JANUMET®
MSD
sitagliptina, fosfato + metformina
Hipoglicemiante.
Apresentações.
JANUMET® é apresentado em comprimidos revestidos de 50/500 mg, 50/850 mg ou 50/1.000 mg acondicionados em caixas com 28 ou 56 comprimidos.
Uso Oral.
Uso Adulto.
Composição.
Ingredientes ativos: Cada comprimido revestido de JANUMET® contém 64,25 mg de fosfato de sitagliptina e cloridrato de metformina, equivalente a: 50 mg de sitagliptina como base livre e 500 mg de cloridrato de metformina (JANUMET® 50 mg/500 mg), 850 mg de cloridrato de metformina (JANUMET® 50 mg/850 mg) ou 1.000 mg de cloridrato de metformina (JANUMET® 50 mg/1.000 mg). Ingredientes inativos: Cada comprimido revestido de JANUMET® contém: celulose microcristalina, polivinilpirrolidona, laurilsulfato de sódio, estearil fumarato de sódio. Além disso, o revestimento contém os seguintes ingredientes inativos: álcool polivinílico, polietilenoglicol, talco, dióxido de titânio, óxido de ferro vermelho e óxido de ferro preto.
Informações técnicas.
CARACTERÍSTICAS QUÍMICAS
JANUMET® contém fosfato de sitagliptina e cloridrato de metformina.
Fosfato de sitagliptina
O fosfato de sitagliptina é descrito quimicamente como: fosfato de 7-[(3R)-3-amino-1-oxo-4-(2,4,5-trifluorfenil)butil]-5,6,7,8-tetrahidro-3-(trifluormetil)-1,2,4-triazol[4,3-a] pirazina (1:1) monohidratado.
A sitagliptina é um pó cristalino, não higroscópico, branco a esbranquiçado. O composto é solúvel em água e N,N-dimetilformamida, discretamente solúvel em metanol, muito pouco solúvel em etanol, acetona e acetonitrila e insolúvel em isopropanol e acetato de isopropila.
Cloridrato de metformina
O cloridrato de metformina (N,N-cloridrato de dimetilimidodicarbonimidico diamida) não é quimicamente ou farmacologicamente relacionado a qualquer outra classe de agentes antihiperglicêmicos orais. O cloridrato de metformina é um composto cristalino branco a esbranquiçado.
O cloridrato de metformina é livremente solúvel em água e praticamente insolúvel em acetona, éter e clorofórmio. O pKa da metformina é 12,4. O pH da solução aquosa a 1% do cloridrato de metformina é 6,68.
Indicações.
JANUMET® é indicado como adjuvante à dieta e à prática de exercícios para melhorar o controle glicêmico de pacientes com diabetes mellitus tipo 2 não adequadamente controlados com metformina ou sitagliptina isoladamente ou para pacientes que já começaram o tratamento combinado com sitagliptina e metformina.
Resultados de eficácia.
Em estudos clínicos, a co-administração de sitagliptina e metformina proporcionou significativa melhora do controle glicêmico em pacientes com diabetes tipo 2 inadequadamente controlados com metformina. Não foram conduzidos estudos clínicos de eficácia com JANUMET®, entretanto foi demonstrada a bioequivalência de JANUMET® com os comprimidos de sitagliptina e cloridrato de metformina co-administrados.
Dois estudos clínicos duplo-cegos, controlados por placebo, que envolveram pacientes com diabetes mellitus tipo 2 avaliaram a segurança e a eficácia da co-administração de sitagliptina e metformina. Em ambos os estudos, os pacientes com controle glicêmico inadequado sob doses estáveis de metformina 1.500 mg foram distribuídos de modo randômico para receber 100 mg de sitagliptina por dia ou placebo em adição ao tratamento com metformina.
