ISENTRESS™
MSD
raltegravir
Antiviral.
Apresentações.
ISENTRESS™ é apresentado na forma de comprimidos revestidos acondicionados em frascos com 60 comprimidos.
USO ORAL USO ADULTO E/OU PEDIÁTRICO (acima de 16 anos de idade)
Composição.
Ingrediente(s) Ativo(s). Cada comprimido revestido de ISENTRESS™ contém 434,4 mg de raltegravir potássico (como sal), equivalente a 400 mg de raltegravir (sem fenol). Ingredientes Inativos. Cada comprimido revestido de ISENTRESS™ contém os seguintes ingredientes inativos: celulose microcristalina, lactose monoidratada, fosfato de cálcio dibásico anidro, hipromelose 2208, poloxâmero 407 (contém 0,01% de hidroxitolueno butilado como antioxidante), estearil fumarato de sódio, estearato de magnésio. Além disso, o filme de revestimento contém os seguintes ingredientes inativos: álcool polivinílico, dióxido de titânio, polietileno glicol 3350, talco e óxido de ferro preto.
Indicações.
ISENTRESS™ é indicado em combinação com outros agentes anti-retrovirais para o tratamento de infecção pelo vírus da imunodeficiência humana (HIV-1).
Resultados de eficácia.
Descrição dos Estudos Clínicos
As evidências de eficácia duradoura do ISENTRESS™ baseiam-se nas análises dos dados de 48 semanas de 2 estudos randômicos, em andamento, duplo-cegos e controlados por placebo: BENCHMRK 1 e BENCHMRK 2 (Protocolos 018 e 019), em pacientes adultos infectados por HIV-1 já tratados com agentes anti-retrovirais e na análise dos dados de 48 semanas de um estudo em andamento, randômico, duplo-cego, controlado por agente ativo, STARTMRK (P021). Esses resultados de eficácia têm base na análise de 48 semanas de um estudo randômico, duplo-cego, controlado, de definição de intervalo de dose: protocolo 005, em pacientes adultos infectados pelo HIV-1 e já tratados com anti-retrovirais e na análise de 96 semanas de um estudo de definição de intervalo de dose, randômico, duplo-cego, controlado, Protocolo 004, de sujeitos adultos infectados pelo HIV-1 não tratados com anti-retroviral.
Pacientes Já Tratados
O BENCHMRK 1 e o BENCHMRK 2 são estudos Fase III para avaliar a segurança e a atividade anti-retroviral de ISENTRESS™ 400 mg 2x/dia em combinação com um Tratamento Otimizado de Base (OBT), versus a OBT isoladamente, em pacientes infectados pelo HIV, com 16 anos de idade ou mais, com resistência documentada a pelo menos 1 fármaco de cada uma das 3 classes (ITRNs, ITRNNs, IPs) de tratamento anti-retroviral. A distribuição randômica foi estratificada por grau de resistência ao IP (1 IP vs. > 1 IP) e uso de enfuvirtida na OBT. Antes da distribuição randômica, a OBT foi selecionada pelo pesquisador com base no teste de resistência genotípica/fenotípica e no histórico anterior de ART.
A tabela 1 mostra as características demográficas entre os pacientes do grupo que recebeu ISENTRESS™ 400 mg 2x/dia e os pacientes do grupo que recebeu placebo.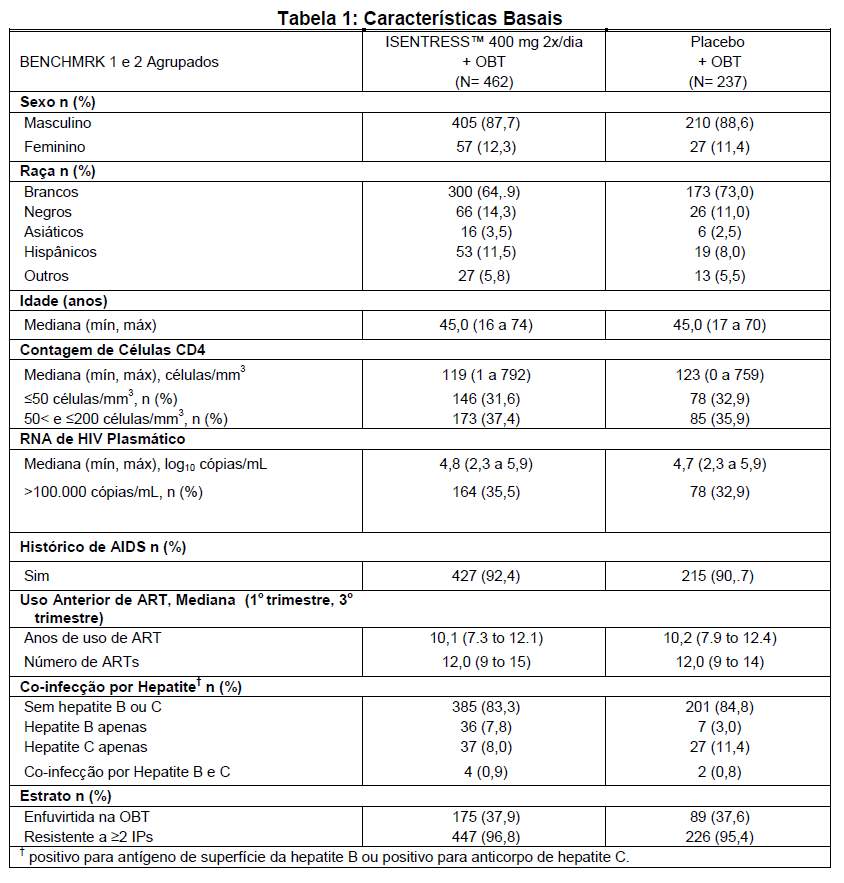
A tabela 2 compara as características dos pacientes sob Tratamento Otimizado de Base no período basal que receberam ISENTRESS™ 400 mg 2x/dia versus as características dos pacientes do grupo de controle.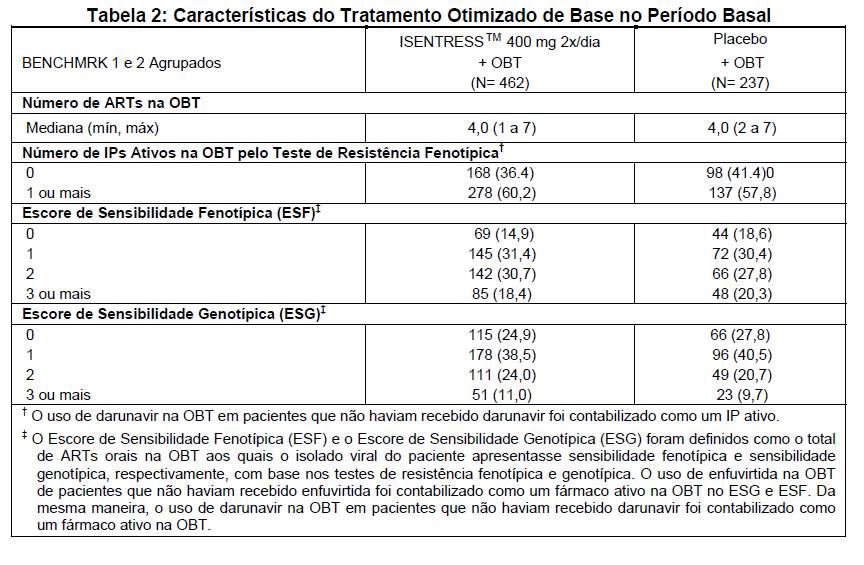
Os resultados da 48a semana para 699 pacientes distribuídos de modo randômico e tratados com a dose recomendada de 400 mg de ISENTRESS™ 2x/dia ou o agente comparador dos estudos BENCHMRK 1 e 2 agrupados são mostrados na tabela 3.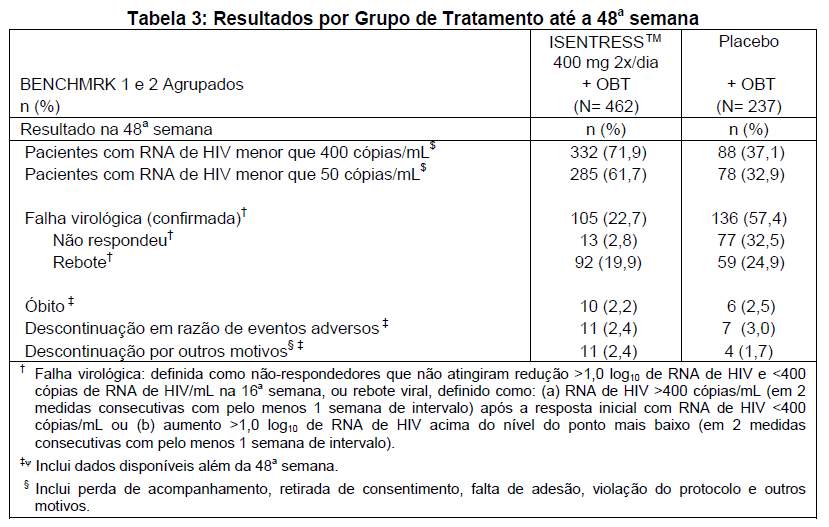
As alterações médias no RNA de HIV-1 plasmático em relação ao período basal foram de -1,71 log10 cópias/mL no grupo de ISENTRESS™ 400 mg 2x/dia e -0,78 log10 cópias/mL no grupo de controle. O aumento médio a partir do período basal na contagem de células CD4+ foi maior no grupo que recebeu ISENTRESS™ 400 mg 2x/dia (109 células/mm3) do que no grupo controle (45 células/mm3).
