
DOCEGLENNU
GLENMARK
docetaxel
Antineoplásico.
Apresentações.
Concentrado para infusão 20 mg/1,0 mL (solução para administração parenteral após diluição): embalagem com 1 frasco-ampola com 1,0 mL de solução injetável.
Concentrado para infusão 80 mg/4,0 mL (solução para administração parenteral após diluição): embalagem com 1 frasco-ampola com 4,0 mL de solução injetável.
USO INTRAVENOSO (IV)
USO ADULTO
Composição.
Doceglennu 20 mg/ 1,0 mL:
Cada frasco-ampola contém 20mg de docetaxel anidro
Excipientes: polissorbato 80 e álcool etílico (anidro).
Doceglennu 80 mg/4,0 mL:
Cada frasco-ampola contém 80mg de docetaxel anidro
Excipientes: polissorbato 80 e álcool etílico (anidro).
Informações técnicas.
.INDICAÇÕES
Este medicamento é destinado aos tratamentos de:
- Câncer de mama
Câncer de mama adjuvante
Doceglennu em associação com doxorrubicina e ciclofosfamida é indicado para o tratamento adjuvante de pacientes com câncer de mama operável, com linfonodo positivo.
Doceglennu em associação com doxorrubicina e ciclofosfamida é indicado para o tratamento adjuvante de pacientes com câncer de mama operável, linfonodo-negativo com um ou mais fatores de alto risco: tamanho do tumor > 2 cm, idade < 35 anos, status de receptor hormonal negativo, tumor grau 2 ou 3.
Doxorrubicina e ciclofosfamida seguida de Doceglennu em associação com trastuzumabe (AC- TH) é indicado para o tratamento adjuvante de pacientes com câncer de mama operável cujos tumores superexpressam HER 2.
Doceglennu em associação com trastuzumabe e carboplatina (TCH) é indicado para o tratamento adjuvante de pacientes com câncer de mama operável cujos tumores superexpressam HER2.
Câncer de mama metastático
Doceglennu em associação com doxorrubicina é indicado para o tratamento de pacientes com câncer de mama localmente avançado ou metastático que não receberam terapia citotóxica prévia para esta condição.
Doceglennu em monoterapia é indicado para o tratamento de pacientes com câncer de mama localmente avançado ou metastático após falha de terapia citotóxica. Quimioterapia prévia deve ter incluído a administração de antraciclina ou agente alquilante.
Doceglennu em associação com capecitabina é indicado para o tratamento de pacientes com câncer de mama localmente avançado ou metastático após falha de quimioterapia citotóxica. Terapia prévia deve ter incluído a administração de antraciclina.
Doceglennu em associação com trastuzumabe é indicado para o tratamento de pacientes com câncer de mama metastático cujos tumores superexpressam HER2 e que previamente não receberam quimioterapia para doença metastática.
- Câncer de pulmão de não-pequenas células
Doceglennu é indicado para o tratamento de pacientes com câncer de pulmão de não-pequenas células localmente avançado ou metastático, mesmo após falha de quimioterapia prévia.
Doceglennu em associação com cisplatina é indicado para o tratamento de pacientes com câncer de pulmão de não-pequenas células irressecável, localmente avançado ou metastático que não tenham recebido quimioterapia para esta condição (vide Reações adversas e Características Farmacológicas - Propriedades Farmacodinâmicas).
- Câncer de ovário
Doceglennu é indicado para o tratamento carcinoma metastático de ovário após falha de quimioterapia de primeira linha ou subsequente.
- Câncer de próstata
Doceglennu em associação com prednisona ou prednisolona é indicado para o tratamento de pacientes com câncer de próstata metastático androgênio independente (refratário a hormônio).
- Adenocarcinoma gástrico
Doceglennu em associação com cisplatina e fluoruracila é indicado para o tratamento de pacientes com adenocarcinoma gástrico avançado, incluindo adenocarcinoma da junção gastroesofágica, que não receberam quimioterapia prévia para a doença avançada.
- Câncer de cabeça e pescoço
Doceglennu em associação com cisplatina e fluoruracila é indicado para o tratamento de indução de pacientes com carcinoma de células escamosas de cabeça e pescoço localmente avançado na cavidade oral, orofaringe, hipofaringe e laringe.
2. RESULTADOS DE EFICÁCIA
Os resultados de eficácia estão descritos ao final da bula, após as Instruções de Preparo.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades farmacodinâmicas
O princípio ativo de Doceglennu atua promovendo a agregação das tubulinas na formação de microtúbulos estáveis, inibindo a sua despolimerização, o que promove diminuição expressiva de tubulina livre. A ligação de docetaxel aos microtúbulos não altera o número de protofilamentos.
In vitro, docetaxel mostrou romper a rede de microtúbulos nas células, essencial para as funções celulares vitais durante a intérfase e mitose.
Docetaxel mostrou ser citotóxico contra várias linhagens de células tumorais humanas e murinas in vitro, e contra células tumorais humanas de remoção recente em ensaios clonogênicos. Docetaxel atinge altas concentrações intracelulares, com um longo período de permanência na célula. Docetaxel demonstrou ser ativo em algumas, mas não em todas, as linhagens celulares que superexpressam p-glicoproteína codificada pelo gene de resistência a múltiplos fármacos. In vivo, docetaxel é regime-independente e apresenta um amplo espectro de atividade antitumoral experimental contra tumores murinos e tumores humanos xenotransplantados.
Para o tratamento adjuvante de pacientes com câncer de mama operável com linfonodo positivo: O efeito benéfico de TAC (Doceglennu em associação com doxorrubicina e ciclofosfamida) não foi provado em pacientes com 4 nódulos ou mais (37% da população), embora tenha sido observada uma redução de 18% do risco de recidiva neste grupo de pacientes. O benefício de TAC nessas pacientes não foi inteiramente definido após o acompanhamento de 55 meses do estudo TAX 316.
Propriedades farmacocinéticas
A farmacocinética do docetaxel foi avaliada em pacientes com câncer após administração de 20 a 115 mg/m2 em estudos de Fase I. O perfil farmacocinético do docetaxel é dose-independente e consistente com um modelo farmacocinético tricompartimental com meia-vida para as fases a,b e c de 4 min, 36 min e 11,1 h, respectivamente. A fase tardia é devida, em parte, ao efluxo relativamente lento de docetaxel dos compartimentos periféricos.
Após administração de uma dose de 100 mg/m2 em infusão de 1 hora, obteve-se concentração plasmática média de 3,7 mg/mL com AUC correspondente de 4,6 h.mg/mL. Os valores médios de clearance corpóreo total e volume de distribuição no estado de equilíbrio foram de 21 L/h/m2 e 113 L, respectivamente. A variação interindividual do clearance corpóreo total foi de aproximadamente 50%. A ligação do docetaxel às proteínas plasmáticas é > 95%.
Foi conduzido um estudo realizado com C14- docetaxel em três pacientes com câncer. No período de 7 dias, o docetaxel foi eliminado na urina e nas fezes após sofrer metabolismo oxidativo do grupo éster terc-butila, mediado pelo citocromo P450. A excreção urinária e fecal foi de aproximadamente 6% e 75% da radioatividade administrada, respectivamente. Aproximadamente 80% da radioatividade recuperada nas fezes é excretada durante as primeiras 48 horas na forma de um metabólito principal inativo, três metabólitos secundários inativos e uma quantidade muito pequena do fármaco inalterado.