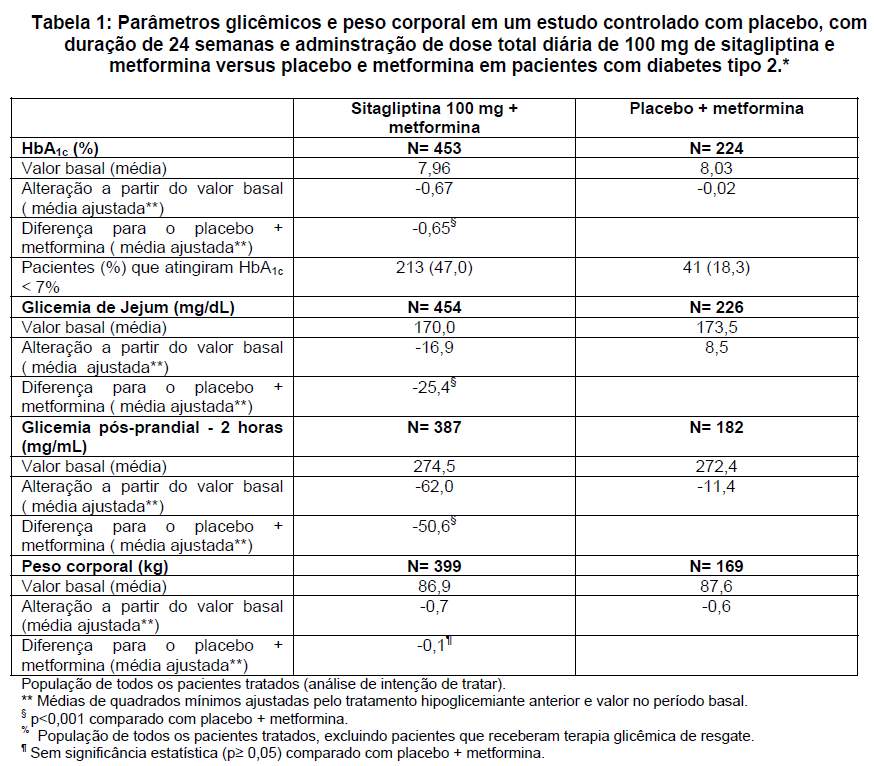
Em um estudo, 701 pacientes receberam 100 mg de sitagliptina ou placebo uma vez ao dia durante 24 semanas. A adição de sitagliptina ao tratamento com metformina, em comparação à adição de placebo ao tratamento com metformina, melhorou significativamente os níveis de HbA1c (-0,65%), a glicemia de jejum (-25,4 mg/dL) e a glicemia pós-prandial de 2 horas (-50,6 mg/mL) (veja figura 1 e Tabela 1). Essa melhora na HbA1c não foi afetada pelo valor basal de HbA1c, pelo tratamento hipoglicemiante prévio, sexo, idade, valor basal do IMC, duração do diabetes, presença de síndrome metabólica ou pelos índices padrões de resistência a insulina (HOMA-IR) ou de secreção de insulina (HOMA-). Observou-se diminuição semelhante do peso corporal em ambos os grupos de tratamento.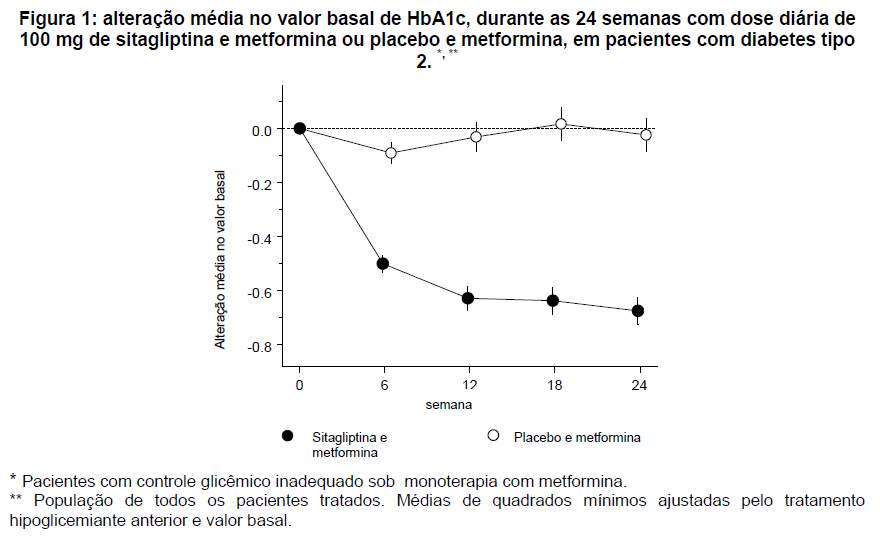
Em um estudo separado, foi avaliada a glicose plasmática nas 24 horas. Vinte e oito pacientes receberam 50 mg de sitagliptina ou placebo duas vezes ao dia durante 4 semanas em adição a um esquema com metformina (duas vezes ao dia). Após 4 semanas de tratamento, a diferença na eficácia na diminuição da glicose foi avaliada como média ponderada da glicose (MPG) nas 24 horas, com base em múltiplas coletas de sangue, incluindo aquelas obtidas antes e após as refeições e durante a noite. A co-administração de sitagliptina 50 mg e metformina, duas vezes ao dia, diminui significativamente a MPG nas 24 horas (-32,8 mg/dL) em comparação com o placebo co-administrado com metformina. Além disso, a co-administração de sitagliptina com metformina, em comparação com a administração de placebo e metformina, diminuiu substancialmente as concentrações de glicose em jejum e demonstrou picos de glicose menores após as três refeições (veja Figura 2). Nas avaliações da glicose coletada pelo paciente, a co-administração de sitagliptina e metformina também propiciou reduções significativas na média da glicemia em jejum (-20,3 mg/dL), média da glicemia em 7 pontos (-28,0 mg/dL) e na concentração de glicose pós-prandial de 2 horas (-36,6 mg/dL), em comparação com a administração de placebo e metformina.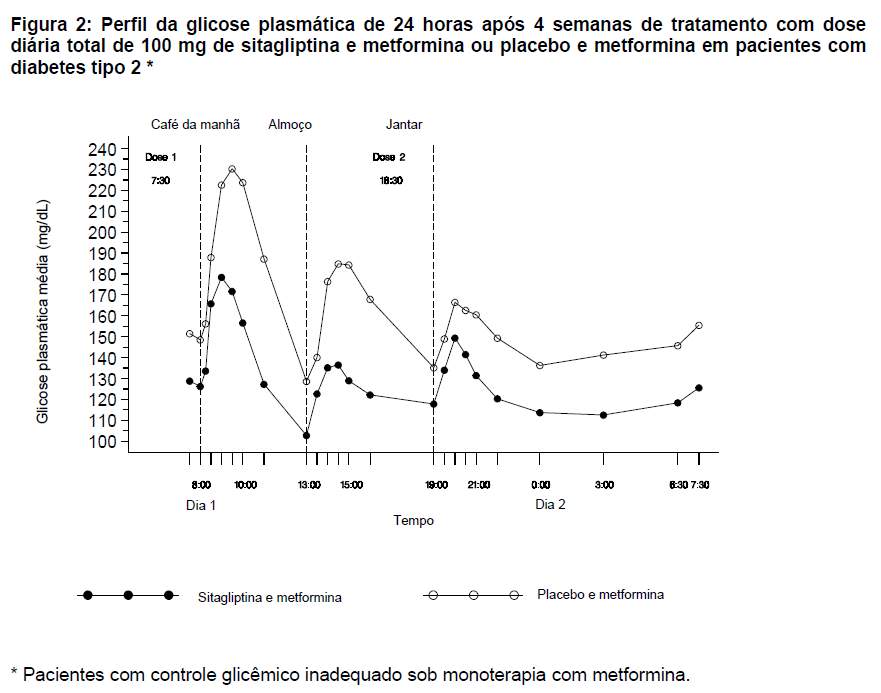
Cloridrato de metformina
O UKPDS, um estudo randômico e prospectivo, estabeleceu os benefícios a longo prazo do controle intensivo da glicemia no diabetes tipo 2. A análise dos resultados de pacientes com sobrepeso que receberam metformina após insucesso da dieta isoladamente, mostrou:
- significativa redução do risco absoluto de qualquer complicação relacionada ao diabetes no grupo da metformina (29,8 eventos/1.000 paciente-anos) versus somente dieta (43,3 eventos/1.000 paciente-anos), p= 0,0023 e versus o grupo que recebeu a combinação de sulfoniluréias e monoterapia com insulina (40.1 eventos/1.000 paciente-anos), p= 0,0034
- significativa redução do risco absoluto de morte relacionada ao diabetes: metformina, 7,5 eventos/1.000 paciente-anos e somente dieta, 12,7 eventos/1.000 paciente-anos, p= 0,017.
