LONSURF
SERVIER
trifluridina + tipiracila, cloridrato
Antineoplásico. Antimetabólito.
Apresentações.
Comprimidos revestidos em embalagem com 20 ou 60 comprimidos, contendo 15mg/7,065mg e 20mg/9,420mg de trifluridina + cloridrato de tipiracila.
USO ORAL
USO ADULTO
Composição.
Cada comprimido de LONSURF® 15mg/7,065mg contém: trifluridina 15mg, cloridrato de tipiracila 7,065mg. Correspondente a 6,14 mg de tipiracila. Excipiente q.s.p. 1 comprimido revestido.
Cada comprimido de LONSURF® 20mg/9,420mg contém: trifluridina 20mg, cloridrato de tipiracila 9,420mg. Correspondente a 8,19 mg de tipiracila. Excipiente q.s.p. 1 comprimido revestido.
Excipientes: lactose monoidratada, amido pré-gelatinizado, ácido esteárico, hipromelose, macrogol, dióxido de titânio, estearato de magnésio, goma laca, óxido de ferro vermelho, óxido de Ferro amarelo, laca de alumínio índigo carmim, cera carnaúba e talco.
Informações técnicas.
1. INDICAÇÕES
LONSURF® é indicado para o tratamento de pacientes adultos com câncer colorretal metastático (CCR) que tenham sido tratados previamente com, ou não são considerados candidatos para, terapias disponíveis incluindo quimioterapia à base de fluoropirimidina, oxaliplatina e irinotecano, terapia anti-VEGF e, se for RAS do tipo selvagem, uma terapia anti-EGFR.
2. RESULTADOS DE EFICÁCIA
Eficácia e segurança clínicas
A segurança e eficácia clínicas de LONSURF® foram avaliadas num estudo internacional de fase III (RECOURSE), duplo-cego, controlado com placebo, randomizado, realizado em pacientes com câncer colorretal metastático previamente tratados. O desfecho primário foi a sobrevida global (OS-overall survival), e os desfechos secundários de eficácia foram a sobrevida livre de progressão da doença (PFS-progression free survival), taxa de resposta global (ORR-overall response rate) e a taxa de controle da doença (DCR-disease control rate).
No total, 800 pacientes foram randomizados numa proporção de 2:1 e receberam LONSURF® (N = 534) associado aos melhores cuidados de suporte (BSC) ou placebo (N = 266) associado ao BSC. A dose de LONSURF® foi baseada na área de superfície corporal com uma dose inicial de 35 mg/m2/dose. Durante o estudo, o tratamento foi administrado por via oral duas vezes ao dia, após o café da manhã e o jantar durante 5 dias com intervalo de 2 dias sem terapêutica na semana, durante 2 semanas seguidas, seguido por um período de 14 dias sem terapêutica, este ciclo repetiu-se a cada 4 semanas. Os pacientes continuaram a terapêutica até a progressão da doença ou até ocorrer uma toxicidade inaceitável (ver seção 8. Posologia e Modo de usar).
A idade média dos 800 pacientes randomizados era de 63 anos, sendo 61% do sexo masculino, 58% caucasianos/raça branca, 35% asiáticos/orientais, 1% negros/afro-americanos e todos os pacientes tinham, ao início do estudo, um Performance Status (PS)/ECOG de 0 ou 1. O sítio primário da doença era o cólon (62%) ou o reto (38%). No início do estudo o status KRAS era selvagem em 49% dos pacientes e mutante em 51%. O número médio de linhas de tratamento prévio para a doença metastásica foi 3. Todos os pacientes receberam tratamento prévio de quimioterapia com fluoropirimidina, oxaliplatina e irinotecano. Todos, com exceção de um paciente, recebeu bevacizumabe, e todos, à exceção de 2 pacientes com tumores KRAS selvagem, receberam panitumumabe ou cetuximabe. Os dois grupos de tratamento foram comparáveis em relação as características demográficas e basais da doença.
Uma análise da sobrevida global (OS) do estudo, realizada como previsto em 72% (N = 574) dos casos, demonstrou um benefício de sobrevida clínica e estatisticamente significativo de LONSURF® associado aos BSC, em comparação com placebo associado aos BSC (risco relativo [HR]: 0,68; 95% intervalo de confiança [IC] [0.58 a 0.81]; p < 0.0001) e sobrevida global média de 7.1 meses vs 5.3 meses, respectivamente; com taxas de sobrevida a 1ano de 26.6% e 17.6%, respectivamente. A PFS melhorou significativamente nos pacientes que administraram LONSURF® associado aos BSC (HR: 0.48; IC 95% [0.41 to 0.57]; p < 0.0001 (ver Tabela 1, Figura 1 e Figura 2).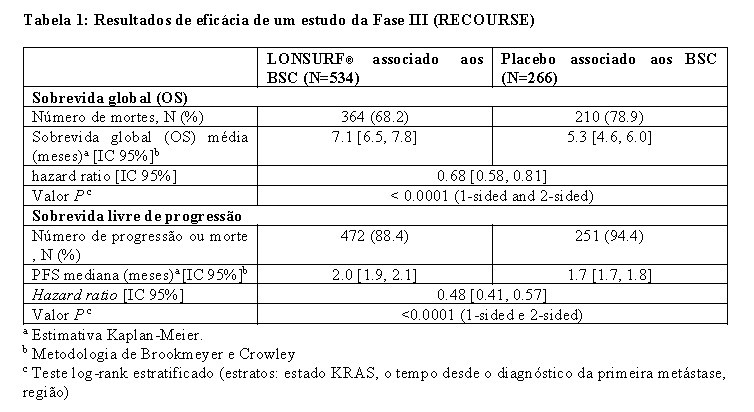
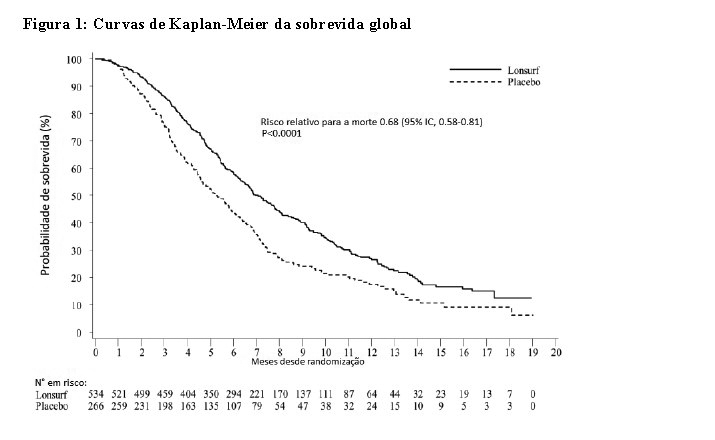