Uma análise populacional farmacocinética foi realizada em 577 pacientes que receberam docetaxel. Os parâmetros farmacocinéticos estimados neste modelo foram muito próximos daqueles obtidos nos estudos de Fase I. Os parâmetros farmacocinéticos do docetaxel não sofreram alteração com a idade ou o sexo do paciente. Em um pequeno número de pacientes (n=23) com dados bioquímicos e clínicos indicadores de alteração leve a moderada da função hepática (TGP, TGO ≥ 1,5 vezes o limite superior da normalidade, associado com fosfatase alcalina ≥ 2,5 vezes o limite superior da normalidade), o clearance total diminuiu em média 27% (vide Posologia e Modo de usar - Instruções de preparo). O clearance do docetaxel não foi alterado em pacientes com retenção hídrica leve a moderada; não existem informações disponíveis em pacientes com retenção hídrica severa.
Quando utilizado em associação, docetaxel não influencia o clearance da doxorrubicina e os níveis plasmáticos do doxorrubicinol (um metabólito da doxorrubicina). Por outro lado, o clearance do docetaxel é aumentado enquanto sua eficácia é mantida.
As farmacocinéticas de docetaxel, doxorrubicina e ciclofosfamida estudadas em 30 pacientes com câncer de mama não foram influenciadas por suas administrações concomitantes.
Avaliando o efeito da capecitabina na farmacocinética do docetaxel e o efeito do docetaxel na farmacocinética da capecitabina nos estudos de fase I, não foi observado nenhum efeito da capecitabina na farmacocinética do docetaxel (Cmax e AUC) e nenhum efeito do docetaxel na farmacocinética do 5'DFUR (o metabólito mais importante da capecitabina).
O clearance do docetaxel na terapia associada com cisplatina ou carboplatina foi semelhante àquele observado após a monoterapia com docetaxel. O perfil farmacocinético da cisplatina administrada logo após a infusão de docetaxel é semelhante àquele observado com a cisplatina isolada.
O efeito da prednisona na farmacocinética do docetaxel administrado com pré-medicação padrão de dexametasona foi estudado em 42 pacientes. Não foi observado nenhum efeito da prednisona na farmacocinética do docetaxel. A administração combinada de docetaxel, cisplatina e fluoruracila nos 12 pacientes com tumores sólidos não apresentaram influência na farmacocinética de cada droga individualmente.
Dados de segurança pré-clínica
Carcinogênese
O potencial carcinogênico do Doceglennu ainda não foi estudado.
Mutagenicidade
Docetaxel mostrou ser mutagênico em testes in vitro de micronúcleo e de aberrações cromossômicas em células CHO-K1 e em testes in vivo de micronúcleo em camundongo. Contudo, docetaxel não induziu mutagenicidade no teste de Ames ou no ensaio de mutação gênica CHO/HGPRT. Estes dados são compatíveis com a atividade farmacológica do docetaxel.
Alteração de fertilidade
Estudos de toxicidade em roedores demonstraram efeitos adversos nos testículos, sugerindo que o docetaxel pode prejudicar a fertilidade masculina.
4. CONTRAINDICAÇÕES
Doceglennu é contraindicado:
- Em pacientes com história de reações de hipersensibilidade severas ao docetaxel ou ao polissorbato 80;
- Em pacientes com contagem neutrofílica basal < 1.500 células/mm3;
- Quando houver contraindicações a outros fármacos, estas também são aplicáveis quando associados com docetaxel.
Este medicamento é contraindicado para uso por pacientes pediátricos.
Este medicamento é contraindicado para uso em mulheres grávidas;
Este medicamento é contraindicado para uso em pacientes com insuficiência hepática severa;
Categoria de risco na gravidez: D. Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
5. ADVERTÊNCIAS E PRECAUÇÕES
Doceglennu deve ser administrado somente sob supervisão médica com experiência na utilização de agentes quimioterápicos. Deverão estar disponíveis recursos de suporte apropriados, devido à possibilidade da ocorrência de reações de hipersensibilidade. Durante a infusão, recomenda-se a realização de cuidadosa monitorização das funções vitais.
Um corticosteroide oral (veja a seguir para câncer de próstata), como dexametasona 16 mg/dia (por exemplo: 8 mg, 2 vezes ao dia) durante 3 dias, com início no dia anterior à administração de Doceglennu, a menos que contraindicado, pode reduzir a incidência e a severidade da retenção hídrica, assim como a severidade das reações de hipersensibilidade.
O regime de pré-tratamento para câncer de próstata é dexametasona oral 8 mg, 12 horas, 3 horas e 1 hora antes da infusão de Doceglennu.
Para o tratamento adjuvante de pacientes com câncer de mama operável com linfonodo positivo: Na análise de acompanhamento de 55 meses do estudo clínico TAX 316, o hazard ratio (HR) para período livre de doença de TAC comparado com FAC foi de 0,72 (IC = 0,59 - 0,88; p=0,0010) para a população global do estudo. Na análise de subgrupos nas pacientes com 1 a 3 linfonodos acometidos, o HR foi de 0,61 (IC = 0,46 - 0,82; p=0,0009), enquanto que para 4 linfonodos ou mais, o HR foi de 0,82 (IC = 0,63 - 1,08; p=0,1629). Pacientes com 4 linfonodos ou mais: O efeito benéfico de TAC não foi provado em pacientes com 4 linfonodos ou mais (37% da população), embora tenha sido observada uma redução de 18% do risco de recidiva neste grupo de pacientes. O benefício de TAC nessas pacientes não foi inteiramente definido após o acompanhamento de 55 meses do estudo TAX 316.
Reações de hipersensibilidade
Os pacientes devem ser rigorosamente observados quanto à ocorrência de reações de hipersensibilidade, especialmente durante a primeira e a segunda infusão. Podem ocorrer reações de hipersensibilidade minutos após o início da infusão de Doceglennu, sendo que devem estar disponíveis recursos para o tratamento da hipotensão e broncoespasmo. Foram relatadas, em pacientes que receberam pré-medicação, reações severas, tais como rash/eritema generalizados, hipotensão severa, broncoespasmo ou muito raramente anafilaxia fatal. Reações de hipersensibilidade requerem descontinuação imediata de docetaxel e terapia apropriada. Pacientes que desenvolveram reações de hipersensibilidade severa não devem ser retratados com docetaxel.
Pacientes que, previamente, apresentaram reações de hipersensibilidade com paclitaxel, podem desenvolver reações de hipersensibilidade potencialmente fatais ao docetaxel.
Neutropenia
O nadir neutrofílico ocorreu com uma mediana de 7 dias, porém este intervalo pode ser menor em pacientes extensivamente pré-tratados. Deve-se realizar frequente monitorização do hemograma completo de todos os pacientes que estejam recebendo Doceglennu. Os pacientes devem ser novamente tratados com Doceglennu somente quando a contagem de neutrófilos retomar um nível ≥ 1.500 células/mm3 (vide Posologia e Modo de usar - Instruções de preparo).
