INVANZ®
MSD
ertapeném
Antibiótico de amplo espectro.
Apresentações.
Pó para Solução para Infusão Intravenosa ou Injeção Intramuscular.
Uso Intravenoso ou Intramuscular.
INVANZ® é apresentado em caixas com 1 frasco-ampola contendo 1,046 g de ertapeném sódico, equivalente a 1 g de ertapeném.
USO ADULTO E PEDIÁTRICO (crianças acima de 3 meses).
Composição.
Cada frasco-ampola de INVANZ® contém 1,046 g de ertapeném sódico como princípio ativo, equivalente a 1 g de ertapeném. Ingredientes inativos: Cada frasco-ampola de INVANZ® contém os seguintes ingredientes inativos: bicarbonato de sódio e hidróxido de sódio (para ajustar o pH).
Informações técnicas.
CARACTERÍSTICAS QUÍMICAS
INVANZ® é o sal monossódico do ácido [4R-[3(3S*,5S*),4a,56 (R*)]]-3-[[5-[[(3-carboxifenil)amino]carbonil]-3-pirrolidinil]tio]-6-(1-hidroxietil)-4-metil-7-oxo-1-azabiciclo[3.2.0]hept-2-ene-2-carboxílico.
A fórmula molecular do ertapeném sódico é C22H24N3O7SNa e a estrutural: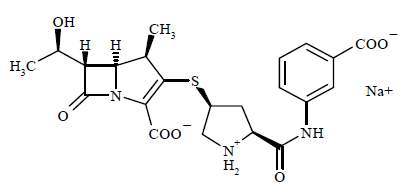
O ertapeném sódico é um pó branco a quase branco, higroscópico, pouco cristalino, com peso molecular de 497,50. É solúvel em água e em solução de cloreto de sódio a 0,9%, praticamente insolúvel em etanol e insolúvel em acetato de isopropila e tetraidrofurano.
INVANZ® é um pó liofilizado estéril para infusão intravenosa (IV) após reconstituição com um diluente apropriado (veja MODO DE USAR E CUIDADOS DE CONSERVAÇÃO DEPOIS DE ABERTO, INSTRUÇÕES DE USO) e diluição em 50 mL de cloreto de sódio a 0,9%, injetável ou, para injeção intramuscular (IM), após reconstituição em cloridrato de lidocaína a 1% ou 2% (sem epinefrina). Cada frasco-ampola contém 1,046 g de ertapeném sódico, equivalente a 1 g de ertapeném e 137 mg (aproximadamente 6,0 mEq) de sódio.
Indicações.
INVANZ® é indicado para o tratamento de pacientes com infecções moderadas a graves causadas por cepas sensíveis dos microorganismos e para o tratamento empírico inicial anterior à identificação do patógeno causador das infecções relacionadas a seguir:
- Infecções Intra-abdominais Complicadas;
- Infecções Complicadas de Pele e Anexos (incluindo pé diabético);
- Pneumonia Adquirida na Comunidade;
- Infecções Complicadas do Trato Urinário (incluindo pielonefrite);
- Infecções Pélvicas Agudas (incluindo endomiometrite pós-parto, aborto séptico e infecções ginecológicas pós-cirúrgicas);
- Septicemia Bacteriana.
Resultados de eficácia.
Pacientes Adultos
Infecções Intra-abdominais Complicadas
O ertapeném foi avaliado em 665 adultos com infecções intra-abdominais complicadas em um estudo clínico controlado, randômico, multicêntrico, duplo-cego, que comparou o ertapeném (1 g IV uma vez ao dia) com a associação piperacilina/tazobactama (3,375 g IV a cada 6 horas) durante 5 a 14 dias. No período basal, os pacientes foram estratificados em dois grupos: apendicite localizada complicada (estrato 1) e qualquer outra infecção intra-abdominal complicada, incluindo infecções do cólon, do intestino delgado, das vias biliares e peritonite generalizada (estrato 2). Uma a duas semanas após o tratamento, as taxas de sucesso clínico e microbiológico foram de 89,6% (190/212) para o ertapeném e de 82,7% (162/196) para a piperacilina/tazobactama; 4 a 6 semanas após o tratamento (teste de cura), as taxas de sucesso foram de 86,7% (176/203) para o ertapeném e de 81,3% (157/193) para a piperacilina/tazobactama. No teste de cura para os pacientes do estrato 1, as taxas de sucesso foram de 90,4% (85/94) para o ertapeném e de 90,1% (82/91) para a piperacilina/tazobactama e, para os pacientes do estrato 2, foram de 83,5% (91/109) para o ertapeném e de 73,5% (75/102) para a piperacilina/tazobactama. As taxas de sucesso clínico no teste de cura para cada patógeno entre os pacientes passíveis de avaliação microbiológica são apresentadas na tabela 4.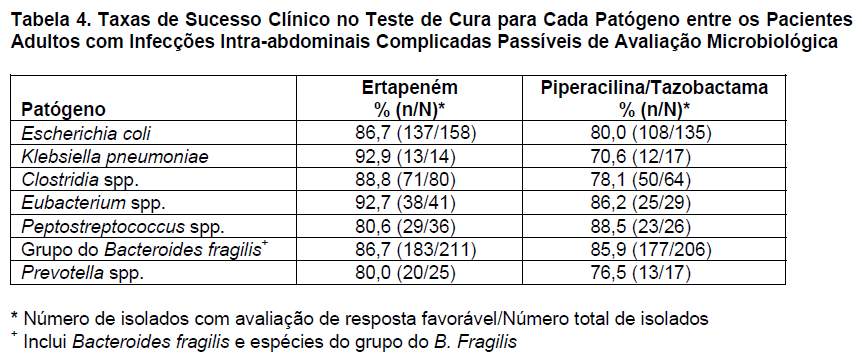
Em pacientes com bacteremia por E. coli, 100% (3/3) foram tratados com sucesso com o ertapeném.