- significativa redução do risco absoluto de mortalidade total: metformina, 13,5 eventos/1.000 paciente-anos versus somente dieta, 20,6 eventos/1.000 paciente-anos (p= 0,011) e versus a combinação de sulfoniluréias e monoterapia com insulina 18,9 eventos/1.000 paciente-anos (p= 0,021).
- significativa redução no risco absoluto de infarto do miocárdio: metformina, 11 eventos/1.000 paciente-anos, somente dieta, 18 eventos/1.000 paciente-anos (p= 0,01).
Caract. farmacológicas.
Mecanismo de Ação
JANUMET® combina dois agentes antidiabéticos com mecanismos de ação complementares, para melhorar o controle da glicemia em pacientes com diabetes tipo 2: fosfato de sitagliptina, um inibidor da dipeptidil peptidase 4 (DPP-4) e cloridrato de metformina, membro da classe das biguanidas.
Fosfato de sitagliptin
O fosfato de sitagliptina é membro de uma classe de agentes hipoglicemiantes orais denominada inibidores da dipeptidil peptidase 4 (DPP-4), que melhoram o controle glicêmico em pacientes com diabetes tipo 2 por meio do aumento dos níveis de hormônios incretina ativos. Os hormônios incretina, inclusive o peptídeo-1 semelhante ao glucagon (GLP-1) e o peptídeo insulinotrópico dependente de glicose (GIP), são liberados pelo intestino ao longo do dia e seus níveis aumentam em resposta a uma refeição. As incretinas são parte de um sistema endógeno envolvido na regulação fisiológica da homeostase da glicose. Quando as concentrações sanguíneas de glicose estão normais ou elevadas,
o GLP-1 e o GIP aumentam a síntese e a liberação de insulina das células betapancreáticas por meio de vias sinalizadoras intracelulares que envolvem o AMP cíclico. O tratamento com GLP-1 ou com inibidores da DPP-4 em modelos animais com diabetes tipo 2 demonstrou melhorar a responsividade das células beta à glicose e estimular a biossíntese e a liberação de insulina. Com níveis de insulina mais altos, a captação tecidual de glicose é aumentada. Além disso, o GLP-1 diminui a secreção de glucagon pelas células alfa pancreáticas. A redução das concentrações de glucagon, associada a níveis mais altos de insulina, resulta em redução da produção hepática de glicose e conseqüente redução da glicemia. Quando as concentrações sanguíneas de glicose estão baixas, não são observadas estimulação da liberação de insulina e supressão da secreção de glucagon pelo GLP-1. O GLP-1 e o GIP não impedem a resposta normal do glucagon à hipoglicemia. A atividade do GLP-1 e do GIP é limitada pela enzima DPP-4, que hidrolisa rapidamente os hormônios incretina para produzir produtos inativos. A sitagliptina evita a hidrólise dos hormônios incretina pela DPP-4, aumentando conseqüentemente as concentrações plasmáticas das formas ativas de GLP-1 e GIP. Ao aumentar os níveis de incretina ativa, a sitagliptina aumenta a liberação de insulina e diminui os níveis de glucagon de forma dependente da glicose. Esse mecanismo dependente de glicose é diferente do mecanismo observado com as sulfoniluréias, pelo qual a insulina é liberada mesmo quando os níveis de glicose são baixos, o que pode causar hipoglicemia em pacientes com diabetes tipo 2 e em indivíduos normais. Em pacientes com diabetes tipo 2 com hiperglicemia, essas alterações nos níveis de insulina e de glucagon resultam em níveis mais baixos de hemoglobina A1c (HbA1c) e concentrações mais baixas da glicemia de jejum e pós-prandial. Embora a sitagliptina seja um inibidor potente e altamente seletivo da enzima DPP-4, ela não inibe as enzimas estreitamente relacionadas DPP-8 ou DPP-9. A inibição da DPP-8 ou da DPP-9, mas não da DPP-4, está associada à toxicidade nos modelos animais pré-clínicos e a alteração da função imunológica in vitro.
