NINLARO
TAKEDA
ixazomibe
Antineoplásico.
Apresentações.
Cápsulas duras contendo 4 mg, 3 mg ou 2,3 mg de ixazomibe: embalagens com 3 cápsulas.
USO ORAL
USO ADULTO
Composição.
NINLARO® 4 mg Cada cápsula contém 5,7 mg de citrato de ixazomibe, equivalente a 4 mg de ixazomibe. Excipientes: celulose microcristalina, estearato de magnésio e talco. Componentes da cápsula: gelatina, dióxido de titânio, óxido de ferro amarelo e óxido de ferro vermelho. Tinta de impressão: shellac, propilenoglicol, hidróxido de potássio e óxido de ferro preto.
NINLARO® 3 mg Cada cápsula contém 4,3 mg de citrato de ixazomibe equivalente a 3 mg de ixazomibe. Excipientes: celulose microcristalina, estearato de magnésio e talco. Componentes da cápsula: gelatina, dióxido de titânio, óxido de ferro preto. Tinta de impressão: shellac, propilenoglicol, hidróxido de potássio e óxido de ferro preto.
NINLARO® 2,3 mg Cada cápsula contém 3,3 mg de citrato de ixazomibe equivalente a 2,3 mg de ixazomibe. Excipientes: celulose microcristalina, estearato de magnésio e talco. Componentes da cápsula: gelatina, dióxido de titânio, óxido de ferro vermelho. Tinta de impressão: shellac, propilenoglicol, hidróxido de potássio e óxido de ferro preto
Informações técnicas.
1. INDICAÇÕES
NINLARO® é indicado, em combinação com lenalidomida e dexametasona, para o tratamento de pacientes com mieloma múltiplo que receberam pelo menos um tratamento anterior.
2. RESULTADOS DE EFICÁCIA
A eficácia e a segurança do ixazomibe em combinação com a lenalidomida e a dexametasona foram avaliadas em um estudo de superioridade, internacional, randomizado, duplo-cego, controlado por placebo, multicêntrico, de fase 3, em pacientes com mieloma múltiplo recidivado e/ou refratário, que haviam recebido pelo menos um tratamento anterior. Um total de 722 pacientes foram randomizados na proporção de 1:1 para receber a combinação de ixazomibe, lenalidomida e dexametasona (N=360; esquema do ixazomibe) ou placebo, lenalidomida e dexametasona (N=362; esquema do placebo) até a progressão da doença ou toxicidade inaceitável. A randomização foi estratificada de acordo com o número de linhas de tratamento anteriores (1 versus 2 ou 3), Sistema Internacional de Estadiamento (ISS) do mieloma (estadio I ou II versus III) e tratamento anterior com inibidor de proteassoma (exposto ou virgem).
Os pacientes admitidos no estudo tinham mieloma múltiplo que era mensurável pela presença de paraproteína no soro, urina ou pela medição da cadeia leve livre e cuja doença era refratária, incluindo refratariedade primária (isto é, nunca respondeu ao tratamento anterior), recidiva após tratamento anterior ou recidiva e refratariedade a qualquer tratamento anterior. Os pacientes que mudaram de tratamento antes da progressão da doença também foram elegíveis para o recrutamento, assim como aqueles com condições cardiovasculares controladas. Pacientes refratários a inibidores de proteassoma ou a lenalidomida foram excluídos do estudo. Tromboprofilaxia era recomendada para todos os pacientes em ambos os grupos de tratamento de acordo com a bula da lenalidomida. Medicamentos concomitantes, tais como antieméticos, antivirais e anti-histamínicos foram administrados aos pacientes a critério médico, como profilaxia e/ou para controle dos sintomas.