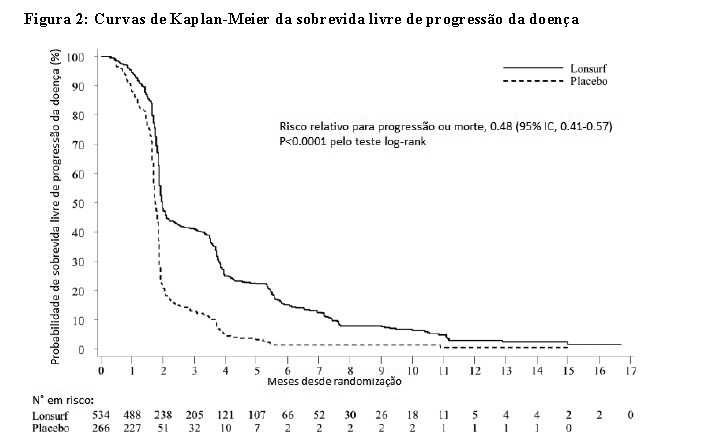
Uma análise atualizada da OS, realizada em 89% (N = 712) dos casos, confirmou o benefício clínico e estatisticamente significativo de LONSURF® associado aos BSC em comparação com placebo associado aos BSC (hazard ratio: 0,69; 95% IC [0,59 a 0,81]; p < 0.0001) e a OS média foi de 7.2 meses vs 5.2 meses; com taxas de sobrevida a 1ano de 27.1% e 16.6%, respectivamente.
Os benefícios do OS e PFS foram consistentemente observados, em todos os subgrupos relevantes préespecificados, incluindo raça, região geográfica, idade ( < 65 anos; ≥ 65 anos), gênero, ECOG PS, a mutação de KRAS, o tempo desde o diagnóstico da primeira metástase, o número de locais metastizados e local do tumor primário. O benefício de LONSURF® na sobrevida manteve-se após o ajuste de todos os fatores de prognóstico significativos, como o tempo desde o diagnóstico da primeira metástase, ECOG PS e o número de locais metastizados (hazard ratio: 0,69, IC 95% [0,58 a 0,81]).
Sessenta e um por cento (61%, N = 485) de todos os pacientes randomizados receberam fluoropirimidina como parte do seu último regime de tratamento antes da randomização, dos quais 455 (94%) eram refratários à fluoropirimidina nessa altura. Nestes pacientes o benefício de OS com LONSURF® foi mantido (hazard ratio: 0.75, IC 95% [0.59 a 0.94]).
Dezoito por cento (18%, N = 144) de todos os pacientes randomizados receberam regorafenibe anteriormente à randomização. Entre estes pacientes, o benefício OS com LONSURF® foi mantido (hazard ratio: 0,69, 95% IC [0,45 a 1,05]). O efeito também se manteve nos pacientes que nunca receberam regarofenibe (hazard ratio: 0,69, 95% IC [0,57 a 0,83]).
A DCR (resposta completa ou resposta parcial ou doença estável) foi significativamente mais elevada nos pacientes tratados com LONSURF® (44% vs 16%, p < 0,0001).
O tratamento com LONSURF® associado aos BSC resultou no prolongamento estatisticamente significativo do PS < 2 em comparação com o placebo associado aos BSC. O tempo mèdio para o PS ≥ 2 para o grupo LONSURF® e para o grupo placebo foi 5.7 meses e 4.0 meses, respectivamente, com uma hazard ratio de 0,66 (95% IC: [0,56, 0,78]), p < 0,0001.
Idosos
Os dados existentes em pacientes entre 75 a 84 anos de idade são limitados (N=60). Não houveram pacientes com 85 anos de idade ou mais no ensaio clínico RECOURSE e no ensaio japonês de fase 2. O efeito no OS foi similar em pacientes < 65 anos e ≥ 65 anos de idade.
Referências Bibliográficas: Mayer RJ, Van CE, Falcone A, Yoshino T, Garcia-Carbonero R, Mizunuma N, Yamazaki K, Shimada Y, Tabernero J, Komatsu Y, Sobrero A, Boucher E, Peeters M, Tran B, Lenz HJ, Zaniboni A, Hochster H, Cleary JM, Prenen H, Benedetti F, Mizuguchi H, Makris L, Ito M, Ohtsu A. Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N Engl J Med 2015; 372(20): 1909-19
3. CARACTERÍSTICAS FARMACOLÓGICAS
Grupo farmacoterapêutico: medicamentos antineoplásicos, antimetabolitos.
Propriedades farmacodinâmicas:
Mecanismo de ação:
LONSURF® é composto pela trifluridina, um antineoplásico análogo da timidina um nucleosídeo-base, e o cloridrato de tipiracila, um inibidor da timidina fosforilase (TPase), numa razão molar de 1:0,5 (relação peso, 1:0,471). Após ser captado pelas células cancerígenas, a trifluridina, é fosforilada pela timidina quinase, depois metabolizada nas células num substrato do ácido desoxirribonucleico (DNA), e incorporada diretamente no DNA, interferindo assim com a função do DNA para prevenir a proliferação da célula. Contudo, como a trifluridina é rapidamente degradada pela TPase e prontamente metabolizada pelo efeito de primeira passagem após a administração oral, por esse motivo tem-se a inclusão de um inibidor de TPase, o cloridrato de tipiracila.
Em estudos não clínicos, a trifluridina e o cloridrato de tipiracila demonstraram uma ação antitumoral contra ambas as linhas celulares de câncer colorretal resistentes e sensíveis ao 5-Fluorouracil (5-FU). A atividade citotóxica da trifluridina/cloridrato de tipiracila versus diversos xenoenxertos de tumor humano altamente correlacionados com a quantidade de trifluridina incorporada no DNA, que sugere este como o mecanismo de ação primário.
