LAMZEDE
CHIESI
velmanase alfa
Terapia de reposição enzimática (TRE).
Apresentações.
Pó para solução para infusão
Cada frasco-ampola contém 10 mg de alfavelmanase. Cartuchos com 1, 5 ou 10 frasco-ampolas.
USO INTRAVENOSO
USO ADULTO E PEDIÁTRICO
Composição.
Cada frasco-ampola contém: alfavelmanase 10 mg*. Excipientes: fosfato de sódio dibásico di-hidratado, fosfato de sódio monobásico di-hidratado, manitol e glicina.
* Após reconstituição 1 mL da solução contém 2 mg de alfavelmanase (10 mg/5 mL).
A alfavelmanase é produzida em células mamíferas de ovário de hamster chinês (CHO) utilizando a tecnologia de DNA recombinante.
Informações técnicas.
1. INDICAÇÕES
LAMZEDE® é indicado para terapia de reposição enzimática (TRE) para o tratamento de manifestações não neurológicas em pacientes com alfa-manosidose leve a moderada.
2. RESULTADOS DE EFICÁCIA
Um total de 33 pacientes (20 homens e 13 mulheres com idades entre 6 e 35 anos) foram expostos à alfavelmanase em 5 estudos clínicos. Os pacientes foram diagnosticados com base na atividade da alfa-manosidase < 10% da atividade normal nos leucócitos do sangue. Os pacientes com o fenótipo mais grave de progressão rápida (com uma deterioração no período de um ano e envolvimento do sistema nervoso central) foram excluídos. Com base neste critério, foram incluídos os pacientes com formas leves a moderadas da doença, que apresentavam uma gravidade heterogênea com a capacidade de realizar provas de resistência, uma grande variabilidade de manifestações clínicas e idade de manifestação da doença.
Os efeitos gerais do tratamento foram avaliados nos domínios da farmacodinâmica (redução dos oligossacarídeos séricos), funcional [prova de subida de escada em três minutos (3MSCT), prova de caminhada de seis minutos (6MWT) e capacidade vital forçada (CVF)] % prevista e qualidade de vida [índice de incapacidade pelo questionário de avaliação da saúde em crianças (childhood health assessment questionnaire - CHAQ)] e dor pelo CHAQ VAS (escala analógica visual).