Os pacientes receberam 4 mg de ixazomibe ou placebo nos Dias 1, 8 e 15 mais lenalidomida (25 mg) nos Dias 1 a 21 e dexametasona (40 mg) nos Dias 1, 8, 15 e 22 de um ciclo de 28 dias.
Os pacientes com comprometimento renal receberam uma dose inicial de lenalidomida de acordo com a bula do medicamento. O tratamento continuou até a progressão da doença ou toxicidades inaceitáveis.
A Tabela 1 resume as características dos pacientes e da doença no momento inicial do estudo. As características demográficas e da doença no momento inicial eram equilibradas e comparáveis entre os esquemas do estudo.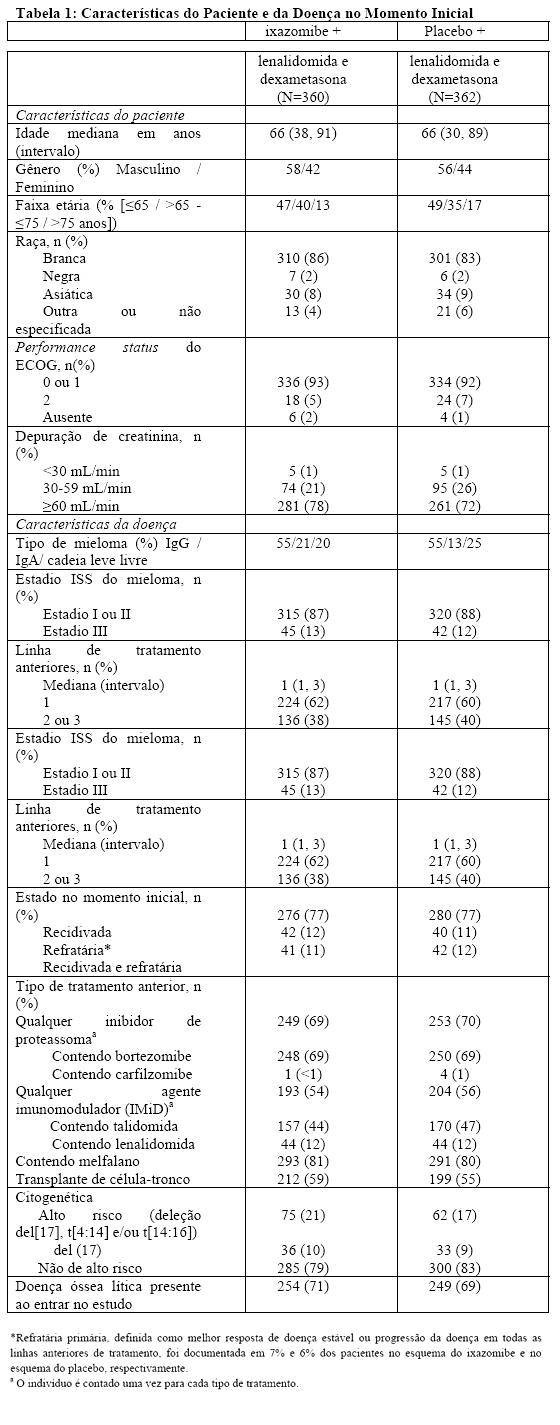
O desfecho primário era a sobrevida livre de progressão (SLP) de acordo com os Critérios de Resposta do Grupo de Trabalho Internacional sobre Mieloma (IMWG) de 2011, avaliado por um comitê de revisão independente (CRI), com base nos resultados do laboratório central. A resposta foi avaliada a cada quatro semanas até a progressão da doença. Na primeira análise interina, aproximadamente 40% dos pacientes tiveram um evento de SLP (286 pacientes de 722 pacientes, os detalhes para cada grupo de tratamento estão na Tabela 2 e na Figura 1). Esta primeira análise pré-especificada tornou-se a análise final para propósitos de testes estatísticos (mediana de seguimento de 14,7 meses e mediana do número de ciclos igual a 13), o esquema do ixazomibe demonstrou resultados significantemente superiores, com uma melhora na mediana da SLP de aproximadamente 6 meses. Nesta análise, os pacientes que estavam recebendo o esquema do ixazomibe viveram significantemente mais tempo sem piora de sua doença em comparação aos pacientes no esquema do placebo. A melhora na SLP no esquema do ixazomibe foi sustentada por melhora nas taxas de resposta global. A SLP e as taxas de respostas estão resumidas na Tabela 2.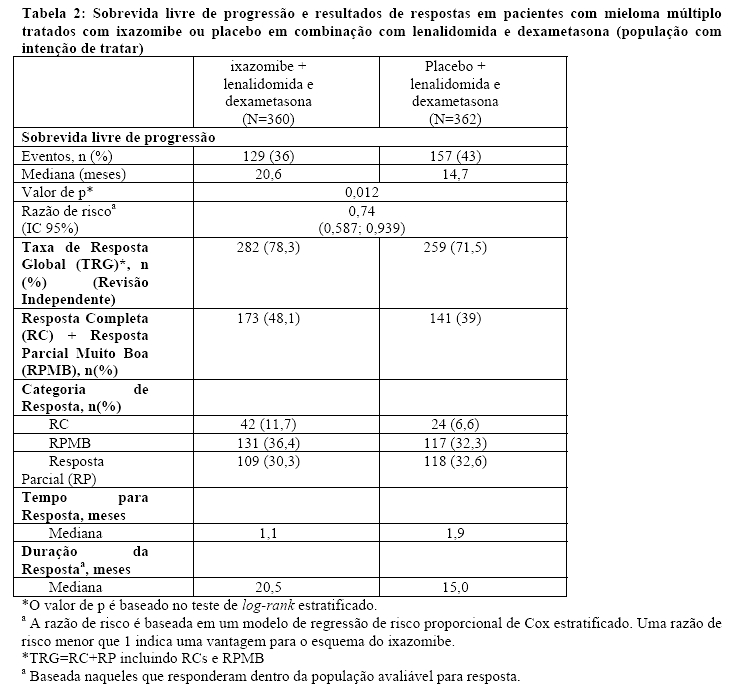
O esquema com ixazomibe demonstrou uma melhora estatisticamente significante na mediana da sobrevida livre de progressão em comparação ao esquema do placebo, como observado abaixo na Figura 1.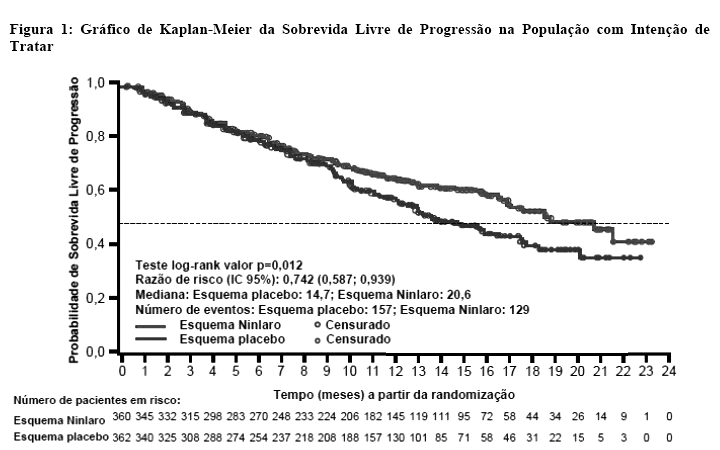
Uma análise interina planejada da SG em um seguimento mediano de 23 meses foi realizada com 35% do número necessário de mortes para a análise final de SG; houve 81 mortes no esquema com ixazomibe e 90 mortes no esquema com placebo. A sobrevida global (SG) mediana não foi alcançada em qualquer um dos regimes. Ao mesmo tempo, foi realizada uma análise exploratória de SLP não-inferencial. Os resultados (número estimado de SLP mediana foi de 20 meses no esquema com ixazomibe e 15,9 meses no esquema com placebo) foram consistentes com a conclusão de efeito positivo do tratamento, como demonstrado na análise primária de SLP.
Cento e trinta e sete pacientes com anomalias citogenéticas de alto risco, del(17), t(4:14), t(14:16), foram inscritos no estudo de Fase 3. Sessenta e nove destes pacientes tinham del(17). O efeito do tratamento foi positivo em pacientes com anormalidades citogenéticas de alto risco. No momento da análise primária para SLP, a SLP mediana em pacientes com del (17) foi de 21,4 meses no regime de ixazomibe em comparação com 9,7 meses no regime de placebo. A SLP mediana na população geral de alto risco (del (17), t(04:14) e/ou t(14:16)) também foi de 21,4 meses no regime de ixazomibe em comparação com 9,7 meses no regime de placebo.