Efeitos farmacodinâmicos: LONSURF® não teve efeito clinicamente relevante no prolongamento do QT/QTc em comparação com o placebo num estudo aberto realizado em pacientes com tumores sólidos em estado avançado.
Propriedades Farmacocinéticas:
Absorção: Após a administração oral de LONSURF® com [14C]-trifluridina, pelo menos 57% da trifluridina administrada foi absorvida e só 3% da dose foi excretada pelas fezes. Após a administração oral de LONSURF® com [14C]-cloridrato de tipiracila, pelo menos 27% do cloridrato de tipiracila administrado foi absorvido e 50% da dose total radioativa foram quantificados nas fezes, o que sugere a absorção gastrointestinal moderada do cloridrato de tipiracila.
Após a administração de uma dose única de LONSURF® (35 mg/m2) em pacientes com tumores sólidos em estágio avançado, o tempo médio para o pico de concentrações plasmáticas (tmax) de trifluridina e cloridrato de tipiracila foi de cerca de 2 e 3 horas, respectivamente.
Nas análises farmacocinéticas (PK) da administração de doses múltiplas de LONSURF® (35 mg/m2/dose, duas vezes por dia, durante 5 dias por semana, com 2 dias sem terapêutica, durante 2 semanas seguidas de intervalo terapêutico de 14 dias, ciclo repetido todas as 4 semanas), a área sob a curva de concentração da trifluridina de 0 à última concentração medida (AUC0-last) foi aproximadamente 3 vezes superior e a concentração máxima (Cmax) foi aproximadamente 2 vezes superior após a administração da dose múltipla de LONSURF® (Dia 12 do Ciclo 1) quando comparado a dose única (Dia 1 do Ciclo 1).
No entanto, não houve acúmulo de cloridrato de tipiracila, e nem acúmulo adicional de trifluridina com os sucessivos ciclos (Dia 12 dos Ciclos 2 e 3) de administração de LONSURF®. Após a administração de doses múltiplas de LONSURF® (35 mg/m2/dose duas vezes por dia) em pacientes com tumores sólidos em estágio avançado, o tempo médio para o pico de concentrações plasmáticas (tmax) de trifluridina e cloridrato de tipiracila foi de cerca 2 e 3 horas, respectivamente.
Contribuição do cloridrato de tipiracila:
A administração de uma dose única de LONSURF® (35 mg/m2/dose) aumentou a média AUC0-last de trifluridina 37 vezes e o Cmax 22 vezes com uma variabilidade reduzida em comparação com a trifluridina administrada isoladamente (35 mg/m2/dose).
O efeito dos alimentos:
Quando LONSURF® foi administrado na dose única de 35 mg/m2 em 14 pacientes com tumores sólidos após uma refeição padronizada com elevado teor de gorduras e calorias, a área sob a curva de concentração (AUC) da trifluridina não se alterou, mas o Cmax de trifluridina e o Cmax e a AUC do cloridrato de tipiracila diminuíram aproximadamente 40% em comparação com os pacientes que estavam em jejum. Nos estudos clínicos LONSURF® foi administrado cerca de 1 hora após o café da manhã e o jantar (ver seção 8. Posologia e Modo de usar).
Distribuição:
A ligação da trifluridina às proteínas plasmáticas humanas foi superior a 96% e a trifluridina liga-se principalmente à albumina do soro humano. A ligação às proteínas do plasma do cloridrato de tipiracila foi inferior a 8%. Após uma dose única de LONSURF® (35 mg/m2) em pacientes com tumores sólidos em estágio avançado, o volume aparente de distribuição (Vd/F) para a trifluridina e para o cloridrato de tipiracila foi de 21 L e 333 L, respectivamente.
Biotransformação:
A trifluridina foi eliminada principalmente pelo metabolismo via TPase para formar um metabólito inativo, o FTY. A trifluridina absorvida foi metabolizada e excretada na urina como FTY e isômeros glucoronídeos de trifluridina. Outros metabolitos menores, 5-carboxyuracil e 5-carboxy-2'-deoxyuridina, foram detectados, mas seus níveis no plasma e na urina foram baixos ou residuais. O cloridrato de tipiracila não foi metabolizado na fração S9 do fígado humano ou nos hepatócitos humanos criopreservados. O cloridrato de tipiracila foi o principal componente e o 6-hidroximetiluracilo foi o mais consistente metabólito no plasma humano, urina e fezes.
Eliminação:
Após a administração de dose múltipla de LONSURF® na dose e regime recomendados, a meia-vida de eliminação média (t1/2) da trifluridina no Dia 1 do Ciclo 1 e no Dia 12 do Ciclo1 foram 1,4 horas e 2,1 horas, respectivamente. Os valores médios de t1/2 para o cloridrato de tipiracila no Dia 1 do Ciclo 1 e no Dia 12 do Ciclo 1 foram 2,1 horas e 2,4 horas, respectivamente. Após uma dose única de LONSURF® (35 mg/m2) em pacientes com tumores sólidos em estágio avançado, o 'clearance' oral (CL/F) para a trifluridina e para o cloridrato de tipiracila foi 10.5 L/hr e 109 L/h, respectivamente. Após a administração oral da dose única de LONSURF® com [14C]-trifluridina, a excreção cumulativa total de radioatividade foi 60% da dose administrada. A maior parte da radioatividade recuperada foi eliminada na urina (55% da dose) em 24 horas, e a excreção nas fezes e o ar expirado foi inferior a 3% para ambos. Após a administração oral da dose única de LONSURF® com [14C]-cloridrato de tipiracila, a radioatividade recuperada foi 77% da dose, o que correspondeu a 27% de excreção urinária e 50% de excreção fecal.
Linearidade / não-linearidade:
Num estudo de determinação da dose (15 a 35 mg/m2 duas vezes ao dia), o AUC de 0 a 10 horas (AUC0-10) da trifluridina tendeu a aumentar mais do que se esperava tendo por base o aumento da dose; no entanto, o 'clearance' oral (CL/F) e o volume aparente de distribuição (Vd/F) da trifluridina foram em geral constantes no intervalo de dose de 20 a 35mg/m2. Quanto aos outros parâmetros de exposição da trifluridina e do cloridrato de tipiracila, estes pareceram ser proporcionais à dose.