No estudo de referência rhLAMAN 05 de fase 3, multicêntrico, duplo-cego, aleatorizado, controlado com placebo, em grupos paralelos, foram avaliadas a eficácia e segurança de administrações repetidas de alfavelmanase durante 52 semanas, a uma dose de 1 mg/kg, administrada semanalmente, por infusão intravenosa. Foram incluídos um total de 25 pacientes, incluindo 12 pacientes pediátricos (intervalo de idades: 6 a 17 anos; média: 10,9 anos) e 13 pacientes adultos (intervalo de idades: 18 a 35 anos; média: 24,6). Todos os pacientes, com exceção de um, não tinham sido tratados previamente com alfavelmanase. No total, 15 pacientes (7 crianças e 8 adultos) receberam o tratamento ativo e 10 pacientes receberam placebo (5 crianças e 5 adultos). Os resultados (concentração sérica de oligossacarídeos, 3MSCT, 6MWT e CVF%) são apresentados na tabela 1. Foi demonstrado um efeito farmacodinâmico com uma diminuição estatisticamente significativa dos oligossacarídeos séricos em comparação com o placebo. Os resultados observados em pacientes com menos de 18 anos de idade mostraram uma melhora. Em pacientes com mais de 18 anos de idade foi demonstrada uma estabilização. A melhoria numérica da maioria dos critérios de avaliação clínicos em relação ao placebo (2 a 8%) observada no ano de observação pode ser sugestiva da capacidade da alfavelmanase de retardar a progressão da doença existente.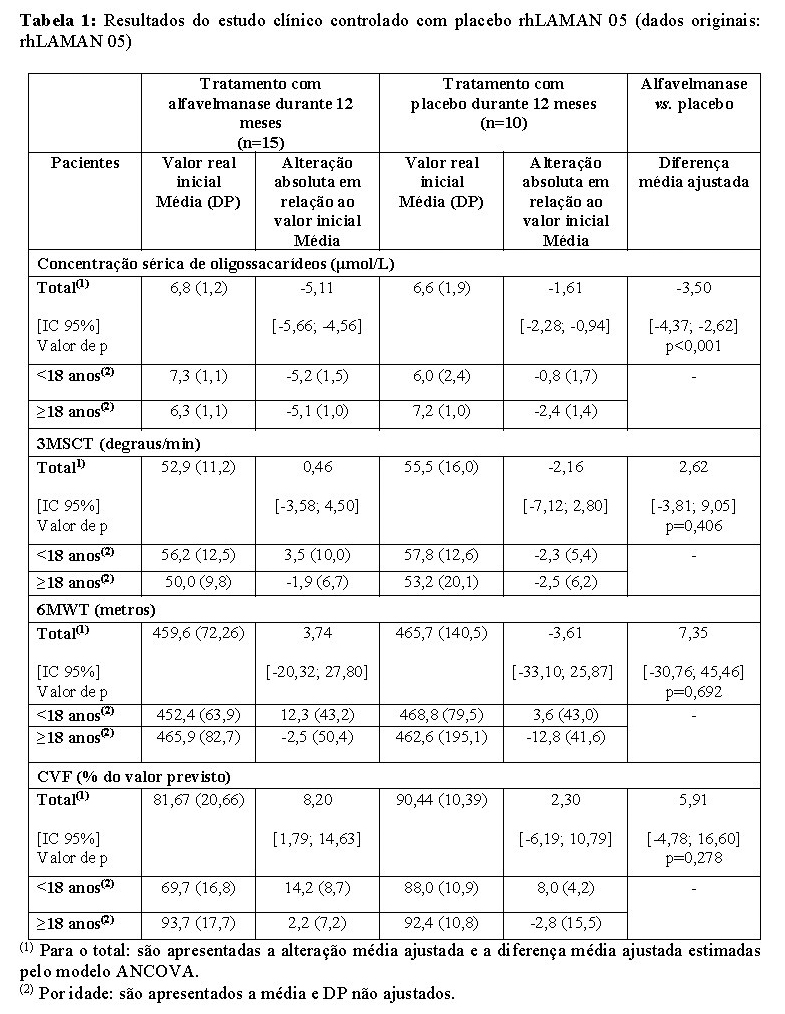
A eficácia e segurança a longo prazo da alfavelmanase foram investigadas no estudo clínico rhLAMAN 10 de fase 3, não controlado, aberto, em 33 indivíduos (19 crianças e 14 adultos, de 6 a 35 anos no início do tratamento) que participaram anteriormente de estudos da alfavelmanase. Uma base de dados integrada foi criada através do agrupamento de bases de dados cumulativas de todos os estudos com alfavelmanase. Melhorias estatisticamente significativas foram detectadas nos níveis séricos de oligossacarídeos, 3MSCT, função pulmonar, IgG sérica e EQ 5D 5L (dimensões euro da qualidade de vida 5 (euro quality of life 5 dimensions)) ao longo do tempo até à última observação (tabela 2). Os efeitos da alfavelmanase foram mais evidentes em pacientes com menos de 18 anos de idade.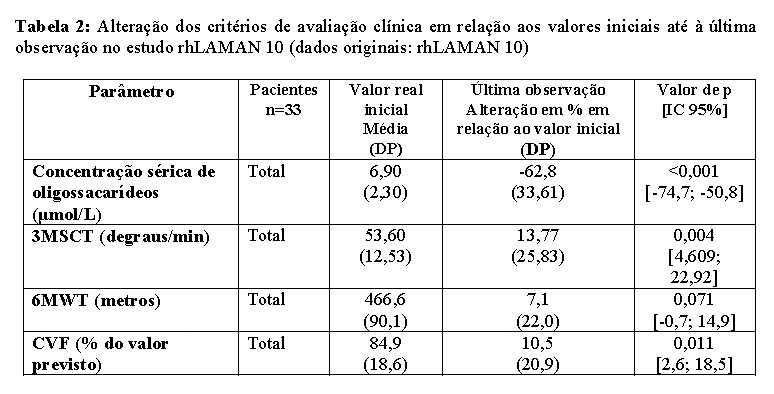
Os dados sugerem que os efeitos benéficos do tratamento com alfavelmanase diminuem com o aumento da carga da doença e das infecções respiratórias relacionadas com a doença.
