EZETROL™
ORGANON BRASIL
ezetimiba
Hipolipidemiante.
Apresentações.
EZETROL™ é apresentado em caixas com 10 e 30 comprimidos de 10 mg.
USO ORAL.
USO ADULTO.
Composição.
Ingrediente Ativo: Cada comprimido de EZETROLTM para administração oral contém 10 mg de ezetimiba. Ingredientes Inativos: Cada comprimido de 10 mg contém croscarmelose sódica, lactose monoidratada, estearato de magnésio, celulose microcristalina, povidona e laurilsulfato de sódio.
Indicações.
Hipercolesterolemia Primária
EZETROLTM, administrado em associação com um inibidor da enzima HMG-CoA redutase (vastatina) ou isoladamente, é indicado como terapia adjuvante à dieta para a redução dos níveis elevados de colesterol total (C total), de colesterol da lipoproteína de baixa densidade (LDL-C), da apolipoproteína B (apo B) e dos triglicérides (TG) e para aumentar o colesterol da lipoproteína de alta densidade (HDL-C) em pacientes com hipercolesterolemia primária (familiar heterozigótica e não familiar).
EZETROL™, administrado em combinação com o fenofibrato, é indicado como terapia adjuvante à dieta para redução de níveis elevados de colesterol total, LDL-C, Apo B, e não-HDL-C em pacientes com hiperlipidemia mista.
Hipercolesterolemia Familiar Homozigótica (HFHo)
EZETROLTM administrado em associação com uma vastatina é indicado para a redução dos níveis elevados de colesterol total e do LDL-C em pacientes com HFHo. Os pacientes também poderão receber tratamentos adjuvantes (por exemplo, aférese de LDL).
Sitosterolemia Homozigótica (Fitosterolemia)
EZETROLTM é indicado para a redução dos níveis elevados de sitosterol e campesterol em pacientes com sitosterolemia familiar homozigótica.
Resultados de eficácia.
Hipercolesterolemia Primária
Monoterapia
Em dois estudos multicêntricos, duplo-cegos, controlados com placebo, com 12 semanas de duração e que envolveram 1.719 pacientes com hipercolesterolemia primária, EZETROLTM 10 mg reduziu de forma significativa os níveis de colesterol total, LDL-C, apo B e TG e aumentou os níveis de HDL-C em comparação com o placebo (Tabela 1). A redução do LDL-C foi uniforme em todas as idades, sexos, etnias e níveis basais de LDL-C. Além disso, EZETROLTM não exerceu efeito sobre a concentração plasmática das vitaminas lipossolúveis A, D e E ou sobre o tempo de protrombina e não comprometeu a produção de hormônios esteróides adrenocorticais.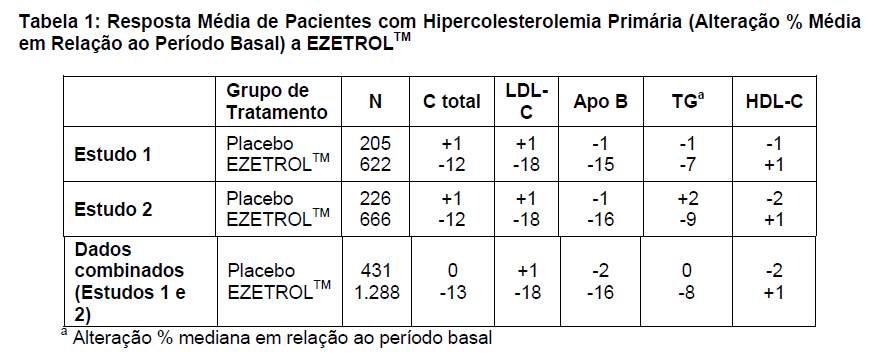