Farmacocinética em populações especiais
Idade, gênero e raça
Com base na análise farmacocinética (PK) da população, a idade, o gênero ou a raça não tiveram efeito estatisticamente significativo na farmacocinética da trifluridina e do cloridrato de tipiracila.
Comprometimento renal
Dos 533 pacientes no estudo RECOURSE que administraram LONSURF®, 306 (57%) pacientes tinham função renal normal (CrCl≥90 mL/min), 178 (33%) comprommetimento renal leve (CrCl 60 a 89 mL/min) e 47 (9%) comprometimento renal moderado (CrCl 30 a 59 mL/min), estando em falta os dados relativos a 2 pacientes. Os pacientes com comprometimento renal grave não foram incluídos no estudo.
Tendo por base a análise farmacocinética da população, a exposição de LONSURF® nos pacientes com comprometimento renal leve (CrCl=60 a 89 mL/min) foi semelhante à dos pacientes com função renal normal (CrCl≥ 90mL/min). Uma exposição maior de LONSURF® foi observada em pacientes com comprometimento renal moderado (CrCl=30 a 59 mL/min). A estimada (CrCl) foi uma co-variável significativa para a CL/F em ambos os modelos de trifluridina e cloridrato do tipiracila. A proporção relativa média da AUC em pacientes com comprometimento renal leve (n=38) e moderado (n=16) em comparação com os pacientes com função renal normal (n=84) foi 1,31 e 1,43 para a trifluridina, respectivamente, e 1,34 e 1,65 para o cloridrato de tipiracila, respectivamente.
Em um estudo dedicado, a farmacocinética da trifluridina e do cloridrato de tipiracila foi avaliada em pacientes com câncer com função renal normal (CrCl ≥90 mL/min, N=12), insuficiência renal leve (CrCl =60 a 89 mL/min, N=12), insuficiência renal moderada (CrCl=30 a 59 mL/min, N=11) ou insuficiência renal grave (CrCl =15 a 29 mL/min, N= 8). Os pacientes com insuficiência renal grave receberam uma dose inicial ajustada de 20 mg/m2 duas vezes ao dia (reduzida para 15 mg/ m2 duas vezes ao dia com base na segurança e tolerabilidade individuais). O efeito da insuficiência renal após administração repetida foi um aumento de 1,6 e 1,4 vezes na exposição total à trifluridina em pacientes com insuficiência renal moderada e grave, respectivamente, em comparação com pacientes com função renal normal; Cmax permaneceu semelhante. A exposição total do cloridrato de tipiracila em pacientes com insuficiência renal moderada e grave após administração repetida foi 2,3 e 4,1 vezes maior, respectivamente, em comparação com pacientes com função renal normal; isso está associado a uma depuração mais reduzida com aumento da insuficiência renal. A farmacocinética da trifluridina e do cloridrato de tipiracila não foi estudada em pacientes com doença renal em fase terminal (ClCr < 15 mL/min ou necessitando de diálise) (ver seção 5. Advertências e Precauções e seção 8. Posologia).
Comprometimento hepático
Com base na análise PK da população, os parâmetros da função hepática, incluindo a fosfatase alcalina (ALP, 36-2322 U/L), o aspartato aminotransferase (AST, 11-197 U/L), a alanina aminotransferase (ALT, 5-182 U/L), e a bilirrubina total (0,17-3,20 mg/dL) não foram co-variáveis significativas para os parâmetros farmacocinéticos da trifluridina ou do cloridrato de tipiracila. A depuração da trifluridina foi significativamente afetada pela albumina sérica com uma correlação negativa. Para valores baixos de albumina de 2,2 a 3,5g/dL os valores da depuração correspondente situaram-se entre 4,2 a 3,1 L/h.
Em um estudo de farmacocinética de trifluridina e cloridrato de tipiracila foram avaliados pacientes com comprometimento hepático leve e moderado (National Cancer Institute [NCI] grupos de critério B e C, respectivamente) e pacientes com função hepática normal. Com base em dados limitados, com uma variabilidade considerável, nenhuma diferença estatiscamente significante foi observada na farmacocinética dos pacientes com função hepática normal versus pacientes com comprometimento leve ou moderado. Nenhuma correlação foi observada para trifluridina ou cloridrato de tipiracila, entre os parâmetros de PK e AST e/ou total de bilirrubna sanguínea. A meia vida (t1/2) e a taxa de acúmulo de trifluridina e cloridrato de tipiracila foram muito similares entre pacientes com comprometimento hepático moderado, leve e função hepática normal. Não há necessidade para uma dose inicial de ajuste para os pacientes com compromentimento hepático leve (ver seção 8 Posologia).
Gastrectomia
A influência da gastrectomia sobre os parâmetros farmacocinéticos não foi possível ser avaliada na análise farmacocinética da população devido ao número reduzido de pacientes submetidos a gastrectomia (1% do total).
Estudos de interação in vitro
A trifluridina é um substrato da TPase, mas não é metabolizada pelo citocromo P450 (CYP). O cloridrato de tipiracila não é metabolizado nem na fração S9 no fígado humano nem nos hepatócitos criopreservados.
Estudos in vitro mostraram que a trifluridina, cloridrato de tipiracila e o FTY (metabólito inativo da trifluridina) não inibem a atividade das isoformas do CYP testadas (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 e CYP3A4/5). A avaliação in vitro mostrou que a trifluridina, cloridrato de tipiracila e FTY não induzem efeito nas isoformas do CYP1A2, CYP2B6 ou CYP3A4/5. Assim, não se espera que o trifluridina e o cloridrato de tipiracila causem ou sejam sujeitos a uma interação significativa do medicamento mediada pelo CYP.
A avaliação in vitro da trifluridina e do cloridrato de tipiracila foi realizada utilizando transportadores de absorção humana e efluentes (trifluridina com MDR1, OATP1B1, OATP1B3 e BCRP; cloridrato de tipiracila com OAT1, OAT3, OCT2, MATE1, MDR1 e BCRP). Nem a trifluridina nem o cloridrato de tipiracila foram inibidores ou substratos para transportadores de absorção humana e efluentes com base em estudos in vitro, com exceção de OCT2 e MATE1. O cloridrato de tipiracila foi um inibidor in vitro de OCT2 e MATE1, mas a concentrações substancialmente maiores do que a Cmax de plasma humano no estado de equilíbrio. Assim, é pouco provável que cause uma interação com outros medicamentos, em doses recomendadas, devido à inibição de OCT2 e MATE1. O transporte de cloridrato de tipiracila por OCT2 e MATE1 pode ser afetado quando o Lonsurf é administrado concomitantemente com inibidores de OCT2 e MATE1.