Nos pacientes tratados com Doceglennu em associação com cisplatina e fluoruracila (TCF) ocorreram neutropenia febril e/ou infecção neutropênica nos índices mais baixos quando pacientes receberam G- CSF profilático. Os pacientes tratados com TCF devem receber G-CSF profilático para aliviar o risco de neutropenia complicada (neutropenia febril, neutropenia prolongada ou infecção neutropênica). Os pacientes recebendo TCF devem ser rigorosamente monitorizados.
Em pacientes tratados com Doceglennu em associação com doxorrubicina e ciclofosfamida (TAC), neutropenia febril e/ou infecção neutropênica ocorreram em níveis mais baixos quando os pacientes receberam G-CSF profilático primário. Profilaxia primária com G-CSF deve ser considerada em pacientes que recebem terapia adjuvante com TAC para câncer de mama de modo a minimizar o risco de neutropenia complicada (neutropenia febril, neutropenia prolongada ou infecção neutropênica). Pacientes recebendo TAC devem ser rigorosamente monitorizados (vide Posologia e Modo de Usar - Instruções de preparo e Reações adversas).
Reações gastrointestinais
Recomenda-se cuidado para pacientes com neutropenia, particularmente para o risco de desenvolver complicações gastrointestinais. Pode-se desenvolver enterocolite a qualquer momento, podendo levar à morte logo no início do tratamento. Os pacientes devem ser cuidadosamente monitorados para manifestações recentes de toxicidade gastrointestinal severa.
Reações cutâneas
Observou-se eritema cutâneo localizado nas extremidades (palma das mãos e planta dos pés), com edema seguido por descamação.
Sistema nervoso
O desenvolvimento de sinais e/ou sintomas neurossensoriais severos tem sido observado e requer uma redução de dose (vide Posologia e Modo de usar - Instruções de preparo).
Foram relatados graves sintomas neurossensoriais, tais como parestesia, disestesia, dor, podendo ser necessário a redução da dose ou interrupção do tratamento.
Toxicidade cardíaca
Foi observada insuficiência cardíaca em pacientes que receberam Doceglennu em associação com trastuzumabe, particularmente após quimioterapia contendo antraciclina (doxorrubicina ou epirrubicina). Esta pôde ser moderada a severa e foi associada com morte (vide Reações Adversas).
Arritmia ventricular incluindo taquicardia ventricular (algumas vezes fatal) foi relatada em pacientes tratados com docetaxel em combinação com tratamentos incluindo doxorrubicina, fluoruracila e/ou ciclofosfamida (vide Reações Adversas). Recomenda-se avaliação cardíaca basal.
Distúrbios oculares
Edema Macular Cistóide (EMC) tem sido reportado em pacientes tratados com docetaxel, bem como com outros taxanos. Pacientes com visão comprometida devem ser submetidos a um exame oftalmológico completo. Em caso de diagnóstico de EMC, o tratamento com docetaxel deve ser descontinuado e tratamento apropriado deve ser iniciado.
Leucemia
No tratamento adjuvante do câncer de mama, o risco de mielodisplasia tardia ou leucemia mieloide requer acompanhamento hematológico (vide Reações Adversas).
Excipientes
A quantidade de etanol no Doceglennu pode ser prejudicial em pacientes que sofrem de alcoolismo e também deve ser considerada em mulheres grávidas ou que estejam amamentando, em crianças e nos pacientes do grupo de risco, como pacientes com insuficiência hepática ou com epilepsia.
Devem ser considerados possíveis efeitos sobre o Sistema Nervoso Central.
A quantidade de etanol no Doceglennu pode alterar o efeito de outros medicamentos.
A quantidade de etanol no Doceglennu pode prejudicar a capacidade de dirigir veículos ou operar máquinas.
Gravidez e lactação
Docetaxel mostrou ser embriotóxico e fetotóxico em coelhos e ratos; além de reduzir a fertilidade de ratos. Docetaxel pode causar dano fetal quando administrado a mulheres grávidas. Portanto, Doceglennu não deve ser utilizado durante a gravidez. Mulheres em idade fértil que estejam em tratamento com Doceglennu devem ser aconselhadas a evitarem a gravidez e a informarem imediatamente o médico caso isto ocorra (vide Contraindicações).
Não se sabe se docetaxel é excretado no leite materno. Devido às potenciais reações adversas do docetaxel em lactentes, a amamentação deve ser descontinuada durante o tratamento com Doceglennu.
A quantidade de etanol no Doceglennu pode ser prejudicial em mulheres grávidas ou que estejam amamentando.
Alteração na capacidade de dirigir veículos e/ou operar máquinas
Não foram conduzidos estudos para avaliar os efeitos sobre a capacidade de dirigir veículos ou operar máquinas.
A quantidade de etanol no Doceglennu e os efeitos colaterais do produto podem prejudicar a capacidade de dirigir veículos ou operar máquinas. Portanto, os pacientes devem ser avisados sobre o potencial impacto dos efeitos colaterais do produto na capacidade de dirigir ou operar máquinas, e devem ser avisados para não conduzirem veículos ou operarem máquinas se eles apresentarem esses efeitos colaterais durante o tratamento.
Populações especiais
Pacientes pediátricos
Eficácia não foi estabelecida em crianças.
Pacientes idosos
Uma análise de dados de segurança em pacientes com 60 anos de idade ou mais, tratados com a associação de Doceglennu e capecitabina mostraram um aumento na incidência de eventos adversos grau 3 e 4 relacionados ao tratamento, eventos adversos sérios relacionados ao tratamento e exclusão precoce do tratamento foram devido aos eventos adversos comparados aos de pacientes com menos de 60 anos de idade.
A proporção de pacientes idosos foi de 5,5% e 6,6% nos regimes AC-TH e TCH, respectivamente e é muito limitado para permitir conclusões a respeito dos eventos adversos por idade ( < 65 anos versus ≥ 65 anos).
No estudo conduzido em pacientes com NSCLC que não receberam quimioterapia prévia (TAX 326), 148 pacientes no grupo docetaxel + cisplatina tinham 65 anos de idade ou mais e 15 pacientes tinham 75 anos de idade ou mais; não foi observada nenhuma diferença total na efetividade quando pacientes mais idosos foram comparados aos pacientes mais jovens. Nos pacientes idosos no grupo docetaxel + cisplatina, houve uma maior tendência à diarreia e neurotoxicidade de grau 3/4 (ambas mais frequentes e severas) em comparação ao grupo vinorelbina + cisplatina.
De 333 pacientes tratados com Doceglennu a cada 3 semanas no estudo de câncer de próstata (TAX 327), 209 pacientes tinham 65 anos de idade ou mais e 68 pacientes tinham mais que 75 anos. Não foram identificadas diferenças na eficácia entre pacientes idosos e mais jovens. Em pacientes tratados com Doceglennu a cada 3 semanas, a incidência de anemia, infecção, alterações nas unhas, anorexia, perda de peso ocorreu em proporção ≥ 10% maior que em pacientes com 65 anos ou mais comparados a pacientes mais jovens.
Dentre os 221 pacientes tratados com Doceglennu em associação com cisplatina e fluoruracila no estudo do câncer gástrico (TAX 325), 54 tinham 65 anos de idade ou mais e 2 pacientes tinham mais de 75 anos de idade. Neste estudo, o número de pacientes que tinham 65 anos de idade ou mais foi insuficiente para determinar se eles reagem diferentemente dos pacientes mais jovens. Entretanto, a incidência de eventos adversos sérios foi mais elevada nos pacientes idosos comparada aos pacientes mais jovens. A incidência dos seguintes eventos adversos (todos os graus): letargia, estomatite, diarreia, neutropenia febril/infecção neutropênica ocorreram nos valores ≥ 10% mais elevado em pacientes que tinham 65 anos de idade ou mais comparado aos pacientes mais jovens. Os pacientes idosos tratados com TCF devem ser rigorosamente monitorizados.