Co-Administração com uma Vastatina
EZETROLTM Iniciado Concomitantemente com uma Vastatina
Em quatro estudos multicêntricos, duplo-cegos, controlados com placebo, com 12 semanas de duração e que envolveram 1.187 pacientes com hipercolesterolemia, EZETROLTM 10 mg foi administrado isoladamente ou com várias doses de atorvastatina, sinvastatina, pravastatina ou lovastatina. Em geral, o efeito aumentado sobre a redução de LDL-C foi independente da dose ou vastatina específica utilizada. Além disso, a redução do LDL-C com EZETROLTM co-administrado com a dose mais baixa testada (10 mg) de qualquer uma das vastatinas foi semelhante ou maior que a redução do LDL-C observada com a dose mais alta testada da vastatina correspondente administrada isoladamente (Tabela 2).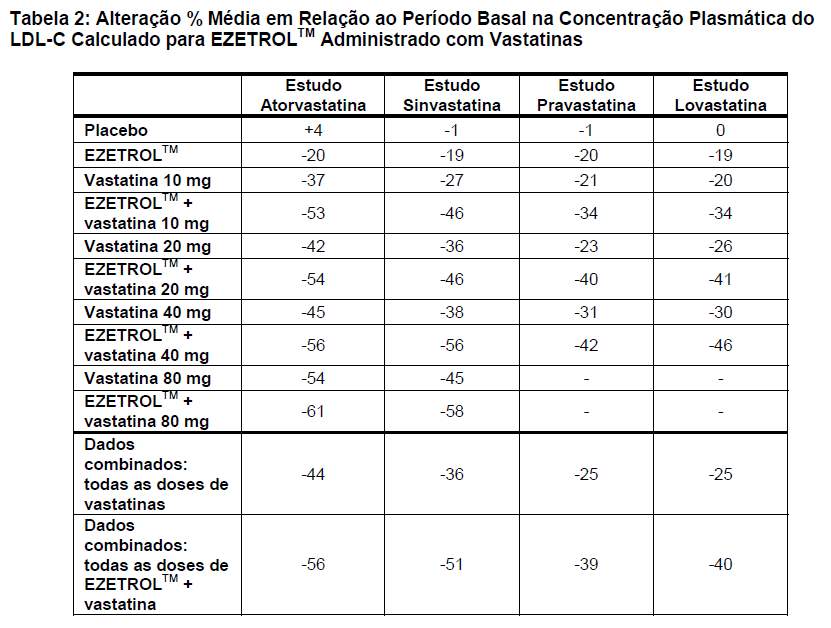
Em uma análise combinada de EZETROLTM + todas as doses de vastatina, EZETROLTM exerceu efeito benéfico sobre o colesterol total, a apo B, os TG e o HDL-C (Tabela 3).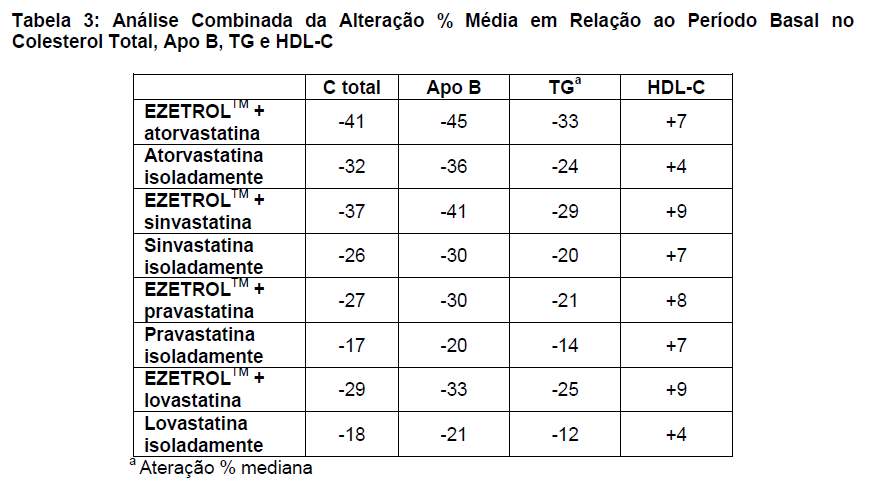
EZETROLTM Adicionado ao Tratamento Preexistente com Vastatina
Em um estudo multicêntrico, duplo-cego, controlado com placebo, com 8 semanas de duração, 769 pacientes com hipercolesterolemia que já recebiam monoterapia com vastatina e cujos níveis de LDL-C estavam fora das metas estabelecidas pelo National Cholesterol Education Program (NCEP) (meta de LDL-C de 100 a 160 mg/dL, dependendo das características no período basal) foram distribuídos de modo randômico para receber EZETROLTM 10 mg ou placebo, além do tratamento já em andamento com vastatina.
Entre os pacientes que recebiam vastatina e cujos níveis de LDL-C estavam fora da meta no período basal (~82%), 72% e 19% dos pacientes distribuídos de modo randômico para EZETROLTM e placebo, respectivamente, atingiram a meta no final do estudo.
EZETROLTM adicionado ao tratamento preexistente com vastatina reduziu significativamente os níveis de colesterol total, LDL-C, apo B e TG e aumentou o nível de HDL-C em comparação com o placebo (Tabela 4). A redução de LDL-C foi uniforme entre todas as vastatinas.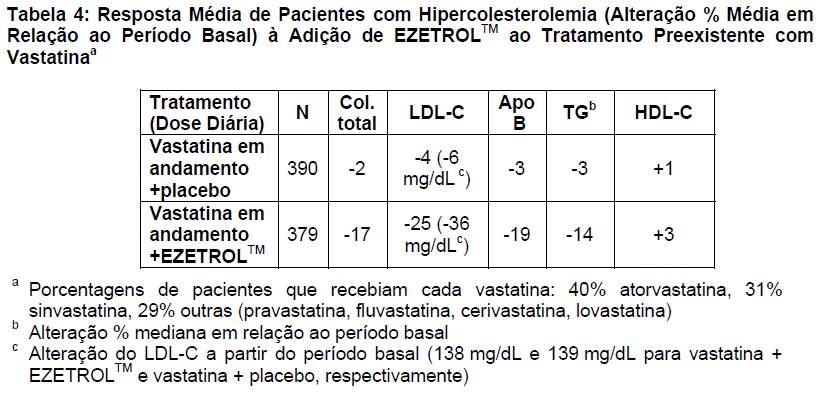
EZETROLTM ou placebo adicionados ao tratamento com vastatina reduziram a proteína C reativa em 10% ou 0% em relação ao período basal, respectivamente (valores medianos).