Relacionamento farmacocinético/farmacodinâmico
A eficácia e segurança do Lonsurf foi comparada entre um grupo de alta exposição ( > mediana) e um grupo de baixa exposição (≤mediana) com base no valor médio de AUC da trifluridina. O OS pareceu mais favorável no grupo de AUC elevado em comparação com o grupo de AUC baixo (OS médio de 9,3 versus 8,1 meses, respectivamente). Todos os grupos de AUC apresentaram melhor desempenho do que o placebo ao longo do período de seguimento. As incidências de neutropenia Grau ≥ 3 foram maiores no grupo AUC de alta trifluridina (47,8%) em comparação com o grupo AUC de baixa trifluridina (30,4%).
Dados de segurança pré-clínica
Toxicidade por dose repetida
A avaliação toxicológica da trifluridina/cloridrato do tipiracila foi realizada em ratos, cães e macacos. Os órgãos alvo identificados foram os sistemas linfático e hematopoiético e o trato gastrointestinal. Todas as alterações, isto é, leucopenia, anemia, hipoplasia da medula óssea, alterações atróficas nos tecidos linfáticos e hematopoiéticos e o trato gastrointestinal, foram reversíveis no período de 9 semanas após a suspensão da terapêutica. Foi observado branqueamento, quebra e mal oclusão nos dentes dos ratos tratados com trifluridina e o cloridrato de tipiracila, os quais são considerados roedores específicos e por isso não relevantes para o ser humano.
Carcinogenicidade e mutagenicidade
Não foram realizados estudos a longo prazo para avaliar o potencial carcinogênico da trifluridina/ cloridrato de tipiracila em animais. A trifluridina demonstrou ser genotóxica num ensaio de mutação reversa em bactérias, num ensaio de aberrações cromossômicas em células de mamíferos em cultura e num teste de micronúcleos em camundongos. Assim, LONSURF® deve ser considerado como potencialmente carcinogênico.
Toxicidade reprodutiva
Os resultados dos estudos em animais não demonstraram um efeito da trifluridina e do cloridrato de tipiracila sobre a fertilidade dos ratos machos e fêmeas. O aumento do corpo lúteo e dos embriões implantados observados em ratos fêmea em doses elevadas não foi considerado adverso (ver seção 5. Advertências e Precauções).
LONSURF® mostrou causar letalidade e toxicidade fetal-embrionária em fêmeas de ratos grávidas quando administrado em doses inferiores às clinicamente utilizadas. Não foram efetuados estudos de toxicidade perinatais e pós-natais.
4. CONTRAINDICAÇÕES
Hipersensibilidade às substâncias ativas ou a qualquer um dos componentes da fórmula.
Este medicamento é contraindicado para menores de 18 anos.
Categoria D: Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
5. ADVERTÊNCIAS E PRECAUÇÕES
Supressão da medula óssea
LONSURF® provoca um aumento na incidência de mielosupressão incluindo anemia, neutropenia, leucopenia e trombocitopenia. Antes do início do tratamento um hemograma completo deve ser realizado e sempre que necessitar de monitorar a toxicidade, mas no mínimo antes de cada ciclo de tratamento. O tratamento não pode ser iniciado se o número de neutrófilos for < 1,5 x 109/L, se o número de plaquetas for < 75 x 109/L, ou se o paciente desenvolver toxicidade não hematológica de Grau 3 ou 4 clinicamente relevante de terapias anteriores.
Foram notificadas infecções graves após o tratamento com LONSURF® (ver seção 9. Reações Adversas). Uma vez que a maioria foi notificada num contexto de supressão da função da medula, a situação clínica do paciente deve ser monitorada frequentemente e tomadas medidas apropriadas, tais como a administração de antimicrobianos e de fatores de crescimento de granulócitos (G-CSF), como clinicamente indicadas. No estudo RECOURSE, 9,4% dos pacientes incluídos no grupo LONSURF® receberam G-CSF predominantemente para uso terapêutico.
Toxicidade gastrointestinal
LONSURF® provoca um aumento na incidência de toxicidade gastrointestinal incluindo náuseas, vômitos e diarreia.
Pacientes com náuseas, vômitos, diarreia e outras toxicidades gastrointestinais devem ser cuidadosamente monitorados e devem ser administrados como clinicamente indicado antieméticos, antidiarreicos e outras medidas tais como líquidos/eletrólitos de substituição. Se necessário deve ser efetuada modificação da dose (atraso e/ou redução) (ver seção 5. Advertências e Precauções).
Comprometimento da função renal
Não se recomenda o uso de LONSURF® em pacientes com doença renal em fase terminal (depuração da creatinina CrCl < 15 ml/min ou que necessitem de diálises) uma vez que que LONSURF® não foi estudado nestes pacientes (ver seção 3. Características Farmacológicas).
A incidência global de eventos adversos (EAs) é semelhante nos subgrupos de função renal normal (CrCl
≥ 90 mL / min), leve (CrCl = 60 a 89 mL/min) ou moderada (CrCl = 30 a 59 mL/min). No entanto, a incidência de EA sérios, graves, levando à modificação da dose, tende a aumentar com o aumento dos níveis de insuficiência renal.
Também foi observada uma maior exposição de trifluridina e cloridrato tipiracila em pacientes com comprometimento renal moderado, comparado com pacientes com função renal normal ou pacientes com comprometimento renal leve (ver seção 3. Características Farmacológicas).
Pacientes com insuficiência renal grave (CrCl = 15 a 29 mL/min) e dose inicial ajustada de 20 mg/m2 duas vezes ao dia tinham um perfil de segurança consistente com o perfil de segurança de Lonsurf em pacientes com função renal normal ou insuficiência renal leve. A sua exposição à trifluridina foi semelhante à de pacientes com função renal normal e a sua exposição ao cloridrato de tipiracila foi aumentada em comparação com pacientes com função renal normal, comprometimento renal leve e moderado (ver seções 5 e 3).