O percentual (IC 95%) de pacientes que atingiram RNA de HIV < 50 cópias/mL em função do tempo é apresentado na Figura 1.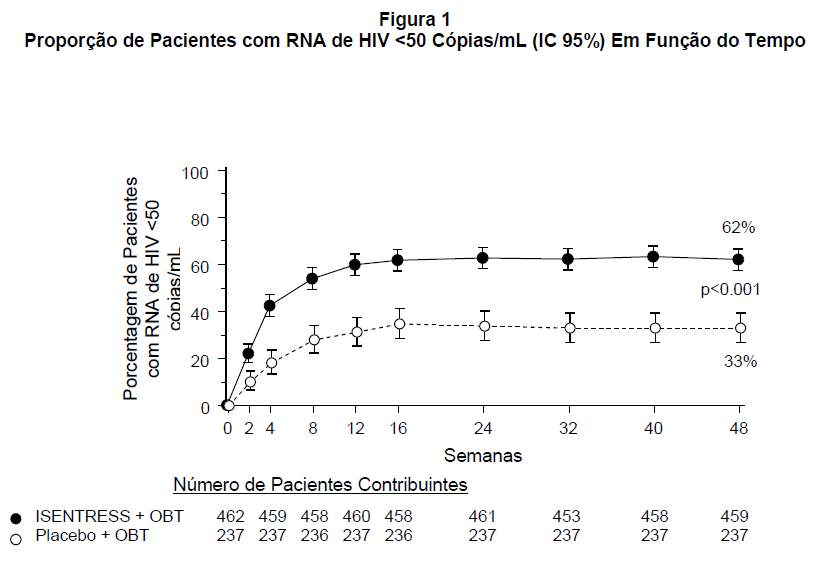
As respostas virológicas na 48a semana por escore de sensibilidade genotípica e fenotípica no período basal são mostradas na tabela 4.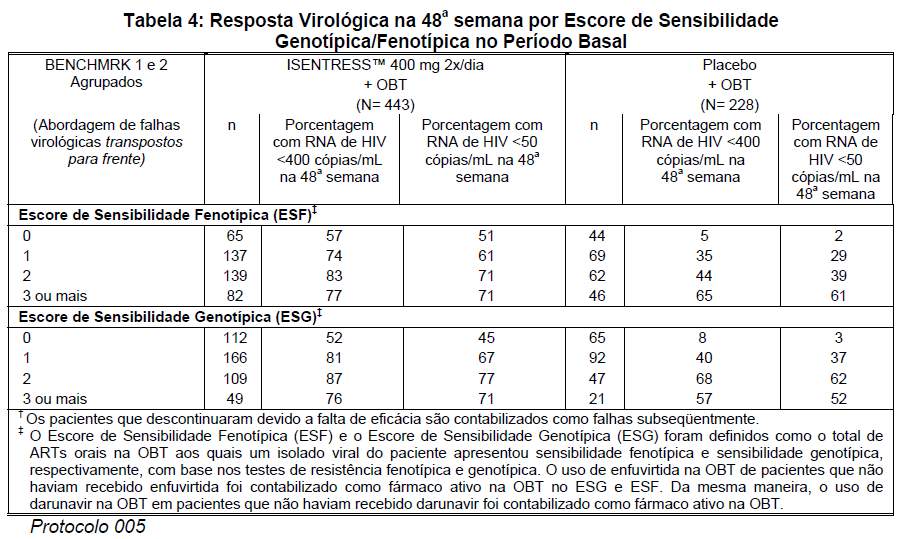
Dados adicionais sobre a eficácia de ISENTRESS™ 400 mg 2x/dia foram obtidos de pacientes que já haviam sido tratados e participaram do estudo randômico, duplo-cego, controlado, de definição de intervalo de dose, Protocolo 005, para avaliar ISENTRESS™ em combinação com OBT versus OBT isoladamente em pacientes infectados pelo HIV, com 18 anos ou mais. De um total de 178 pacientes distribuídos de maneira randômica e tratados, 45 receberam ISENTRESS™ 400 mg 2x/dia e 45 pacientes receberam placebo. Cerca de 36% dos pacientes receberam enfuvirtida na OBT, incluindo 25% que não haviam recebido enfuvirtida. Com exceção da enfuvirtida, não houve ART ativo na OBT de 72% desses pacientes com base nos testes de resistência genotípica (ESG= 0) e 48% com base nos testes de resistência fenotípica (ESF= 0). As respostas virológicas na 24ª e 48ª semana são mostradas na tabela 5.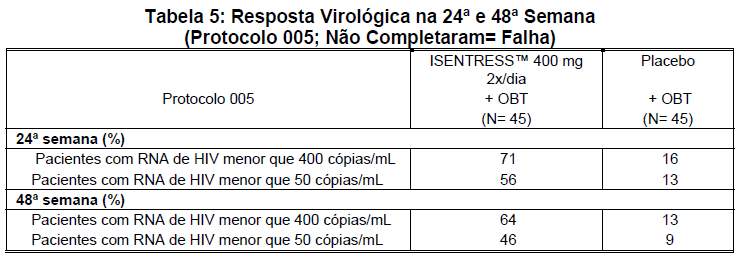
Pacientes Não Tratados Previamente
STARTMRK é um estudo Fase III para avaliar a segurança e a atividade anti-retroviral de ISENTRESS™ 400 mg 2x/dia + entricitabina (+) tenofovir versus efavirenz + entricitabina (+) tenofovir em pacientes infectados por HIV não tratados previamente e com RNA de HIV > 5000 cópias/mL. A distribuição randômica foi estratificada pelo nível de RNA de HIV na triagem (≤50.000 cópias/mL; e > 50.000 cópias/mL) e pela presença de hepatite.