Infecções Complicadas da Pele e Anexo - (incluindo pé diabético)
O ertapeném foi avaliado em 540 adultos com infecções complicadas de pele e anexos em um estudo clínico controlado, randômico, multicêntrico, duplo-cego, que comparou o ertapeném (1 g IV uma vez ao dia) com a associação piperacilina/tazobactama (3,375 g IV a cada 6 horas) durante 7 a 14 dias. Foram incluídos pacientes com pé diabético, abscesso profundo de tecido mole, infecção de ferida pós-trauma e celulite com drenagem purulenta. A taxa de sucesso clínico, 10 a 21 dias após o tratamento (teste de cura), foi de 82,2% (152/185) para o ertapeném e de 84,5% (147/174) para a piperacilina/tazobactama. As taxas de sucesso clínico, por tipo de infecção, para o ertapeném e para a piperacilina/tazobactama no teste de cura, respectivamente, foram: pé diabético, 65,7% (23/35) e 73,3% (22/30); abscesso profundo de tecido mole, 96,7% (29/30) e 94,4% (34/36); infecção de ferida pós-trauma, 83,3% (25/30) e 84,6% (22/26); e celulite com drenagem purulenta, 93,1% (27/29) e 87,5% (21/24). As taxas de sucesso clínico no teste de cura para cada patógeno entre os pacientes passíveis de avaliação microbiológica são apresentadas na tabela 5.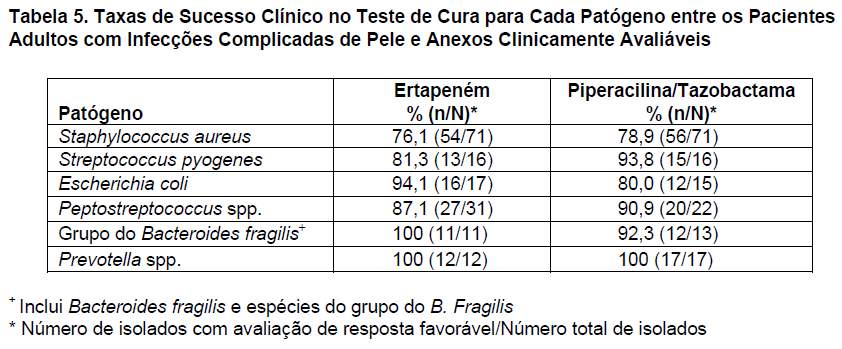
Pneumonia Adquirida na Comunidade
O ertapeném foi avaliado em 866 adultos com pneumonia adquirida na comunidade em dois estudos clínicos controlados, randômicos, multicêntricos, duplo-cegos, que compararam o ertapeném (1 g por via parenteral/dia) com a ceftriaxona (1 g por via parenteral/dia). Foi permitido trocar os antibióticos parenterais por amoxicilina/clavulanato por via oral para completar 10 a 14 dias de tratamento (parenteral e oral). As taxas de sucesso clínico (estudos agrupados) depois de 7 a 14 dias de tratamento (teste de cura) foram de 92,0% (335/364) para o ertapeném e de 91,8% (270/294) para a ceftriaxona. As taxas de sucesso clínico no teste de cura para cada patógeno entre os pacientes passíveis de avaliação microbiológica agrupados são apresentadas na tabela 6.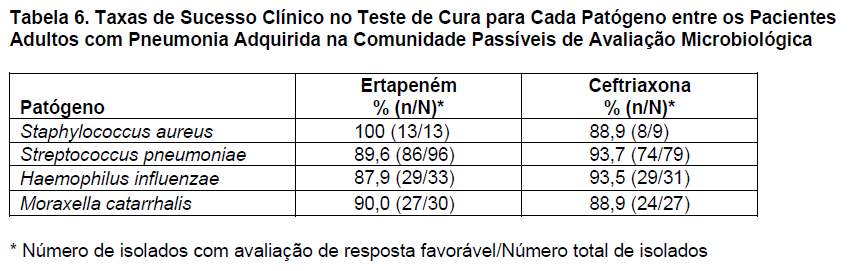
Dos pacientes com bacteremia por S. pneumoniae, 88,9% (16/18) foram tratados com sucesso com o ertapeném; e nenhum desses pacientes apresentava bacteremia persistente documentada.
Infecções Complicadas do Trato Urinário - (incluindo pielonefrite)
O ertapeném foi avaliado em 850 adultos com infecções complicadas do trato urinário (incluindo pielonefrite), em dois estudos clínicos controlados, randômicos, multicêntricos, duplo-cegos, que compararam o ertapeném (1 g por via parenteral/dia) com a ceftriaxona (1 g por via parenteral/dia). Foi permitido trocar os antibióticos parenterais por ciprofloxacino oral (500 mg 2 vezes ao dia) para 10 a 14 dias de tratamento (parenteral e oral). As taxas de sucesso microbiológico (estudos agrupados) depois de 5 a 9 dias de tratamento (teste de cura) foram de 89,5% (229/256) para o ertapeném e de 91,1% (204/224) para a ceftriaxona. No período basal, os pacientes foram estratificados em dois grupos: pielonefrite e qualquer outra infecção complicada do trato urinário. No estrato da pielonefrite, as taxas de sucesso microbiológico (estudos agrupados) foram de 91,3% (116/127) para o ertapeném e de 93,4% (99/106) para a ceftriaxona. As taxas de erradicação (combinadas) no teste de cura para cada patógeno entre os pacientes passíveis de avaliação microbiológica são apresentadas na tabela 7.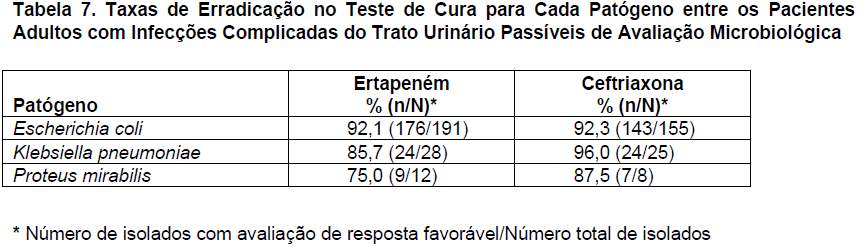
Dos pacientes com bacteremia por E. coli, 91,7% (22/24) foram tratados com sucesso com o ertapeném; nenhum desses pacientes apresentava bacteremia persistente documentada.