Entre 174 e 251 pacientes que receberam tratamento de indução com Doceglennu em associação com cisplatina e fluoruracila (TPF) para SCCHN nos estudos TAX 323 e 324, somente 18 (10%) e 32 (13%) dos pacientes tinham 65 anos de idade ou mais, respectivamente. O número de pacientes idosos que receberam esse regime não foi suficiente para determinar se pacientes idosos responderam diferentemente dos pacientes mais jovens.
Outros grupos de risco
Retenção hídrica
Pacientes com retenção hídrica severa como efusão pleural, efusão pericárdica e ascite devem ser rigorosamente monitorizados.
Pacientes com insuficiência hepática
Pacientes tratados com 100 mg/m2 de docetaxel em monoterapia, com níveis plasmáticos de transaminases (TGP e/ou TGO) > 1,5 vezes o limite superior da normalidade simultaneamente a níveis plasmáticos de fosfatase alcalina > 2,5 vezes o limite superior da normalidade, apresentam maior risco de desenvolver reações adversas severas como toxicidade fatal incluindo sepse e hemorragia gastrintestinal, neutropenia febril, infecções, trombocitopenia, estomatite e astenia.
A dose recomendada de Doceglennu em pacientes com níveis elevados nos parâmetros de função hepática é de 75 mg/m2.
Deve-se realizar monitorização da função hepática no estado basal e antes do início de cada ciclo.
Em pacientes com níveis plasmáticos de bilirrubina maiores que o limite superior da normalidade e/ou TGP e TGO > 3,5 vezes o limite superior da normalidade, simultaneamente aos níveis plasmáticos de fosfatase alcalina > 6 vezes o limite superior da normalidade, não se recomenda a realização de ajuste posológico e Doceglennu não deve ser utilizado, salvo se estritamente indicado.
Não existem dados disponíveis em pacientes com insuficiência hepática tratados com Doceglennu em associação.
A quantidade de etanol no Doceglennu deve ser considerada quando for administrado a pacientes com insuficiência hepática.
6. INTERAÇÕES MEDICAMENTOSAS
Medicamento-medicamento:
Estudos in vitro mostraram que o metabolismo do docetaxel pode ser modificado pela administração
concomitante de fármacos que induzem, inibem ou são metabolizados pelo citocromo P450-3A, tais como ciclosporina, terfenadina, cetoconazol, eritromicina e troleandomicina. Como consequência, deve-se ter cautela quando da administração concomitante destas substâncias, visto que existe potencial para uma interação significativa.
O uso concomitante de Doceglennu com potentes inibidores da CYP3A4 (como por exemplo: cetoconazol, itraconazol, claritromicina, indinavir, nefazodona, nelfinavir, ritonavir, saquinavir, telitromicina e voriconazol) deve ser evitado. No caso de coadministração com inibidores da CYP3A4, a ocorrência de reações adversas de Doceglennu pode aumentar, como uma consequência da redução do metabolismo. Se o uso concomitante de um potente inibidor do CYP3A4 (por exemplo, cetoconazol, itraconazol, claritromicina, indinavir, nefazodona, nelfinavir, ritonavir, saquinavir, telitromicina e voriconazol) não pode ser evitado, é necessária uma supervisão médica rigorosa e ajuste de dose de Doceglennu pode ser necessário durante o tratamento com o potente inibidor de CYP3A4. Em um estudo farmacocinético com 7 pacientes, a coadministração de docetaxel com o cetoconazol, potente inibidor da CYP3A4, levou a uma redução significativa do clearance de docetaxel em 49%.
O docetaxel liga-se altamente às proteínas plasmáticas ( > 95%). Embora a possibilidade de interação in vivo de docetaxel com medicamentos administrados concomitantemente não tenha sido investigada formalmente, fármacos com alta ligação às proteínas in vitro, tais como eritromicina, difenidramina, propranolol, propafenona, fenitoína, salicilato, sulfametoxazol e valproato de sódio, não afetaram a ligação do docetaxel às proteínas plasmáticas. Além disto, a dexametasona não afetou a ligação do docetaxel às proteínas plasmáticas. O docetaxel não influenciou a ligação da digitoxina às proteínas plasmáticas.
Outros medicamentos, incluindo aqueles obtidos sem prescrição médica, podem interferir no efeito do Doceglennu. O Doceglennu ou a outra medicação podem ter sua eficácia reduzida e o paciente pode estar mais susceptível a eventos adversos.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Doceglennu deve ser mantido em sua embalagem original, em temperatura abaixo de 25°C, protegido da luz. O congelamento não afeta adversamente o produto.
Prazo de validade:
Doceglennu 20 mg/1,0 mL: 24 meses a partir da data de fabricação. Doceglennu 80 mg/4,0 mL: 24 meses a partir da data de fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Não é recomendado o contato do concentrado não diluído com equipamento ou dispositivos plastificantes de PVC utilizados para preparar soluções para infusão.
Uma vez adicionada, conforme recomendações, à bolsa de infusão, a solução para infusão de Doceglennu, se armazenada em temperatura abaixo de 25°C e luminosidade normal, é estável por 6 horas. A solução deve ser utilizada dentro de 6 horas (incluindo 1 hora de infusão intravenosa).
Adicionalmente, a estabilidade física e química da solução para infusão em uso, preparada conforme recomendado, foi demonstrada em bolsas que não contêm PVC por até 48 horas quando armazenada entre 2° e 8°C e luminosidade normal.
A solução para infusão de Doceglennu é supersaturada, desta forma, pode cristalizar com o tempo. Se aparecerem cristais, a solução não deve mais ser utilizada, devendo ser descartada.
Características físicas e organolépticas
Doceglennu é uma solução amarela pálida a amarela acastanhada, livre de partículas estranhas.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
Instruções de preparo
Recomendações para o manuseio seguro
Doceglennu é um agente antineoplásico, e assim como com outros compostos potencialmente tóxicos, deve-se ter cautela na manipulação e no preparo das soluções de Doceglennu. É recomendado o uso de luvas.
Caso a solução de Doceglennu concentrado, solução pré-mistura ou solução para infusão entre em contato com a pele, lave a região imediata e completamente com água e sabão. Caso a solução de Doceglennu concentrado, solução pré-mistura ou solução para infusão entre em contato com membranas mucosas, lave-as imediata e completamente com água.
Preparo da solução para administração intravenosa
A) Preparo da Solução para Infusão:
Pode ser necessário mais do que um frasco de Doceglennu concentrado para infusão para se obter a dose necessária ao paciente. Retire assepticamente a quantidade necessária de solução concentrada (20 mg/mL) utilizando uma seringa calibrada com agulha de 21G.
Transfira este volume, através de uma injeção única, para uma bolsa ou frasco de infusão com 250 mL de solução glicosada a 5% ou solução de cloreto de sódio 0,9%.