Uma análise da resposta multiparamétrica post-hoc dá suporte ao benefício de um tratamento mais longo com a alfavelmanase em 87,9% dos respondedores em, pelo menos, 2 domínios na última observação (tabela 3).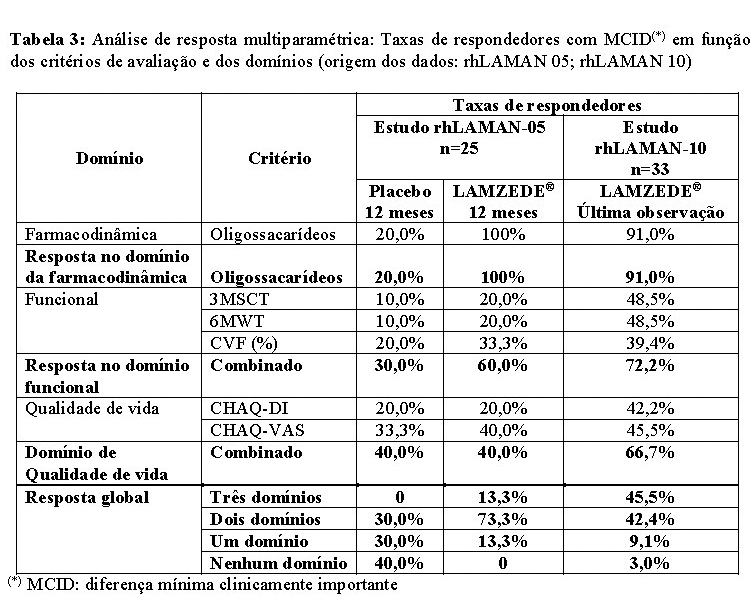
População pediátrica
O uso de alfavelmanase na faixa etária de 6 a 17 anos é apoiado por evidências obtidas nos estudos clínicos em pacientes pediátricos (19 de um total de 33 pacientes) e adultos. Não existem dados clínicos disponíveis em crianças com menos de 6 anos de idade.
(1) Harmatz P et al. Enzyme replacement therapy with velmanase alfa (human recombinant alpha-mannosidase): Novel global treatment response model and outcomes in patients with alpha-mannosidosis. Mol Genet Metab. 2018 Jun;124(2):152-160.
(2) Lund AM et al. Comprehensive long-term efficacy and safety of recombinant human alpha-mannosidase (velmanase alfa) treatment in patients with alpha-mannosidosis. J Inherit Metab Dis. 2018 Nov;41(6):1225-1233.
(3) Borgwardt L et al. Efficacy and safety of Velmanase alfa in the treatment of patients with alpha-mannosidosis: results from the core and extension phase analysis of a phase III multicentre, double-blind, randomised, placebo-controlled trial. J Inherit Metab Dis. 2018 Nov;41(6):1215-1223.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades Farmacodinâmicas
Grupo farmacoterapêutico: outros medicamentos para o trato digestivo e metabolismo, enzimas.
Código ATC: A16AB15.
Mecanismo de ação
A alfavelmanase, a substância ativa de LAMZEDE®, é uma forma recombinante da alfa-manosidase humana. A sequência de aminoácidos da proteína monomérica é idêntica à enzima humana de ocorrência natural, a alfa-manosidase.
A alfavelmanase destina-se a suplementar ou a substituir a alfa-manosidase natural, uma enzima que catalisa a degradação sequencial de oligossacarídeos ricos em manose, complexos e híbridos, nos lisossomos, reduzindo a quantidade de oligossacarídeos ricos em manose acumulados.
Propriedades Farmacocinéticas
Não se observaram diferenças farmacocinéticas aparentes relacionadas ao gênero dos pacientes com a doença alfa-manosidose.
Absorção: LAMZEDE® é administrado por infusão intravenosa. No estado de equilíbrio após a administração semanal de 1 mg/kg de alfavelmanase por infusão, a concentração plasmática máxima média foi de cerca de 8 mg/mL e foi atingida 1,8 horas após o início da administração, correspondendo ao tempo de duração médio da infusão.
Distribuição: como é esperado para uma proteína deste tamanho, o volume de distribuição no estado de equilíbrio foi baixo (0,27 L/kg), indicando uma distribuição limitada ao plasma. A depuração da alfavelmanase do plasma (média de 6,7 mL/h/kg) é consistente com uma captação celular rápida da alfavelmanase através dos receptores da manose.
Biotransformação: prevê-se que a via metabólica da alfavelmanase seja semelhante à de outras proteínas naturais que se degradam em pequenos peptídeos e, finalmente, em aminoácidos.
Eliminação: após o final da infusão, as concentrações plasmáticas de alfavelmanase diminuíram de maneira bifásica com uma meia-vida de eliminação terminal média de cerca de 30 horas.
Linearidade/(não) linearidade: a alfavelmanase exibiu um perfil farmacocinético linear (isto é, de primeira ordem) e a Cmax e a AUC aumentaram proporcionalmente à dose com doses que variaram entre 0,8 e 3,2 mg/kg (correspondendo a 25 e 100 unidades/kg).