A Tabela 6 mostra as características demográficas entre os pacientes do grupo que recebeu ISENTRESS™ 400 mg 2x/dia e os pacientes do grupo que recebeu efavirenz.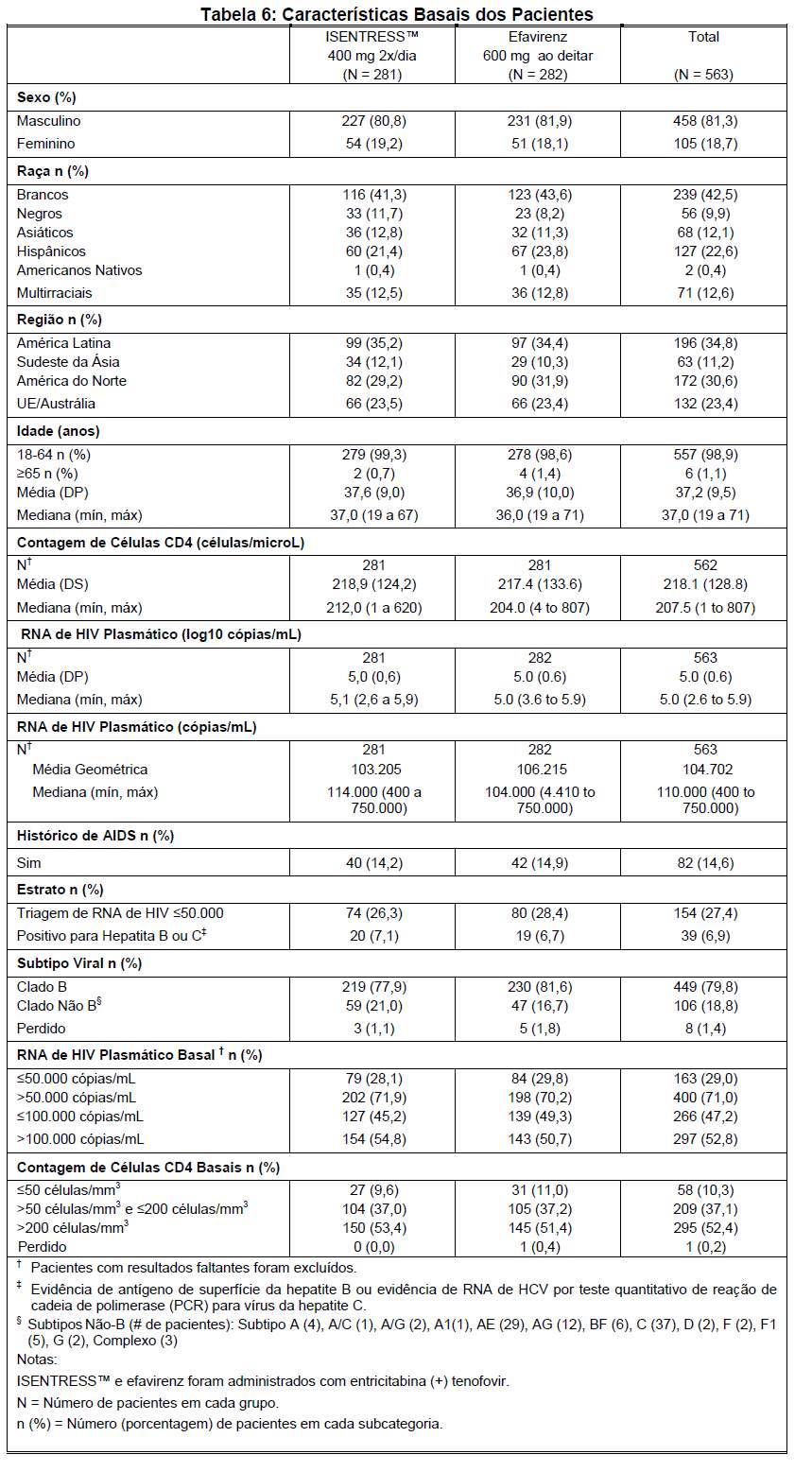
Em relação ao endpoint primário de eficácia, a proporção (%) de pacientes com RNA de HIV < 50 cópias/mL na 48ª semana foi de 241/280 (86,1%) no grupo que recebeu ISENTRESS™ e 230/281 (81,9%) no grupo que recebeu efavirenz. A diferença de tratamento (ISENTRESS™ - efavirenz) foi 4,2% a favor de ISENTRESS™, com IC de 95% associado de (-1,92, 10,32), estabelecendo que ISENTRESS™ é não-inferior ao efavirenz (valor de P para não-inferioridade < 0,001).
Os resultados da 48ª semana do STARTMRK são mostrados na Tabela 7.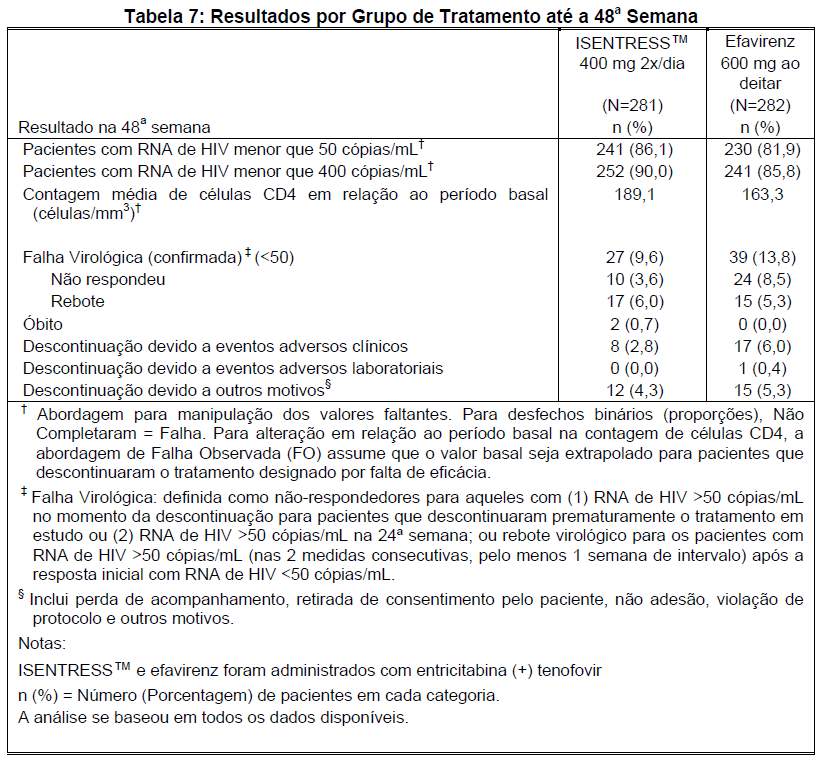
Eficácia pelos Subtipos Virais
Observou-se eficácia consistente de ISENTRESS™ em todos os subtipos de HIV com 90,3% (186/206) e 96,3% (52/54) dos pacientes com os subtipos B e não B, respectivamente, atingindo RNA de HIV < 50 cópias/mL na 48ª semana (abordagem FO).
A Figura 2 apresenta a proporção de pacientes com RNA de HIV plasmático < 50 cópias/mL em função do tempo por grupo de tratamento. Os pacientes que receberam ISENTRESS obtiveram supressão viral (RNA de HIV < 50 cópias/mL) mais cedo do que os que receberam EFV. Durante 48 semanas de tratamento, 86% do grupo que recebeu ISENTRESS™ 400 mg 2x/dia e 82% no grupo comparador, obteve RNA de HIV < 50 cópias/mL.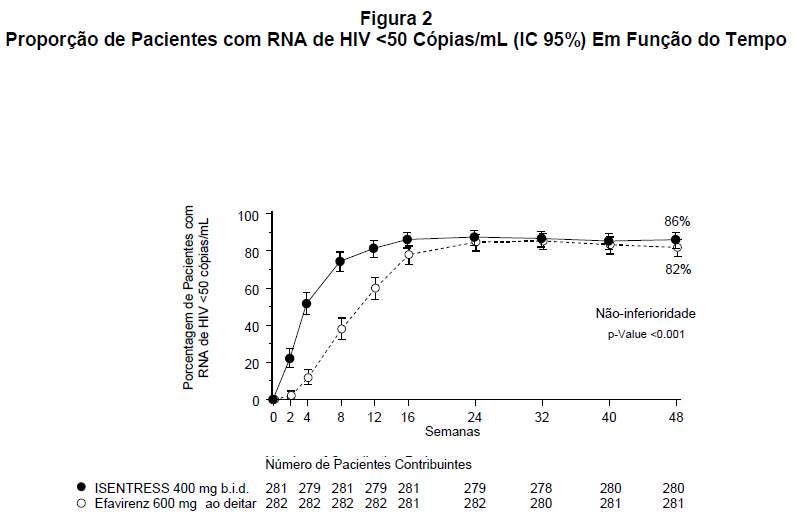
Os pacientes que receberam ISENTRESS™ atingiram supressão viral (RNA de HIV < 50 cópias/mL) mais cedo do que os que receberam efavirenz, ambos em combinação com entricitabina (+) tenofovir.
Os dados de eficácia prolongada de ISENTRESS™ 400 mg 2x/dia de até 96 semanas em pacientes não tratados previamente estão disponíveis no estudo de definição de dose Fase II (Protocolo 004). Durante 96 semanas de tratamento, 84% dos pacientes do grupo que recebeu ISENTRESS™ 400 mg 2x/dia mantiveram RNA de HIV-1 < 400 cópias/mL e 83% também mantiveram RNA de HIV-1 < 50 cópias/mL. O aumento médio em relação ao período basal na contagem de células CD4+ foi de +221 células/mm3 no grupo que recebeu ISENTRESS™.
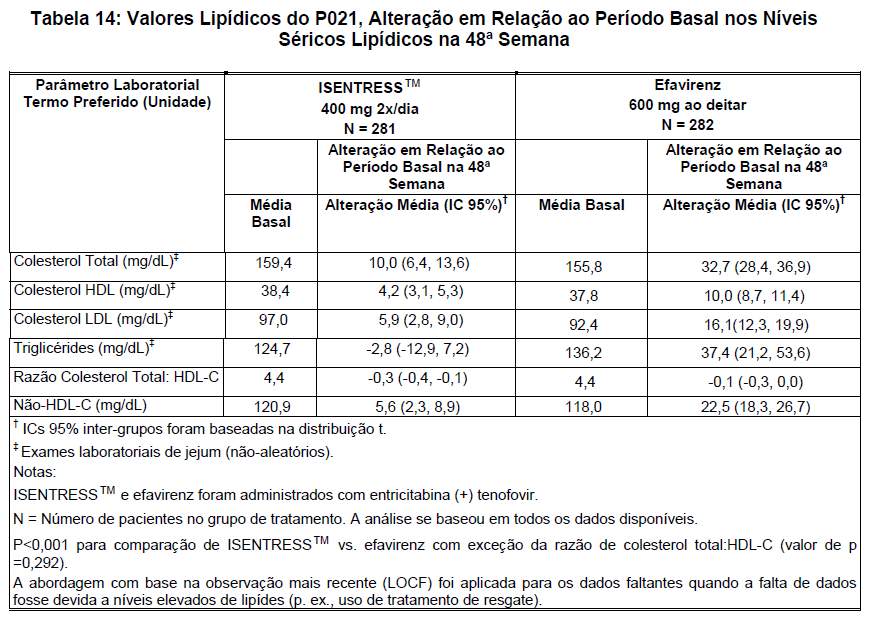
Durante 48 semanas de tratamento, ISENTRESS™ demonstrou efeitos mínimos sobre os lípides séricos, com pequenos aumentos dos níveis de colesterol total e de LDL-colesterol e redução nos níveis séricos de triglicérides. O grupo tratado com efavirenz apresentou alteração média significativamente maior em relação ao período basal dos níveis de colesterol total, triglicérides, colesterol não-HDL-C e LDL-C. Foram observados aumentos modestos de HDL em ambos os grupos, significativamente maiores para efavirenz (ACHADOS EM EXAMES LABORATORIAIS, Lipídeos, Alteração em Relação ao Período Basal).