Infecções Pélvicas Agudas - (incluindo endomiometrite pós-parto, aborto séptico e infecções ginecológicas pós-cirúrgicas)
O ertapeném foi avaliado em 412 pacientes com infecções pélvicas agudas (inclusive 350 com infecções obstétricas/pós-parto e 45 com aborto séptico) em um estudo clínico controlado, randômico, multicêntrico, duplo-cego, que comparou o ertapeném (1 g IV por dia) com a associação piperacilina/tazobactama (3,375 g IV a cada 6 horas) durante 3 a 10 dias. As taxas de sucesso clínico depois de 2 a 4 semanas de tratamento (teste de cura) foram de 93,9% (153/163) para o ertapeném e de 91,5% (140/153) para a piperacilina/tazobactama. As taxas de sucesso clínico no teste de cura para cada patógeno entre os pacientes passíveis de avaliação microbiológica são apresentadas na tabela 8.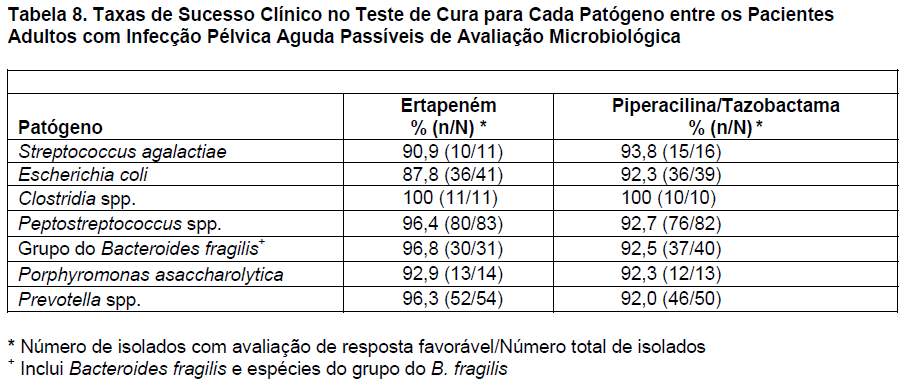
Dos pacientes com bacteremia por E. coli, 100% (6/6) foram tratados com sucesso com o ertapeném.
Pacientes Pediátricos
O ertapeném foi avaliado em pacientes pediátricos de 3 meses a 17 anos de idade em dois estudos multicêntricos, randômicos. O primeiro estudo envolveu 404 pacientes e comparou ertapeném (15 mg/kg IV a cada 12 horas em pacientes de 3 meses a 12 anos de idade, e 1 g IV uma vez ao dia em pacientes de 13 a 17 anos de idade) com ceftriaxona (50 mg/kg/dia IV divididos em duas doses em pacientes de 3 meses a 12 anos de idade e uma dose única diária de 50 mg/kg/dia IV em pacientes pediátricos de 13 a 17 anos de idade) para o tratamento de infecções urinárias complicadas (ITU), infecções da pele e dos tecidos moles e ou pneumonia adquirida da comunidade (PAC). Ambos esquemas permitiram a opção de troca para amoxicilina/clavulanato via oral durante um total de até 14 dias de tratamento (parenteral e oral). A análise das taxas de sucesso microbiológico avaliáveis por protocolo em pacientes com ITU tratados foram de 87,0% (40/46) para ertapeném e 90,0% (18/20) para ceftriaxona. A análise das taxas de sucesso clínico avaliáveis por protocolo em pacientes com infecções de pele e tecidos moles tratados foram de 95,5% (64/67) para ertapeném e 100% (26/26) para ceftriaxona, e em pacientes com PAC foram 96,1% (74/77) para ertapeném e 96,4% (27/28) para ceftriaxona.
O segundo estudo envolveu 112 pacientes e comparou o ertapeném (15 mg/kg IV a cada 12 horas em pacientes com 3 meses a 12 anos de idade, e 1 g IV uma vez ao dia em pacientes de 13 a 17 anos de idade) com ticarcilina/clavulanato (50 mg/kg para pacientes com peso < 60 kg ou 3,0 g para pacientes com peso > 60 kg, 4 ou 6 vezes/dia) por até 14 dias para o tratamento de infecções intra-abdominais complicadas e infecções pélvicas agudas (IPA). Para os pacientes com infecções intra-abdominais (primariamente pacientes com apendicite perfurada ou complicado), as taxas de sucesso clínico foram de 83,7% (36/43) para o ertapeném e de 63,6% (7/11) para a ticarcillina/clavulanato na análise avaliável por protocolo. Para as pacientes com IPA (endomiometrite obstétrica espontânea ou pós-cirúrgica ou aborto séptico) as taxas de sucesso clínico foram de 100% (23/23) para o ertapeném e de 100% (4/4) para a ticarcillina/clavulanato na análise avaliável por protocolo.
TOXICOLOGIA ANIMAL
Toxicidade Aguda
A DL50 aproximada do ertapeném após dose única IV em camundongos e ratos foi maior do que as doses mais altas estudadas (700 mg/kg em ratos e 2.000 mg/kg em camundongos). Não ocorreu nenhuma morte entre as duas espécies e observou-se diminuição temporária da atividade em camundongos que receberam 2.000 mg/kg.
Toxicidade Crônica
Avaliou-se o potencial tóxico do ertapeném em uma série de estudos de toxicidade IV em dose diária e com até 6 meses de duração realizados em macacos e ratos. Não houve achados que impedissem a administração no nível posológico terapêutico.
Carcinogênese
Não foram realizados estudos a longo prazo em animais para avaliar o potencial carcinogênico do ertapeném.
Mutagênese
O ertapeném não foi nem mutagênico nem genotóxico nos seguintes ensaios in vitro: ensaio de eluição alcalina/hepatócitos de ratos, ensaio de aberração cromossômica em células de ovário de hamster chinesa e ensaio de mutagênese em células linfoblastóides humanas TK6; além disso, o ertapeném também não foi nem mutagênico nem genotóxico no ensaio de micronúcleo de camundongo in vivo.
Reprodução
Em camundongos e ratos, doses IV de até 700 mg/kg/dia (para camundongos, cerca de 3 vezes a dose recomendada de 1 g para humanos com base na área de superfície corpórea e, para ratos, aproximadamente 1,2 vez a exposição humana à dose recomendada de 1 g com base nas AUCs plasmáticas) não provocaram efeitos sobre desempenho no acasalamento, na fecundidade, fertilidade ou sobrevida embrionária.