Em um estudo multicêntrico, duplo-cego, com 14 semanas de duração, 621 pacientes com hipercolesterolemia que recebiam 10 mg/dia de atorvastatina e cujos níveis de LDL-C estavam acima de 130 mg/dL foram distribuídos de modo randômico para receber 20 mg de atorvastatina ou 10 mg de EZETROLTM adicionado ao tratamento com atorvastatina 10 mg. A dose de atorvastatina poderia ser titulada até 80 mg no grupo da atorvastatina e até 40 mg no grupo da co-administração de EZETROLTM mais atorvastatina, com base nos pacientes que não atingiram a meta de LDL-C ( < 100 mg/dL). A média de LDL-C no período basal foi de 187 mg/dL e aproximadamente 60% dos pacientes apresentavam hipercolesterolemia familiar heterozigótica (HFHe). Ao final do estudo, houve diferença significativa na obtenção da meta de LDL-C entre os pacientes que receberam a coadministração de EZETROLTM (22%) e os que receberam monoterapia com atorvastatina (7%). Na 4ª semana houve diferença significativa nas reduções de LDL-C entre os pacientes que receberam a co-administração (24%; EZETROLTM + atorvastatina 10 mg) e os que receberam monoterapia (9%; atorvastatina 20 mg). No subgrupo de pacientes com HFHe, foram obtidos resultados semelhantes em termos de obtenção da meta de LDL-C e de redução dos níveis de LDL-C.
Em um estudo de desenho semelhante que envolveu 100 pacientes com hipercolesterolemia que recebiam 20 mg de sinvastatina e cujos níveis de LDL-C estavam fora da meta, a adição de EZETROLTM 10 mg associada à titulação das doses da sinvastatina em comparação com a titulação da sinvastatina isoladamente resultou em vantagens semelhantes às observadas no estudo da atorvastatina descrito acima. Por exemplo, foram obtidas diferenças significativas em relação à obtenção da meta de LDL-C (27% para EZETROLTM + sinvastatina vs. 3% para sinvastatina isoladamente) e à redução de LDL-C (24% para EZETROLTM + sinvastatina vs. 11% para sinvastatina isoladamente).
Co-administração com Fenofibratos
Em um estudo clínico multicêntrico, duplo-cego, controlado com placebo, em pacientes com hiperlipidemia mista, 625 pacientes foram tratados por até 12 semanas e 576 por até 1 ano. Os pacientes foram distribuídos de forma randômica para receber placebo, EZETROLTM apenas, 160 mg de fenofibrato apenas, ou EZETROLTM e 160 mg de fenofibrato.
EZETROLTM co-administrado com fenofibrato diminuiu significativamente os níveis de colesterol total, LDL-C, apo B, e não-HDL-C em comparação com a administração de fenofibrato apenas. A redução percentual de TG e o aumento percentual de HDL-C para EZETROLTM co-administrado com fenofibrato foram comparáveis aos da administração de fenofibrato apenas (Tabela 5).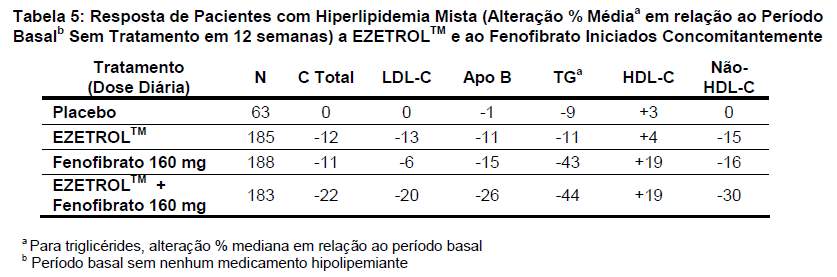
A melhora nos desfechos lipídicos após 1 ano de tratamento foram compatíveis com os dados de 12 semanas apresentados acima.
Hipercolesterolemia Familiar Homozigótica (HFHo)
Foi conduzido um estudo para avaliar a eficácia de EZETROLTM no tratamento da HFHo. Nesse estudo duplo-cego, randômico, de 12 semanas de duração, foram admitidos 50 pacientes com diagnóstico clínico e/ou genotípico de HFHo, submetidos ou não à aférese concomitante de LDL, que já recebiam atorvastatina ou sinvastatina (40 mg). Os pacientes foram distribuídos de modo randômico para um de três grupos de tratamento: atorvastatina ou sinvastatina (80 mg), EZETROLTM 10 mg administrado com atorvastatina ou sinvastatina (40 mg), ou EZETROLTM 10 mg administrado com atorvastatina ou sinvastatina (80 mg). Os resultados são apresentados na tabela 6. A coadministração de EZETROLTM e atorvastatina (40 mg ou 80 mg) ou sinvastatina (40 mg ou 80 mg) reduziu significativamente o LDL-C em comparação com a titulação da dose da sinvastatina ou da atorvastatina em monoterapia (de 40 mg para 80 mg).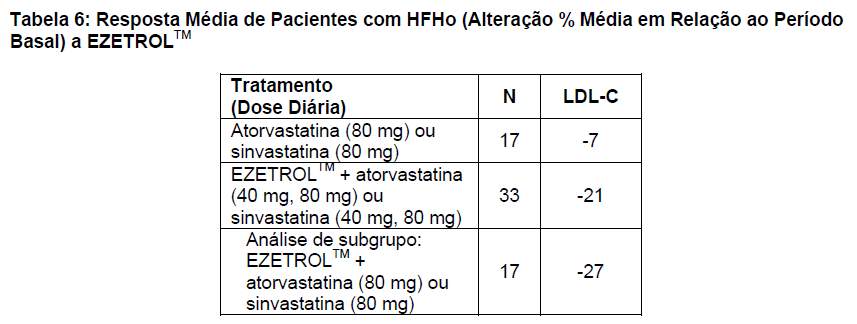
Sitosterolemia Homozigótica (Fitosterolemia)
Foi conduzido um estudo para avaliar a eficácia de EZETROLTM no tratamento de sitosterolemia homozigótica. Neste estudo multicêntrico, duplo-cego, controlado com placebo, de 8 semanas de duração, 37 pacientes com sitosterolemia homozigótica foram distribuídos de modo randômico para EZETROLTM 10 mg (n= 30) ou placebo (n= 7). EZETROLTM reduziu de forma significativa os dois principais fitosteróis - o sitosterol e o campesterol - em 21% e 24% em relação ao período basal, respectivamente. Em contrapartida, os pacientes que receberam placebo apresentaram aumento do nível de sitosterol e de campesterol de 4% e 3% em relação ao período basal, respectivamente. Quanto aos pacientes que receberam EZETROLTM, a redução dos níveis de fitosteróis foi progressiva ao longo do estudo.