Cloridrato de metformina
A metformina é um agente antidiabético que melhora a tolerância à glicose em pacientes com diabetes tipo 2, diminuindo a glicose basal e pós-prandial. Farmacologicamente, o mecanismo de ação é diferente de outras classes de agentes antidiabéticos orais. A metformina diminui a produção hepática de glicose, diminui a absorção da glicose pelo intestino e melhora a sensibilidade à insulina, aumentando a captação e utilização periféricas da glicose. Diferentemente das sulfoniluréias, a metformina não causa hipoglicemia em pacientes com diabetes tipo 2 ou indivíduos normais (exceto em circunstâncias especiais, veja ADVERTÊNCIAS, Cloridrato de metformina) e não causa hiperinsulinemia. O tratamento com metformina não altera a secreção de insulina, enquanto os níveis de insulina em jejum e a resposta de insulina ao longo do dia podem, na verdade, diminuir.
Farmacocinética
JANUMET®
Os resultados de um estudo definitivo de bioequivalência em indivíduos saudáveis demonstraram que os comprimidos combinados de JANUMET® 50 mg/500 mg e 50 mg/1.000 mg são bioequivalentes à co-administração das doses correspondentes de fosfato de sitagliptina e cloridrato de metformina em comprimidos individuais.
Como a bioequivalência é demonstrada por meio da combinação de comprimidos da mínima e máxima dose disponível, a bioequivalência é conferida para a combinação de dose fixa de 50 mg/850 mg.
Absorção
Fosfato de sitagliptina
A biodisponibilidade absoluta da sitagliptina é de aproximadamente 87%. A co-administração de uma refeição rica em gordura com fosfato de sitagliptina não exerceu efeito na farmacocinética.
Cloridrato de metformina
A biodisponibilidade do comprimido de 500 mg de cloridrato de metformina administrado em jejum é de aproximadamente 50-60%. Estudos que utilizaram comprimidos de 500 mg a 1.500 mg e 850 mg a 2.550 mg de cloridrato de metformina indicaram que há falta de proporcionalidade da dose com doses crescentes, o que se deve à absorção diminuída, em vez de alternância na eliminação. O alimento diminui a magnitude e atrasa ligeiramente a absorção da metformina, como mostrado por diminuição de aproximadamente 40 % na média do pico de concentração plasmática (Cmáx), e diminuição de 25% na área sob a curva da concentração plasmática versus tempo e prolongamento de 35 minutos do tempo para atingir as concentrações plasmáticas máximas após a administração de um único comprimido de 850 mg de metformina com alimento, quando comparado com administração em jejum de um comprimido com a mesma concentração. A importância clínica desses decréscimos não é conhecida.
Distribuição
Fosfato de sitagliptina
Após uma dose única de 100 mg de sitagliptina administrada por via intravenosa a indivíduos saudáveis, o volume médio de distribuição no estado de equilíbrio é de aproximadamente 198 litros. A fração da sitagliptina que se liga reversivelmente às proteínas plasmáticas é pequena (38%).
Cloridrato de metformina
O volume de distribuição da metformina após dose única de um comprimido de 850 mg de cloridrato de metformina administrado via oral foi de 654 358 L, em média. A ligação da metformina às proteínas plasmáticas é insignificante, em contraste com as sulfoniluréias, cuja porcentagem de ligação protéica é de mais de 90%. A metformina se distribui nos eritrócitos mais provavelmente em função do tempo. Com os esquemas posológicos e doses clínicas usuais dos comprimidos de cloridrato de metformina, a concentração plasmática da metformina no estado de equilíbrio é alcançada em 24-48 horas e é geralmente < 1 mcg/mL. Durante estudos clínicos controlados, os níveis máximos de metformina no plasma não excederam 5 mcg/mL, mesmo com as doses máximas.
Metabolismo
Fosfato de sitagliptina
A sitagliptina é eliminada principalmente de forma inalterada na urina; o metabolismo é uma via de menor importância. Aproximadamente 79% da sitagliptina é excretada inalterada na urina.
Após uma dose oral de [14C]sitagliptina, aproximadamente 16% da radioatividade foi excretada na forma de metabólitos de sitagliptina. Seis metabólitos foram detectados em níveis-traço e não se espera que contribuam para a atividade inibitória de DPP-4 plasmática da sitagliptina. Estudos in vitro indicaram que a principal enzima responsável pelo metabolismo limitado da sitagliptina foi a CIP3A4, com contribuição da CIP2C8.
Cloridrato de metformina
Um estudo com dose única por via intravenosa em indivíduos normais demonstrou que a metformina é excretada inalterada na urina e não é metabolizada no fígado (nenhum metabólito foi identificado em humanos) nem excretada na bile.
Eliminação
Fosfato de sitagliptina
Após a administração de uma dose de [14C]sitagliptina por via oral a indivíduos saudáveis, aproximadamente 100% da radioatividade administrada foi eliminada nas fezes (13%) ou na urina (87%), uma semana após a administração. A t½ terminal aparente após uma dose de 100 mg de sitagliptina por via oral foi de aproximadamente 12,4 horas e a depuração renal foi de aproximadamente 350 mL/min. A eliminação da sitagliptina ocorre principalmente por excreção renal e envolve secreção tubular ativa. A sitagliptina é um substrato para o transportador-3 aniônico orgânico humano (hOAT-3), que pode estar envolvido na eliminação renal da sitagliptina. A relevância clínica do hOAT-3 no transporte da sitagliptina não foi estabelecida. A sitagliptina também é um substrato da p-glicoproteína, que também pode estar envolvida na mediação da eliminação renal da sitagliptina. No entanto, a ciclosporina, um inibidor da p-glicoproteína, não reduziu a depuração renal da sitagliptina.
Cloridrato de metformina
A depuração renal é aproximadamente 3,5 vezes maior que a depuração da creatinina, o que indica que a secreção tubular é a principal rota de eliminação da metformina. Após a administração de uma dose oral, aproximadamente 90% do fármaco absorvido é eliminado por via renal nas primeiras 24 horas, com meia-vida de eliminação plasmática de aproximadamente 6,2 horas. No sangue, a meia-vida de eliminação é de aproximadamente 17,6 horas, sugerindo que a massa de eritrócitos pode ser um compartimento de distribuição.