Desenvolvimento
Em camundongos e ratos que receberam doses IV de até 700 mg/kg/dia (para camundongos, cerca de 3 vezes a dose recomendada de 1 g para humanos com base na área de superfície corpórea e, para ratos, aproximadamente 1,2 vez a exposição humana à dose recomendada de 1 g com base nas AUCs plasmáticas), não houve evidências de toxicidade de desenvolvimento, de acordo com exame externo, das vísceras e dos esqueletos dos fetos; entretanto, em camundongos que receberam 700 mg/kg/dia, observou-se pequena diminuição do peso médio dos fetos, com diminuição associada do número médio de vértebras sacrocaudais ossificadas. Em ratos, o ertapeném atravessa a barreira placentária.
Caract. farmacológicas.
FARMACOLOGIA CLÍNICA
Mecanismo de Ação
O ertapeném apresenta atividade in vitro contra um amplo espectro de bactérias Gram-positivas e Gram-negativas, aeróbias e anaeróbias. A atividade bactericida do ertapeném resulta da inibição da síntese da parede celular e é mediada pela ligação do ertapeném às proteínas ligadoras de penicilina (PBPs); o ertapeném apresenta alta afinidade pelas PBPs 1a, 1b, 2, 3, 4 e 5 da Escherichia coli, com preferência pelas PBPs 2 e 3. O ertapeném é significativamente estável à hidrólise pela maioria das classes de betalactamases, incluindo as penicilinases, as cefalosporinases e as betalactamases de espectro estendido, mas não as metalobetalactamases.
Farmacocinética
Absorção
O ertapeném reconstituído com cloridrato de lidocaína a 1% injetável USP (em solução fisiológica sem epinefrina) é bem absorvido após administração IM da dose recomendada de 1 g. A biodisponibilidade média é de cerca de 92%. Após a administração de 1 g/dia IM, as concentrações plasmáticas máximas médias (Cmáx) são atingidas em cerca de 2 horas (Tmáx).
Distribuição
A taxa de ligação do ertapeném às proteínas plasmáticas humanas é elevada. Em adultos jovens saudáveis, a taxa de ligação do ertapeném às proteínas diminui à medida que as concentrações plasmáticas aumentam -de cerca de 95% em concentrações plasmáticas aproximadas de < 100 mcg/mL a cerca de 85% em concentrações plasmáticas aproximadas de 300 mcg/mL.
A tabela 1 apresenta as concentrações plasmáticas médias (mcg/mL) do ertapeném após infusão IV de dose única de 1 g ou 2 g durante 30 minutos e após administração IM de dose única de 1 g a adultos jovens saudáveis.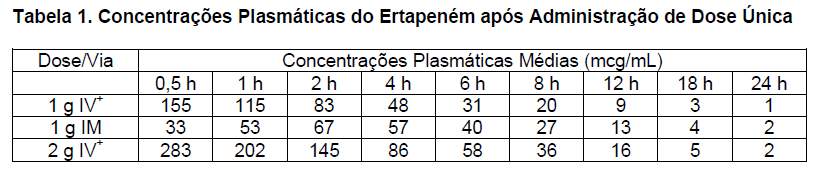
A área sob a curva de concentração plasmática (AUC) do ertapeném em adultos aumenta quase que proporcionalmente à dose na faixa posológica de 0,5 g a 2 g.
Não há acúmulo do ertapeném em adultos após doses múltiplas IV de 0,5 g a 2 g/dia ou doses IM de 1 g/dia.
A concentração plasmática média (mcg/mL) de ertapeném em pacientes pediátricos são apresentados na Tabela 2.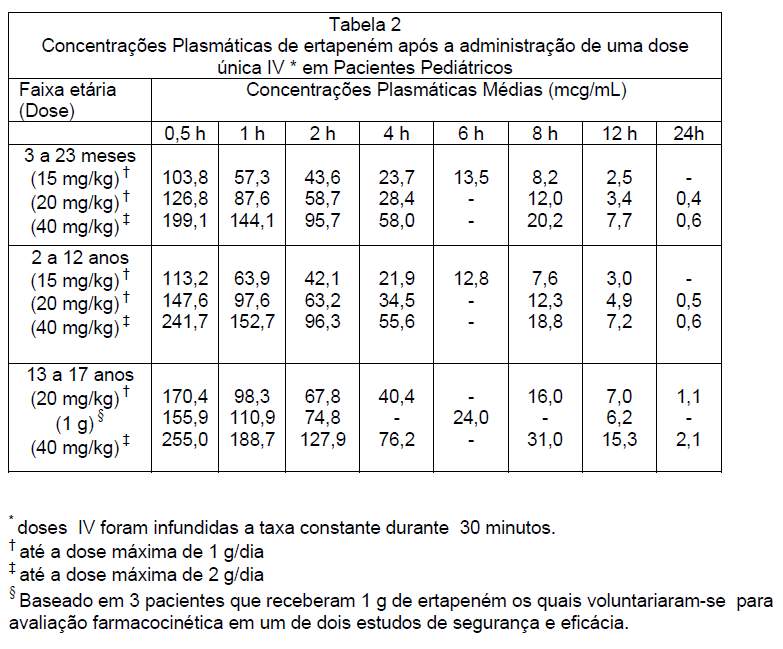
O volume de distribuição (Vdss) do ertapeném em adultos é de cerca de 8 litros (0,11 litros/kg), aproximadamente 0,2 litros/kg em pacientes pediátricos de 3 meses a 12 anos de idade e aproximadamente 0,16 litros/kg em pacientes pediátricos de 13 a 17 anos de idade.
O ertapeném penetra as vesículas cutâneas induzidas por sucção. A tabela 3 apresenta as concentrações do ertapeném obtidas no fluido de vesículas cutâneas a cada ponto de amostragem no terceiro dia de administração IV de 1 g em dose única. A proporção da AUC no fluido da vesícula cutânea para a AUC no plasma é de 0,61.