No momento da análise interina planejada de SG, mais mortes ocorreram em pacientes com del(17) recebendo o esquema de placebo (45 %) em comparação com pacientes que receberam o esquema de ixazomibe (25 %). Além disso, mais mortes ocorreram em pacientes com anomalias citogenéticas de alto risco que receberam o esquema de placebo (39%) em comparação com pacientes que receberam o esquema de ixazomibe (20%).
A qualidade de vida, avaliada pela pontuação da saúde global (EORTC QLQ- C30 e MY- 20), foi mantida durante o tratamento e foi semelhante em ambos os regimes de tratamento.
Referências bibliográficas:
1. Moreau P, Masszi T, Grzasko N, et al. Oral Ixazomib, Lenalidomide, and Dexamethasone for Multiple Myeloma. N Engl J Med. 2016;374(17):1621-1634.
2. Leleu X, Masszi T, Bahlis NJ, et al. Patient-reported quality of life with ixazomib-lenalidomide-dexamethasone (IRd) vs placebo-Rd in relapsed/refractory multiple myeloma patients in the global, placebo-controlled TOURMALINE-MM1 study. Haematologica. 2016;101(s1):261 (abstract P660).
3. Hou J, Jin J, Xu Y, et al. Ixazomib plus lenalidomide-dexamethasone (IRd) vs placebo-Rd in patients (pts) with relapsed/refractory multiple myeloma (RRMM): China Continuation of TOURMALINE-MM1. Haematologica. 2016;101(s1):540 (abstract E1305).
4. Mateos M-V, Masszi T, Grzasko N, et al. Efficacy and safety of oral ixazomib-lenalidomide-dexamethasone (IRd) vs placebo-Rd in relapsed/refractory multiple myeloma patients: impact of prior therapy in the phase 3 TOURMALINE-MM1 study. Haematologica. 2016;101(s1):527 (abstract E1276).
5. Avet-Loiseau H, Bahlis NJ, Chng WJ, et al. Impact of cytogenetic risk status on efficacy and safety of ixazomiblenalidomide-dexamethasone (IRd) vs placebo-Rd in relapsed/refractory multiple myeloma patients in the global TOURMALINE-MM1 study. Haematologica. 2016;101(s1):80 (abstract P269).
6. Garderet L, Laubach JP, Stoppa AM, et al. Longer Time to Best Response and Depth of Response Are Associated with Improved Duration of Best Achieved Response and Progression-Free Survival (PFS): Post-Hoc Analysis of Phase 3 Tourmaline-MM1 Trial in Relapsed/Refractory Multiple Myeloma (RRMM). Blood. 2016;128(22):2134 (abstract 2134).
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades farmacodinâmicas
Mecanismo de ação
Ixazomibe é um inibidor oral de proteassoma, reversível e altamente seletivo. Ixazomibe preferencialmente ligase e inibe a atividade do tipo da quimiotripsina da subunidade beta 5 do proteassoma 20S.
Ixazomibe induziu apoptose de diversos tipos de células tumorais in vitro. O ixazomibe demonstrou citotoxicidade in vitro contra células de mieloma de pacientes que haviam apresentado recidiva depois de vários tratamentos anteriores, incluindo bortezomibe, lenalidomida e dexametasona. A combinação de ixazomibe e lenalidomida demonstrou efeitos citotóxicos sinérgicos em linhagens de células de mieloma múltiplo. In vivo, ixazomibe demonstrou atividade antitumoral em vários modelos de enxerto de tumor, incluindo modelos de mieloma múltiplo.
O ixazomibe também altera o microambiente da medula óssea. In vitro, ixazomibe inibiu a proliferação de células de mieloma múltiplo cultivadas simultaneamente com células estromais da medula óssea. Ixazomibe demonstrou um efeito antiangiogênico em um ensaio in vitro de formação de tubo capilar. Ixazomibe promoveu osteoblastogênese e atividade osteoblástica e inibiu a osteoclastogênese e a reabsorção de osteoclastos in vitro. Adicionalmente, ixazomibe impediu a perda óssea em um modelo in vivo de mieloma múltiplo em camundongos.
Eletrofisiologia cardíaca
Ixazomibe não prolonga o intervalo QTc em exposições clinicamente relevantes com base nos resultados de uma análise farmacocinética-farmacodinâmica de dados de 245 pacientes. Na dose de 4 mg, a variação média no QTcF desde o momento basal é estimada em 0,07 ms (IC 90%; -0,22, 0,36) pela análise baseada no modelo.
Não houve nenhuma relação perceptível entre a concentração de ixazomibe e o intervalo RR, sugerindo que não houve efeito clinicamente significativo do ixazomibe na frequência cardíaca.
Propriedades farmacocinéticas
Absorção
Após a administração oral, o pico das concentrações plasmáticas de ixazomibe é atingido em aproximadamente uma hora após a administração. A biodisponibilidade oral absoluta média é 58%. A ASC (área sob a curva) do ixazomibe aumenta de maneira proporcional à dose em um intervalo de dose de 0,2-10,6 mg.
A administração com uma refeição de alto teor de gordura diminuiu a ASC de ixazomibe em 28% comparado com a administração após uma noite de jejum (veja POSOLOGIA E MODO DE USAR).
Distribuição
Ixazomibe apresenta 99% de ligação às proteínas plasmáticas e se distribui nos glóbulos vermelhos com uma razão da ASC do sangue para o plasma igual a 10. O volume de distribuição no estado de equilíbrio é 543L.
Eliminação
Ixazomibe exibe um perfil de disposição multiexponencial. Baseado na análise da farmacocinética populacional, a depuração sistêmica foi aproximadamente 1,86 L/h, com variabilidade interindividual de 44%. A meia-vida terminal (t1/2) do ixazomibe foi de 9,5 dias. Acúmulo de aproximadamente duas vezes foi observado com a administração oral semanal no Dia 15.