Pacientes com insuficiência renal devem ser monitorados de perto ao serem tratados com Lonsurf;
Pacientes com comprometimento renal moderado ou grave devem ser mais frequentemente monitorados no que se refere a toxicidade hematológica.
Comprometimento hepático
Não se recomenda o uso de LONSURF® em pacientes com comprometimento hepático moderado ou grave (Nacional Cancer Institute [NCI] Critério de Grupo C e D definido pela bilirrubina total > 1.5 ULN) uma vez que uma maior incidência de hiperbilirrubinemia grau 3 ou 4 foi observada em pacientes com comprometimento hepático avançado ao início do tratamento, contudo estes são baseados em dados muito limitados (ver seção 3 características farmacológicas).
Proteinuria
É recomendada a monitoração da proteinúria por análise da urina através de uma fita de análise antes de iniciar e durante o tratamento (ver seção 9. Reações Adversas).
Intolerância à lactose
LONSURF® contém lactose. Pacientes com problemas hereditários raros de intolerância à galactose, deficiência de lactase total ou má absorção de glucose-galactose não devem administrar este medicamento.
Atenção: Este medicamento contém açúcar (lactose), portanto, deve ser usado com cautela em portadores de Diabetes.
Fertilidade, Gravidez e Aleitamento
Mulheres com potencial para engravidar/contracepção em homens e mulheres
Baseado nos resultados em animais a trifluridina pode causar danos fetais se for administrada em mulheres grávidas. As mulheres devem evitar engravidar enquanto administram LONSURF® e até 6 meses após o fim do tratamento. Assim, as mulheres com potencial para engravidar têm de utilizar métodos contraceptivos extremamente eficazes durante o tratamento com LONSURF® e nos 6 meses seguintes após a descontinuação do tratamento. Desconhece-se se o LONSURF® pode reduzir a eficácia dos contraceptivos hormonais, portanto se a mulher utiliza um contraceptivo hormonal deve associá-lo a um método contraceptivo de barreira.
Homens com parceiras com potencial para engravidar têm de utilizar métodos contraceptivos durante o tratamento com LONSURF® e até 6 meses após a descontinuação do tratamento.
Gravidez
Não existem dados disponíveis do uso do LONSURF® em mulheres grávidas. Com base no mecanismo de ação, deve considerar-se que a trifluridina pode provocar má formações congênitas quando administrado durante a gravidez. Os estudos em animais revelaram toxicidade reprodutiva (ver seção 3. Características Farmacológicas). LONSURF® não deve ser utilizado durante a gravidez a menos que o estado clínico da mulher exija tratamento com LONSURF®.
Amamentação
É desconhecido se LONSURF® ou os seus metabólitos são excretados no leite humano. Em estudos em animais encontrou-se trifluridina e cloridrato de tipiracila e/ou os seus metabólitos no leite (ver seção 3. Características Farmacológicas). O risco de passar para a criança pelo leite materno não pode ser excluído. A amamentação deverá ser descontinuada durante o tratamento com LONSURF®.
Fertilidade
Não existem dados disponíveis sobre os efeitos do LONSURF® na fertilidade humana. Resultados de estudos efetuados em animais não mostraram qualquer efeito do LONSURF® na fertilidade masculina ou feminina (ver seção 3. Características Farmacológicas).
Categoria D: Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
Efeitos sobre a capacidade de conduzir e utilizar máquinas
LONSURF® tem influência ligeira na capacidade de conduzir e utilizar máquinas. Pode ocorrer fadiga, tonturas ou mal-estar durante o tratamento (ver seção 9. Reações Adversas).
6. INTERAÇÕES MEDICAMENTOSAS
Estudos in vitro mostraram que a trifluridina, cloridrato de tipiracila e o 5-[trifluorometil] uracil (FTY) não inibem a atividade das isoformas do citocromo P450 (CYP). A avaliação in vitro mostrou que a trifluridina, cloridrato de tipiracila e FTY não induzem efeito nas isoformas do CYP (ver seção 3. Características Farmacológicas).
Estudos in vitro mostraram que a trifluridina é um substrato para os transportadores de nucleosídeos CNT1, ENT1 e ENT2. Contudo é necessário precaução quando se utilizam medicamentos que interagem com estes transportadores. O cloridrato de tipiracila foi um substrato para o OCT2 e MATE1, portanto a concentração pode ser aumentada quando o LONSURF® é administrado concomitantemente com inibidores do OCT2 ou MATE1.
Associações que necessitam de precauções de uso
Deve ter-se precaução quando se usam medicamentos que atuam como substratos da timidina quinase humana, por exemplo, a zidovudina. Tais medicamentos, se usados concomitantemente com LONSURF®, podem competir com a substância ativa, trifluridina, para ativação da via timidina quinase. Portanto, quando se usam medicamentos antivirais que são substratos da timidina quinase humana, deve-se monitorar uma possível diminuição da eficácia dos medicamentos antivirais, e considerar a substituição para um medicamento antiviral que não seja um substrato da timidina quinase humana, tal como a lamivudina, didanosina e abacavir (ver seção 3. Características Farmacológicas).
Não se conhece se LONSURF® pode reduzir a eficácia dos contraceptivos hormonais. Portanto, as mulheres que usam contraceptivos hormonais devem também usar um método anticonceptivo de barreira.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
LONSURF® deve ser guardado na sua embalagem original. Conservar em temperatura ambiente (entre 15 e 30°C). Proteger da umidade. Nestas condições, este medicamento possui prazo de validade de 24 (vinte e quatro) meses, a partir da data de fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
CARACTERÍSTICAS FÍSICAS E ORGANOLÉPTICAS
LONSURF® 15mg/7,065mg apresenta-se sob forma de comprimidos revestidos redondos brancos, com diâmetro de 7.1mm e espessura de 2.7mm, gravados "15" em uma face, e "102" e "15mg" na outra face na cor acinzentada.
LONSURF® 20mg/9,420mg apresenta-se sob forma de compridos revestidos redondos de cor vermelho claro, com diâmetro de 7.6mm e espessura de 3.2mm, gravados "20" em uma face, e "102" e "20mg" na outra face na cor acinzentada.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
Uso Oral
LONSURF® deve ser prescrito por médicos com experiência na utilização de medicamentos anticancerígenos.