O nível de ertapeném no leite de 5 nutrizes foi determinado aleatoriamente ao longo das 24 horas, durante 5 dias consecutivos, após a administração da última dose de 1 g IV. A concentração de ertapeném no leite materno determinada no último dia de tratamento (5 a 14 dias após o parto) nas 5 mulheres foi < 0,38 mcg/mL; as concentrações máximas não foram avaliadas. No quinto dia após a descontinuação do tratamento, o nível de ertapeném não foi detectado no leite de 4 mulheres e foram detectados níveis mínimos ( < 0,13 mcg/mL) em uma delas.
Estudos in vitro indicam que o ertapeném não inibe o transporte da digoxina ou da vimblastina mediado pela glicoproteína P e que ele também não é substrato para esse transporte (veja INTERAÇÕES MEDICAMENTOSAS).
Metabolismo
Após infusão IV de 1 g de ertapeném marcado com substância radioativa, a radioatividade plasmática consiste principalmente de ertapeném (94%) em adultos jovens saudáveis. O principal metabólito do ertapeném é o derivado de anel aberto, formado pela hidrólise do anel betalactâmico.
Estudos in vitro em microssomos hepáticos humanos indicam que o ertapeném não inibe o metabolismo mediado pelas seis principais isoenzimas do citocromo P450 (CYP): 1A2, 2C9, 2C19, 2D6, 2E1 e 3A4 (veja INTERAÇÕES MEDICAMENTOSAS).
Eliminação
O ertapeném é eliminado principalmente pelos rins. A meia-vida plasmática média em adultos jovens saudáveis e pacientes de 13 a 17 anos de idade é de cerca de 4 horas e aproximadamente 2,5 horas em pacientes pediátricos de 3 meses a 12 anos de idade.
Após a administração IV de 1 g de ertapeném marcado com substância radioativa a adultos jovens saudáveis, cerca de 80% da dose é recuperada na urina (38% na forma inalterada e 37% como o metabólito de anel aberto, aproximadamente) e 10% nas fezes.
Em adultos jovens saudáveis que receberam 1 g IV, as concentrações urinárias médias de ertapeném excederam 984 mcg/mL até 2 horas após a dose e 52 mcg/mL 12 a 24 horas após a dose.
Características nos Pacientes
Sexo
As concentrações plasmáticas do ertapeném são comparáveis em homens e mulheres.
Idosos
Após administração IV de 1 g e 2 g de ertapeném, as concentrações plasmáticas são um pouco mais altas (aproximadamente 39% e 22%, respectivamente) em adultos idosos (65 anos de idade) do que em adultos jovens ( < 65 anos de idade). Não há necessidade de ajuste posológico para pacientes idosos.
Pacientes Pediátricos
As concentrações plasmáticas de ertapeném são comparáveis em pacientes pediátricos de 13 a 17 anos de idade e adultos após uma dose IV de 1 g ao dia.
Após uma dose de 20 mg/kg (até a dose máxima de 1 g), os valores do parâmetro farmacocinético em pacientes de 13 a 17 anos de idade foram geralmente comparáveis àqueles de pacientes adultos jovens saudáveis. Três de seis pacientes entre 13 a 17 anos de idade receberam uma dose menor do que 1g. A fim de gerar dados farmacocinéticos estimados, caso todos os pacientes dessa faixa etária tenham recebido uma dose de 1 g, os dados farmacocinéticos foram calculados ajustando para uma dose de 1 g, assumindo linearidade. A comparação dos resultados mostra que uma dose de 1 g de ertapeném diariamente apresenta perfil farmacocinético em pacientes de 13 a 17 anos de idade comparável ao de adultos.
As taxas para AUC (13 a 17 anos de idade/adultos), a concentração no final da infusão e a concentração no ponto médio do intervalo da dose foram, respectivamente, 0,99, 1,20, e 0,84.
As concentrações plasmáticas no ponto médio do intervalo da dose após uma dose única IV de 15 mg/kg de ertapeném em pacientes de 3 meses a 12 anos de idade são comparáveis às concentrações plasmáticas do ponto médio do intervalo da dose após uma dose de 1g IV diariamente em adulto (veja Distribuição). A depuração plasmática (mL/min/kg) de ertapeném em pacientes de 3 meses a 12 anos de idade é aproximadamente 2 vezes maior quando comparada à de adultos. O valor da AUC (duplicado para o modelo de um esquema de duas doses ao dia,ou seja, exposição a 30 mg/kg/dia) de uma dose de 15 mg/kg, em pacientes de 3 meses a 12 anos de idade foi comparável ao valor da AUC em pacientes adultos jovens saudáveis recebendo uma dose de 1 g IV de ertapeném.
Insuficiência Hepática
A farmacocinética do ertapeném em pacientes com insuficiência hepática ainda não foi estabelecida. Como o ertapeném é pouco metabolizado no fígado, não é esperado que sua farmacocinética seja alterada pela insuficiência hepática; portanto, não há necessidade de ajuste posológico para pacientes com insuficiência hepática.
Insuficiência Renal
Em comparação com a AUC em indivíduos saudáveis (com 25 a 82 anos de idade), a AUC após uma dose única IV de 1 g de ertapeném: em pacientes adultos com insuficiência renal leve (Clcr 60-90 mL/min/1,73 m2) é semelhante; em pacientes adultos com insuficiência renal moderada (Clcr 31-59 mL/min/1,73 m2) é cerca de 1,5 vez maior; em pacientes adultos com insuficiência renal avançada (Clcr 5-30 mL/min/1,73 m2) é aproximadamente 2,6 vezes maior; e em pacientes com insuficiência renal terminal (Clcr < 10 mL/min/1,73 m2) é cerca de 2,9 vezes maior. Após administração IV de uma dose única de 1 g imediatamente antes da sessão de hemodiálise, aproximadamente 30% da dose é recuperada no dialisado. Não há dados em pacientes pediátricos com insuficiência renal.
Recomenda-se ajuste posológico para pacientes adultos com insuficiência renal em estágio avançado ou terminal (veja POSOLOGIA E ADMINISTRAÇÃO).