Metabolismo
Após a administração oral de uma dose marcada radioativamente, 70% do material total relacionado com a droga no plasma foi representado pelo ixazomibe. O metabolismo por várias enzimas CYP e proteínas não CYP é esperado como o principal mecanismo de depuração de ixazomibe. Em concentrações clinicamente relevantes de ixazomibe, estudos in vitro usando isoenzimas do citocromo P450 expressas em cDNA humano indicaram que nenhuma isoenzima CYP específica contribui predominantemente para o metabolismo de ixazomibe e que as proteínas não CYP contribuem para o metabolismo global. Em concentrações que excederam aquelas observadas clinicamente, ixazomibe foi metabolizado por múltiplas isoformas do CYP, com contribuições relativas estimadas de 3A4 (42,3%), 1A2 (26,1%), 2B6 (16,0%), 2C8 (6,0%), 2D6 (4,8%), 2C19 (4,8%) e 2C9 ( < 1%).
Excreção
Após a administração de uma dose oral única de 14C-ixazomibe a 5 pacientes com câncer avançado, 62% da radioatividade administrada foi excretada na urina e 22% nas fezes. Ixazomibe inalterado representou < 3,5% da dose administrada recuperada na urina.
Populações Especiais
Idade, Gênero, Raça
Não houve efeito clinicamente significativo da idade (23-91 anos), sexo, área da superfície corporal (1,2-2,7 m2) ou raça na depuração de ixazomibe, com base nos resultados de uma análise da farmacocinética populacional.
Função hepática comprometida
A farmacocinética de ixazomibe é similar em pacientes com função hepática normal e em pacientes com comprometimento hepático leve (bilirrubina total ≤ limite superior da normalidade (LSN) e aspartato aminotransferase (AST) > LSN ou bilirrubina total > 1-1,5 x LSN e qualquer AST) com base nos resultados de uma análise da farmacocinética populacional.
A farmacocinética de ixazomibe foi caracterizada em pacientes com função hepática normal em 4 mg (N=12), comprometimento hepático moderado em 2,3 mg (bilirrubina total > 1,5-3 x LSN, N=13) ou comprometimento hepático grave em 1,5 mg (bilirrubina total > 3 x LSN, N=18). A ASC normalizada para a dose não ligada foi 27% maior em pacientes com comprometimento hepático moderado ou grave em comparação aos pacientes com função hepática normal (veja POSOLOGIA E MODO DE USAR).
Função renal comprometida
A farmacocinética de ixazomibe é similar em pacientes com função renal normal e em pacientes com comprometimento renal leve ou moderado (depuração de creatinina ≥30 mL/min), com base nos resultados de uma análise da farmacocinética populacional.
A farmacocinética de ixazomibe foi caracterizada em 3 mg em pacientes com função renal normal (depuração de creatinina > 90 mL/min, N=18), comprometimento renal grave (depuração de creatinina < 30 mL/min, N=14) ou estágio final de doença renal exigindo diálise (N=6). A ASC não ligada foi 38% maior em pacientes com comprometimento renal grave ou estágio final de doença renal exigindo diálise em comparação aos pacientes com função renal normal. As concentrações de ixazomibe antes e depois do dialisador, mensuradas durante a sessão de hemodiálise, eram semelhantes, sugerindo que ixazomibe não é dialisável (veja POSOLOGIA E MODO DE USAR).
Interações Medicamentosas
Efeito de outros fármacos no ixazomibe
Indutores fortes de CYP3A4
A administração concomitante de ixazomibe com rifampicina diminuiu a Cmáx de ixazomibe em 54% e a ASC em 74% (veja INTERAÇÕES MEDICAMENTOSAS).
Inibidores fortes de CYP3A4
A administração concomitante de ixazomibe com claritromicina não resultou em alteração clinicamente significante na exposição sistêmica de ixazomibe. A Cmáx de ixazomibe foi reduzida em 4% e a ASC aumentou em 11% (veja INTERAÇÕES MEDICAMENTOSAS).
Inibidores fortes da CYP1A2
A administração concomitante de ixazomibe com inibidores fortes da CYP1A2 não resultou em alteração clinicamente significativa na exposição sistêmica de ixazomibe, com base em uma análise da farmacocinética populacional (veja INTERAÇÕES MEDICAMENTOSAS).
Efeito de ixazomibe em outros fármacos
Não é esperado que ixazomibe produza interações medicamentosas através da inibição ou indução do CYP. Ixazomibe não é um inibidor reversível ou dependente do tempo das isoenzimas CYPs 1A2, 2B6, 2C8, 2C9, 2C19, 2D6 ou 3A4/5. Ixazomibe não induziu a atividade da CYP1A2, CYP2B6 e CYP3A4/5 ou os níveis correspondentes de proteína imunorreativa.
Interações baseadas em transportadores
Não é esperado que ixazomibe cause interações medicamentosas mediadas por transportadores. Ixazomibe é um substrato de P-gp de baixa afinidade. Ixazomibe não é um substrato de BCRP, MRP2 e OATPs hepáticas. Ixazomibe não é inibidor de gp-P, BCRP, MRP2, OATP1B1, OATP1B3, transportador de cátion orgânico (OCT)2, transportador de ânion orgânico (OAT)1, OAT3, MATE1 ou MATE2-K.
Dados não-clínicos de segurança
Carcinogênese, Mutagênese, Comprometimento da Fertilidade
Ixazomibe não foi mutagênico em um teste de mutação bacteriana reversa (teste de Ames) nem foi clastogênico em um teste de micronúcleo de medula óssea em camundongos. Ixazomibe foi considerado positivo em um teste de clastogenicidade in vitro em linfócitos do sangue periférico humano. Entretanto, ixazomibe foi negativo em um teste cometa (comet test) in vivo em camundongos, no qual o DNA foi avaliado no estômago e no fígado. Portanto, o balanço das evidências mostra que ixazomibe não apresenta risco de genotoxicidade. Não foram realizados estudos de carcinogenicidade com ixazomibe.
Ixazomibe causou toxicidade embrio-fetal em ratas e coelhas grávidas apenas em doses maternas tóxicas e em exposições que foram ligeiramente maiores que as observadas em pacientes que receberam as doses recomendadas. Estudos de fertilidade e desenvolvimento embrionário inicial e toxicologia pré-natal e pós-natal não foram conduzidos com ixazomibe, mas a avaliação dos tecidos reprodutores foi conduzida nos estudos gerais de toxicologia. Não houve efeitos devido ao tratamento com ixazomibe nos órgãos reprodutores masculinos ou femininos em estudos de até 6 meses de duração em ratos e até 9 meses de duração em cães.