A redução dos níveis de sitosterol e de campesterol foi consistente entre os pacientes que receberam EZETROLTM concomitantemente com seqüestrantes de ácidos biliares (n= 8) e os pacientes que não receberam esses agentes (n= 21).
Caract. farmacológicas.
FARMACOLOGIA CLÍNICA
Mecanismo de Ação
EZETROLTM pertence a uma nova classe de compostos hipolipemiantes que inibem de forma seletiva a absorção intestinal de colesterol e de fitosteróis relacionados.
EZETROLTM é ativo e potente por via oral e apresenta mecanismo de ação exclusivo, que difere de outras classes de compostos redutores do colesterol (por exemplo, vastatinas, seqüestrantes de ácidos biliares [resinas], derivados do ácido fíbrico e fitosteróis). A meta molecular da ezetimiba é o transportador de esterol, Niemann-Pick C1-Like 1 (NPC1L1), responsável pela captação intestinal de colesterol e de fitosteróis.
A ezetimiba localiza-se na borda em escova dos enterócitos do intestino delgado, onde inibe a absorção do colesterol, promovendo redução do aporte de colesterol do intestino para o fígado. Isto leva à redução do estoque de colesterol hepático e ao aumento da depuração do colesterol sangüíneo. A ezetimiba não aumenta a excreção de ácido biliar (como os seqüestrantes de ácidos biliares) e não inibe a síntese hepática de colesterol (como as vastatinas).
Em um estudo clínico com duração de 2 semanas que envolveu 18 pacientes hipercolesterolêmicos, EZETROLTM inibiu a absorção intestinal de colesterol em 54% em comparação ao placebo; ao inibir a absorção do colesterol intestinal, a ezetimiba reduz o aporte de colesterol para o fígado. As vastatinas reduzem a síntese hepática de colesterol. Juntos, esses mecanismos distintos promovem redução complementar do colesterol. Administrado com uma vastatina, EZETROLTM reduz o colesterol total (C total), o colesterol da lipoproteína de baixa densidade (LDL-C), a apolipoproteína B (apo B) e os triglicérides (TG) e aumenta o colesterol da lipoproteína de alta densidade (HDL-C) em pacientes com hipercolesterolemia mais do que cada tratamento isoladamente. A administração de EZETROLTM com fenofibrato é eficaz na melhora dos níveis séricos de colesterol total, LDL-C, apo B, TG, HDL-C e não-HDL-C em pacientes com hiperlipidemia mista.
Estudos clínicos demonstram que níveis elevados de colesterol total, LDL-C e apo B - o principal constituinte protéico da LDL - promovem a aterosclerose humana. Além disso, níveis reduzidos de HDL-C estão associados ao desenvolvimento de aterosclerose. Estudos epidemiológicos estabeleceram que a morbidade e a mortalidade cardiovasculares variam diretamente conforme o nível de colesterol total e de LDL-C e inversamente conforme o nível de HDL-C. A exemplo do LDL, lipoproteínas ricas em TG e enriquecidas com colesterol, incluindo as lipoproteínas de densidade muito baixa (VLDL) e as lipoproteínas de densidade intermediária (IDL) e remanescentes também podem causar aterosclerose.
Inúmeros estudos pré-clínicos foram realizados para determinar a seletividade da ezetimiba na inibição da absorção do colesterol. A ezetimiba inibiu a absorção do [14C]-colesterol sem exercer efeito sobre a absorção de TG, ácidos graxos, ácidos biliares, progesterona, etinilestradiol ou vitaminas lipossolúveis A e D.
Farmacocinética
Absorção: após administração oral, a ezetimiba é rapidamente absorvida e extensivamente conjugada a um glicuronídeo fenólico farmacologicamente ativo (glicuronídeo da ezetimiba), cuja concentração plasmática máxima (Cmáx) média ocorre em 1 a 2 horas; já para a ezetimiba, essa concentração é atingida em 4 a 12 horas. A biodisponibilidade absoluta da ezetimiba não pode ser determinada, já que o composto é praticamente insolúvel em meios aquosos próprios para injeção.