Populações especiais: a alfavelmanase é uma proteína e prevê-se que seja degradada metabolicamente em aminoácidos. Proteínas superiores a 50.000 Da, como a alfavelmanase, não são eliminadas por via renal. Consequentemente, não se espera que os comprometimentos hepático e renal afetem a farmacocinética da alfavelmanase. Como não foram identificados pacientes com mais de 41 anos de idade em toda a Europa, não se espera uso relevante em pacientes idosos.
Dados de segurança pré-clínica
Os dados não clínicos não revelam riscos especiais para o ser humano, segundo estudos convencionais de farmacologia de segurança, toxicidade de dose repetida, toxicidade juvenil e toxicidade reprodutiva e do desenvolvimento.
4. CONTRAINDICAÇÕES
Reação alérgica grave à substância ativa ou a qualquer um dos excipientes mencionados no item Composição.
Gravidez e Lactação
Gravidez
Não existem dados sobre a utilização de alfavelmanase em mulheres grávidas. Os estudos em animais não indicam efeitos nefastos diretos ou indiretos em relação à gravidez, desenvolvimento embrionário/fetal, parto ou desenvolvimento pós-natal (ver item 3 - Dados de segurança pré-clínica). Como o objetivo da alfavelmanase é normalizar os níveis da alfa-manosidase nos pacientes com alfa-manosidose, LAMZEDE® só deve ser utilizado durante a gravidez quando estritamente necessário.
Lactação
Não se sabe se a alfavelmanase ou os seus metabolitos são excretados no leite humano. No entanto, a absorção de qualquer leite ingerido que contenha alfavelmanase pela criança amamentada é considerada mínima e, portanto, não são previstos efeitos indesejáveis. LAMZEDE® pode ser utilizado durante a amamentação.
Categoria C de risco na gravidez.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
5. ADVERTÊNCIAS E PRECAUÇÕES
Os efeitos do tratamento com alfavelmanase devem ser periodicamente avaliados e a descontinuação do tratamento deve ser considerada nos casos em que não foram observados benefícios claros.
À medida que o acúmulo de danos nos órgãos-alvo progride ao longo do tempo, é mais difícil para o tratamento reverter os danos ou mostrar melhorias. Tal como acontece com outras terapias de reposição enzimática (TRE), a alfavelmanase não atravessa a barreira hematoencefálica. Deve ser considerado pelo médico assistente que a administração de alfavelmanase não afeta as complicações irreversíveis (ou seja, deformidades esqueléticas, disostose múltipla, manifestações neurológicas e comprometimento da função cognitiva).
Hipersensibilidade
Foram relatadas reações de hipersensibilidade em pacientes em estudos clínicos. Suporte médico adequado deve estar prontamente disponível quando a alfavelmanase for administrada. Se ocorrerem reações anafiláticas ou alérgicas graves, recomenda-se a descontinuação imediata de alfavelmanase e devem ser seguidos os padrões médicos atuais para tratamento de emergência.
Reação relacionada à infusão (RRI)
A administração de alfavelmanase pode resultar em uma RRI, incluindo uma reação anafilactóide (ver item 9. Reações Adversas). As RRIs observadas em estudos clínicos de alfavelmanase foram caracterizadas por um rápido início dos sintomas e a sua gravidade foi leve a moderada.
O controle e tratamento das RRIs devem basear-se na gravidade da reação e incluem a diminuição da taxa de infusão, tratamento com medicamentos como anti-histamínicos, antipiréticos e/ou corticosteroides e/ou interrupção e retomada do tratamento com um tempo de infusão mais longo. O pré-tratamento com anti-histamínicos e/ou corticosteroides pode prevenir reações subsequentes nos casos em que foi necessário tratamento sintomático. Os pacientes não foram rotineiramente pré-medicados antes da infusão de alfavelmanase durante os estudos clínicos.
Se ocorrerem sintomas como angioedema (edema da língua ou garganta), obstrução das vias aéreas superiores ou hipotensão durante ou imediatamente após a infusão, deve-se suspeitar de anafilaxia ou de uma reação anafilactóide. Neste caso, o tratamento com um anti-histamínico e corticosteroides deve ser considerado adequado. Nos casos mais graves, devem ser seguidas os padrões médicos atuais para o tratamento de emergência.
O paciente deve ser mantido sob observação por uma hora ou mais após a infusão para descartar a possibilidade de RRI, de acordo com o critério do médico assistente.
Imunogenicidade
Os anticorpos podem desempenhar um papel nas reações relacionadas ao tratamento observadas com o uso de alfavelmanase. Para uma avaliação mais aprofundada da relação, em casos de desenvolvimento de RRIs graves ou de ausência ou perda de efeito do tratamento, os pacientes devem ser submetidos a testes para detecção de presença de anticorpos anti-alfavelmanase. No caso de deterioração do estado do paciente durante a TRE, deve-se considerar a interrupção do tratamento.