Posologia
A dose inicial recomendada de LONSURF® em adultos é de 35 mg/m2/dose (com base no ativo trifluridina), administrada oralmente duas vezes ao dia, nos dias 1 a 5 e nos dias 8 a 12 de cada ciclo de 28 dias, desde que se observe benefício ou até que ocorra toxicidade inaceitável (ver seção 5. Advertências e Precauções).
A dose é calculada de acordo com a área de superficie corporal (ASC) (ver Tabela 1). Cada administração não deve exceder 80 mg/dose.
Se as doses forem esquecidas ou suspensas o paciente não deve compensar as doses esquecidas.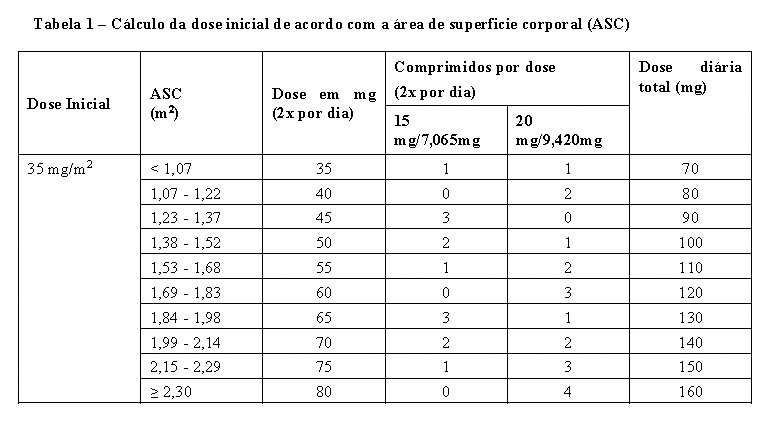
Recomendações para o ajuste de dose
Podem ser necessários ajustes da dose baseados na segurança e tolerabilidade individual.
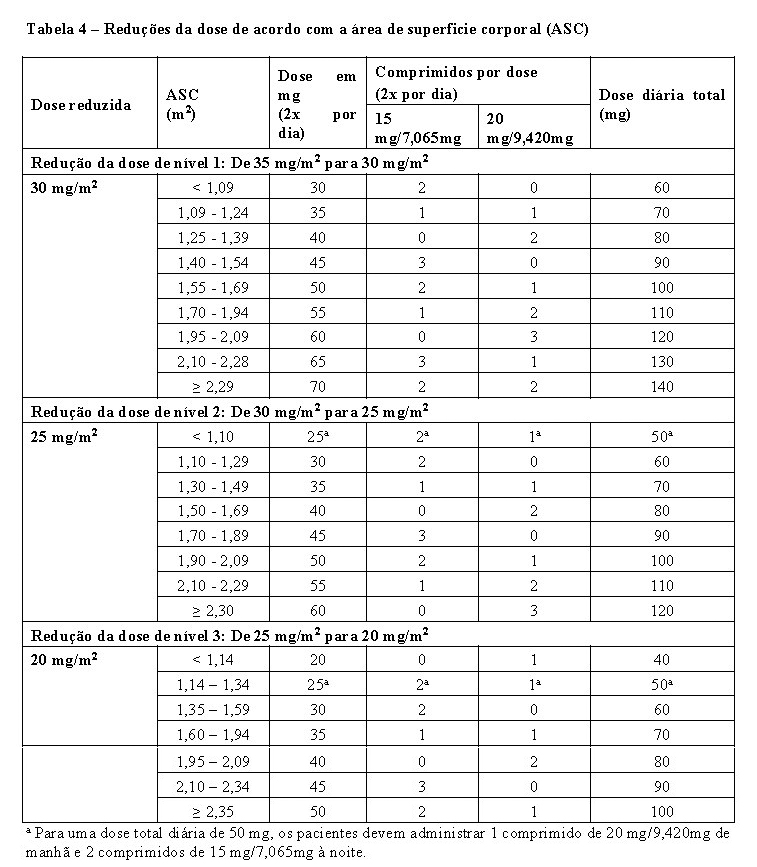
No máximo, são permitidas 3 reduções de dose até a dose mínima de 20 mg/m2 duas vezes ao dia. O aumento da dose não é permitido após esta ter sido reduzida.
Em caso de toxicidade hematológica e/ou não hematológica devem ser seguidos os critérios de interrupção, recomeço e redução da dose indicados nas tabela 2, 3 e 4.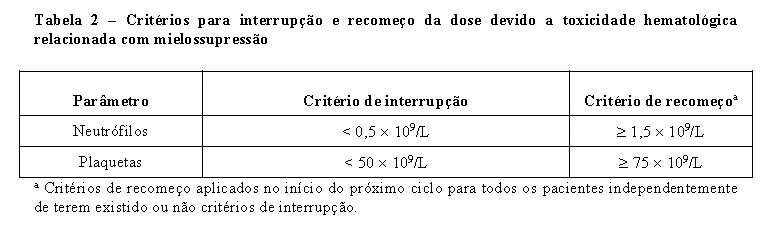

Populações especiais
Comprometimento renal
-Comprometimento renal leve (Depuração de creatinina de 60 a 89 ml/min) ou comprometimento renal moderado (Depuração de creatinina de 30 a 59 ml/min)
Nenhum ajuste de dose é recomendado na dose inicial em pacientes com comprometimento renal leve ou moderado (ver seções 3. Características Farmacológicas e 5. Advertências e Precauções)
-Comprometimento renal grave (Depuração da creatinina CrCl entre 15 a 29 mL/min) Para pacientes com insuficiência renal grave, recomenda-se uma dose inicial de 20 mg/m2 duas vezes por dia (ver seção 5. Advertências e seção 3. Propriedades Farmacocinéticas). É permitida uma redução de dose para uma dose mínima de 15 mg/m2, duas vezes ao dia, com base na segurança e tolerabilidade individuais (consulte a Tabela 5). O escalonamento da dose não é permitida depois da redução.