CLASSE TERAPÊUTICA
INVANZ® é um 1- metilcarbapenem sintético, de ação prolongada e estruturalmente relacionada aos antibióticos betalactâmicos (como as penicilinas e as cefalosporinas), disponível em formulação estéril para uso parenteral e com atividade contra um amplo espectro de bactérias Gram-positivas e Gram-negativas, aeróbias e anaeróbias.
MICROBIOLOGIA
O ertapeném é ativo in vitro contra um amplo espectro de bactérias Gram-positivas e Gram-negativas, aeróbias e anaeróbias. A atividade bactericida do ertapeném resulta da inibição da síntese da parede celular e é mediada pela ligação do ertapeném às proteínas ligadoras da penicilina (PBPs); o ertapeném apresenta alta afinidade pelas PBPs 1a, 1b, 2, 3, 4 e 5 da Escherichia coli, com preferência pelas PBPs 2 e 3. O ertapeném é significativamente estável à hidrólise pela maioria das classes de betalactamases, incluindo as penicilinases, as cefalosporinases e as betalactamases de espectro estendido, mas não as metalobetalactamases.
Demonstrou-se que INVANZ® é ativo in vitro e em infecções clínicas contra a maioria das cepas dos seguintes microorganismos (veja INDICAÇÕES):
GRAM-POSITIVOS AERÓBIOS E ANAERÓBIOS FACULTATIVOS:
Staphylococcus aureus (inclusive cepas produtoras de penicilinase);
Streptococcus agalactiae;
Streptococcus pneumoniae;
Streptococcus pyogenes.
Obs.: estafilococos resistentes à meticilina são resistentes a INVANZ®. Muitas cepas de Enterococcus faecalis e a maioria das cepas de Enterococcus faecium são resistentes.
GRAM-NEGATIVOS AERÓBIOS E ANAERÓBIOS FACULTATIVOS:
Escherichia coli;
Haemophilus influenzae (inclusive cepas produtoras de betalactamase);
Klebsiella pneumoniae;
Moraxella catarrhalis;
Proteus mirabilis.
ANAERÓBIOS:
Bacteroides fragilis e outras espécies do grupo do B. fragilis;
Clostridium spp. (excluindo C. difficile);
Eubacterium spp.;
Peptostreptococcus spp.;
Porphyromonas asaccharolytica;
Prevotella spp.
Os seguintes dados in vitro estão disponíveis, porém não se sabe qual a sua significância clínica.
In vitro, as concentrações inibitórias mínimas (CIMs) de INVANZ® são 1 mcg/mL contra a maioria (90%) das cepas de Streptococcus spp. (inclusive Streptococcus pneumoniae), 0,5 mcg/mL contra a maioria (90%) das cepas de Haemophilus spp., 2 mcg/mL contra a maioria (90%) das cepas de outros microorganismos anaeróbios facultativos e aeróbios e 4 mcg/mL contra a maioria (90%) dascepas dos microorganismos estritamente anaeróbios relacionados a seguir (a segurança e a eficácia de INVANZ® para o tratamento das infecções clínicas causadas por esses microorganismos, entretanto, não foram estabelecidas em estudos clínicos adequados e bem controlados):
GRAM-POSITIVOS AERÓBIOS E ANAERÓBIOS FACULTATIVOS:
Staphylococcus spp., coagulase-negativas, sensíveis à meticilina;
Streptococcus pneumoniae resistente à penicilina;
Streptococcus viridans.
Obs.: estafilococos resistentes à meticilina são resistentes a INVANZ®. Muitas cepas de Enterococcus faecalis e a maioria das cepas de Enterococcus faecium são resistentes.
GRAM-NEGATIVOS AERÓBIOS E ANAERÓBIOS FACULTATIVOS:
Citrobacter freundii;
Enterobacter aerogenes;
Enterobacter cloacae;
Escherichia coli produtora de ESBLs;
Haemophilus parainfluenzae;
Klebsiella oxytoca;
Klebsiella pneumoniae produtora de ESBLs;
Morganella morganii;
Proteus vulgaris;
Serratia marcescens;
Obs.: muitas cepas dos microorganismos mencionados acima, resistentes a vários outros antibióticos (por exemplo, penicilinas, cefalosporinas [inclusive as de terceira geração]) e aminoglicosídeos) são sensíveis a INVANZ®.
ANAERÓBIOS:
Fusobacterium spp.
Contraindicações.
INVANZ® é contra-indicado para pacientes com hipersensibilidade conhecida a qualquer um de seus componentes ou a outros medicamentos da mesma classe ou para pacientes que já tenham apresentado reações anafiláticas a betalactâmicos.
Em razão do cloridrato de lidocaína ser utilizado como diluente, INVANZ® IM é contra-indicado para pacientes com hipersensibilidade conhecida a anestésicos locais do tipo amida e para pacientes com choque ou bloqueio cardíaco grave (consulte a bula do cloridrato de lidocaína).
Advertências.
Há relatos de reações de hipersensibilidade (anafiláticas) graves e eventualmente fatais em pacientes tratados com betalactâmicos; essas reações são mais prováveis em indivíduos com histórico de sensibilidade a múltiplos alérgenos. Há relatos de indivíduos com histórico de hipersensibilidade à penicilina que apresentaram reações graves de hipersensibilidade quando tratados com outro betalactâmico. Antes de iniciar o tratamento com INVANZ®, deve-se fazer um levantamento minucioso das reações de hipersensibilidade a penicilinas, cefalosporinas, outros betalactâmicos e outros alérgenos. Se ocorrer reação alérgica a INVANZ®, este deve ser descontinuado imediatamente. Reações anafiláticas graves exigem tratamento de emergência.
A exemplo do que ocorre com outros antibióticos, o uso prolongado de INVANZ® pode resultar em supercrescimento de microorganismos não-sensíveis. A avaliação contínua da condição do paciente é fundamental. Na ocorrência de superinfecção durante o tratamento, deve-se adotar as condutas adequadas.
Há relatos de colite pseudomembranosa com praticamente todos os agentes antibacterianos, incluindo o ertapeném, cuja gravidade pode variar de leve a potencialmente fatal; portanto, é importante considerar esse diagnóstico em pacientes que apresentarem diarréia posterior à administração de agentes antibacterianos. Os estudos indicam que a toxina produzida pelo Clostridium difficile é uma das principais causas de "colite associada a antibiótico".