Existe um potencial para imunogenicidade. Nos estudos clínicos a qualquer momento durante o tratamento, 8 dos 33 pacientes (24%) desenvolveram anticorpos da classe IgG contra a alfavelmanase. Não se observou uma correlação clara entre os títulos de anticorpos (nível de anticorpos IgG anti-alfavelmanase) e a redução da eficácia, ocorrência de anafilaxia ou outras reações de hipersensibilidade.
O desenvolvimento de anticorpos não demonstrou afetar a eficácia ou a segurança clínica.
Gravidez
Não existem dados sobre a utilização de alfavelmanase em mulheres grávidas. Os estudos em animais não indicam efeitos nefastos diretos ou indiretos em relação à gravidez, desenvolvimento embrionário/fetal, parto ou desenvolvimento pós-natal (ver item 3. Dados de segurança pré-clínica). Como o objetivo da alfavelmanase é normalizar os níveis da alfa-manosidase nos pacientes com alfa-manosidose, LAMZEDE® só deve ser utilizado durante a gravidez quando estritamente necessário.
Lactação
Não se sabe se a alfavelmanase ou os seus metabolitos são excretados no leite humano. No entanto, a absorção de qualquer leite ingerido que contenha alfavelmanase pela criança amamentada é considerada mínima e, portanto, não são previstos efeitos indesejáveis. LAMZEDE® pode ser utilizado durante a amamentação.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
Fertilidade
Não existem dados clínicos sobre os efeitos da alfavelmanase na fertilidade. Estudos em animais não mostram evidências de comprometimento da fertilidade.
Efeitos sobre a capacidade de dirigir e utilizar máquinas
Os efeitos de LAMZEDE® sobre a capacidade de dirigir e utilizar máquinas são nulos ou desprezíveis.
Uso em idosos, crianças e outros grupos de risco:
Não há dados disponíveis e nem se encontra descrito uso relevante em pacientes idosos.
Não existe experiência em pacientes com menos de 6 anos de idade.
6. INTERAÇÕES MEDICAMENTOSAS
Não foram realizados estudos de interação.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
LAMZEDE® é um pó branco a esbranquiçado para solução para infusão, fornecido num frasco-ampola para injetáveis de vidro.
LAMZEDE® deve ser conservado sob refrigeração (entre 2°C e 8°C), protegido da luz, na sua embalagem de origem.
O prazo de validade é de 36 meses.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Solução para infusão reconstituída
A estabilidade física e química em uso foi demonstrada por 24 horas a 2°C - 8°C.
Do ponto de vista microbiológico, o medicamento deve ser utilizado imediatamente. Se não for usado imediatamente, os tempos e condições de armazenamento antes do uso são de responsabilidade do usuário e, normalmente, não seriam mais que 24 horas entre 2°C a 8°C.
A solução deve estar límpida e não deve ser utilizada se se observarem partículas opacas ou se a solução estiver descolorida. Devido à natureza do medicamento, a solução reconstituída pode conter ocasionalmente algumas partículas proteicas na forma de filamentos finos brancos ou de fibras translúcidas que serão removidas pelo filtro em linha durante a infusão.
Antes de usar, observe o aspecto do medicamento.
Após reconstituição, o medicamento deve ser utilizado imediatamente. Se não for usado imediatamente, a solução reconstituída deve ser armazenada durante um período máximo de 24 horas entre 2°C e 8°C.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
O tratamento deve ser supervisionado por um médico com experiência no tratamento de pacientes com alfa-manosidose ou na administração de outras terapias de reposição enzimática (TRE) em doenças lisossomais de depósito. A administração de LAMZEDE® deve ser realizada por um profissional de saúde com a capacidade de gerenciar TRE e emergências médicas.
Posologia
O regime posológico recomendado é de 1 mg/kg de peso corporal administrado uma vez por semana, por infusão intravenosa a uma velocidade controlada. Para a taxa de infusão consulte a seção "Modo de administração".
Populações especiais
Insuficiência renal ou hepática
Não são necessários ajustes posológicos em pacientes com insuficiência renal ou hepática.
Idosos
Não há dados disponíveis e nem se encontra descrito uso relevante em pacientes idosos.
População pediátrica
Não são necessários ajustes posológicos na população pediátrica.
Modo de administração
Apenas para infusão por via intravenosa.
Para instruções sobre a reconstituição do medicamento antes da administração, ver item 8. Posologia e Modo de Usar (Instruções para reconstituição e administração).