Caract. farmacológicas.
CARACTERÍSTICAS FARMACOLÓGICAS
ISENTRESS™ é um inibidor da integrase, enzima responsável pela transferência do filamento de DNA viral do HIV, ativo contra o vírus da imunodeficiência humana (HIV-1).
FARMACOLOGIA CLÍNICA
Mecanismo de Ação
O raltegravir inibe a atividade catalítica da integrase do HIV, uma enzima codificada pelo HIV que é necessária para replicação viral. A inibição da integrase evita a inserção ou integração covalente do genoma do HIV no genoma da célula hospedeira durante a fase inicial da infecção. Os genomas do HIV que não conseguem se integrar, não conseguem dirigir a produção de novas partículas infecciosas virais e dessa forma, a inibição da integração impede a propagação da infecção viral. O raltegravir não inibiu de forma significativa as fosforiltransferases humanas, incluindo as polimerases a, b, e c do DNA.
Absorção
O raltegravir é rapidamente absorvido em jejum com um Tmáx de aproximadamente 3 horas após a dose. A AUC e a Cmáx do raltegravir aumentam de forma proporcional à dose no intervalo de dose de 100 mg a 1.600 mg. A C12h do raltegravir aumenta proporcionalmente com a dose no intervalo de dose de 100 a 800 mg e aumenta um pouco menos proporcionalmente à dose no intervalo de dose de 100 mg a 1.600 mg. Com a administração duas vezes ao dia, o estado de equilíbrio farmacocinético é atingido rapidamente, aproximadamente nos primeiros 2 dias de administração. Existe pouco ou nenhum acúmulo de AUC e de Cmáx e evidências de discreto acúmulo de C12h. A biodisponibilidade absoluta do raltegravir ainda não foi estabelecida.
Em pacientes que recebem monoterapia com 400 mg duas vezes ao dia, as exposições ao raltegravir foram caracterizadas por uma AUC0-12h média geométrica de 14,3 m•h e C12h de 142 nM.
Efeito da Presença de Alimentos sobre a Absorção Oral
ISENTRESS™ pode ser administrado com ou sem alimentos. O raltegravir foi administrado independentemente da presença de alimentos nos principais estudos de segurança e eficácia em pacientes HIV-positivos. Em voluntários sadios foi determinado, na farmacocinética de raltegravir, o efeito de refeições com baixo, moderado e alto teor de gorduras. A administração de doses mútiplas de raltegravir após uma refeição com teor moderado de gorduras não afetou a AUC do raltegravir em um grau clinicamente significativo com um aumento de 13% em relação ao jejum. A C12 h do raltegravir foi 66% maior e a Cmáx foi 5% maior após uma refeição com teor moderado de gorduras em comparação ao jejum. A administração de raltegravir após uma refeição rica em gorduras aumentou a AUC e a Cmáx do raltegravir em aproximadamente 2 vezes e a C12h em 4,1 vezes. A administração de raltegravir após uma refeição com baixo teor de gorduras diminuiu a AUC e a Cmáx em 46% e 52%, respectivamente; a C12h ficou basicamente inalterada. A presença de alimentos parece aumentar a variabilidade farmacocinética em relação ao jejum.
Distribuição
O raltegravir apresenta taxa de ligação a proteínas plasmáticas de aproximadamente 83% no intervalo de concentração de 2 a 10 mM. O raltegravir atravessa prontamente a placenta de ratas, porém não penetrou o cérebro em grau perceptível.
Metabolismo e Eliminação
A meia-vida terminal aparente do raltegravir é de aproximadamente 9 horas, com uma meia-vida de fase a mais curta (~1 hora) respondendo pela maior parte da AUC. Após a administração de uma dose oral de raltegravir radiomarcado, aproximadamente 51% e 32% da dose foram excretados nas fezes e urina, respectivamente. Nas fezes, apenas o raltegravir estava presente, e a maioria provavelmente era derivada da hidrólise do glicuronídeo de raltegravir na bile, conforme observado nas espécies pré-clínicas. Dois componentes, raltegravir e glicuronídeo de raltegravir, foram detectados na urina e responderam por cerca de 9% e 23% da dose, respectivamente. A principal entidade circulante era o raltegravir, representando aproximadamente 70% da radioatividade total; a radioatividade restante no plasma foi atribuída ao glicuronídeo de raltegravir. Estudos que utilizaram inibidores químicos seletivos da isoforma e as UDP-glicuronosiltransferases (UGT) expressas pelo cDNA mostram que a UGT1A1 é a principal enzima responsável pela formação do glicuronídeo de raltegravir. Assim, os dados indicam que o principal mecanismo de clearance do raltegravir em humanos é a glicuronidação mediada por UGT1A1.
Características em Pacientes
Gênero
Um estudo sobre a farmacocinética do raltegravir foi realizado em homens e mulheres jovens saudáveis. Além disso, o efeito do gênero na resposta ao tratamento foi avaliado em uma análise combinada dos dados farmacocinéticos de 103 indivíduos saudáveis e 28 pacientes com HIV que receberam monoterapia com raltegravir administrada em jejum. Essa hipótese também foi avaliada em uma análise da farmacocinética populacional (PK) dos dados de concentração de 80 indivíduos saudáveis e pacientes com HIV que recebem apenas raltegravir ou raltegravir em combinação com outros fármacos, na presença ou não de alimentos. Não houve nenhuma diferença farmacocinética clinicamente importante decorrente do gênero. Não é necessário nenhum ajuste de dose.
Idade
O efeito da idade sobre a farmacocinética do raltegravir foi avaliado na análise combinada e na análise de farmacocinética populacional. Não houve nenhum efeito clinicamente significativo decorrente da idade sobre a farmacocinética do raltegravir. Não é necessário ajuste de dose.
Pacientes Pediátricos
A farmacocinética do raltegravir em pacientes pediátricos com menos de 16 anos de idade não foi estabelecida.
Raça
O efeito da raça sobre a farmacocinética do raltegravir foi avaliado na análise combinada. Não houve efeito clinicamente significativo da raça sobre a farmacocinética do raltegravir. Não é necessário nenhum ajuste de dose.
Índice de Massa Corporal (IMC)
A análise combinada avaliou o efeito do IMC sobre a farmacocinética do raltegravir. Não houve efeito clinicamente significativo do IMC sobre a farmacocinética do raltegravir. Além disso, nenhum efeito clinicamente significativo do peso corporal sobre a farmacocinética do raltegravir foi identificado na análise da farmacocinética populacional. Não é necessário nenhum ajuste de dose.
Insuficiência Hepática
O raltegravir é eliminado principalmente por glicuronidação no fígado. Um estudo da farmacocinética do raltegravir foi realizado em pacientes com insuficiência hepática moderada. Além disso, a insuficiência hepática foi avaliada na análise farmacocinética combinada. Não houve diferenças farmacocinéticas clinicamente importantes entre os pacientes com insuficiência hepática moderada e os indivíduos saudáveis. Não é necessário nenhum ajuste de dose para pacientes com insuficiência hepática leve a moderada. O efeito da insuficiência hepática grave sobre a farmacocinética do raltegravir não foi estudado.
Insuficiência Renal
O clearance renal do fármaco inalterado é uma via de eliminação de menor importância. Um estudo da farmacocinética do raltegravir foi realizado em pacientes com insuficiência renal grave. Além disso, a insuficiência renal foi avaliada na análise farmacocinética combinada. Não houve diferenças farmacocinéticas clinicamente relevantes entre os pacientes com insuficiência renal grave e os indivíduos saudáveis. Não é necessário nenhum ajuste de dose. Como o grau em que o ISENTRESS™ pode ser dialisável é desconhecido, a administração prévia à uma sessão de diálise deve ser evitada.