Farmacodinâmica
Fosfato de sitagliptina
Geral
Em pacientes com diabetes tipo 2, a administração de doses únicas de sitagliptina por via oral leva à inibição da atividade enzimática da DPP-4 por um período de 24 horas, o que resulta em aumento de 2 a 3 vezes nos níveis circulantes de GLP-1 e GIP ativos, aumento dos níveis plasmáticos de insulina e de peptídeo-C, redução das concentrações de glucagon, redução da glicemia de jejum e redução dos picos de glicose após uma sobrecarga oral de glicose ou refeição.
Nos estudos clínicos fase III, com duração de 18 e 24 semanas, o tratamento com 100 mg/dia de sitagliptina em pacientes com diabetes tipo 2 melhorou significativamente a função da célula beta, conforme avaliada por vários marcadores, inclusive HOMA-b (Avaliação de um Modelo de Homeostase-), razão pró-insulina:insulina e medidas de responsividade da célula beta ao teste de tolerância à refeição amostrado freqüentemente. Nos estudos fase II, a administração de 50 mg de sitagliptina duas vezes ao dia proporcionou eficácia glicêmica semelhante em comparação com a dose de 100 mg uma vez ao dia.
Em um estudo randômico, controlado com placebo, duplo cego, duplo-mascarado, com quatro períodos cruzados de dois dias, que envolveu indivíduos adultos saudáveis, os efeitos nas concentrações plasmáticas das formas ativas e total de GLP-1 e nas concentrações de glicose após co-administração de sitagliptina e metformina foram comparados ao observados depois da administração da sitapliptina, metformina ou placebo isoladamente. A média ponderada das concentrações de GLP-1 ativo incrementadas 4 horas após alimentação aumentou aproximadamente 2 vezes depois da administração de sitagliptina isoladamente ou da metformina isoladamente, em comparação com o placebo. O efeito nas concentrações de GLP 1 ativo após co-administração da sitagliptina e metformina foi aditivo e essas concentrações aumentaram aproximadamente 4 vezes em comparação com o placebo. A sitagliptina isoladamente aumentou as concentrações de GLP-1 ativo, provavelmente refletindo a inibição da DPP-4, enquanto a metformina isoladamente aumentou à uma mesma extensão as concentrações de GLP-1 ativo e total, sugerindo um mecanismo diferente para esse aumento, principalmente em conseqüência do aumento das concentrações de GLP-1 total. Os resultados do estudo também demonstraram que a sitagliptina, mas não a metformina, aumenta as concentrações de GIP ativo. Nos estudos com indivíduos saudáveis, a sitagliptina não diminuiu a glicemia ou causou hipoglicemia, sugerindo que as ações insulinotrópicas e supressoras de glucagon do fármaco são dependentes da glicose.
Efeitos na Pressão Arterial
Em um estudo cruzado, randômico, controlado com placebo, conduzido em pacientes hipertensos que recebiam um ou mais anti-hipertensivos (inclusive inibidores da enzima conversora de angiotensina, antagonistas da angiotensina II, bloqueadores dos canais de cálcio, betabloqueadores e diuréticos), a co-administração com sitagliptina foi geralmente bem tolerada. Nesses pacientes, a sitagliptina exerceu efeito redutor discreto na pressão arterial; em comparação com o placebo, o tratamento com 100 mg/dia de sitagliptina reduziu a pressão arterial sistólica ambulatorial média de 24 horas em aproximadamente 2 mmHg. Não foram observadas reduções em indivíduos normotensos.
Eletrofisiologia Cardíaca
Em um estudo cruzado, randômico, controlado com placebo, 79 indivíduos saudáveis receberam uma dose única de 100 mg ou de 800 mg de sitagliptina (8 vezes a dose recomendada) por via oral e placebo. A dose recomendada de 100 mg não exerceu efeito no intervalo QTc na concentração plasmática máxima ou em qualquer outro ponto de tempo durante o estudo. Após a dose de 800 mg, o aumento máximo da alteração média do intervalo QTc corrigida pelo placebo em relação ao período basal três horas após a dose foi de 8,0 milissegundos; este pequeno aumento não foi considerado clinicamente significativo. As concentrações plasmáticas máximas de 800 mg de sitagliptina foram aproximadamente 11 vezes mais altas do que as concentrações máximas após uma dose de 100 mg.
Os pacientes com diabetes tipo 2 que receberam diariamente 100 mg (N= 81) ou 200 mg de sitagliptina (N= 63) não apresentaram alterações significativas no intervalo QTc com base nos dados de ECG obtidos no momento da concentração plasmática máxima esperada.
Contraindicações.
JANUMET® é contra-indicado para pacientes com:
1. Nefropatia ou disfunção renal, por exemplo, níveis de creatinina sérica 1,5 mg/dL (homens), 1,4 mg/dL (mulheres), ou depuração anormal de creatinina, que podem também resultar de choque cardiovascular, infarto agudo do miocárdio e septicemia, por exemplo.
2. Insuficiência cardíaca congestiva que exija o uso de agentes farmacológicos.
3. Hipersensibilidade conhecida ao fosfato de sitagliptina, ao cloridrato de metformina ou a qualquer outro componente de JANUMET® (veja ADVERTÊNCIAS, Fosfato de sitagliptina, Reações de hipersensibilidade e REAÇÕES ADVERSAS, Experiência Póscomercialização).