A administração concomitante de alimentos (com altos teores de gorduras ou sem gordura) não exerceu efeito sobre a biodisponibilidade oral da ezetimiba presente nos comprimidos de 10 mg de EZETROLTM. EZETROLTM pode ser administrado com ou sem alimentos.
Distribuição: a ezetimiba e o glicuronídeo da ezetimiba estão 99,7% e 88% a 92% ligados às proteínas plasmáticas de seres humanos, respectivamente.
Metabolismo: a ezetimiba é metabolizada principalmente no intestino delgado e no fígado, por meio da conjugação do glicuronídeo (uma reação de fase II) e da excreção biliar subseqüente. Observou-se metabolismo oxidativo mínimo (uma reação de fase I) em todas as espécies avaliadas. A ezetimiba e o glicuronídeo da ezetimiba são os principais derivados do fármaco detectados no plasma, constituindo aproximadamente 10% a 20% e 80% a 90% do total, respectivamente. Tanto a ezetimiba quanto o glicuronídeo da ezetimiba são eliminados lentamente do plasma, com evidência de recirculação êntero-hepática significativa. A meia-vida da ezetimiba e do glicuronídeo da ezetimiba é de aproximadamente 22 horas.
Eliminação: após administração oral de [14C]-ezetimiba 20 mg a seres humanos, a ezetimiba total respondeu por cerca de 93% da radioatividade plasmática total. Aproximadamente 78% e 11% da carga radioativa administrada foi recuperada nas fezes e na urina, respectivamente, ao longo de um período de coleta de 10 dias. Após 48 horas, os níveis plasmáticos de radioatividade eram indetectáveis.
Contraindicações.
Hipersensibilidade a qualquer componente desta medicação. Quando EZETROLTM for administrado com uma vastatina, ou com fenofibrato, a bula desse medicamento em particular deverá ser consultada.
Advertências.
Quando EZETROLTM for administrado com uma vastatina ou com o fenofibrato, a bula desse medicamento em particular deverá ser consultada.
Enzimas Hepáticas Em estudos controlados para avaliar a co-administração de EZETROLTM e uma vastatina, foram observadas elevações consecutivas das transaminases (3 vezes o limite superior da normalidade [LSN]). Quando EZETROLTM for co-administrado com uma vastatina, deverão ser realizados testes de função hepática no início do tratamento e de acordo com as recomendações para a vastatina (veja REAÇÕES ADVERSAS).
Músculo esquelético
Em estudos clínicos, não foi verificado excesso de miopatia ou rabdomiólise associados a EZETROL™ em comparação com o braço controle (placebo ou vastatina isoladamente). Entretanto, miopatia e rabdomiólise são reações adversas conhecidas das vastatinas e de outros fármacos redutores de lipídes. Em estudos clínicos, a incidência de CPK > 10 vezes o LSN foi de 0,2% para EZETROL™ versus 0,1% para o placebo, e de 0,1% para EZETROL™ co- administrado com uma vastatina versus 0,4% para as vastatinas isoladamente.
Na experiência pós-comercialização com EZETROLTM, foram relatados casos de miopatia e rabdomiólise, independentemente da causalidade. A maioria dos pacientes que desenvolveram rabdomiólise estava recebendo uma vastatina antes de iniciar o tratamento com EZETROLTM. No entanto, a rabdomiólise foi relatada muito raramente com a monoterapia com EZETROLTM ou com a adição de EZETROLTM a agentes conhecidamente associados a risco aumentado de rabdomiólise. Todos os pacientes que iniciam tratamento com EZETROLTM devem ser alertados sobre o risco de miopatia e instruídos a relatar imediatamente qualquer dor, sensibilidade ou fraqueza muscular inexplicada. EZETROLTM e qualquer vastatina que o paciente esteja tomando concomitantemente devem ser imediatamente descontinuados se houver suspeita de ou for comprovada a miopatia. A presença desses sintomas e um nível de creatina fosfoquinase (CPK) > 10 vezes o LSN indica miopatia.
Insuficiência Hepática
Uma vez que os efeitos da maior exposição à ezetimiba em pacientes com insuficiência hepática moderada ou grave são desconhecidos, EZETROLTM não é recomendado para esses pacientes (veja USO EM IDOSOS, CRIANÇAS E OUTROS GRUPOS DE RISCO).
Fibratos
A co-administração da ezetimiba com fibratos -exceto o fenofibrato - não foi estudada. Portanto, a co-administração de EZETROLTM e fibratos (exceto o fenofibrato) não é recomendada (veja INTERAÇÕES MEDICAMENTOSAS).
Fenofibrato
Se houver suspeita de colelitíase em um paciente que esteja recebendo EZETROLTM e fenofibrato, são indicados estudos da vesícula biliar e um tratamento hipolipemiante alternativo deve ser considerado (veja REAÇÕES ADVERSAS e a bula do produto de fenofibrato).
Ciclosporina
Deve-se ter cautela ao prescrever ezetimiba para pacientes que estejam utilizando ciclosporina; a concentração de ciclosporina deve ser monitorada nesses pacientes (veja INTERAÇÕES MEDICAMENTOSAS).