Polimorfismo da UGT1A1
Não há evidências de que os polimorfismos comuns da UGT1A1 alterem a farmacocinética do raltegravir em um grau clinicamente significativo. Em uma comparação de 7 indivíduos com genótipo *28/*28 (associado a atividade reduzida da UGT1A1) com 27 indivíduos com genótipo do tipo selvagem, a razão da média geométrica (IC 90%) da AUC foi de 1,41 (0, 96; 2,09).
Farmacodinâmica
Microbiologia
O raltegravir em concentrações de 31 ± 20 nM resultou em inibição de 95% (CI95) da disseminação viral (em relação a uma cultura não tratada infectada por vírus) em culturas de células linfóides T humanas infectadas com a linhagem de célula adaptada HIV-1 variante H9IIIB. Além disso, o raltegravir em concentrações de 6 a 50 nM resultou em inibição de 95% da disseminação viral em culturas de células mononucleares de sangue periférico humano ativadas por mitógeno infectadas por isolados clínicos primários diversos de HIV-1, incluindo isolados de 5 subtipos não-B, e isolados resistentes a inibidores da transcriptase reversa e a inibidores da protease. Em um ensaio de infecção de ciclo único, o raltegravir inibiu a infecção de 23 isolados de HIV representando 5 subtipos não-B e 5 formas recombinantes circulantes com valores de CI50 variando de 5 a 12 nM. O raltegravir também inibiu a replicação de um isolado de HIV-2 quando testado em células CEMx174 (CI95= 6 nM). Foi observada atividade anti-retroviral aditiva a sinérgica quando células linfóides T humanas infectadas com H9IIIB variante do HIV-1 foram incubadas com o raltegravir em combinação com inibidores nucleosídeos análogos da transcriptase reversa (zidovudina, zalcitabina, estavudina, abacavir, tenofovir, didanosina ou lamivudina); inibidores não-nucleosídeos da transcriptase reversa (efavirenz, nevirapina ou delavirdina); inibidores da protease (indinavir, saquinavir, ritonavir, amprenavir, lopinavir, nelfinavir ou atazanavir); ou com o inibidor de entrada do HIV em células (enfuvirtida).
Resistência ao Medicamento
As mutações observadas na HIV-1 integrase que contribuíram para a resistência ao raltegravir (desenvolvida tanto in vitro como em pacientes tratados com o raltegravir) geralmente incluíram substituição tanto em Q148 (alterado para H, K, ou R) como em N155 (alterado para H) acrescida de uma ou mais mutações adicionais (por exemplo, L74I/M, E92Q, E138A/K, G140A/S, ou V151I). A substituição aminoácida no Y143C/H/R é outra via para resistência ao raltegravir.
Os vírus recombinantes com uma única mutação primária (Q148H, K ou R, ou N155H) apresentaram capacidade de replicação diminuída e sensibilidade reduzida ao raltegravir in vitro. As mutações secundárias diminuem ainda mais a sensibilidade ao raltegravir e algumas vezes agiram como mutações compensatórias para capacidade de replicação viral.
Eletrofisiologia Cardíaca
Em um estudo randômico, controlado com placebo e cruzado, 31 indivíduos saudáveis receberam uma dose única oral supraterapêutica de 1.600 mg de raltegravir e placebo. Não houve efeito sobre o intervalo QTc. Os picos das concentrações plasmáticas de raltegravir foram aproximadamente 4 vezes mais altos dos que os picos das concentrações após uma dose de 400 mg.
Contraindicações.
ISENTRESS™ é contra-indicado para pacientes com hipersensibilidade conhecida a qualquer componente deste produto.
Advertências e precauções.
Gravidez Categoria de risco: C
Este medicamento não deve ser utilizado por mulheres grávidas ou que possam ficar grávidas durante o tratamento.
Os estudos sobre desenvolvimento de toxicidade foram realizados em coelhos (em doses de até 1.000 mg/kg/dia) e ratos (em doses de até 600 mg/kg/dia). As doses mais altas nestes estudos produziram exposições sistêmicas nessas espécies aproximadamente 3 a 4 vezes acima da exposição à dose recomendada para humanos. Não foram observadas alterações externas, viscerais ou esqueléticas relacionadas ao tratamento em coelhos. Foram observados aumentos relacionados ao tratamento em relação aos controles na incidência de costelas supranumerárias em ratos na dose de 600 mg/kg/dia (exposições 4,4 vezes acima da exposição à dose humana recomendada). Tanto em coelhos como em ratos, não se observou efeitos relacionados ao tratamento na sobrevida embrio/fetal ou no peso dos fetos.
Em ratos, a dose administrada a fêmeas prenhes foi de 600 mg/kg/dia, e as concentrações plasmáticas médias do fármaco no plasma de fetos foram aproximadamente 1,5 a 2,5 vezes maiores que no plasma materno 1 hora e 24 horas pós-dose, respectivamente. Em coelhos, uma dose de 1.000 mg/kg/dia foi administrada a fêmeas prenhes, e as concentrações médias do fármaco no plasma fetal foram aproximadamente 2% da concentração materna média tanto 1 como 24 horas após a dose. Estudos toxicocinéticos demonstraram transferência placentária do fármaco em ambas as espécies.
Não existem estudos adequados e bem-controlados em mulheres grávidas; portanto, a segurança do ISENTRESS™ em mulheres grávidas não é conhecida. A exemplo de outros agentes anti-retrovirais, o uso de ISENTRESS™ durante a gravidez não é recomendado.
Lactação
Não se sabe se o raltegravir é secretado no leite humano. No entanto, o raltegravir é secretado no leite de ratas lactantes. Em ratas que receberam doses de 600 mg/kg/dia, as concentrações médias do fármaco no leite foram aproximadamente 3 vezes maiores do que no plasma materno. A amamentação não é recomendada durante o tratamento com ISENTRESS™. Além disso, recomenda-se que mães infectadas pelo HIV não amamentem seus bebês para evitar o risco de transmissão pós-natal do HIV.
PRECAUÇÕES
Síndrome de Reconstituição Imunológica
Durante a fase inicial do tratamento, os pacientes que respondem ao tratamento anti-retroviral podem desenvolver resposta inflamatória a infecções oportunistas indolentes ou residuais (como o complexo Mycobacterium avium, citomegalovírus, pneumonia por Pneumocystis jiroveci e tuberculose, ou reativação do vírus de varicela zoster), que podem precisar de avaliação e tratamento adicionais.
Interações Medicamentosas
Deve-se ter cautela ao se co-administrar ISENTRESS™ com fortes indutores da uridina difosfato glicuronosiltransferase (UGT) 1A1 (p. ex., rifampicina) em razão da redução de concentração plasmática do raltegravir (veja INTERAÇÕES MEDICAMENTOSAS).
USO EM IDOSOS, CRIANÇAS E OUTROS GRUPOS DE RISCO
Uso Pediátrico
A segurança e a eficácia de ISENTRESS™ ainda não foram estabelecidas em crianças com menos de 16 anos de idade.
Uso em Idosos
Estudos clínicos com ISENTRESS™ não incluíram número suficiente de pacientes com 65 anos ou mais para determinar se eles respondem de forma diferente de pacientes mais jovens. Outras experiências clínicas relatadas não identificaram diferença de resposta entre pacientes idosos e jovens. Em geral, a seleção da dose para um paciente idoso deve ser feita com cautela, refletindo a freqüência mais alta de insuficiência hepática, renal ou cardíaca e de doenças concomitantes ou outros tratamentos medicamentosos.
Interações medicamentosas.
O raltegravir não é um substrato das enzimas do citocromo P450 (CYP) e não inibe (CI50 > 100 mM) a CYP1A2, a CYP2B6, a CYP2C8, a CYP2C9, a CYP2C19, a CYP2D6 ou a CYP3A in vitro. Além disso, in vitro, o raltegravir não induziu a CYP3A4. Um estudo de interação medicamentosa do midazolam confirmou a baixa propensão do raltegravir para alterar a farmacocinética dos agentes metabolizados pela CYP3A4 in vivo pela demonstração da falta de efeito significativo do raltegravir sobre a farmacocinética do midazolam, um substrato sensível à CYP3A4.
Da mesma maneira, o raltegravir não é um inibidor (CI50 > 50 mM) das UDP-glicuronosiltransferases (UGTs) testadas (UGT1A1, UGT2B7) e não inibe o transporte mediado pela P-glicoproteína. Com base nesses dados, não se espera que ISENTRESS™ afete a farmacocinética dos medicamentos substratos dessas enzimas ou da P-glicoproteína (por exemplo, inibidores da protease, ITRNNs, metadona, analgésicos opióides, vastatinas, antifúngicos azóis, inibidores da bomba de próton e agente[s] para o tratamento da disfunção erétil).