4. Acidose metabólica aguda ou crônica, incluindo cetoacidose diabética, com ou sem coma.
JANUMET® deve ser descontinuado temporariamente em pacientes que serão submetidos a estudos radiológicos com administração de material de contraste iodado, porque o uso de tais produtos podem resultar em alteração aguda da função renal (veja ADVERTÊNCIAS, cloridrato de metformina).
Advertências.
Gerais
JANUMET® não deve ser utilizado por pacientes com diabetes tipo 1 ou para o tratamento de cetoacidose diabética.
Monitoramento da função renal: sabe-se que a metformina e a sitagliptina são substancialmente excretadas pelos rins. O risco de acúmulo de metformina e de acidose láctica aumenta de acordo com o grau de disfunção renal. Assim, pacientes com níveis de creatinina acima do limite normal para suas idades não devem receber JANUMET®. Em pacientes com idade avançada, JANUMET® deve ser titulado cuidadosamente para estabelecer a dose mínima com efeito adequado sobre a glicemia, porque o envelhecimento pode estar associado com redução da função renal. Em pacientes idosos, particularmente ≥80 anos de idade, a função renal deve ser monitorada regularmente.
Antes de iniciar o tratamento com JANUMET® e ao menos anualmente a seguir, a função renal deve ser avaliada e considerada normal. Nos casos em que é previsto o desenvolvimento de disfunção renal, esta avaliação deve ser mais freqüente e JANUMET® deve ser descontinuado se houver evidência de comprometimento renal.
Fosfato de sitagliptina
Hipoglicemia: nos estudos clínicos de sitagliptina como monoterapia e sitagliptina como parte do tratamento combinado com metformina ou pioglitazona, as taxas de hipoglicemia relatadas com sitagliptina foram semelhantes às observadas em pacientes que recebiam placebo. O uso de sitagliptina em combinação com medicamentos que sabidamente causam hipoglicemia, como as sulfoniluréias ou a insulina, não foi adequadamente estudado.
Reações de hipersensibilidade: após a comercialização, houve relatos de reações de hipersensibilidade graves em pacientes que receberam sitagliptina, um dos componentes de JANUMET®. Essas reações incluem anafilaxia, angioedema e afecções cutâneas exfoliativas, inclusive síndrome de Stevens-Johnson. Uma vez que essas reações foram relatadas voluntariamente por uma população de tamanho incerto, em geral não é possível estimar de forma confiável sua freqüência ou estabelecer uma relação causal com a exposição ao medicamento. O início dessas reações ocorreu nos primeiros 3 meses após o início do tratamento com sitagliptina e em alguns relatos, após a primeira dose. Se houver suspeita de uma reação de hipersensibilidade, deve-se descontinuar o uso de JANUMET®, avaliar outras possíveis causas para o evento e instituir um outro tratamento para o diabetes (veja CONTRA-INDICAÇÕES e REAÇÕES ADVERSAS, Experiência Pós-comercialização).
Cloridrato de metformina
Acidose láctica: a acidose láctica é uma complicação metabólica rara, porém grave, que pode ocorrer por acúmulo de metformina durante o tratamento com JANUMET® (fosfato de sitagliptina /cloridrato de metformina); quando ocorre é fatal em aproximadamente 50% dos casos. A acidose láctica também pode ocorrer em associação com várias situações fisiopatológicas, incluindo diabetes mellitus, e sempre que houver hipoperfusão e hipoxemia tecidual. A acidose láctica é caracterizada pela elevação dos níveis de lactato no sangue ( > 5 mmol/L), diminuição do pH, distúrbios eletrolíticos com aumento do intervalo aniônico e aumento da relação lactato/piruvato. Quando a metformina é considerada a causa da acidose láctica, são encontrados no plasma níveis de metformina geralmente > 5 mcg/mL.
A incidência relatada de acidose láctica em pacientes que recebem cloridrato de metformina é muito baixa (aproximadamente 0,03 casos/1.000 paciente-anos, com aproximadamente 0,015 casos fatais/1.000 paciente-anos). Em mais de 20.000 paciente-anos de exposição à metformina em estudos clínicos, não houve relatos de acidose láctica. Os casos relatados ocorreram principalmente em pacientes com diabetes e insuficiência renal significativa, incluindo doença renal intrínseca e hipoperfusão renal, freqüente em cenários de múltiplos problemas médico/cirúrgicos e múltiplas medicações concomitantes. Pacientes com insuficiência cardíaca congestiva exigindo manejo farmacológico, em particular com insuficiência cardíaca congestiva aguda ou instável, sob risco de hipoperfusão e hipoxemia, têm risco aumentado de acidose láctica; esse risco aumenta com o grau de disfunção renal e a idade do paciente. O risco de acidose láctica pode, portanto, ser significativamente diminuído por meio da monitoração regular da função renal em pacientes em uso de metformina e com o uso da dose mínima efetiva. Em particular, o tratamento dos pacientes idosos deve ser acompanhado com monitoração cuidadosa da função renal. O tratamento com metformina não deve ser iniciado em pacientes com ≥80 anos de idade a menos que as medidas da depuração de creatinina demonstrem que a função renal não está diminuída, pois esses pacientes são mais susceptíveis ao desenvolvimeneto de acidose láctica. Além disso, a metformina deve ser imediatamente descontinuada na presença de qualquer condição associada à hipoxemia, desidratação ou septicemia. Um vez que a função hepática comprometida pode limitar significativamente a capacidade de depurar o lactato, a metformina geralmente deve ser evitada em pacientes com evidência clínica ou laboratorial de hepatopatia. Os pacientes devem ser advertidos contra a ingesta excessiva de álcool, seja aguda ou crônica, quando estiverem tomando metformina, pois o álcool potencializa os efeitos do cloridrato de metformina no metabolismo do lactato. Além disso, a metformina deve ser temporariamente descontinuada antes de qualquer estudo com utilização de radiocontraste intravascular e de qualquer procedimento cirúrgico.