Toxicologia e/ou Farmacologia Animal
Em estudos gerais de toxicidade de ciclos múltiplos conduzidos em ratos e cães, os principais órgãos alvo incluíram o trato gastrointestinal (GI), tecidos linfoides e o sistema nervoso. Os achados gastrointestinais incluíram emese e/ou diarreia, aumentos nos parâmetros de leucócitos e alterações microscópicas (inflamação, hiperplasia epitelial, infiltração neutrofílica, necrose de célula isolada e erosão/ulceração). No sistema linfoide a toxicidade foi caracterizada por depleção/necrose linfóide (incluindo a medula óssea), infiltração neutrofílica e necrose de célula isolada. Os efeitos no sistema nervoso foram observados primariamente em cães, em doses orais ≥0,1 mg/kg (2 mg/m2) e incluíram achados microscópicos de degeneração neuronal mínima a leve dos gânglios simpáticos, da raiz dorsal, autonômicos periféricos (glândula salivar) e de órgão terminal, e degeneração secundária mínima de fibra axonal/nervosa dos nervos periféricos e tratos ascendentes nas colunas dorsais da medula espinhal. No estudo de 9 meses (10 ciclos) em cães, onde o esquema de administração se assemelha ao esquema clínico (ciclo de 28 dias), os efeitos neuronais microscópicos foram, em geral, mínimos quanto a natureza e observados apenas em 0,2 mg/kg (4 mg/m2; ASC 168 1940 h*ng/mL). A maioria dos achados em órgãos alvo demonstrou recuperação parcial a completa após a descontinuação do tratamento, com exceção dos achados neuronais no gânglio da raiz dorsal lombar e na coluna dorsal. A ausência de degeneração neuronal em andamento nos gânglios periféricos e a presença apenas de alterações degenerativas secundárias nas fibras nervosas e axônios é consistente com a ausência de toxicidade persistente.
Com base em estudos em animais, ixazomibe não cruza a barreira hemato-encefálica. Adicionalmente, os estudos não clínicos de farmacologia de segurança e as avaliações tanto in vitro (em canais hERG) como in vivo não demonstraram efeitos do ixazomibe nas funções cardiovascular e respiratória.
4. CONTRAINDICAÇÕES
Não há contraindicações para o uso de NINLARO®.
5. ADVERTÊNCIAS E PRECAUÇÕES
Trombocitopenia
Trombocitopenia foi relatada com ixazomibe, com ocorrência típica de nadir de plaquetas durante os dias 14-21 de cada ciclo de 28 dias de tratamento e recuperação para o valor basal no início do próximo ciclo. Três por cento dos pacientes no esquema do ixazomibe e 1% dos pacientes no esquema do placebo apresentaram contagem de plaquetas ≤10.000/mm3 durante o tratamento. Menos de 1% dos pacientes em ambos os regimes tiveram contagem de plaquetas ≤ 5.000/mm3 durante o tratamento. A trombocitopenia resultou em descontinuação de um ou mais dos três medicamentos em < 1% dos pacientes no esquema de ixazomibe e 2% dos pacientes no esquema do placebo. A trombocitopenia não resultou em aumento de eventos hemorrágicos ou da transfusão de plaquetas.
Monitorar a contagem de plaquetas pelo menos uma vez por mês durante o tratamento com ixazomibe. Considerar
o monitoramento mais frequente durante os três primeiros ciclos de acordo com a bula de lenalidomida. Controlar a trombocitopenia com modificações da dose (veja POSOLOGIA E MODO DE USAR) e transfusões de plaquetas conforme as diretrizes médicas padrão.
Toxicidades gastrointestinais
Diarreia, náusea e vômito foram relatados com ixazomibe e, ocasionalmente, exigiram o uso de medicamentos antieméticos e antidiarreicos, e cuidados de suporte. A diarreia resultou em descontinuação de um ou mais dos três medicamentos em 1% dos pacientes no esquema do ixazomibe e < 1% nos pacientes no esquema do placebo. Ajustar a dose para sintomas graves (Grau 3-4) (veja POSOLOGIA E MODO DE USAR).
Hepatotoxicidade
Lesões hepáticas induzidas por fármacos, lesões hepatocelulares, esteatose hepática, hepatite colestática e hepatotoxicidade têm sido relatadas raramente com NINLARO® (citrato de ixazomibe). As enzimas hepáticas devem ser monitorizadas regularmente e a dose deve ser ajustada quando os sintomas atingirem grau 3 ou 4.
Tromboprofilaxia
O tromboembolismo é uma reação adversa que pode ser observada em pacientes que fazem uso de lenalidomida. Portanto, a tromboprofilaxia é recomendada em pacientes a serem tratados com NINLARO® (citrato de ixazomibe) em combinação com lenalidomida e dexametasona e deve basear-se numa avaliação dos riscos subjacentes e do estado clínico do paciente.
Uso durante a gravidez e a lactação
Categoria C de Risco na Gravidez - Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Gravidez
Ixazomibe pode causar dano fetal quando administrado a mulheres grávidas com base no seu mecanismo de ação e achados em animais. Mulheres com potencial reprodutivo devem ser avisadas para evitar a gravidez durante o tratamento com ixazomibe. Se ixazomibe for usado durante a gravidez ou se a paciente se tornar grávida durante o tratamento com ixazomibe, a paciente deve ser informada do risco potencial para o feto. Avisar mulheres com potencial reprodutivo de que elas devem utilizar um método contraceptivo eficaz durante o tratamento com ixazomibe e durante 90 dias após a dose final. Mulheres que usam contraceptivos hormonais devem utilizar adicionalmente um método contraceptivo de barreira (ver Dados não-clínicos de segurança).