A solução reconstituída de LAMZEDE® deve ser administrada utilizando um conjunto de infusão equipado com uma bomba e um filtro em linha de 0,22 mm, com baixa ligação às proteínas. A duração da infusão deve ser calculada individualmente, considerando uma taxa máxima de infusão de 25 mL/hora para controlar a carga proteica. A duração da infusão deve ser de no mínimo de 50 minutos. Uma taxa de infusão mais lenta pode ser prescrita quando clinicamente apropriado, de acordo com o critério do médico, por exemplo no início do tratamento ou no caso de reações relacionadas à infusão (RRIs) anteriores.
Para o cálculo da taxa de infusão e do tempo de infusão com base no peso corporal, consulte a tabela no item 8. Posologia e Modo de Usar (Instruções para reconstituição e administração).
O paciente deve ser observado para detecção de RRI por pelo menos uma hora após a infusão, de acordo com as condições clínicas e o critério do médico. Para mais instruções consulte o item 5. Advertências e Precauções.
LAMZEDE® necessita ser reconstituído e destina-se apenas a infusão intravenosa.
Cada frasco-ampola é para uso único.
Instruções para reconstituição e administração
LAMZEDE® deve ser reconstituído e administrado por um profissional de saúde.
Utilizar uma técnica asséptica durante a preparação. Não se podem utilizar agulhas com filtro durante a preparação.
a) O número de frasco-ampolas a serem utilizados deve ser calculado com base no peso de cada paciente individualmente. A dose recomendada de 1 mg/kg é determinada usando o seguinte cálculo:
- Peso do paciente (kg) × dose (mg/kg) = dose do paciente (em mg)
- Dose do paciente (em mg) dividida por 10 mg/frasco-ampola (conteúdo de um frasco-ampola) = número de frasco-ampolas para reconstituir. Se o número de frasco-ampolas calculados incluir uma fração, ele deverá ser arredondado para o próximo número inteiro.
- Aproximadamente 30 minutos antes da reconstituição, o número necessário de frasco-ampolas deve ser retirado da geladeira. Os frasco-ampolas devem atingir a temperatura ambiente (entre 15°C e 30°C) antes da reconstituição.
Cada frasco-ampola é reconstituído injetando lentamente 5 mL de água para injetáveis ao longo da parede interior de cada frasco-ampola. Cada mL de solução reconstituída contém 2 mg de alfavelmanase. Apenas o volume que corresponde à dose recomendada deve ser administrado.
Exemplo:
- Peso do paciente (44 kg) × dose (1 mg/kg) = dose do paciente (44 mg)
- 44 mg divididos por 10 mg/frasco-ampola para injetáveis = 4,4 frasco-ampolas; portanto, devem ser reconstituídos 5 frasco-ampolas.
- Do volume total reconstituído, apenas devem ser administrados 22 mL (correspondentes a 44 mg).
b) O pó deve ser reconstituído no frasco-ampola com uma adição gota-a-gota de água para injetáveis ao longo da parede interior do frasco-ampola e não diretamente sobre o pó liofilizado. Deve evitar-se a expulsão enérgica da água da seringa sobre o pó a fim de minimizar a formação de espuma. Os frasco-ampolas reconstituídos devem repousar sobre a mesa durante cerca de 5 a 10 minutos. Em seguida, cada frasco-ampola deve ser inclinado e rolado suavemente durante 15 a 20 segundos para aprimorar o processo de dissolução. O frasco-ampola não deve ser invertido, rodado vigorosamente ou agitado.
c) Após a reconstituição, deve ser realizada imediatamente uma inspeção visual da solução para detecção de partículas e descoloração. A solução deve estar límpida e não deve ser utilizada se forem observadas partículas opacas ou se a solução estiver descolorida. Devido à natureza do medicamento, a solução reconstituída pode ocasionalmente conter algumas partículas proteicas sob a forma de filamentos finos brancos ou de fibras translúcidas que serão removidas pelo filtro em linha durante a infusão (ver item e).
d) A solução reconstituída é extraída lentamente de cada frasco-ampola com cuidado para evitar a formação de espuma na seringa. Se o volume da solução exceder a capacidade de uma seringa, deverá ser preparado o número necessário de seringas para substituir rapidamente a seringa durante a infusão.
e) A solução reconstituída deve ser administrada utilizando um conjunto de infusão equipado com uma bomba e um filtro em linha de 0,22 mm, com baixa ligação às proteínas.