O início da acidose láctica freqüentemente é sutil e acompanhado somente de sintomas inespecíficos, tais como: mal-estar, mialgias, desconforto respiratório, sonolência crescente e desconforto abdominal inespecífico. Pode haver hipotermia, hipotensão e bradiarritmias associadas quando a acidose for mais acentuada. O paciente e o seu médico devem estar cientes da importância de tais sintomas e o paciente deve ser instruído a avisar o médico imediatamente se eles ocorrerem. A metformina deve ser descontinuada até a situação ser esclarecida. Eletrólitos séricos, cetonas, glicemia e, se indicado, pH sangüíneo, níveis de lactato e mesmo os níveis de metformina sangüíneos podem ser úteis. Uma vez que o paciente esteja estabilizado com qualquer dose de metformina, os sintomas gastrointestinais relacionados à metformina que comumente ocorrem no início do tratamento são improváveis. A ocorrência tardia de sintomas gastrointestinais pode ser causada pela acidose láctica ou por outra doença grave.
Níveis de lactose plasmática venosa em jejum, acima do limite superior da normalidade, porém menores que 5 mmol/L em pacientes que recebem metformina não indicam necessariamente acidose láctica iminente e podem ser explicados por outros mecanismos, tais como diabetes mal controlado ou obesidade, atividade física vigorosa ou problemas técnicos no manuseio das amostras.
Deve-se suspeitar de acidose láctica em qualquer paciente com diabetes e acidose metabólica sem evidência de cetoacidose (cetonúria e cetonemia).
A acidose láctica é uma emergência médica que deve ser tratada em ambiente hospitalar. Em pacientes com acidose láctica que estejam recebendo metformina, o fármaco deve ser descontinuado imediatamente e medidas de suporte gerais devem ser prontamente instituídas. Uma vez que o cloridrato de metformina é dialisável (com depuração de até 170 mL/min sob boas condições hemodinâmicas), a hemodiálise imediata é recomendada para correção da acidose e para remover a metformina acumulada. Tais medidas freqüentemente resultam em pronta reversão dos sintomas e recuperação (veja CONTRA-INDICAÇÕES).
Hipoglicemia: a hipoglicemia não ocorre em pacientes que utilizem somente metformina sob circunstâncias normais, mas pode ocorrer quando a ingesta calórica for deficiente, quando exercícios vigorosos não forem compensados por suplementação calórica, durante uso concomitante de outros agentes hipoglicemiantes (tais como sulfoniluréias e insulina) ou etanol. Idosos, debilitados ou pacientes mal nutridos e aqueles com insuficiência adrenal ou pituitária ou intoxicação alcoólica são particularmente susceptíveis a efeitos hipoglicêmicos. A hipoglicemia pode ser mal reconhecida em idosos e em pessoas que estão tomando fármacos bloqueadores -adrenérgicos.
Uso concomitante com medicamentos que podem afetar a função renal ou a disposição da metformina: medicamento(s) concomitante(s) que podem afetar a função renal ou resultar em alterações hemodinâmicas significativas ou que podem interferir com a disposição da metformina, tais como compostos catiônicos eliminados por secreção tubular renal (veja INTERAÇÕES MEDICAMENTOSAS, Cloridrato de metformina) devem ser utilizados com cuidado.
Estudos radiológicos que envolvem o uso intravascular de materiais de contraste iodado (como por exemplo, urografia excretória, colangiografia intravenosa, angiografia e tomografia de varredura computadorizada (TC) com contraste intravascular): estudos com contrastes intravasculares de material iodado podem causar alteração aguda da função renal e foram associados à acidose láctica em pacientes que recebem metformina (veja CONTRA-INDICAÇÕES). Conseqüentemente, quando um desses estudos estiver planejado, JANUMET® deve ser temporariamente descontinuado antes ou no momento do procedimento, suspenso durante as 48 horas subseqüentes e reinstituído somente após a função renal ter sido reavaliada e considerada normal.
Estados hipóxicos: choque cardiovascular de qualquer causa, insuficiência cardíaca congestiva, infarto agudo do miocárdio e outras afecções caracterizadas por hipoxemia foram associadas à acidose láctica e também podem causar azotemia pré-renal. Se tais eventos ocorrerem em pacientes que recebem JANUMET®, o fármaco deve ser imediatamente descontinuado.
Procedimentos cirúrgicos: o uso de JANUMET® deve ser temporariamente suspenso para qualquer procedimento cirúrgico (exceto procedimentos menores, não associados à restrição de alimentos ou líquidos) e não deve ser reiniciado até que a ingesta oral tenha sido retomada e a função renal avaliada e considerada normal.
Ingestão de álcool: sabe-se que o álcool potencializa o efeito da metformina no metabolismo do lactato. Consequentemente, os pacientes devem ser advertidos contra a ingestão excessiva de álcool, aguda ou crônica, enquanto estiverem recebendo JANUMET®.
Comprometimento da função hepática: uma vez que a função hepática diminuída foi associada com alguns casos de acidose láctica, JANUMET® deve geralmente ser evitado por pacientes com evidências clínicas ou laboratoriais de doença hepática.
Níveis de vitamina B12: em estudos clinicos controlados de metformina com 29 semanas de duração, foi observada diminuição para níveis abaixo do normal de níveis séricos previamente normais da vitamina B12, sem manifestação clinica, em aproximadamente 7% dos pacientes. Tal diminuição, possivelmente por interferência na absorção de B12 do complexo de fator intrínseco-B12, é, entretanto, muito raramente associada à anemia e parece ser rapidamente reversível com a descontinuação de metformina ou da suplementação de vitamina B12. Recomenda-se avaliação anual dos parâmetros hematológicos para pacientes que recebem JANUMET® e qualquer anormalidade aparente deve ser apropriadamente investigada e manejada.