Caso seja necessária uma dose maior que 200 mg de docetaxel, utilize um volume superior de veículo de infusão, visando não exceder a concentração de 0,74 mg/mL de docetaxel.
Misture o conteúdo da bolsa ou frasco de infusão manualmente, utilizando movimento oscilante.
A solução para infusão de Doceglennu deve ser administrada assepticamente por via intravenosa dentro de um período de 6 horas (incluindo 1 hora de infusão), em condições de temperatura abaixo de 25°C e luminosidade normal. Doceglennu deve ser administrado separadamente de outros medicamentos.
A solução para infusão de Doceglennu é supersaturada, portanto, pode cristalizar com o tempo. Se aparecerem cristais, a solução não deve mais ser utilizada, devendo ser descartada.
Todos os materiais utilizados na diluição e administração devem ser descartados, seguindo procedimentos padrões.
Não é recomendado o contato do concentrado não diluído com equipamento ou dispositivos plastificantes de PVC utilizados para preparar soluções para infusão.
POSOLOGIA
Posologia recomendada
Informações Gerais:
Uma pré-medicação com corticosteroide (veja a seguir para câncer de próstata) como 16 mg/dia (por
exemplo 8 mg duas vezes ao dia) de dexametasona oral durante 3 dias, com início no dia anterior à administração de Doceglennu, a menos que contraindicada, pode ser utilizada (vide Advertências e Precauções).
Para câncer de próstata, determinado o uso associado de prednisona ou prednisolona, o regime de pré- medicação recomendado é dexametasona oral 8 mg, 12 horas, 3 horas e 1 hora antes da infusão de Doceglennu.
Pode ser utilizado o tratamento profilático com G-CSF para abrandar o risco de toxicidades hematológicas.
Doceglennu é administrado por infusão de 1 hora a cada 3 semanas.
Câncer de mama
Câncer de mama adjuvante
No tratamento adjuvante do câncer de mama operável de linfonodo positivo e de linfonodo negativo, a posologia recomendada de Doceglennu é de 75 mg/m2, administrada 1 hora após a administração de doxorrubicina 50 mg/m2 e ciclofosfamida 500 mg/m2, a cada 3 semanas durante 6 ciclos (regime TAC) (vide Posologia e Modo de usar - Ajuste posológico durante o tratamento).
No tratamento adjuvante de pacientes com câncer de mama operável cujos tumores superexpressam HER2, a dose recomendada de Doceglennu é a seguinte:
AC-TH:
AC (ciclos 1 - 4): doxorrubicina (A) 60 mg/m2 seguida por ciclofosfamida (C) 600 mg/m2 administrada a cada três semanas por 4 ciclos.
TH (ciclos 5 - 8): Doceglennu (T) 100 mg/m2 administrada a cada três semanas por 4 ciclos e trastuzumabe (H) administrada semanalmente conforme descrito abaixo:
• Ciclo 5 (iniciando três semanas após o último ciclo de AC): Dia 1: trastuzumabe 4 mg/kg (dose de ataque)
Dia 2: Doceglennu 100 mg/m2
Dias 8 e 15: trastuzumabe 2 mg/kg
• Ciclos 6 - 8:
Dia 1: Doceglennu 100 mg/m2 e trastuzumabe 2 mg/kg
Dias 8 e 15: trastuzumabe 2 mg/kg
Três semanas após dia 1 do ciclo 8: trastuzumabe 6 mg/kg é administrado a cada três semanas. Trastuzumabe é administrado por um total de duração de 1 ano.
TCH:
TCH (ciclos 1 - 6): Doceglennu (T) 75 mg/m2 e carboplatina (C) com AUC de 6 mg/mL/min administrado a cada três semanas e trastuzumabe (H) administrado semanalmente conforme descrito abaixo:
• Ciclo 1:
Dia 1: trastuzumabe 4 mg/kg (dose de ataque)
Dia 2: Doceglennu 75 mg/m2 e carboplatina com AUC de 6 mg/mL/min Dias 8 e 15: trastuzumabe 2 mg/kg
•Ciclos 2 - 6:
Dia 1: Doceglennu 75 mg/m2 seguido de carboplatina com AUC de 6 mg/mL/min e trastuzumabe 2 mg/kg
Dias 8 e 15: trastuzumabe 2 mg/kg
Três semanas após dia 1 do ciclo 6: trastuzumabe 6 mg/kg é administrado a cada três semanas. Trastuzumabe é administrado por um total de duração de 1 ano.
Câncer de mama metastático
Em tratamento de primeira linha do câncer de mama, a posologia recomendada de Doceglennu é de 75mg/m2 na terapia associada com doxorrubicina 50 mg/m2.
Para a associação de Doceglennu e trastuzumabe, a posologia recomendada de Doceglennu é de 100 mg/m2 a cada três semanas, com trastuzumabe administrado semanalmente. Para a dosagem e administração de trastuzumabe, veja a bula do fabricante do produto à base de trastuzumabe.
Em tratamento de segunda linha do câncer de mama, a posologia recomendada de Doceglennu é de 100 mg/m2 em monoterapia.
Para pacientes em tratamento de câncer de mama, a posologia recomendada de Doceglennu em monoterapia é de 100 mg/m2, administrada em infusão de 1 hora, a cada 3 semanas.
A dose recomendada de Doceglennu é de 75 mg/m2 a cada três semanas, quando associada com capecitabina administrada por via oral a 1250 mg/m2 2 vezes ao dia (dentro de 30 minutos após a refeição) durante 2 semanas seguida por um período de 1 semana de descanso. Para a dose de capecitabina calculada de acordo com a área da superfície corpórea, veja as instruções recomendadas em bula pelo fabricante do produto à base de capecitabina. Em caso de terapia combinada, a posologia recomendada de Doceglennu é de 75 mg/m2 em associação com doxorrubicina (50 mg/m²) (vide Posologia e Modo de usar - Instruções de preparo).
Câncer de pulmão de não-pequenas células
Para os pacientes em tratamento de câncer de pulmão de não-pequenas células, a posologia recomendada de Doceglennu é de 75 a 100 mg/m2 em monoterapia, e de no máximo 75 mg/m2 em caso de associação com derivados de platina, administrada em infusão de 1 hora, a cada 3 semanas.
Câncer de ovário
Para pacientes em tratamento de câncer de ovário, a posologia recomendada de Doceglennu é de 100 mg/m2, administrada em infusão de 1 hora, a cada 3 semanas (vide Posologia e Modo de usar - Instruções de preparo). Os pacientes devem ser rigorosamente monitorados principalmente durante a primeira e a segunda infusões de Doceglennu, devido ao risco de reações de hipersensibilidade (vide Advertências e Precauções).
Câncer de próstata
Para câncer de próstata, a dose recomendada de Doceglennu é 75 mg/m2 a cada 3 semanas. Prednisona ou prednisolona 5 mg, via oral, duas vezes ao dia é administrada continuamente.
Adenocarcinoma gástrico
Para adenocarcinoma gástrico, a dose recomendada de Doceglennu é 75 mg/m2 com 1 hora de infusão, seguida por cisplatina 75 mg/m2, com 1 a 3 horas de infusão (ambos somente no dia 1), seguida por fluoruracila 750 mg/m2 por dia administrado com infusão contínua de 24 horas por 5 dias, iniciando no final da infusão da cisplatina. O tratamento é repetido a cada três semanas. Os pacientes devem receber pré-medicação com antieméticos e hidratação apropriada para a administração de cisplatina. O G-CSF profilático deve ser utilizado para aliviar o risco de toxicidades hematológicas (vide Posologia e Modo de usar - Ajuste posológico durante o tratamento).