Anticoagulantes
Se EZETROLTM for acrescentado ao tratamento com varfarina, outro anticoagulante cumarínico ou fluindiona, a Razão Normalizada Internacional (International Normalized Ratio - INR) deve ser adequadamente monitorada (veja INTERAÇÕES MEDICAMENTOSAS).
Uso durante a Gravidez e a Lactação Gravidez Categoria de Risco de Gravidez: C
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica.
Não há dados clínicos disponíveis sobre a exposição durante a gravidez. Estudos em animais sobre a administração isolada de ezetimiba não indicam efeitos nocivos, diretos ou indiretos, no que diz respeito à gravidez, ao desenvolvimento embrionário/fetal, ao parto ou ao desenvolvimento pós-natal, entretanto deve-se ter cautela ao prescrever o medicamento a gestantes.
Quando se administrou ezetimiba com lovastatina, sinvastatina, pravastatina ou atorvastatina, não foram observados efeitos teratogênicos em estudos de desenvolvimento embriofetal conduzidos em ratas prenhas. Em coelhas prenhas, observou-se incidência baixa de malformações esqueléticas.
Quando a ezetimiba for administrada com uma vastatina, a bula dessa vastatina em particular deverá ser consultada.
Nutrizes
Estudos conduzidos em ratas demonstraram que a ezetimiba é excretada no leite. Não se sabe se a ezetimiba é excretada no leite de seres humanos; portanto, EZETROLTM não deverá ser administrado a nutrizes a não ser que o potencial benefício justifique o provável risco para o lactente.
USO EM IDOSOS, CRIANÇAS E OUTROS GRUPOS DE RISCO
Pacientes Pediátricos
A absorção e o metabolismo da ezetimiba são semelhantes entre crianças, adolescentes (10 a 18 anos) e adultos. Com base na ezetimiba total, não existem diferenças farmacocinéticas entre adolescentes e adultos. Não estão disponíveis dados farmacocinéticos na população pediátrica < 10 anos de idade. A experiência clínica em pacientes pediátricos e adolescentes (idade entre 9 e 17 anos) se limitou a pacientes com HFHo ou sitosterolemia.
Pacientes Idosos
A concentração plasmática da ezetimiba total é, aproximadamente, 2 vezes mais elevada nos indivíduos idosos (≥ 65 anos de idade) em relação aos jovens (18 a 45 anos de idade). A redução de LDL-C e o perfil de segurança são comparáveis em indivíduos idosos e jovens que recebem EZETROLTM. Não é necessário, portanto, ajuste posológico para pacientes idosos.
Insuficiência Hepática
Após uma dose única de 10 mg de ezetimiba, a média da área sob a curva (AUC) para ezetimiba total aumentou aproximadamente 1,7 vez em pacientes com insuficiência hepática leve (escore Child-Pugh de 5 ou 6), em comparação com indivíduos saudáveis. Em um estudo de doses múltiplas de 14 dias de duração (10 mg/dia) em pacientes com insuficiência hepática moderada (escore de Child-Pugh de 7 a 9), a AUC média para ezetimiba total aumentou aproximadamente 4 vezes no 1° e no 14° dia em comparação com indivíduos saudáveis. Não é necessário nenhum ajuste de dose para pacientes com insuficiência hepática leve. Em razão dos efeitos desconhecidos da exposição aumentada à ezetimiba em pacientes com insuficiência hepática moderada ou grave (escore de Child-Pugh > 9), a ezetimiba não é recomendada para esses pacientes (veja ADVERTÊNCIAS).
Insuficiência Renal
Após uma dose única de 10 mg de ezetimiba em pacientes com doença renal grave (n=8; CrCl médio ≤ 30 mL/min/1,73 m2), a AUC média para ezetimiba total aumentou aproximadamente 1,5 vez, em comparação com indivíduos saudáveis (n= 9). Esse resultado não é considerado clinicamente significativo. Não é necessário nenhum ajuste de dose para pacientes com insuficiência renal.
Um outro paciente desse estudo (pós-transplante renal e sob administração de múltiplos medicamentos, incluindo ciclosporina) teve exposição 12 vezes maior à ezetimiba total.
Sexo
A concentração plasmática da ezetimiba é um pouco maior ( < 20%) em mulheres do que em homens. A redução do LDL-C e do perfil de segurança é comparável entre homens e mulheres tratados com ezetimiba. Portanto, não é necessário nenhum ajuste de dose com base no sexo.
Raça
Com base em uma metanálise de estudos farmacocinéticos, não houve diferenças farmacocinéticas entre negros e caucasianos.
Interações medicamentosas.
Em estudos pré-clínicos, demonstrou-se que a ezetimiba não induz enzimas de metabolização do citocromo P-450. Não foram observadas interações farmacocinéticas clinicamente relevantes entre a ezetimiba e os medicamentos reconhecidamente metabolizados pelos citocromos P-450 1A2, 2D6, 2C8, 2C9 e 3A4 ou N-acetiltransferase.
A ezetimiba não exerceu efeito sobre a farmacocinética dos seguintes compostos: dapsona, dextrometorfano, digoxina, anticoncepcionais orais (etinilestradiol e levonorgestrel), glipizida, tolbutamida ou midazolam durante a co-administração. A cimetidina, co-administrada com a ezetimiba, não exerceu efeito sobre a biodisponibilidade da ezetimiba.