Certos indivíduos (aqueles com ingestão ou absorção inadequada de vitamina B12 ou cálcio) parecem ser predispostos ao desenvolvimento de níveis abaixo do normal de vitamina B12. Nestes pacientes, pode ser útil a avaliação rotineira da vitamina B12, a intervalos de 2-3 anos.
Mudança no estado clínico de pacientes com diabetes tipo 2 previamente controlado: um paciente com diabetes tipo 2 bem controlado previamente com JANUMET® que desenvolve anormalidades laboratoriais ou doenças clínicas (especialmente doenças vagas ou obscuras) deve ser avaliado imediatamente quanto à evidência de cetoacidose ou acidose láctica. A avaliação deve incluir eletrólitos e cetonas séricos, glicemia e, se indicado, pH sangüíneo, lactato, piruvato e níveis de metformina. Se ocorrer qualquer forma de acidose, JANUMET® deve ser imediatamente interrompido e outras medidas apropriadas de correção devem ser iniciadas.
Descontrole da glicemia: quando um paciente estabilizado em qualquer esquema antidiabético é exposto a situações de estresse, tais como febre, trauma, infecção ou cirurgia, pode ocorrer descontrole temporário do controle glicêmico. Nessas ocasiões, pode ser necessário interromper a administração de JANUMET® e administrar temporariamente insulina. JANUMET® pode ser reinstituído após a resolução do episódio agudo.
Gravidez
Categoria de risco: B
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Não existem estudos adequados e bem controlados conduzidos em mulheres grávidas com JANUMET® ou seus componentes; portanto, não se conhece a segurança de JANUMET® nessa população. O uso de JANUMET®, assim como o de outros agentes hipoglicemiantes orais, não é recomendado na gravidez.
Não existem estudos em animais com os componentes combinados de JANUMET® para avaliar efeitos na reprodução. Os seguintes dados são baseados nos achados de estudos com sitagliptina ou metformina individualmente.
Fosfato de sitagliptina
A sitagliptina não foi teratogênica para ratos em doses orais de até 250 mg/kg ou para coelhos que receberam até 125 mg/kg durante a organogênese (até 32 e 22 vezes, respectivamente, a exposição humana com base na dose diária recomendada de 100 mg/dia para humanos adultos). Em ratos, observou-se discreto aumento da incidência de malformações das costelas fetais (ausência, hipoplasia e costelas flutuantes) com doses orais de 1.000 mg/kg/dia (aproximadamente 100 vezes a exposição em humanos com base na dose diária recomendada de 100 mg/dia para humanos adultos). Na prole de ratos que receberam doses orais de 1.000 mg/kg/dia, foram observadas discretas reduções dos pesos corporais médios pré-desmame em ambos os sexos e ganhos de peso corporal pós-desmame em machos. No entanto, estudos de reprodução animal nem sempre são preditivos da resposta humana.
Cloridrato de metformina
A metformina não foi teratogênica em ratos e coelhos com doses de até 600 mg/kg/dia. Essa exposição representa cerca de 2-6 vezes a dose diária máxima recomendada para humanos de 2.000 mg, com base nas comparações da área de superfície corporal de ratos e coelhos, respectivamente. A determinação das concentrações fetais demonstraram barreira placentária parcial à metformina.
Lactação
Não foram conduzidos estudos em animais lactantes com os componentes combinados de JANUMET®. Em estudos conduzidos com os componentes individuais, a sitagliptina e a metformina foram excretados no leite de ratas lactantes. Não se sabe se a sitagliptina e/ou a metformina são excretadas no leite humano. Portanto, JANUMET® não deve ser utilizado por uma mulher que esteja amamentando.
USO EM IDOSOS, CRIANÇAS E OUTROS GRUPOS DE RISCO
Uso Pediátrico
A segurança e a eficácia de JANUMET® em pacientes pediátricos não foram estabelecidas.
Uso em Idosos
Uma vez que a sitagliptina e a metformina são substancialmente excretadas pelos rins e porque o envelhecimento pode estar associado com a redução da função renal, JANUMET® deve ser usado com cautela à medida que a idade aumenta. A seleção das doses deve ser feita com cautela e baseada no monitoramento cuidadoso e regular da função renal (veja ADVERTÊNCIAS, Monitoramento da Função Renal).
Fosfato de sitagliptina
Nos estudos clínicos, a segurança e a eficácia de JANUMET® em idosos (65 anos de idade, N= 409) foram comparáveis às observadas em pacientes mais jovens ( < 65 anos de idade).
Cloridrato de metformina
Estudos clínicos controlados com a metformina não incluíram número suficiente de pacientes idosos para determinar se eles respondem diferentemente dos pacientes mais jovens, embora outros experimentos clínicos relatados não tenham mostrado diferenças na resposta entre pacientes idosos e mais jovens. Sabe-se que a metformina é substancialmente excretada pelos rins e, uma vez que o risco de reações adversas graves com esse fármaco é maior em pacientes com função renal diminuída, a metformina deve ser utilizada somente por pacientes com função renal normal (veja CONTRA-INDICAÇÕES).
Insuficiência hepática
Fosfato de sitagliptina
Em pacientes com insuficiência hepática moderada (escore de Child-Pugh de 7 a 9), a AUC média e a Cmáx da sitagliptina aumentaram aproximadamente 21% e 13%, respectivamente, em comparação aos controles pareados saudáveis, após administração de uma dose única de 100 mg de fosfato