Não há dados disponíveis em humanos com relação ao potencial efeito de ixazomibe na gravidez ou no desenvolvimento do embrião ou feto. Entretanto, estudos embrio-fetais em animais demonstraram que ixazomibe tem potencial para causar letalidade embrio-fetal. As mulheres devem ser informadas sobre o potencial risco para o feto e para evitar a gravidez enquanto estiverem sob tratamento com ixazomibe. Ixazomibe não é recomendado durante a gravidez e em mulheres em idade fértil que não utilizam métodos contraceptivos.
Os pacientes do sexo masculino e feminino em idade fértil devem usar medidas contraceptivas eficazes durante o tratamento e por 90 dias após o seu término. Quando ixazomibe é administrado em conjunto com a dexametasona, que é conhecida por ser um indutor fraco a moderado do CYP3A4, bem como de outras enzimas e transportadores, o risco de diminuição da eficácia de contraceptivos orais deve ser considerado. Mulheres que usam contraceptivos hormonais orais devem utilizar adicionalmente um método contraceptivo de barreira.
Em estudos de determinação da dose em ratas (0,6 mg/kg; 3,6 mg/m2) e coelhas (1,0 mg/kg; 12 mg/m2) grávidas, houve redução do peso fetal, uma tendência para diminuição da viabilidade fetal e/ou aumento de perdas pósimplantação. No entanto, estes achados não foram reproduzidos claramente em estudos definitivos e foram observados apenas em doses tóxicas para as mães (doses que causaram redução do peso corporal e/ou do consumo de alimentos). No estudo definitivo em coelhos foram observados aumentos nas variações/anormalidades do esqueleto fetal (vértebras caudais, número de vértebras lombares e costelas supranumerárias completas) em doses ≥0,3 mg/kg (3,6 mg/m2), que também foram associadas com toxicidade materna. Uma dose de 0,1 mg/kg (1,2 mg/m2) não resultou em toxicidade materna ou causou efeitos nos embriões/fetos.
Amamentação
Não se sabe se ixazomibe ou seus metabólitos são excretados no leite humano. Muitos fármacos são excretados no leite humano e, como resultado, pode haver um potencial para eventos adversos em lactentes. A amamentação deve ser descontinuada.
Fertilidade
Não foram conduzidos estudos de fertilidade com ixazomibe; entretanto, em estudos não clínicos em ratos e cães não foram observados efeitos nos órgãos reprodutores de machos ou fêmeas (veja "Dados não-clínicos de segurança").
Efeitos na capacidade de dirigir veículos e operar máquinas
Não há dados sobre o efeito de ixazomibe na capacidade de dirigir ou operar máquinas. Foram observadas fadiga e tonturas em estudos clínicos. Os pacientes devem ser aconselhados a não dirigir veículos ou utilizar máquinas se sentirem algum destes sintomas.
6. INTERAÇÕES MEDICAMENTOSAS
Indutores fortes de CYP3A4
A administração concomitante de indutores fortes de CYP3A4 (como rifampicina, fenitoína, carbamazepina eerva de São João) com ixazomibe não é recomendada (veja PROPRIEDADES FARMACOCINÉTICAS).
Inibidores fortes de CYP3A4
Não é necessário modificar a dose de ixazomibe na administração concomitante com inibidores fortes de CYP3A4 (veja PROPRIEDADES FARMACOCINÉTICAS).
Inibidores fortes de CYP1A2
Não é necessário modificar a dose de ixazomibe na administração concomitante com inibidores fortes de CYP1A2 (veja PROPRIEDADES FARMACOCINÉTICAS).
Interação com alimentos
A administração com uma refeição de alto teor de gordura diminuiu a ASC do ixazomibe em 28% comparado com a administração após uma noite de jejum. Ixazomibe deve ser tomado pelo menos uma hora antes ou duas horas depois da ingestão de alimentos (veja POSOLOGIA E MODO DE USAR).
7. CONDIÇÕES DE ARMAZENAMENTO DO MEDICAMENTO
NINLARO® deve ser conservado em temperatura ambiente (15°C a 30°C).
A cápsula não deve ser removida até pouco antes da administração.
Este medicamento tem validade de 36 meses a partir da data de sua fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
As cápsulas de NINLARO® são cor de laranja claro (4 mg), cinza claro (3 mg) ou rosa claro (2,3 mg).
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
Posologia
NINLARO® (citrato de ixazomibe) em combinação com lenalidomida e dexametasona
A dose inicial recomendada de NINLARO® é uma cápsula de 4 mg administrada por via oral, uma vez por semana nos Dias 1, 8 e 15 de um ciclo de tratamento de 28 dias. A dose inicial recomendada de lenalidomida é 25 mg, administrada diariamente nos Dias 1 até 21 de um ciclo de tratamento de 28 dias. A dose inicial recomendada de dexametasona é 40 mg, administrada nos Dias 1, 8, 15 e 22 de um ciclo de tratamento de 28 dias.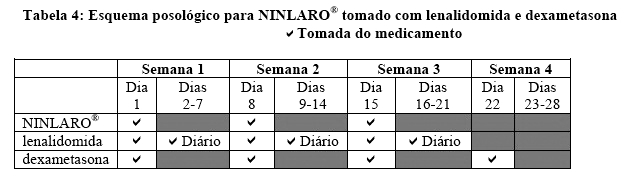
Para informações adicionais relacionadas à lenalidomida e à dexametasona consulte as respectivas bulas.
Antes de iniciar um novo ciclo de tratamento: ·
· A contagem absoluta de neutrófilos deve ser ≥1.000/mm3.
· A contagem de plaquetas deve ser ≥75.000/mm3.
· As toxicidades não hematológicas devem, a critério médico, estar em geral recuperadas para a condição do paciente no momento basal ou ≤ Grau 1.
O tratamento deve ser mantido até a progressão da doença ou toxicidade inaceitável.
Doses atrasadas ou esquecidas
Caso haja atraso ou esquecimento de uma dose de NINLARO®, a dose deve ser tomada apenas se a próxima dose estiver programada para depois de 72 horas ou mais. Uma dose esquecida não deve ser tomada nas 72 horas que antecedem a próxima dose programada. Uma dose duplicada não deve ser tomada para compensar a dose esquecida.
Se o paciente vomitar após a tomada da dose, ele não deve repetir a dose. O paciente deve prosseguir com a administração na hora programada para a próxima dose.