O volume total de infusão é determinado pelo peso do paciente e deve ser administrado por um período mínimo de 50 minutos. No caso de pacientes com um peso inferior a 18 kg, e aos quais serão administrados menos de 9 mL de solução reconstituída, a taxa de infusão deverá ser calculada para que o tempo de infusão seja ≥ 50 minutos. A taxa máxima de infusão é de 25 mL/hora (ver item 8. Posologia e Modo de Usar). O tempo de infusão pode ser calculado com base na seguinte tabela: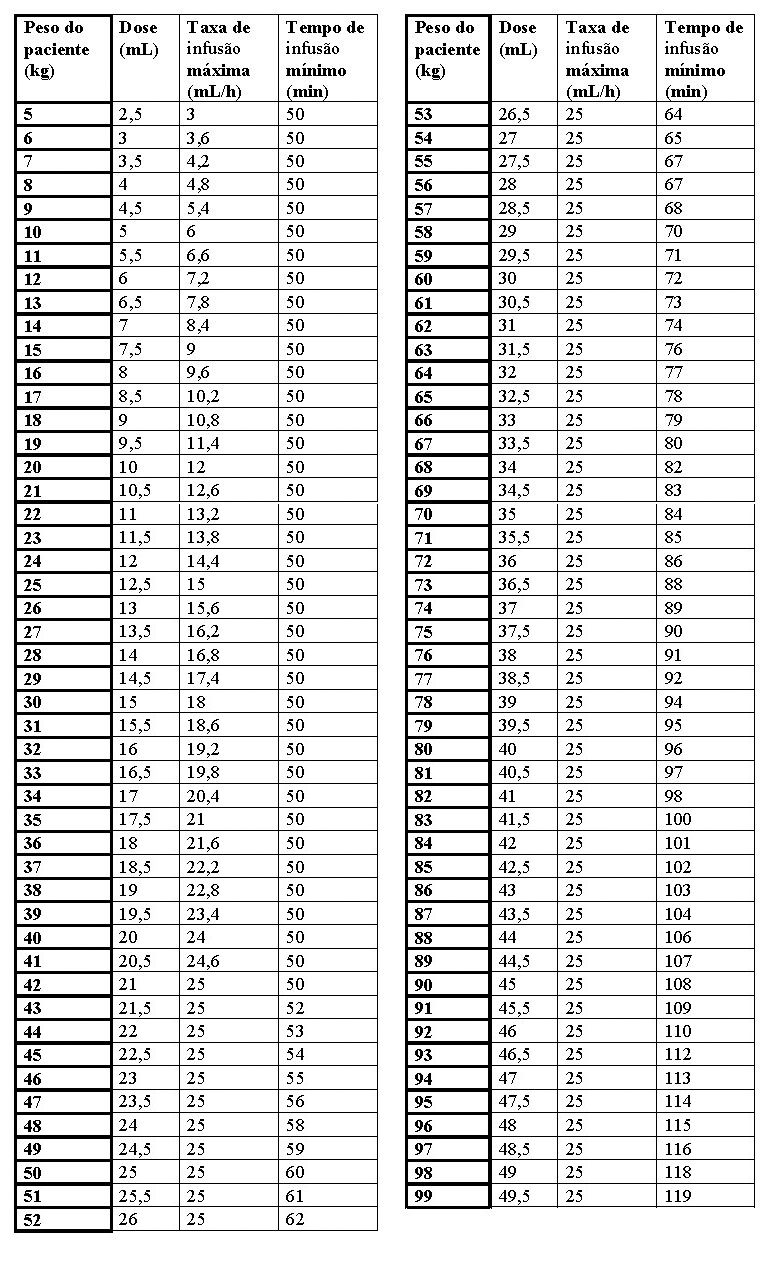
f) Quando a última seringa estiver vazia, a seringa de administração é substituída por uma seringa de 20 mL cheia com uma solução injetável de cloreto de sódio a 9 mg/mL (0,9%). Um volume de 10 mL de solução de cloreto de sódio deve ser administrado através do sistema de infusão para infundir a fração restante de LAMZEDE® na linha do paciente.
Descarte: qualquer medicamento não utilizado ou resíduos devem ser descartados de acordo com os requisitos locais.
9. REAÇÕES ADVERSAS
As reações adversas mais comuns observadas foram aumento de peso (18%), RRP (9%), diarreia (12%), cefaleias (9%), artralgia (9%), aumento do apetite (6%) e dor nas extremidades (6%).
Nenhuma destas reações adversas foi grave. As RRP incluem hipersensibilidade em 3 pacientes e reação anafilactóide em 1 paciente. Essas reações não foram graves e de intensidade leve a moderada.
Observou-se um total de 2 reações adversas graves (perda de consciência em 1 paciente e insuficiência renal aguda em 1 paciente). Em ambos os casos, os pacientes se recuperaram sem sequelas.