No caso de toxicidade hematológica e/ou não hematológica, os pacientes devem seguir os critérios de interrupção, retomada e redução da dose, apresentados na Tabela 2, Tabela 3 e Tabela 5.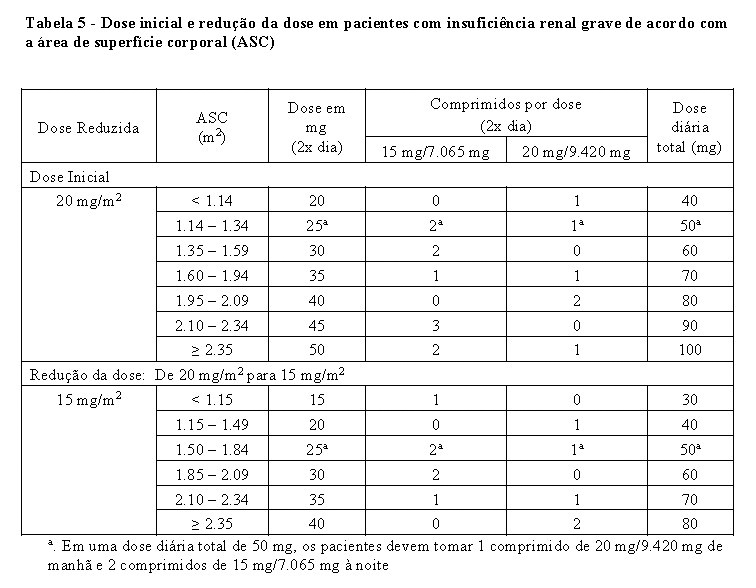
Doença renal em estágio terminal (ClCr abaixo de 15 mL/min ou necessitando de diálise)
Não se recomenda a administração em pacientes com doença renal em fase terminal uma vez que não existem dados disponíveis para estes pacientes (ver seção 5. Advertências e Precauções).
Comprometimento hepático
-Comprometimento hepático leve
Não se recomenda ajuste da dose inicial em pacientes com comprometimento hepático leve (ver seção 3. Características Farmacológicas).
-Comprometimento hepático moderado ou grave
A administração não é recomendada em pacientes com comprometimento hepático moderado ou grave (National Cancer Institute [NCI] grupos de critério C e D definifdo pelo total de bilirrubina > 1.5 ULN) uma vez uma maior incidência de hiperbilirrubinemia grau 3 e 4 é observada em pacientes com comprometimento hepático moderado. Contudo isto é baseado em dados limitados (ver seções 3. Características Farmacológicas e 5. Advertências e Precauções)
Idosos
Não é necessário ajuste da dose inicial em pacientes com idade ≥ 65 anos (ver seções 3. Características Farmacológicas e 9. Reações Adversas).
Os dados de segurança e eficácia em pacientes com mais de 75 anos são limitados.
População pediátrica
Não existe utilização relevante de LONSURF® na população pediátrica para a indicação de câncer colorretal metastático.
Raça
Não é necessário ajuste da dose inicial baseado na raça do paciente (ver seções 3. Características Farmacológicas). Os dados em pacientes negros/afro-americanos são limitados, mas não existe racional biológico para esperar qualquer diferença entre este subgrupo e a população geral.
Modo de administração
LONSURF® é administrado por via oral. Os comprimidos devem ser engolidos com um copo de água no espaço de 1 hora após a refeição da manhã e da noite.
Este medicamento não deve ser partido, aberto ou mastigado.
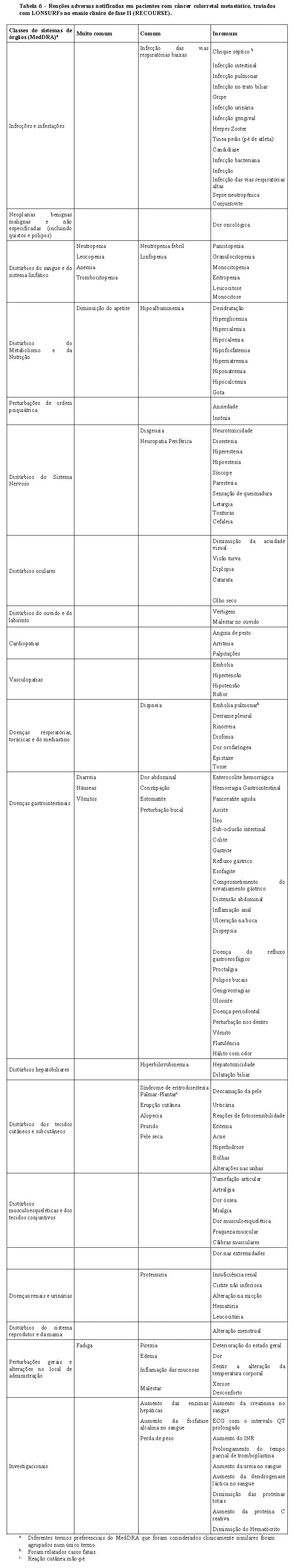
9. REAÇÕES ADVERSAS
Resumo do perfil de segurança
As reações adversas mais graves observadas em pacientes que administraram LONSURF® são supressão da medula óssea e toxicidade gastrointestinal (ver seção 5. Advertências e Precauções).
As reações adversas medicamentosas mais frequentemente observadas (≥ 30%) em pacientes recebendo Lonsurf são neutropenia (53% [34% ≥ Grau 3]), náusea (34% [1% ≥ Grau 3]), fadiga (32% [4% ≥ Grau 3]), anemia (32% [12% ≥ Grau 3]).
As reações adversas mais comuns (≥ 2%) nos pacientes que receberam Lonsurf que resultaram em interrupção do tratamento, redução da dose, atraso na dose ou interrupção da dose foram neutropenia, anemia, leucopenia, fadiga, trombocitopenia, náusea e diarreia.
Tabela com lista das reações adversas:
As reações adversas observadas a partir dos 533 pacientes com câncer colorretal metastático, tratados com uma dose inicial de 35 mg/m2/dose de LONSURF®, no ensaio clínico controlado por placebo de fase III (RECOURSE), são apresentados na Tabela 6. São classificadas de acordo com a Classe de Sistema de Órgãos (SOC) e o Dicionário Médico apropriado para os termos regulamentares (MedDRA) é usado para descrever uma determinada reação adversa e os seus sinônimos e condições relacionadas.
As reações adversas são agrupadas de acordo co