Modificações da dose
Os passos para a redução da dose de NINLARO® são fornecidos na Tabela 5 e as orientações para a modificação da dose estão na Tabela 6.
Uma abordagem de modificação alternada da dose é recomendada para NINLARO® (citrato de ixazomibe) e lenalidomida na presença de toxicidades coincidentes de trombocitopenia e erupção cutânea. Para estas toxicidades, o primeiro passo para a modificação da dose é suspender/reduzir a lenalidomida. Veja na bula do produto os passos para a redução da dose da lenalidomida para estas toxicidades e neutropenia.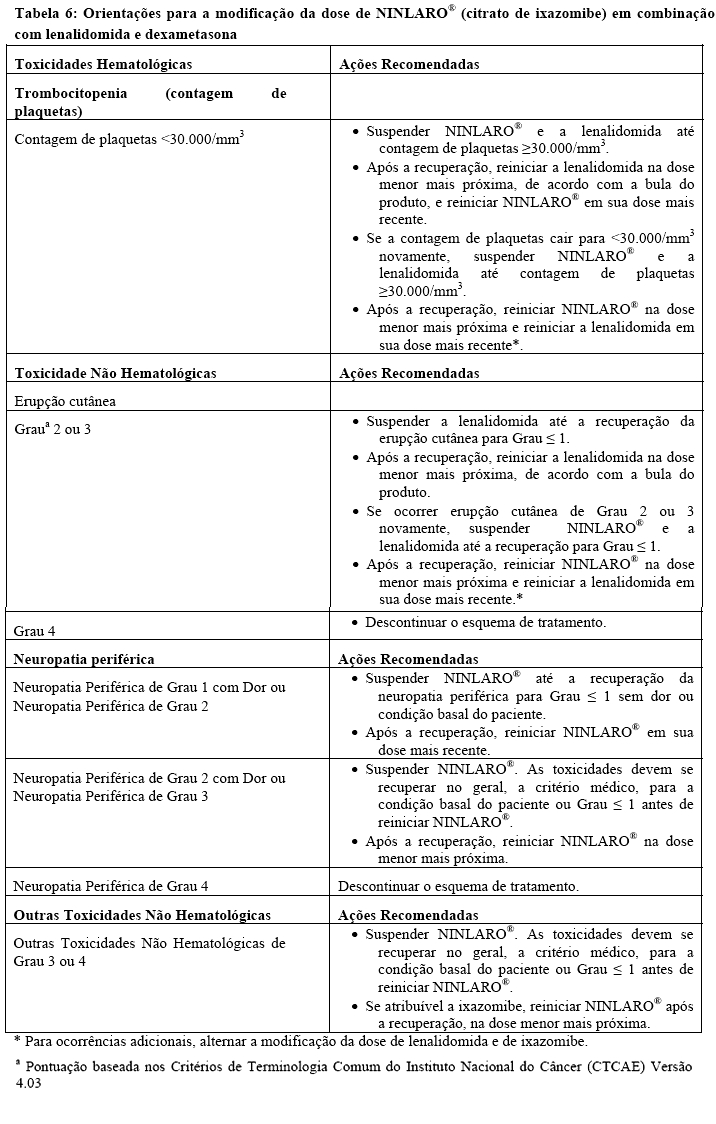
Medicamentos concomitantes
A profilaxia antiviral deve ser considerada em pacientes que estão sendo tratados com NINLARO® para diminuir
o risco de reativação de herpes zoster. Os pacientes incluídos em estudos com NINLARO® que receberam profilaxia antiviral tiveram uma menor incidência de infecção por herpes zoster em comparação com os pacientes que não receberam profilaxia.
Populações de Pacientes Especiais
Pacientes idosos
Não é necessário ajustar a dose de NINLARO® para pacientes acima de 65 anos de idade com base nos resultados da análise da farmacocinética populacional.
Nos estudos de NINLARO®, não foram observadas diferenças clinicamente significantes na segurança e eficácia entre os pacientes com menos de 65 anos de idade e pacientes com 65 anos de idade ou mais velhos.
Pacientes pediátricos
A segurança e a eficácia de NINLARO® não foram estabelecidas em menores de 18 anos de idade. Não há dados disponíveis.
Comprometimento da função hepática
Não é necessário ajustar a dose de NINLARO® para pacientes com comprometimento hepático leve (bilirrubina total ≤ limite superior da normalidade (LSN) e aspartato aminotransferase (AST) > LSN ou bilirrubina total > 11,5 vezes o LSN e qualquer AST) com base nos resultados de uma análise da farmacocinética populacional. Uma dose menor de uma cápsula de 3 mg é recomendada para pacientes com comprometimento hepático moderado (bilirrubina total > 1,5-3 vezes o LSN) ou grave (bilirrubina total > 3 vezes o LSN) com base nos resultados de um estudo de farmacocinética (veja "Propriedades farmacocinéticas").
Comprometimento da função renal
Não é necessário ajustar a dose de NINLARO® para pacientes com comprometimento renal leve ou moderado (depuração de creatinina ≥ 30 mL/min) com base nos resultados de uma análise da farmacocinética populacional. Uma dose menor de uma cápsula de 3 mg é recomendada para pacientes com comprometimento renal grave (depuração de creatinina < 30 mL/min) ou em estágio final da doença renal que necessita de diálise com base nos resultados de um estudo de farmacocinética. NINLARO® não é eliminado durante o procedimento de diálise e, portanto, pode ser administrado independentemente do momento da diálise (veja "Propriedades farmacocinéticas"). Veja na bula do produto as recomendações posológicas da lenalidomida para pacientes com comprometimento renal.
Método de administração
NINLARO® deve ser tomado aproximadamente na mesma hora nos dias 1, 8 e 15, pelo menos uma hora antes ou pelos menos duas horas depois da ingestão de alimentos. A cápsula inteira deve ser deglutida, com água. A cápsula não deve ser esmagada, mastigada ou aberta.
NINLARO® é citotóxico. As cápsulas não devem ser abertas ou trituradas. O contato direto com o conteúdo da cápsula deve ser evitado. Em caso de rompimento da cápsula, evitar contato direto do conteúdo da cápsula com a pele ou olhos. Se ocorrer contato com a pele, lave abundantemente com água e sabão. Se houver contacto com os olhos, lave abundantemente com água.