As reações adversas que refletem a exposição dos 33 pacientes tratados com alfavelmanase em estudos clínicos estão listadas abaixo. As reações adversas são classificadas por classe de sistemas de órgãos e pelo termo preferido, de acordo com a convenção de frequências do MedDRA.
A frequência é definida como muito comum (≥ 1/10), comum (≥ 1/100, < 1/10), incomum (≥ 1/1.000, < 1/100), raros (≥ 1/10.000, < 1/1.000), muito raros ( < 1/10.000) ou desconhecido (não pode ser calculado a partir dos dados disponíveis).
Reação muito comum (≥ 1/10): diarreia; pirexia(1); aumento de peso.
Reação comum (≥ 1/100, < 1/10): hipersensibilidade(1); reação anafilactóide(1); aumento do apetite; comportamento psicótico; insonia inicial; estado confusional; perda de consciência(2); síncope; tremores; tonturas; cefaleia; irritação ocular; edema palpebral; hiperemia ocular; bradicardia; epistaxe; dor abdominal; dor na região superior do abdómen; náuseas(1); vômitos(1); gastrite de refluxo; urticária(1); hiperidrose(1); artralgia; lombalgia; rigidez articular; mialgia; dor nas extremidades; insuficiência renal aguda(2); dor no local do cateter; arrepios(1); sensação de calor(1); fadiga; mal-estar(1); cefaleia resultante do procedimento.
(1) Termos preferidos considerados como RRI, de acordo com o descrito na seção seguinte.
(2) Reação adversa selecionada, de acordo com o descrito na seção seguinte.
Descrição de reações adversas selecionadas
Reação relacionada à infusão
Foram relatadas RRI (incluindo hipersensibilidade, náuseas, vômitos, pirexia, arrepios, sensação de calor, mal-estar, urticária, reação anafilactoide e hiperidrose) em 9% dos pacientes (3 de um total de 33 pacientes) em estudos clínicos. A gravidade de todas as reações foi leve ou moderada e nenhuma foi relatada como um evento adverso grave. Todos os pacientes que tiveram uma RRI se recuperaram.
Insuficiência renal aguda
Nos estudos clínicos, um paciente apresentou insuficiência renal aguda que foi considerada como possivelmente relacionada ao tratamento do estudo. A insuficiência renal aguda foi de gravidade moderada levando a uma descontinuação temporária do tratamento em estudo e totalmente resolvida em um período de 3 meses. O tratamento concomitante prolongado com doses elevadas de ibuprofeno foi apontado como um contribuinte potencialmente causador da ocorrência do evento.
Perda de consciência
Em um paciente foi relatada perda de consciência considerada relacionada ao tratamento do estudo com recuperação após alguns segundos. O paciente recebeu uma infusão de solução salina em ambiente hospitalar e recebeu alta após 6 horas de observação.
Posteriormente, o paciente teve convulsões epiléticas que foram consideradas como não relacionadas.
População pediátrica
O perfil de segurança da alfavelmanase em estudos clínicos que incluíram crianças e adolescentes foi semelhante ao observado em pacientes adultos. No geral, 58% dos pacientes (19 de um total de 33) com alfa manosidose, tratados com alfavelmanase, em estudos clínicos, tinham de 6 a 17 anos de idade no início do estudo.
"Esse medicamento foi registrado por meio de um procedimento especial, conforme previsão da Resolução RDC n° 205, de 28 de dezembro de 2017, considerando a raridade da doença para qual está indicado e a condição séria debilitante que esta representa. Dados complementares e provas adicionais ainda serão submetidos à ANVISA, após a concessão do registro do medicamento. A revisão desses novos dados pela ANVISA poderá implicar a alteração das informações descritas nesta bula ou mesmo a alteração do status do registro do medicamento".
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos pelo Sistema VigiMed, disponível no Portal da ANVISA.
10. SUPERDOSE
Não existe experiência com superdosagem de alfavelmanase. A dose máxima de alfavelmanase em estudos clínicos foi uma administração única de 100 unidades/kg (o que corresponde a aproximadamente 3,2 mg/kg). Durante a infusão com esta dose mais alta, observou-se em um paciente febre de intensidade leve e de curta duração (5 horas). Nenhum tratamento foi administrado.
Para o controle e tratamento de reações adversas, ver itens 5. ADVERTÊNCIAS E PRECAUÇÕES e 9. REAÇÕES ADVERSAS.
Em caso de intoxicação ligue para 0800 722 6001, se você precisar de mais orientações.
Dizeres legais.
USO RESTRITO A HOSPITAIS
VENDA SOB PRESCRIÇÃO MÉDICA
Reg. MS n° 1.0058.0121