Câncer de cabeça e pescoço
Pacientes devem receber pré-medicação com antieméticos e hidratação apropriada (antes e após a administração de cisplatina). A profilaxia para as infecções neutropênicas deve ser administrada. Todos os pacientes no braço contendo Doceglennu dos estudos TAX 323 e TAX 324 receberam antibióticos profiláticos.
• Indução por quimioterápicos seguida por radioterapia (TAX 323)
Para o tratamento de indução de carcinoma de células escamosas de cabeça e pescoço inoperável localmente avançado (SCCHN), a dose recomendada de Doceglennu é 75 mg/m2 por 1 hora de infusão seguida por cisplatina 75 mg/m2 superior a 1 hora no dia 1, seguida por fluoruracila em infusão contínua a 750 mg/m2 por dia por 5 dias. Este regime é administrado a cada 3 semanas por 4 ciclos. Após a quimioterapia, os pacientes devem receber radioterapia.
• Indução por quimioterápicos seguida por quimiorradioterapia (TAX 324)
Para o tratamento de indução em pacientes com SSCHN localmente avançado (não ressecável, cura cirúrgica baixa ou preservação do órgão), a dose recomendada de Doceglennu é 75 mg/m2 por 1 hora de infusão intravenosa no dia 1, seguida de cisplatina 100 mg/m2 administrada por 30 minutos a 3 horas de infusão, seguida por fluoruracila 1000 mg/m2/dia em infusão contínua do dia 1 ao dia 4.
Este regime é administrado a cada 3 semanas por 3 ciclos. Após a quimioterapia, os pacientes devem receber quimiorradioterapia.
Para modificações nas doses de cisplatina e fluoruracila, seguir as instruções da bula recomendadas pelo fabricante do produto.
Não há estudos dos efeitos de Doceglennu administrado por vias não recomendadas. Portanto, por segurança e para garantir a eficácia deste medicamento, a administração deve ser somente por via intravenosa.
Ajuste posológico durante o tratamento Informações Gerais
Doceglennu não deve ser administrado até que a contagem neutrofílica seja ≥ 1500 células/mm3.
Os pacientes que apresentem neutropenia febril, contagem de neutrófilos < 500 células/mm³ durante mais de uma semana, reações cutâneas severas ou cumulativas ou sinais e/ou sintomas neurossensoriais severos durante a terapia com Doceglennu, deverão ter a dose reduzida de 100 mg/m² para 75 mg/m² e/ou de 75 mg/m² para 60 mg/m². Caso o paciente continue a apresentar as mesmas reações com a dose de 60 mg/m², o tratamento deve ser descontinuado.
Alternativamente, pode-se utilizar tratamento profilático com G-CSF em pacientes com neutropenia febril ou infecção severa anteriores, com o intuito de manter a intensidade da dose.
Terapia associada com Doceglennu para câncer de mama
Profilaxia primária com G-CSF deve ser considerada em pacientes que recebem terapia adjuvante com Doceglennu, doxorrubicina e ciclofosfamida (TAC) para câncer de mama. As pacientes que apresentam neutropenia febril e/ou infecção neutropênica devem ter suas doses de Doceglenn
As pacientes que receberam terapia adjuvante com AC-TH ou TCH para câncer de mama operável cujos tumores superexpressam HER2 e que apresentam um episódio de infecção ou neutropenia febril devem receber G-CSF profilático em todos os ciclos subsequentes. Para um segundo episódio de infecção ou neutropenia febril, as pacientes devem continuar com G-CSF profilático e Doceglennu será reduzido de 100 mg/m2 a 75 mg/m2 (no regime AC-TH); Doceglennu será reduzido de 75 mg/m2 a 60 mg/m2 (no regime TCH).
Entretanto, na prática clínica, a neutropenia poderia ocorrer no ciclo 1. Deste modo, G-CSF deve ser utilizado em consideração ao risco neutropênico da paciente nas recomendações atuais. Dependendo do regime de tratamento, as pacientes que apresentam estomatite de Grau 3 ou 4 devem ter sua dose diminuída de 100 mg/m2 para 75 mg/m2 (no regime AC-TH) ou de 75 mg/m2 para 60 mg/m2 (no regime TCH).
Para as alterações na dose de capecitabina quando associada com Doceglennu, veja as instruções recomendadas em bula pelo fabricante do produto à base de capecitabina. Para pacientes desenvolvendo a primeira ocorrência de toxicidade de Grau 2 que persista até o próximo tratamento com Doceglennu
/capecitabina, postergar o tratamento até apresentar toxicidade de Grau 0-1 e retomar 100% da dose original. Para pacientes desenvolvendo a segunda ocorrência de toxicidade de Grau 2 ou a primeira ocorrência de toxicidade de Grau 3, em qualquer período durante o ciclo de tratamento, postergar o tratamento até apresentar toxicidade de Grau 0-1, depois retomar o tratamento com Doceglennu na dose de 55 mg/m2.
Para qualquer ocorrência subsequente de toxicidade ou qualquer toxicidade de Grau 4, descontinuar a dose de Doceglennu.
Para as alterações na dose de Doceglennu devido à insuficiência hepática, vide Advertências e Precauções.
Associação com Doceglennu para câncer de pulmão de não-pequenas células
Para pacientes que receberam inicialmente Doceglennu 75 mg/m2 em combinação com cisplatina e cujo nadir de contagem plaquetária durante o período anterior ao tratamento foi < 25.000 células/mm3 ou em pacientes que apresentaram neutropenia febril, ou em pacientes com toxicidades não-hematológicas sérias, a dose de Doceglennu em ciclos subsequentes deve ser reduzida para 65 mg/m2. Para os ajustes de dose da cisplatina, seguir as instruções da bula recomendadas pelo fabricante do produto.
Terapia associada de Doceglennu com cisplatina e fluoruracila para câncer gástrico ou câncer de cabeça e pescoço
Os pacientes tratados com Doceglennu em associação com cisplatina e fluoruracila devem receber antieméticos e hidratação apropriada de acordo com as normas institucionais atuais. O G-CSF deve ser administrado para aliviar o risco de complicações relacionadas à neutropenia.
Apesar da utilização do G-CSF, se ocorrer um episódio de neutropenia febril, neutropenia prolongada ou infecção neutropênica, a dose de Doceglennu deve ser reduzida de 75 para 60 mg/m2. Caso ocorrerem episódios subsequentes de neutropenia complicada, a dose de Doceglennu deve ser reduzida de 60 para 45 mg/m2. No caso de trombocitopenia grau 4, a dose de Doceglennu deve ser reduzida de 75 para 60 mg/m2. Os pacientes não devem ser tratados novamente com ciclos subsequentes de Doceglennu até que os neutrófilos se restabeleçam para um nível > 1.500 células/mm3 e as plaquetas se restabeleçam para um nível > 100.000 células/mm3. Interromper o tratamento se estas toxicidades persistirem (vide Advertências e Precauções).
Para ajustes na dose de cisplatina e fluoruracila, verificar na bula dos respectivos produtos.