Este medicamento não deve ser partido, aberto ou mastigado.
9. REAÇÕES ADVERSAS
Estudos clínicos
A população de segurança dos estudos clínicos de fase 3, randomizados, duplo-cegos, controlados por placebo incluiu 720 pacientes com mieloma múltiplo recidivado e/ou refratário, que receberam NINLARO® (citrato de ixazomibe) em combinação com lenalidomida e dexametasona (esquema do ixazomibe; N=360) ou placebo em combinação com lenalidomida e dexametasona (esquema do placebo; N=360).
As reações adversas relatadas com maior frequência (≥20%) no esquema de NINLARO® e placebo foram diarreia (42% vs. 36%), constipação (34% vs. 25%), trombocitopenia (28% vs. 14%), neuropatia periférica (28% vs. 21%), náusea (26% vs. 21%), edema periférico (25% vs. 18%), vômito (22% vs. 11%) e dor lombar (21% vs. 16%). Reações adversas graves relatadas em ≥2% dos pacientes incluíram trombocitopenia (2%) e diarreia (2%). Para cada reação adversa, um ou mais dos três medicamentos foram descontinuados em ≤1% dos pacientes no esquema de NINLARO®.
A convenção a seguir é usada para a classificação da frequência de uma reação adversa a medicamento (RAM) e é baseada nas diretrizes do Conselho de Organizações Internacionais de Ciências Médicas (CIOMS): muito comum (≥1/10); comum (≥1/100 a < 1/10), incomum (≥1/1.000 a < 1/100); rara (≥1/10.000 a < 1/1.000); muito rara ( < 1/10.000); não conhecida (não pode ser estimada dos dados disponíveis).
Reação muito comum (≥1/10): infecção do trato respiratório superior como nasofaringite, sinusite e faringite, trombocitopenia*, neuropatias periféricas*, diarreia, constipação, náusea, vômito, erupção cutânea*, dor lombar e edema periférico.
Reação comum (≥1/100 e < 1/10): herpes zoster.
As reações adversas de grau 3 muito comuns incluíram trombocitopenia*. As reações adversas de grau 3 comuns incluíram neuropatias periféricas*, diarreia, vômito, náusea, erupção cutânea*, dor lombar e edema periférico. As reações adversas de grau 3 incomuns incluíram infecção do trato respiratório superior, herpes zoster, constipação e dor lombar. Trombocitopenia* de Grau 4 foi comum.
*representa um agrupamento dos termos preferidos
Erupção cutânea
Erupção cutânea ocorreu em 19% dos pacientes no esquema de NINLARO® comparado a 11% dos pacientes no esquema do placebo. A maioria dos eventos de erupção cutânea era de grau 1 (10 % no esquema de ixazomibe e 7% no esquema de placebo) ou grau 2 (6% no esquema de ixazomibe e 3% no esquema de placebo). Erupção cutânea de grau 3 foi relatada em 3% dos pacientes no esquema de NINLARO® comparado a 1% dos pacientes no esquema do placebo e não houve reações cutâneas de grau 4 ou graves no estudo de fase 3. O tipo mais comum de erupção cutânea relatado em ambos os esquemas incluiu erupção cutânea maculo-papular e macular. A erupção cutânea resultou na descontinuação de um ou mais dos três medicamentos em < 1% dos pacientes em ambos os esquemas.
Eventos cutâneos foram relatados com a lenalidomida e a dexametasona.
Neuropatia periférica
A maioria das reações adversas de neuropatia periférica foi de grau 1 (18 % no esquema de ixazomibe e 14 % no esquema de placebo) e Grau 2 (8 % no esquema de ixazomibe e 5 % no esquema de placebo). Reações adversas de neuropatia periférica de Grau 3 foram relatadas em 2% em ambos os esquemas; não houve reações adversas de Grau 4 ou graves. A reação relatada com maior frequência foi neuropatia periférica sensorial (19% e 14% no esquema de ixazomibe e de placebo, respectivamente). Neuropatia periférica motora não foi relatada comumente em nenhum dos esquemas ( < 1%). A neuropatia periférica resultou em descontinuação de um ou mais dos três medicamentos em 1% dos pacientes em ambos os esquemas.
Distúrbios oculares
Distúrbios oculares foram relatados com muitos termos preferidos diferentes, mas no agregado, a frequência foi de 26% em pacientes no regime de NINLARO® e 16% dos pacientes no regime placebo. As reações adversas mais frequentes foram visão turva (6% no regime NINLARO® e 3% no regime placebo), olho seco (5% no regime de NINLARO® e 1% no regime placebo) e conjuntivite (6% no NINLARO® e 1% no regime placebo). Foram notificadas reações adversas de grau 3 em 2% dos pacientes no regime de NINLARO® e 1% no regime placebo.
Outros Eventos Adversos
Fora do estudo de fase 3, os seguintes eventos adversos graves, para os quais a causalidade não foi estabelecida, foram relatados raramente: dermatose neutrofílica aguda febril (síndrome de Sweet), síndrome de Stevens-Johnson, mielite transversa, síndrome de encefalopatia posterior reversível, síndrome de lise tumoral e púrpura trombocitopênica trombótica.
No estudo pivotal de fase 3, insuficiência hepática, incluindo hepatotoxicidade e alterações enzimáticas, ocorreu com uma taxa similar entre os regimes de NINLARO® (6%) e placebo (5%).
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos pelo Sistema de Notificações em Vigilância Sanitária -NOTIVISA, disponível em www. anvisa.gov.br/hotsite/notivisa/index.htm ou para a Vigilância Sanitária Estadual ou Municipal.
10. SUPERDOSE
Com base em dados limitados, tem sido relatado que a superdose com este medicamento causou eventos como náuseas, vômitos, diarreia, trombocitopenia e neuropatia periférica. Não há um antidoto específico conhecido para a superdose de NINLARO
Em caso de intoxicação, ligue para 0800 722 6001, se você precisar de mais orientações.
Dizeres legais.
MS - 1.0639.0278
VENDA SOB PRESCRIÇÃO MÉDICA