Com base nos estudos in vivo e in vitro, o raltegravir é eliminado principalmente pelo metabolismo via glicuronidação mediada pela UGT1A1.
A co-administração de ISENTRESS™ com medicamentos que são potentes indutores da UGT1A1, como a rifampicina (indutor de várias enzimas metabolizantes de fármacos), reduz as concentrações plasmáticas de ISENTRESS™. Deve-se ter cuidado ao se coadministrar ISENTRESS™ com a rifampicina ou outros fortes indutores da UGT1A1 (veja PRECAUÇÕES). O impacto de outros potentes indutores de enzimas metabolizadoras de fármacos, como fenitoína e fenobarbital, sobre a UGT1A1 é desconhecido. Outros indutores menos potentes (por exemplo, efavirenz, nevirapina, rifabutina, glicocorticóides, erva-de-são-joão, pioglitazona) podem ser utilizados com a dose recomendada de ISENTRESS™.
A co-administração de ISENTRESS™ com medicamentos conhecidos por serem potentes inibidores da UGT1A1 (por exemplo, atazanavir) aumenta os níveis plasmáticos de ISENTRESS™. No entanto, o aumento é discreto e o tratamento combinado com esses inibidores foi bem tolerado nos estudos clínicos, de forma que nenhum ajuste de dose é necessário.
A co-administração de ISENTRESS™ com fármacos conhecidos por aumentar o pH gástrico (p.ex., omeprazol) pode aumentar os níveis plasmáticos de ISENTRESS™ com base no aumento da solubilidade de ISENTRESS™ em um pH mais alto. Em sujeitos que receberam ISENTRESS™ em combinação com os inibidores da bomba de prótons ou bloqueadores de H2 nos Protocolos 018 e 019, perfis de seguranças comparáveis foram observados nesse subgrupo em relação aos sujeitos que não receberam inibidores da bomba de prótons ou bloqueadores de H2. Com base nestes dados, os inibidores da bomba de prótons e os bloqueadores de H2 podem ser coadministrados com ISENTRESS™ sem ajuste de dose.
Efeito do Raltegravir sobre a Farmacocinética de Outros Agentes
Nos estudos de interação medicamentosa, o raltegravir não apresentou efeitos clinicamente significativos sobre a farmacocinética de: contraceptivos hormonais, tenofovir, midazolam e lamivudina. Em um estudo de interação medicamentosa de doses múltiplas, os valores de AUC do etinil estradiol e de norelgestromina foram de 98% e 114%, respectivamente, quando coadministrados com raltegravir em comparação com quando co-administrados sem raltegravir. Em um estudo de interação medicamentosa de doses múltiplas, a AUC e as concentrações de vale do tenofovir quando co-administrado com o raltegravir foram de 90% e 87% dos valores obtidos com a monoterapia com o tenofovir. Em outro estudo de interação medicamentosa, a AUC do midazolam co-administrado foi 92% do valor obtido com o midazolam isoladamente. Em um estudo de Fase II, a farmacocinética da lamivudina foi semelhante em pacientes que receberam combinações com raltegravir versus com efavirenz.
Efeito de Outros Agentes sobre a Farmacocinética do Raltegravir
Nos estudos de interação medicamentosa, atazanavir, efavirenz, ritonavir, tenofovir e tipranavir/ritonavir não apresentaram efeito clinicamente significativo sobre a farmacocinética do raltegravir. A rifampicina, forte indutora das enzimas metabolizadoras de medicamentos, causou redução dos níveis de vale do raltegravir. As interações medicamentosas encontram-se descritas adicionalmente na tabela 8.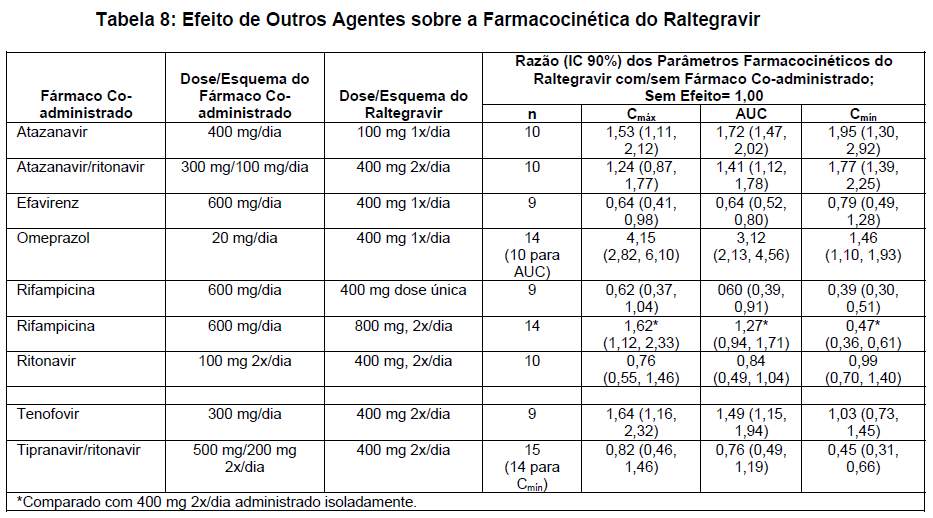
Cuidados de armazenamento.
ISENTRESS™ deve ser armazenado entre 15 e 30°C.
Não tome este medicamento após a expiração da data de validade impressa na embalagem.
TODO MEDICAMENTO DEVE SER MANTIDO FORA DO ALCANCE DAS CRIANÇAS.
Posologia e modo de usar.
Para o tratamento de pacientes com infecção por HIV-1, a posologia de ISENTRESS™ é de 400 mg administrados por via oral, duas vezes ao dia, com ou sem alimentos. ISENTRESS™ deve ser administrado em combinação com outros agentes anti-retrovirais.
Reações adversas.
Reações Adversas Em Pacientes Já Tratados
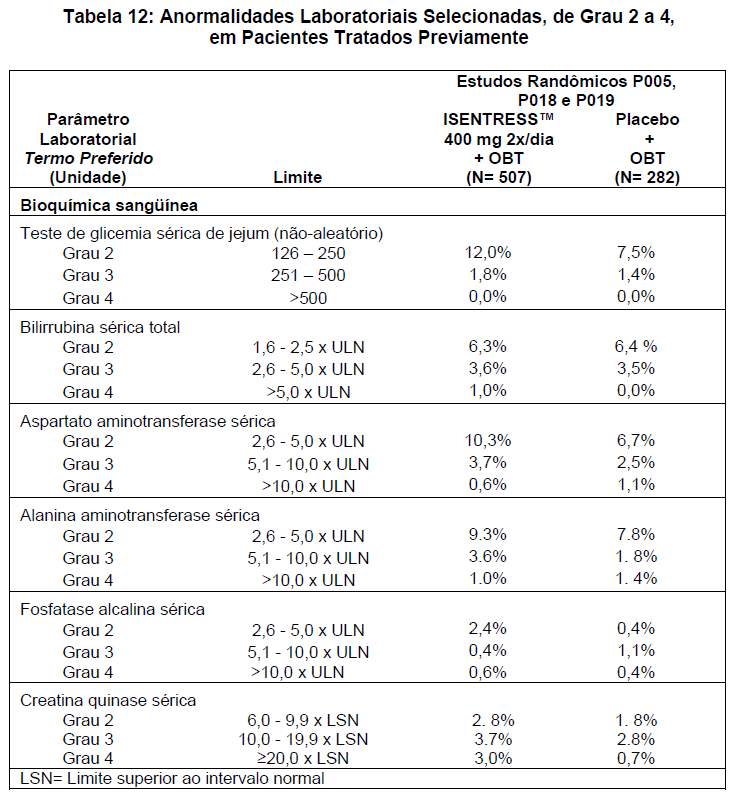
A avaliação da segurança de ISENTRESS™ baseou-se nos relatos de eventos adversos em pacientes que já haviam recebido tratamento desde os estudos clínicos randômicos P005, P018 e P019 nos quais foi utilizada a dose recomendada de ISENTRESS™, 400 mg duas vezes ao dia, em combinação com o Tratamento Otimizado de Base (OBT) em 507 pacientes, em comparação com 282 pacientes que receberam placebo em combinação com OBT. Durante o tratamento duplo-cego, o acompanhamento total foi de 702,8 pacientes-ano do grupo ISENTRESS™ 400 mg 2x/dia e 257,1 pacientes-ano do grupo placebo.
Entre os pacientes do braço de ISENTRESS™ 400 mg 2x/dia + OBT (acompanhamento médio de 72,3 semanas) e do braço para comparação, no qual foi administrado placebo + OBT (acompanhamento médio de 47,6 semanas), na análise agrupada para os estudos P005, P018 e P019, os eventos adversos mais comumente relatados ( > 10% em qualquer um dos grupos) de todas as intensidades e independentemente da causalidade foram: diarréia em 20,3% e 21,3%, náuseas em 12,2% e 15,6%, cefaléia em 10,8% e 7,8%, pirexia em 7,7% e 11,7% dos pacientes, respectivamente. Nessa análise agrupada, a taxa de descontinuação do tratamento em razão de eventos adversos foi de 2,6% entre os pacientes que receberam ISENTRESS™ + OBT e de 3,2% entre os pacientes que receberam placebo + OBT.