Deve-se ter cautela ao administrar INVANZ® por via IM para evitar a injeção inadvertida do medicamento em um vaso sangüíneo (veja POSOLOGIA E ADMINISTRAÇÃO).
O cloridrato de lidocaína é utilizado como diluente da formulação IM de INVANZ®. (Consulte a bula do cloridrato de lidocaína.)
Gravidez Categoria de risco: B
Não há estudos adequados e bem controlados em grávidas. INVANZ® só deve ser utilizado durante a gravidez se o benefício potencial justificar o possível risco para a mãe e para o feto.
Nutrizes
O ertapeném é excretado no leite materno (veja FARMACOLOGIA CLÍNICA, Distribuição); portanto, deve-se ter cautela ao administrar INVANZ® a nutrizes.
USO EM IDOSOS, CRIANÇAS E OUTROS GRUPOS DE RISCO
Uso Pediátrico
A segurança e a eficácia de INVANZ® em pacientes pediátricos entre 3 meses e 17 anos de idade estão sustentados pela evidência de estudos adequados e bem controlados em adultos, dados farmacocinéticos em pacientes pediátricos e dados adicionais de estudos de comparação controlados em pacientes pediátricos entre 3 meses e 17 anos de idade com as seguintes infecções (veja INDICAÇÕES e RESULTADOS DE EFICÁCIA, Pacientes Pediátricos):
- Infecções Intra-abdominais Complicadas;
- Infecções Complicadas de Pele e Anexos;
- Pneumonia Adquirida na Comunidade;
- Infecções Complicadas do Trato Urinário;
- Infecções Pélvicas Agudas;
- Septicemia Bacteriana.
Como não há dados disponíveis em crianças com menos de 3 meses de idade, INVANZ® não é recomendado nessa faixa etária
Uso Em Idosos
Em estudos clínicos, a eficácia e a segurança de INVANZ® em idosos (65 anos de idade) foram comparáveis às observadas em pacientes mais jovens ( < 65 anos de idade).
Após administração IV de 1 e 2 g de ertapeném, as concentrações plasmáticas são um pouco maiores (aproximadamente 39% e 22%, respectivamente) em adultos idosos (65 anos de idade) do que em adultos jovens ( < 65 anos de idade). Não há necessidade de ajuste posológico para pacientes idosos.
Interações medicamentosas.
Quando o ertapeném é administrado com a probenecida, esta compete pela secreção tubular ativa e, desse modo, inibe a excreção renal do ertapeném. Essa competição resulta em aumento pequeno, porém estatisticamente significativo, da meia-vida de eliminação (19%) e do grau de exposição sistêmica (25%). Não há necessidade de ajuste posológico quando o ertapeném for administrado com a probenecida. Uma vez que o efeito sobre a meia-vida é pequeno, não se recomenda a administração concomitante com a probenecida com o objetivo de aumentar a meia-vida do ertapeném.
Estudos in vitro indicam que o ertapeném não inibe o transporte da digoxina ou da vimblastina mediado pela glicoproteína P e que o ertapeném não é substrato desse transporte. Estudos in vitro em microssomos hepáticos humanos indicam que o ertapeném não inibe o metabolismo mediado por nenhuma das seis principais isoenzimas do citocromo P450 (CYP): 1A2, 2C9, 2C19, 2D6, 2E1 e 3A4. É improvável que ocorram interações medicamentosas por inibição do clearance mediado pela glicoproteína P ou pelo CYP (veja FARMACOLOGIA CLÍNICA, Distribuição e Metabolismo).
Com exceção do estudo com a probenecida, não foram conduzidos estudos específicos de interação medicamentosa clínica.
Cuidados de armazenamento.
O pó liofilizado não deve ser armazenado em temperatura acima de 25°C. A solução reconstituída, imediatamente diluída em 0,9 % de solução de cloreto de sódio (veja MODO DE USAR E CUIDADOS DE CONSERVAÇÃO DEPOIS DE ABERTO, INSTRUÇÕES DE USO), pode ser armazenada em temperatura ambiente (25°C) e utilizada em até 6 horas, ou armazenada durante 24 horas sob refrigeração (5°C) e utilizada até 4 horas depois de retirada do refrigerador. As soluções de INVANZ® não devem ser congeladas.
"ATENÇÃO: ESTE PRODUTO É UM NOVO MEDICAMENTO E, EMBORA AS PESQUISAS TENHAM INDICADO EFICÁCIA E SEGURANÇA QUANDO CORRETAMENTE INDICADO, PODEM OCORRER REAÇÕES ADVERSAS IMPREVISÍVEIS AINDA NÃO DESCRITAS OU DESCONHECIDAS. EM CASO DE SUSPEITA DE REAÇÃO ADVERSA, O MÉDICO RESPONSÁVEL DEVE SER NOTIFICADO."
Posologia e modo de usar.
MODO DE USAR E CUIDADOS DE CONSERVAÇÃO DEPOIS DE ABERTO
INSTRUÇÕES DE USO
Pacientes acima de 13 anos de idade:
Preparo para administração intravenosa:
INVANZ® NÃO DEVE SER MISTURADO OU INFUNDIDO COM OUTROS MEDICAMENTOS.
NÃO UTILIZE DILUENTES QUE CONTENHAM DEXTROSE (-D-GLUCOSE).
INVANZ® DEVE SER RECONSTITUÍDO E DILUÍDO ANTES DA ADMINISTRAÇÃO.
1. Reconstitua o conteúdo de um frasco-ampola de 1 g de INVANZ® com 10 mL de um dos seguintes diluentes: Água para Injeção, Cloreto de Sódio a 0,9% Injetável ou Água Bacteriostática para Injeção.
2. Agite bem para dissolver.
3. Retire 10 mL de um frasco de 50 mL de cloreto de sódio a 0,9% Injetável e descarte;
4. Transfira imediatamente o conteúdo do frasco-ampola de 1 g de INVANZ® reconstituído para o frasco de 50 mL de Cloreto de Sódio a 0,9% Injetável.