Antiácidos: a administração concomitante de antiácidos reduziu a taxa de absorção da ezetimiba, embora não tenha exercido efeito sobre a biodisponibilidade. Essa redução da taxa de absorção não é considerada clinicamente relevante.
Colestiramina: a administração concomitante de colestiramina reduziu a AUC média da ezetimiba total (ezetimiba + glicuronídeo de ezetimiba) em aproximadamente 55%. A redução adicional do LDL-C pelo acréscimo da ezetimiba à colestiramina pode ser minimizada por essa interação.
Ciclosporina: em um estudo que envolveu oito pacientes submetidos a transplante renal, com clearance de creatinina > 50 mL/min e que estavam recebendo dose estável de ciclosporina, uma única dose de 10 mg de ezetimiba resultou em aumento de 3,4 vezes (variação de 2,3 a 7,9 vezes) da AUC média da ezetimiba total em comparação com uma população de controle sadia de outro estudo (n= 17). Em um estudo diferente, um paciente submetido a transplante renal com insuficiência renal grave (clearance de creatinina de 13,2 mL/min/1,73 m2) que estava recebendo diversos medicamentos, inclusive ciclosporina, apresentou exposição 12 vezes maior à ezetimiba total em comparação com os controles de comparação. Em um estudo cruzado de dois períodos, a administração diária de 20 mg de ezetimiba durante 8 dias com uma única dose de 100 mg de ciclosporina no 7° dia a 20 indivíduos saudáveis resultou em aumento de 15%, em média, na AUC da ciclosporina (variação de 10% de redução a 51% de aumento) em comparação a uma dose única de 100 mg de ciclosporina isoladamente (veja ADVERTÊNCIAS).
Fibratos: a segurança e a eficácia da ezetimiba co-administrada com fenofibrato foram avaliadas em um estudo clínico (veja REAÇOES ADVERSAS e ESTUDOS CLÍNICOS, Co-administração com Fenofibrato); a co-administração da ezetimiba com outros fibratos não foi estudada. Os fibratos podem aumentar a excreção biliar de colesterol, levando à colelitíase. Em um estudo pré-clínico conduzido em cães, a ezetimiba aumentou as concentrações de colesterol na vesícula biliar. Embora a importância desse achado pré-clínico seja desconhecida no caso do uso em humanos, a co-administração de EZETROLTM com fibratos (exceto fenofibrato) não é recomendada até que o uso em pacientes seja estudado.
Fenofibratos: em um estudo farmacocinético, a administração concomitante do fenofibrato aumentou a concentração total da ezetimiba em aproximadamente 1,5 vez. Esse aumento não é considerado clinicamente significativo.
Genfibrozila: em um estudo farmacocinético, a administração concomitante de genfibrozila aumentou a concentração total de ezetimiba em aproximadamente 1,7 vez. Esse aumento não é considerado clinicamente significativo. Não há dados clínicos disponíveis.
Vastatinas: não foram observadas interações farmacocinéticas clinicamente importantes quando a ezetimiba foi co-administrada com atorvastatina, sinvastatina, pravastatina, lovastatina, fluvastatina ou rosuvastatina.
Anticoagulantes: A administração concomitante de ezetimiba (10 mg em dose única diária) não apresentou efeito significativo sobre a biodisponibilidade da varfarina e sobre o tempo de protrombina em um estudo que incluiu doze adultos saudáveis do sexo masculino. Houve relatos pós-comercialização de Razão Normalizada Internacional (INR) aumentada em pacientes para os quais EZETROL™ foi adicionado à varfarina ou à fluindiona. A maioria destes pacientes também estava recebendo outros medicamentos (veja ADVERTÊNCIAS).
Cuidados de armazenamento.
Mantenha o medicamento acondicionado na embalagem original e em temperatura ambiente (entre 15C - 30°C).
Não tome este medicamento após a expiração da data de validade impressa na embalagem.
TODO MEDICAMENTO DEVE SER MANTIDO FORA DO ALCANCE DAS CRIANÇAS.
Posologia e modo de usar.
MODO DE USAR E CUIDADOS DE CONSERVAÇÃO DEPOIS DE ABERTO
Mantenha o medicamento acondicionado na embalagem original e em temperatura ambiente (entre 15C - 30°C).
POSOLOGIA E ADMINISTRAÇÃO
O paciente deve estar sob dieta redutora de lípides adequada e deve continuá-la durante o tratamento com EZETROLTM.
A dose recomendada de EZETROLTM é de 10 mg uma vez ao dia, isoladamente ou em associação com uma vastatina ou com o fenofibrato. EZETROLTM pode ser administrado em qualquer horário do dia, independentemente do horário de ingestão de alimentos.
Uso em Idosos
Não é necessário ajuste posológico para pacientes idosos (veja USO EM IDOSOS, CRIANÇAS E OUTROS GRUPOS DE RISCO).
Uso Pediátrico
Não é necessário ajuste posológico para crianças e adolescentes com idade 10 anos (veja USO EM IDOSOS, CRIANÇAS E OUTROS GRUPOS DE RISCO). O tratamento com EZETROLTM não é recomendado para crianças com menos de 10 anos de idade.