Eventos Adversos Relacionados ao Medicamento
Os eventos adversos clínicos listados abaixo foram considerados de intensidade moderada a grave pelos pesquisadores. A causa de tais eventos pode ser atribuída a qualquer um dos medicamentos do esquema de combinação (ISENTRESS™/placebo apenas ou em combinação com OBT, ou OBT apenas). Os eventos adversos de intensidade moderada a grave e relacionados ao medicamento que ocorreram em ≥2% dos pacientes adultos que já receberam tratamento são apresentados na tabela 9.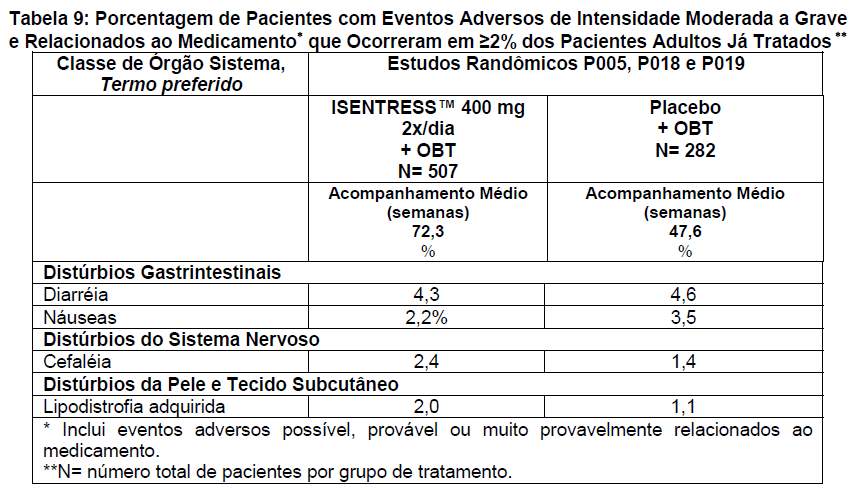
Os eventos adversos de intensidade moderada a grave e relacionados ao medicamento, que ocorreram em menos de 2% dos pacientes já tratados (n= 507) e que haviam recebido ISENTRESS™ + OBT são listados abaixo, por classe de órgão sistema.
[Comuns (≥1/100, < 1/10), Incomuns (≥1/1,000, < 1/100)]
Distúrbios do Sangue e Sistema Linfático
Incomuns: anemia, anemia macrocítica, neutropenia.
Distúrbios Cardíacos
Incomuns: infarto do miocárdio, palpitações, extra-sístoles ventriculares.
Distúrbios do Ouvido e Labirinto
Incomuns: vertigem.
Distúrbios Oculares
Incomuns: distúrbio visual.
Distúrbios Gastrintestinais
Comuns: dor abdominal, vômitos.
Incomuns: distensão abdominal, dor abdominal alta, constipação, dor gastrintestinal, desconforto abdominal, dispepsia, flatulência, gastrite, glossite, doença de refluxo gastroesofágico, boca seca.
Distúrbios Gerais e Condições no Local da Administração
Comuns: astenia, fadiga.
Incomuns: pirexia, desconforto torácico, calafrios, sensação de calor, irritabilidade, intolerância a medicamento, edema de face, liponecrose.
Distúrbios Hepatobiliares
Incomuns: hepatite, hepatomegalia,hiperbilirrubinemia.
Distúrbios do Sistema Imunológico
Incomuns: hipersensibilidade ao medicamento, hipersensibilidade.
Infecções e Infestações
Incomuns: celulite, herpes simplex, herpes genital.
Investigação
Incomuns: redução de peso, aumento de peso.
Lesão, Envenenamento e Complicações de Procedimentos
Incomuns: toxicidade medicamentosa, fratura de compressão
Distúrbios Metabólicos e Nutricionais
Incomuns: diabetes mellitus, obesidade central, dislipidemia, hiperlactacidemia, hiperlipidemia, hipertrigliceridemia, aumento do apetite, diminuição do apetite, lipomatose.
Distúrbios Musculoesqueléticos e do Tecido Conjuntivo
Comuns: artralgia
Incomuns: mialgia, dor nas extremidades, lombalgia, espasmos musculares, dor musculoesquelética, miosite, atrofia muscular, amiotrofia, osteoporose.
Distúrbios do Sistema Nervoso
Comuns: tontura.
Incomuns: neuropatia periférica, alodinia, neuropatia, parestesia polineuropatia, sonolência, cefaléia tensional, tremor, neuropatia sensorial periférica.
Distúrbios Psiquiátricos
Incomuns: depressão, insônia, sonhos anormais, ansiedade.
Distúrbios Renais e Urinários
Incomuns: nefropatia tóxica, síndrome nefrótica, nefrite, nefrite intersticial, nefrolitíase, noctúria, polaciúria, insuficiência renal, insuficiência renal aguda, insuficiência renal crônica, comprometimento renal, necrose tubular renal.
Distúrbios do Sistema Reprodutivo e da Mama
Incomuns: disfunção erétil, ginecomastia.
Distúrbios Respiratórios, Torácicos e do Mediastino
Incomuns: epístaxe.
Distúrbios da Pele e do Tecido Subcutâneo
Incomuns: erupção cutânea, hiperidrose, dermatite acneiforme, eritema, atrofia gordurosa, lipoatrofia, lipo-hipertrofia, sudorese noturna, erupção cutânea macular, erupção cutânea maculopapular, erupção cutânea prurítica, xerodermia, prurigo, emaciação facial.
Eventos Graves
Nos estudos clínicos P005, P018 e P019, foram relatados os seguintes eventos adversos graves relacionados ao medicamento: hipersensibilidade1, anemia, neutropenia, infarto do miocárdio, gastrite, hepatite, hipersensibilidade ao medicamento, nefropatia tóxica e insuficiência renal, herpes genital, superdosagem acidental, insuficiência renal aguda, insuficiência renal crônica e necrose tubular renal.
1 Foi observada hipersensibilidade em 2 pacientes com ISENTRESS™. O tratamento foi interrompido e após a reexposição os pacientes conseguiram retomar o medicamento.
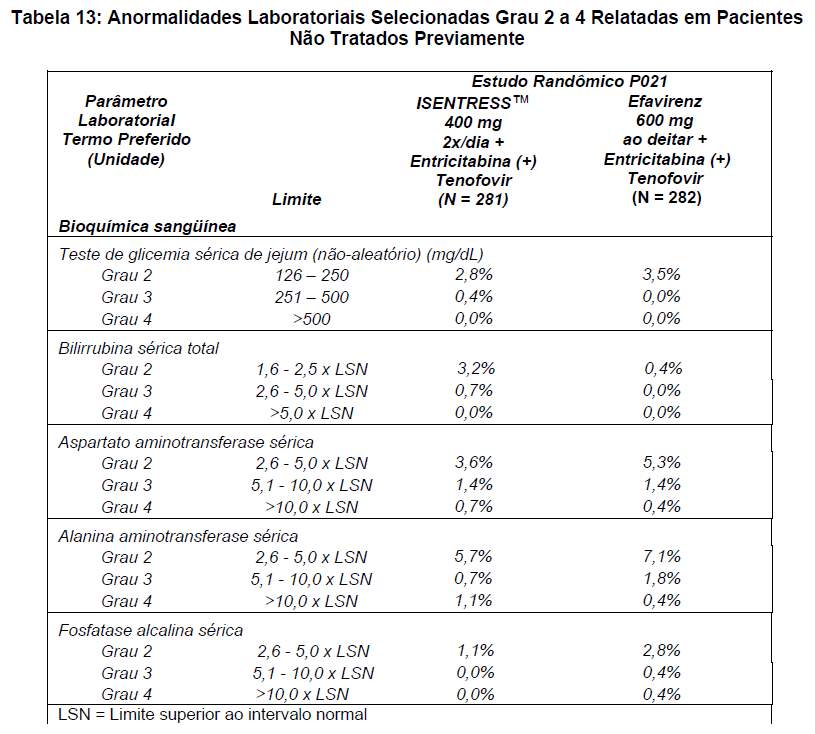
EVENTOS ADVERSOS EM PACIENTES NÃO TRATADOS PREVIAMENTE
A seguinte avaliação da segurança de ISENTRESS™ em pacientes não tratados previamente se baseou no estudo randômico, duplo-cego, controlado com agente ativo que envolveu pacientes não tratados previamente, protocolo 021 (STARTMRK) com ISENTRESS™ 400 mg 2x/dia em combinação com uma dose fixa de entricitabina 200 mg (+) tenofovir 245 mg, (N=281) versus efavirenz (EFV) 600 mg ao deitar em combinação com entricitabina (+) tenofovir (N=282). Durante o tratamento duplo-cego, o acompanhamento total para pacientes com ISENTRESS™ 400 mg 2x/dia + entricitabina (+) tenofovir foi de 333 pacientes-ano e 317 pacientes-ano para pacientes com efavirenz 600 mg ao deitar + entricitabina (+) tenofovir.