5. Finalize a infusão até 6 horas após a reconstituição.
Preparo para administração intramuscular:
INVANZ® DEVE SER RECONSTITUÍDO ANTES DA ADMINISTRAÇÃO.
1. Reconstitua o conteúdo de um frasco-ampola de 1 g de INVANZ® com 3,2 mL de cloridrato de lidocaína a 1,0% ou 2,0% injetável+++ (sem epinefrina). Agite o frasco-ampola energicamente, até formar uma solução.
2. Aspire imediatamente o conteúdo do frasco-ampola e administre por injeção intramuscular profunda em massa muscular grande (como os músculos glúteos ou a parte lateral da coxa).
3. A solução reconstituída IM deve ser utilizada até 1 hora após o preparo.
Obs.: A solução reconstituída não deve ser administrada por via IV.
Pacientes entre 3 meses a 12 anos de idade:
Preparo para administração intravenosa:
INVANZ® NÃO DEVE SER MISTURADO OU INFUNDIDO COM OUTROS MEDICAMENTOS.
NÃO UTILIZE DILUENTES QUE CONTENHAM DEXTROSE (-D-GLUCOSE).
INVANZ® DEVE SER RECONSTITUÍDO E DILUÍDO ANTES DA ADMINISTRAÇÃO.
1. Reconstitua o conteúdo de um frasco-ampola de 1 g de INVANZ® com 10 mL de um dos seguintes diluentes: água para Injeção, cloreto de sódio a 0,9% Injetável ou água bacteriostática para injeção.
2. Agite bem para dissolver e retire imediatamente um volume igual a 15 mg/kg do peso corporal (não exceder 1g/dia) e dilua em cloreto de sódio a 0,9% Injetável para que a concentração final seja equivalente a 20 mg/mL ou menos;
3. Complete a infusão dentro de 6 horas da reconstituição.
Preparo para administração intramuscular:
INVANZ® DEVE SER RECONSTITUÍDO ANTES DA ADMINISTRAÇÃO.
1. Reconstitua o conteúdo de um frasco-ampola de 1 g de INVANZ® com 3,2 mL de cloridrato de lidocaína a 1,0% ou 2,0% injetável+++ (sem epinefrina). Agite o frasco-ampola energicamente, até formar uma solução.
2. Imediatamente retire um volume igual a 15 mg/kg do peso corporal (não exceder 1g/dia) e administre por injeção intramuscular profunda em massa muscular grande (como os músculos glúteos ou a parte lateral da coxa).
3. A solução reconstituída IM deve ser utilizada até 1 hora após o preparo. Obs.: A solução reconstituída não deve ser administrada por via IV.
Os medicamentos destinados a uso parenteral devem ser inspecionados visualmente antes de serem utilizados, sempre que a solução e o frasco permitirem, para verificar a existência de materiais particulados e alteração de cor. As soluções de INVANZ® variam do incolor ao amarelo claro. As variações de cor nessa faixa não afetam a potência do produto.
+++ Consulte a bula do cloridrato de lidocaína
O pó liofilizado não deve ser armazenado em temperatura acima de 25°C. A solução reconstituída, imediatamente diluída em 0,9 % de solução de cloreto de sódio (veja MODO DE USAR E CUIDADOS DE CONSERVAÇÃO DEPOIS DE ABERTO, INSTRUÇÕES DE USO), pode ser armazenada em temperatura ambiente (25°C) e utilizada em até 6 horas, ou armazenada durante 24 horas sob refrigeração (5°C) e utilizada até 4 horas depois de retirada do refrigerador. As soluções de INVANZ® não devem ser congeladas.
POSOLOGIA E ADMINISTRAÇÃO
A dose usual de INVANZ® para pacientes acima de 13 anos de idade é de 1 grama (g), 1 vez ao dia (1x/dia). A dose usual de INVANZ® em pacientes entre 3 meses e 12 anos de idade é de 15 mg/kg duas vezes ao dia (não exceder 1g/dia).
INVANZ® pode ser administrado por infusão intravenosa (IV) com duração superior a 30 minutos ou por injeção intramuscular (IM).
A administração IM de INVANZ® pode ser utilizada como alternativa à administração intravenosa para o tratamento de infecções para as quais a terapia IM é adequada.
A duração usual do tratamento com INVANZ® é de 3 a 14 dias, entretanto varia com o tipo de infecção e patógeno(s) causador(es) (veja INDICAÇÕES). Quando houver indicação clínica e for observada melhora clínica, o paciente pode passar a receber um antimicrobiano adequado por via oral.
Em estudos clínicos controlados, os pacientes foram tratados durante 3 a 14 dias. A duração total do tratamento foi determinada pelo médico responsável, com base no local e na gravidade da infecção e na resposta clínica do paciente. Em alguns estudos, o tratamento passou a ser feito por via oral, a critério do médico responsável, após demonstração de melhora clínica.
Pacientes com Insuficiência Renal: INVANZ® pode ser utilizado para o tratamento de infecções em pacientes adultos com insuficiência renal. Não há necessidade de ajuste posológico para pacientes com clearance de creatinina > 30 mL/min/1,73 m2, enquanto os pacientes adultos com insuficiência renal avançada (clearance de creatinina 30 mL/min/1,73 m2), inclusive aqueles em hemodiálise, devem receber 500 mg/dia. Não há dados clínicos em pacientes pediátricos com insuficiência renal.
Pacientes em Hemodiálise: em um estudo clínico, após a administração de uma dose única IV de 1 g de ertapeném imediatamente antes da sessão de hemodiálise, aproximadamente 30% da dose foi recuperada no dialisado. Quando pacientes adultos em hemodiálise recebem a dose diária recomendada de 500 mg de INVANZ® no período de 6 horas antes da hemodiálise, recomenda-se a administração de uma dose suplementar de 150 mg após a sessão de hemodiálise. Se INVANZ® for administrado no mínimo 6 horas antes da hemodiálise, não há necessidade de dose suplementar. Não há dados de pacientes submetidos a diálise peritoneal ou hemofiltração. Não há dados clínicos em pacientes pediátricos em hemodiálise.
Quando apenas a creatinina sérica estiver disponível, pode-se utilizar a fórmula descrita a seguir para estimar o clearance de creatinina. A creatinina sérica deve