Uso na Insuficiência Hepática
Não é necessário ajuste posológico para pacientes com insuficiência hepática leve (escore de Child-Pugh de 5 a 6). O tratamento com a ezetimiba não é recomendado para pacientes com insuficiência hepática moderada (escore de Child-Pugh de 7 a 9) ou grave (escore de Child-Pugh > 9) (veja ADVERTÊNCIAS e USO EM IDOSOS, CRIANÇAS E OUTROS GRUPOS DE RISCO).
Uso na Insuficiência RenalNão é necessário ajuste posológico para pacientes com disfunção renal (veja USO EM IDOSOS, CRIANÇAS E OUTROS GRUPOS DE RISCO).
Co-administração com Seqüestrantes de Ácidos Biliares EZETROLTM deve ser administrado no mínimo duas horas antes ou no mínimo quatro horas depois da administração de seqüestrantes de ácidos biliares.
Reações adversas.
Estudos clínicos com 8 a 14 semanas de duração, nos quais EZETROLTM 10 mg/dia foi administrado isoladamente, com uma vastatina, ou com fenofibrato, a 3.551 pacientes demonstraram que EZETROLTM foi bem tolerado de modo geral, as reações adversas foram usualmente leves e transitórias, a incidência global das reações adversas relatadas com o uso de EZETROLTM foi semelhante àquela relatada com o placebo e a taxa de descontinuação por reações adversas foi comparável entre EZETROLTM e o placebo.
A seguir, as reações adversas comuns (1/100, < 1/10) relacionadas à medicação, relatadas por pacientes que utilizavam EZETROLTM isoladamente (n= 1.691), com uma vastatina (n= 1.675), ou coadministrada com o fenofibrato (n = 185):
EZETROLTM administrado isoladamente: cefaléia; dor abdominal, diarréia.
EZETROLTM co-administrado com uma vastatina: cefaléia, fadiga; dor abdominal, constipação, diarréia, flatulência, náuseas; aumento de ALT, aumento de AST; mialgia.
EZETROLTM co-administrado com fenofibrato: dor abdominal.
Em um estudo clínico multicêntrico, duplo-cego, controlado com placebo e que incluiu pacientes com hiperlipidemia mista, 625 pacientes foram tratados por até 12 semanas e 576 por até 1 ano. Esse estudo não foi delineado para comparar grupos de tratamento quanto a eventos infreqüentes. As taxas de incidência (IC 95%) para elevações clinicamente importantes ( > 3 X LSN, consecutivas) nas transaminases séricas foram de 4,5% (1,9, 8,8) e 2,7% (1,2, 5,4) para monoterapia com fenofibrato e EZETROLTM co-administrado com fenofibrato, respectivamente, ajustado para exposição ao tratamento. As taxas de incidência correspondentes para colecistectomia foram de 0,6% (0,0, 3,1) e 1,7% (0,6, 4,0) para monoterapia com fenofibrato e EZETROLTM co-administrado com fenofibrato, respectivamente (veja ADVERTÊNCIAS). Não ocorreram elevações de CPK > 10 vezes o LSN em nenhum dos grupos de tratamento desse estudo.
Valores Laboratoriais
Em estudos clínicos controlados nos quais utilizou-se monoterapia, a incidência de aumento clinicamente importante das transaminases séricas (ALT e/ou AST 3x LSN, consecutivas) foi semelhante entre EZETROLTM (0,5%) e placebo (0,3%). Em estudos nos quais utilizou-se coadministração, a incidência foi de 1,3% para pacientes que receberam EZETROLTM em combinação com uma vastatina e de 0,4% para pacientes que receberam vastatina isoladamente. Esses aumentos em geral foram assintomáticos, não associados à colestase e retornaram aos valores do período basal após a descontinuação do tratamento ou mediante tratamento contínuo (veja ADVERTÊNCIAS).
Aumentos clinicamente importantes de CPK (≥10x LSN) em pacientes que receberam EZETROLTM isoladamente ou co-administrado com uma vastatina foram semelhantes aos observados com o placebo ou com uma vastatina administrada isoladamente, respectivamente.
Experiência Pós-comercialização
Após a comercialização foram relatadas as seguintes reações adversas em pacientes que utilizavam EZETROLTM e vastatina, independentemente da determinação de causalidade: reações de hipersensibilidade, incluindo anafilaxia, angioedema, erupções cutâneas e urticária; artralgia; mialgia; aumento de CPK; elevações das transaminases hepáticas; hepatite, trombocitopenia; pancreatite, náuseas; tontura; parestesia; depressão; colelitíase; colecistite; e muito raramente, miopatia/rabdomiólise (veja ADVERTÊNCIAS).
"Atenção: este é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis para comercialização, efeitos indesejáveis e não conhecidos podem ocorrer. Neste caso, informe a seu médico".
Superdose.
A administração de 50 mg/dia de ezetimiba a 15 indivíduos sadios durante até 14 dias ou de 40 mg/dia a 18 pacientes com hipercolesterolemia primária por até 56 dias foi, em geral, bem tolerada. Foram relatados poucos casos de superdosagem com EZETROL™, dos quais a maioria não foi associada a reações adversas. As reações adversas relatadas não foram graves. No caso de superdosagem deverão ser instituídas medidas sintomáticas e de suporte.
Dizeres legais.
Registro MS - 1.0029.0076
VENDA SOB PRESCRIÇÃO MÉDICA.