O número (%) de pacientes com eventos adversos e eventos adversos relacionados ao medicamento no grupo que recebeu ISENTRESS™ foi significativamente menos freqüente que o do grupo que recebeu efavirenz com base nos valores de p nominais (0,002 e < 0,001, respectivamente). Neste estudo, as taxas de descontinuação do tratamento por eventos adversos foram de 3,2% entre os pacientes que receberam ISENTRESS™ + entricitabina (+) tenofovir e 6,4% nos pacientes entre os que receberam efavirenz + entricitabina (+) tenofovir.
Para os pacientes do grupo que recebeu ISENTRESS™ 400 mg 2x/dia + entricitabina (+) tenofovir e do grupo que recebeu o agente comparativo, efavirenz 600 mg ao deitar + entricitabina (+) tenofovir, os eventos adversos mais comumente relatados ( > 10% em qualquer grupo), de todas as intensidades e independentemente da causalidade, são mostrados na Tabela 10.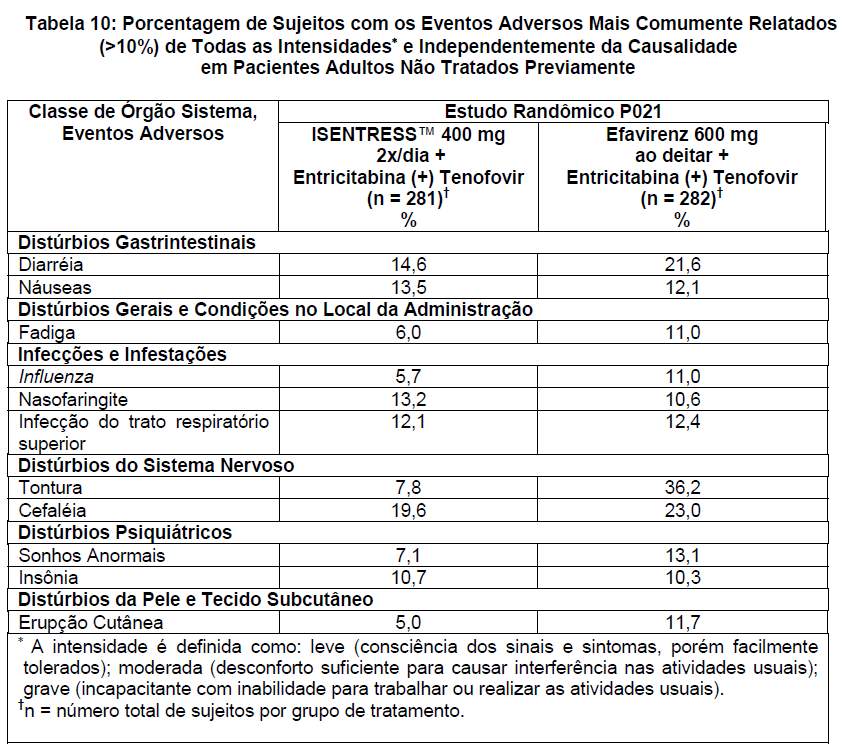
Eventos do SNC
Em pacientes não tratados previamente (P021), os eventos no sistema nervoso central (SNC), medidos pela proporção de pacientes com 1 ou mais sintomas do SNC (descritos abaixo), foram relatados significativamente menos freqüentemente no grupo que recebeu ISENTRESS™ + entricitabina (+) tenofovir em comparação ao grupo que recebeu efavirenz + entricitabina (+) tenofovir, p < 0,001 e < 0,001 para eventos cumulativos até as semanas 8 e 48, respectivamente. No grupo que recebeu ISENTRESS™, a porcentagem de pacientes com 1 ou mais sintomas do SNC foi de 20,3% em comparação com 52,1% no grupo que recebeu efavirenz na 8ª semana e 26,0% em comparação com 58,5% na 48ª semana. Os eventos adversos do SNC para essa análise foram tontura, insônia, comprometimento da concentração, sonolência, depressão, pesadelos, estado confusional, idéias suicidas, distúrbio do sistema nervoso, distúrbio psicótico, sonhos anormais, tentativa suicida, psicose aguda, delírio, nível deprimido de consciência, alucinação, alucinação auditiva, suicídio consumado e depressão importante.
Eventos Adversos Relacionados ao Medicamento
Os pesquisadores consideraram as reações adversas clínicas listadas abaixo de intensidade moderada a grave e de relação causal com qualquer medicamento no regime de combinação (ISENTRESS™/efavirenz apenas ou em combinação com entricitabina (+) tenofovir, ou entricitabina (+) tenofovir apenas).
As reações adversas clínicas de intensidade moderada a grave que ocorreram em ≥2% nos pacientes adultos não tratados previamente estão apresentadas na Tabela 11.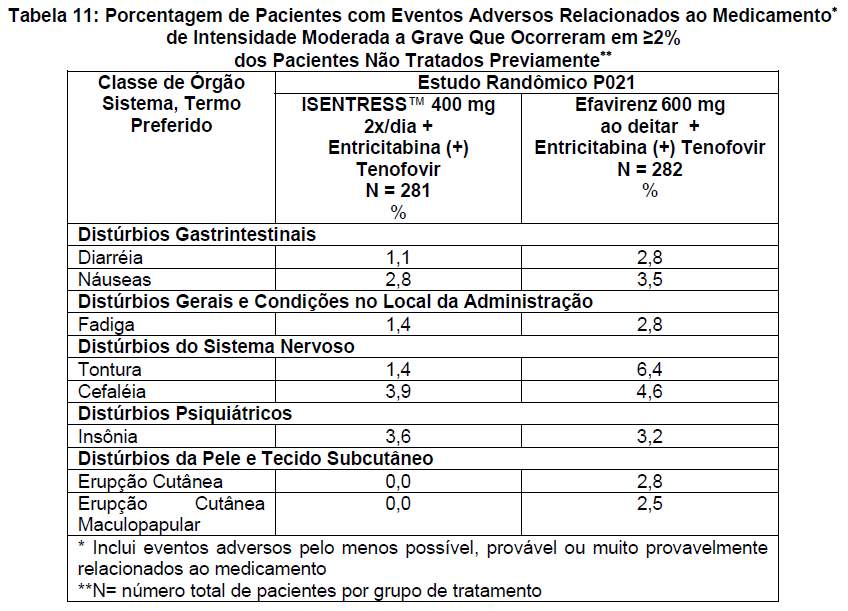
Eventos adversos relacionados ao medicamento, que ocorreram em menos de 2% dos pacientes não tratados previamente (n=281) que receberam ISENTRESS™ + entricitabina (+) tenofovir e de intensidade moderada a grave são listados abaixo por Classe de Órgão Sistema.
[Comuns (≥1/100, < 1/10), Incomuns (≥1/1.000, < 1/100)]
Distúrbios do Sangue e Sistema Linfático
Incomum: dor no linfonodo, neutropenia
Distúrbios do Ouvido e Labirinto
Incomuns: tinido, vertigem
Distúrbios Gastrintestinais
Incomuns: dor abdominal, vômitos, dor abdominal superior
Distúrbios Gerais e Condições no Local da Administração
Comuns: fadiga
Incomuns: astenia
Distúrbios do Sistema Imunológico
Incomuns: síndrome de reconstituição imunológica
Infecções e Infestações
Incomuns: herpes zoster, gastroenterite, foliculite
Distúrbios Metabólicos e Nutricionais
Incomuns: anorexia, diminuição do apetite
Distúrbios Musculoesqueléticos e do Tecido Conjuntivo
Incomuns: artrite, mialgia, espasmos musculares, torcicolo
Distúrbios do Sistema Nervoso
Comuns: tontura
Incomuns: hipersonia, sonolência, comprometimento da memória Distúrbios Psiquiátricos
Comuns: sonhos anormais, pesadelos
Incomuns: ansiedade, distúrbio mental, estado confusional
Distúrbios do Sistema Reprodutivo e da Mama
Incomuns: disfunção erétil
Distúrbios da Pele e Tecido Subcutâneo
Incomuns: acne, alopecia, lesão cutânea
Eventos Graves
Os seguintes eventos adversos graves relacionados ao medicamento foram relatados no estudo clínico P021 em pacientes não t