Populações especiais
Pacientes com insuficiência hepática: com base nos dados farmacocinéticos obtidos com a administração de 100 mg/m2 de Doceglennu em monoterapia, a dose recomendada para pacientes que apresentam simultaneamente aumento de transaminases (TGP e/ou TGO) > 1,5 vezes o limite superior da normalidade e de fosfatase alcalina > 2,5 vezes o limite superior da normalidade é de 75 mg/m2. Em pacientes com nível plasmático de bilirrubina maior do que o limite superior da normalidade e/ou níveis de TGP e TGO > 3,5 vezes o limite superior da normalidade associado a níveis de fosfatase alcalina > 6 vezes o limite superior da normalidade, não se deve realizar ajuste posológico e Doceglennu não deve ser utilizado, a menos que estritamente indicado. Não existem dados disponíveis em pacientes com insuficiência hepática tratados com Doceglennu em terapia combinada.
Crianças: a eficácia e segurança da administração de Doceglennu em crianças ainda não foram estabelecidas.
Idosos: com base na análise farmacocinética desta população, não há necessidade de instruções especiais na administração de Doceglennu em idosos. Para a redução na dose de capecitabina quando associada com docetaxel, ver as instruções recomendadas em bula pelo fabricante do produto à base de capecitabina.
9. REAÇÕES ADVERSAS
As reações adversas consideradas possíveis ou provavelmente relacionadas à administração de Doceglennu foram observadas em pacientes tratados em monoterapia ou em associação, com parâmetros da função hepática normais no estado basal.
A seguinte taxa de frequência é utilizada para as reações adversas a seguir:
Reação muito comum (≥ 10%), comum (≥ 1% e < 10%), incomum (≥ 0,1% e < 1%), rara (≥ 0,01% e < 0,1%), muito rara ( < 0,01%).
Reações hematológicas
Supressão da medula óssea e outras reações adversas hematológicas ao Doceglennu incluem:
• Muito comuns: neutropenia (96,6% dos casos) foi a reação adversa mais frequente em pacientes que não receberam fator estimulador de colônias de granulócitos e mostrou-se reversível e não cumulativa (atingiu-se o nadir em média no sétimo dia e a duração mediana da neutropenia severa (76,4%, < 500 células/mm3) foi de sete dias); neutropenia grau 3/4 (32%) em pacientes tratados com Doceglennu e trastuzumabe; neutropenia febril (11,8%), episódios infecciosos (20%) e anemia ( < 11g/dL): 90,4%) foram relatadas em pacientes tratados com Doceglennu em monoterapia na dose de 100 mg/m2.
• Comuns: infecções severas (4,6%) associadas com a contagem de neutrófilos < 500 células/mm3, trombocitopenia < 100.000 células/mm3 (7,8%), episódios de hemorragia (2,4%) (raramente associada com trombocitopenia severa ( < 50.000 células/mm3), episódios infecciosos severos (5,7%, incluindo sepse e pneumonia, fatal em 1,7%) e anemia severa [(8,9% ( < 8g/dL)] foram relatadas em pacientes tratados com Doceglennu em monoterapia na dose de 100 mg/m2);
• Incomum: trombocitopenia severa (0,2%).
Reações de hipersensibilidade
• Muito comuns: reações de hipersensibilidade (25,9%), ocorrendo geralmente dentro de poucos minutos após o início da infusão de Doceglennu, usualmente são de intensidade leve a moderada. Os sintomas frequentemente relatados com o uso de Doceglennu em monoterapia na dose de 100 mg/m2 foram rubor, rash com ou sem prurido, aperto no peito, dor lombar, dispneia e febre medicamentosa ou calafrio.
• Comuns: reações de hipersensibilidade severas (5,3%) que desapareceram após descontinuação da infusão e emprego de terapia apropriada.
Reações cutâneas
• Muito comuns: alterações nas unhas (27,9%), caracterizadas pela hipo ou hiperpigmentação, dor e onicólise; reações cutâneas reversíveis (56,6%) geralmente consideradas de intensidade leve a moderada.
As reações foram caracterizadas por rash, incluindo erupções localizadas principalmente nos pés, mãos (incluindo síndrome mão e pé severa), mas também nos braços, face ou tórax, e frequentemente associadas com prurido. Geralmente ocorreram erupções dentro de uma semana após a infusão de Doceglennu;
• Comuns: sintomas severos como erupção seguida por descamação, que raramente causaram a interrupção ou descontinuação do tratamento com Doceglennu em monoterapia na dose de 100 mg/m2, foram relatados com menor frequência (5,9%).
Em alguns casos vários fatores como infecções simultâneas, uso concomitante de medicamentos e doenças pré-existentes pode ter contribuído para o desenvolvimento destas reações.
Retenção hídrica
Reações adversas relacionadas à retenção hídrica foram obtidas de 92 pacientes tratados com 100 mg/m2 de Doceglennu em monoterapia, que também receberam 3 dias de administração de pré-medicação, por meio de análise retrospectiva.
•Muito comum: retenção hídrica em 64,1% dos pacientes que receberam 3 dias de pré-medicação;
• Comum: retenção hídrica severa (6,5%) em pacientes que receberam 3 dias de pré-medicação.
Foram relatados eventos como edema periférico e com menor frequência derrame pleural, derrame pericárdico, ascite e aumento de peso. O edema periférico geralmente inicia-se nas extremidades inferiores e pode generalizar-se com um aumento de peso igual ou superior a 3 kg. A retenção hídrica é cumulativa em incidência e severidade (vide Advertências e Precauções).
Em pacientes tratados com 100 mg/m2 de Doceglennu em monoterapia, a dose cumulativa mediana para interrupção do tratamento foi superior a 1.000 mg/m2 e o tempo mediano para a reversibilidade da retenção hídrica foi de 16,4 semanas (intervalo de 0 a 42 semanas). Em pacientes tratados com pré- medicação, o início da retenção moderada e severa é retardado (dose cumulativa mediana: 818,9 mg/m2), quando comparados aos pacientes sem pré-medicação (dose cumulativa mediana: 489,7 mg/m2); contudo, relatou-se retenção hídrica em alguns pacientes durante os primeiros ciclos do tratamento. A retenção hídrica não tem sido acompanhada por episódios agudos de oligúria ou hipotensão.
Reações gastrintestinais
As seguintes reações gastrintestinais foram relatadas em pacientes que receberam Doceglennu em monoterapia na dose de 100 mg/m2:
• Muito comuns: náusea (40,5%), vômito (24,5%), diarreia (40,6%), anorexia (16,8%), estomatite (41,8%), alteração do paladar (10,1%);
• Comuns: náusea severa (4%), vômito severo (3%), diarreia severa (4%), dor abdominal (7,3%, sendo 1% dos casos severa), constipação (9,8%), estomatite severa (5,3%), esofagite (1%) sangramento intestinal (1,4%);
•Incomuns: constipação severa (0,2%), esofagite severa (0,4%), sangramento intestinal severo (0,3%);
• Rara: alteração severa do paladar (0,07%).
