamida, reposição de fluidos e eletrólitos deve ser considerada, principalmente em pacientes idosos e em caso de diarreia grave ou prolongada. A interrupção do tratamento com Phesgo® deve ser considerada se nenhuma melhora na condição do paciente for alcançada. Quando a diarreia está controlada, o tratamento com Phesgo® pode ser reinstaurado.
Mulheres e homens com potencial reprodutivo
Fertilidade
Pertuzumabe
Não foram realizados estudos específicos de fertilidade em animais para avaliar o efeito do pertuzumabe. Não foram observados efeitos adversos nos órgãos reprodutores masculinos e femininos em estudos de toxicidade de dose repetida de pertuzumabe com duração até seis meses em macacos cynomolgus (vide item "3. CARACTERÍSTICAS FARMACOLÓGICAS").
Trastuzumabe
Os estudos de reprodução realizados em macacos cynomolgus com trastuzumabe não revelaram evidência de diminuição da fertilidade nas fêmeas de macacos cynomolgus (vide item "3. CARACTERÍSTICAS FARMACOLÓGICAS").
Uso durante a gravidez e lactação
Phesgo® pode causar danos fetais quando administrado a uma mulher grávida. Em relatórios pós-comercialização, o uso de trastuzumabe intravenoso durante a gravidez resultou em casos de oligoidrâmnio e sequência de oligoidrâmnio que se manifestaram como hipoplasia pulmonar, anormalidades esqueléticas e morte neonatal. Em um estudo de reprodução animal, a administração de pertuzumabe por via intravenosa em macacas cynomolgus prenhes durante o período de organogênese resultou em oligoidrâmnio, atraso no desenvolvimento dos rins fetais e morte embriofetal em exposições 2,5 a 20 vezes a exposição em humanos na dose recomendada, com base na Cmáx.
Informe mulheres grávidas e mulheres com potencial reprodutivo que a exposição ao Phesgo® durante a gravidez ou nos 7 meses anteriores à concepção pode resultar em danos fetais.
Exame de Gravidez
Verifique o status de gravidez de mulheres com potencial reprodutivo antes do início de Phesgo®.
Contracepção
Aconselhe as mulheres com potencial reprodutivo a utilizar métodos contraceptivos eficazes durante o tratamento e por 7 meses após a última dose de Phesgo®.
Gravidez
Categoria de risco na gravidez: D. Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
Phesgo® deve ser evitado durante a gravidez, a menos que o potencial benefício para a mãe supere o risco potencial para o feto.
Notifique o paciente sobre os possíveis riscos a um feto. Existem considerações clínicas se Phesgo® for utilizado durante a gravidez ou no período de 7 meses antes da concepção. Vide informações a seguir.
O risco associado estimado de defeitos congênitos de grande porte e aborto espontâneo para a população indicada é desconhecido. Na população geral dos EUA, o risco associado estimado de defeitos congênitos de grande porte e aborto espontâneo em gestações clinicamente reconhecidas é de 2% - 4% e 15% - 20%, respectivamente.
Considerações Clínicas
Monitore oligoidrâmnio em mulheres que receberam Phesgo® durante a gravidez ou no período de 7 meses antes da concepção. Se ocorrer oligoidrâmnio, realize testes fetais apropriados para a idade gestacional e consistentes com os padrões de tratamento da comunidade.
Relatos de casos descreveram oligoidrâmnio em mulheres grávidas que receberam trastuzumabe isolado ou em combinação com quimioterapia. Em alguns relatos de caso, o índice de líquido amniótico aumentou após trastuzumabe ter sido interrompido. Em um caso, a terapia com trastuzumabe foi retomada após a melhora do índice amniótico, e oligoidrâmnio voltou a ocorrer.
Lactação
Não há informações sobre a presença de pertuzumabe, trastuzumabe ou hialuronidase no leite humano, os efeitos no lactente ou os efeitos na produção de leite. Dados publicados sugerem que a IgG humana está presente no leite humano, mas não entra na circulação neonatal e infantil em quantidades substanciais. Trastuzumabe estava presente no leite de macacas cynomolgus em lactação, mas não esteve associado à toxicidade neonatal. Considere os benefícios de desenvolvimento e saúde do aleitamento materno, juntamente com a necessidade clínica da mãe em relação ao tratamento com Phesgo® e quaisquer efeitos adversos em potencial sobre a criança amamentada quanto ao Phesgo® ou a condição materna subjacente. Essa consideração também deve levar em conta a meia-vida de eliminação do pertuzumabe e o período de eliminação (wash out) de trastuzumabe de 7 meses (Vide item "3. CARACTERÍSTICAS FARMACOLÓGICAS").
Uso em crianças
A segurança e a eficácia de Phesgo® em pacientes pediátricos não foram estabelecidas.
Uso em idosos
Do número total de pacientes no Estudo FeDeriCa (n = 500) tratados com Phesgo®, 11% tinham 65 anos ou mais, enquanto 1,6% tinham 75 anos ou mais.
Os estudos clínicos de Phesgo® não incluíam números suficientes de pacientes com 65 anos ou mais para determinar se eles respondiam de maneira diferente dos pacientes mais jovens. Nos estudos de trastuzumabe intravenoso, o risco de disfunção cardíaca foi maior em pacientes geriátricos do que em pacientes mais jovens, seja naqueles que receberam tratamento para terapia adjuvante ou para doença metastática. Outras diferenças na segurança ou na eficácia não foram observadas entre pacientes idosos e pacientes mais jovens. Nos estudos de pertuzumabe intravenoso em combinação com trastuzumabe, o risco de ocorrência de apetite reduzido, anemia, peso reduzido, astenia, disgeusia, neuropatia periférica e hipomagnesemia foi maior em pacientes com 65 anos de idade ou mais