Reações neurológicas
• Muito comuns: sinais e/ou sintomas neurossensoriais de intensidade leve a moderada ocorreram em 50% dos pacientes no braço de Doceglennu 100 mg/m2 em monoterapia; eventos neuromotores (13,8%) principalmente caracterizados por fraqueza;
•Comuns: sintomas neurossensoriais severos (parestesia, disestesia, dor incluindo ardor) foram observados em 4,1% dos pacientes com câncer de mama metastático, necessitando interrupção do tratamento em 2% dos casos; eventos neuromotores severos (4% dos casos) principalmente caracterizados por fraqueza.
Quando estes sintomas ocorrerem, a dose deve ser ajustada. Em caso de persistência dos sintomas, o tratamento deve ser interrompido (vide Posologia e Modo de usar). Pacientes que apresentaram neurotoxicidade nos estudos clínicos e para os quais a informação de acompanhamento sobre a resolução completa do evento está disponível, apresentaram reversão espontânea dos sintomas com uma média de 81 dias do início (variação: 0 a 741 dias).
Reações cardiovasculares
Os eventos cardiovasculares em pacientes que receberam Doceglennu 100 mg/m2 em monoterapia consistiram em:
· Comuns: hipotensão (3,8%), disritmia (4,1%) e hipertensão (2,4%).
•Incomuns: insuficiência cardíaca (0,5%); insuficiência cardíaca sintomática (2,2% das pacientes que receberam Doceglennu e trastuzumabe comparado a 0% das pacientes tratadas somente com Doceglennu).
No braço com Doceglennu e trastuzumabe, 64% receberam anteriormente uma antraciclina como terapia adjuvante, comparada com 55% no braço com deocetaxel em monoterapia.
Reações Hepáticas
• Comuns: em pacientes tratados com 100 mg/m2 de Doceglennu em monoterapia, foram observados aumentos dos níveis plasmáticos das transaminases (TGP/TGO), bilirrubina e fosfatase alcalina, superiores a 2,5 vezes o limite superior da normalidade, em menos de 5% dos pacientes.
Outros
Em pacientes tratados com Doceglennu 100 mg/m2 em monoterapia, foram relatadas as seguintes reações:
· Muito comuns: alopecia (79%); astenia (62,6% sendo severa em 11,2% dos casos); mialgia (20%); dispneia (16,1%); dor generalizada ou localizada (16,5%);
· Comuns: artralgia (8,6%); dispnéia severa (2,7%); dor torácica (4,5%) sem qualquer envolvimento respiratório ou cardíaco; reações no local de infusão, geralmente leves, ocorreram em 5,6% dos pacientes e consistiram de hiperpigmentação, inflamação, vermelhidão ou secura da pele, flebite ou extravasamento e inchaço da veia;
• Incomuns: alopecia severa (0,5%); dor generalizada ou localizada severa (0,8%); dor torácica severa (0,4%) sem qualquer envolvimento respiratório ou cardíaco.
De uma forma geral, os padrões de eventos adversos observados nos pacientes tratados com Doceglennu em terapia combinada com doxorrubicina são similares àqueles observados em pacientes tratados com Doceglennu em monoterapia.
Terapia combinada com Doceglennu no tratamento adjuvante do câncer de mama operável linfonodo-positivo, e linfonodo-negativo de alto risco - Eventos adversos clinicamente importantes relacionados ao tratamento em pacientes recebendo Doceglennu em associação com doxorrubicina e ciclofosfamida (TAX 316)
Os dados a seguir referem-se a eventos adversos emergentes relacionados ao tratamento (TEAEs) observado em 744 pacientes com câncer de mama linfonodo-positivo que foram tratados com Doceglennu 75 mg/m2 a cada 3 semanas em associação com doxorrubicina e ciclofosfamida (TAX 316). Tais eventos estão classificados em quaisquer eventos e eventos de Grau 3/4 (G3/4
•Muito comuns: anemia (92,1%), neutropenia (71,8%, G3/4: 65,3%), febre na ausência de infecção (36,6%), trombocitopenia (39,5%), infecção (29,2%), neutropenia febril (24,6%), infecção neutropênica (17,3%), edema periférico (26,6%), ganho de peso (12,5%), neuropatia sensorial periférica (23,1%), alopecia (97,7%), alterações cutâneas (16,1%), alterações ungueais (18,4%), náusea (80,4%), estomatite (68,4%), vômito (42,5%), diarreia (30,9%), alteração do paladar (27,3%), constipação (24,5%), anorexia (19,9%), amenorreia (26,2%), astenia (79,2%; G3/4: 11,0%), mialgia (22,8%), artralgia (15,1%), lacrimejamento (10,1%), fogacho (21,4%);
•Comuns: reações de hipersensibilidade (9,0%), anemia G3/4 (4,2%), trombocitopenia G3/4 (2,0%), infecção G3/4 (3,2%), neuropatia periférica motora (2,7%), náusea G3/4 (5,1%), estomatite G3/4 (7,1%), vômito G3/4 (4,3%), diarreia G3/4 (3,2%), anorexia G3/4 (2,2%), dor abdominal (6,5%), tosse (3,0%), arritmia cardíaca (2,8%), hipotensão (1,5%), conjuntivite (3,8%), perda de peso (2,6%), infecção neutropênica G3/4 (3,6%);
· Incomuns: edema periférico G3/4 (0,4%), linfedema (0,3%), perda de peso G3/4 (0,3%), síncope (0,4%), alterações cutâneas G3/4 (0,7%), alterações ungueais G3/4 (0,4%), alteração do paladar G3/4 (0,7%), constipação G3/4 (0,4%), dor abdominal G3/4 (0,5%), arritmia cardíaca G3/4 (0,3%), flebite (0,9%), mialgia G3/4 (0,8%), artralgia G3/4 (0,4%), lacrimejamento G3/4 (0,1%), reações de hipersensibilidade G3/4 (0,9%), sonolência (0,3%), fogacho G3/4 (0,9%).
Febre e Infecção
Foram observadas as seguintes reações adversas nos pacientes do grupo TAC durante o período do estudo:
•Muito comuns: febre na ausência de infecção (36,6%), infecção (29,2%);
•Comuns: infecção G3/4 (3,2%).
Não houve óbito devido à sepse durante o período do estudo.
Eventos gastrintestinais
Além dos eventos gastrintestinais mencionados acima, 7 pacientes apresentaram perfuração intestinal ampla/enterite/colite. Dois desses pacientes requereram descontinuação do tratamento; não houve óbitos devido a esses eventos durante o período do estudo.
Eventos cardiovasculares
Foram relatadas as seguintes reações cardiovasculares emergentes devido ao tratamento durante o período de estudo:
•Comuns: arritmia, todos os graus (6,2%), hipotensão, todos os graus (1,9%) e insuficiência cardíaca congestiva (3,5%). Vinte e seis pacientes do grupo TAC desenvolveram insuficiência cardíaca congestiva durante o período do estudo, sendo a maioria dos casos reportada no período de acompanhamento. Dois pacientes do grupo TAC e 4 pacientes do grupo FAC faleceram devido à insuficiência cardíaca congestiva. O risco de insuficiência cardíaca congestiva foi mais alto no grupo TAC no primeiro ano de uso do medicamento.
Leucemia Mieloide Aguda (LMA)/Síndrome Mielodisplástica
Após 10 anos de acompanhamento do estudo TAX316, LMA ocorreu em 3 dos 744 (0,4%) pacientes que receberam Doceglennu, doxorrubicina e ciclofosfamida e em 1 dos 736 (0,1%) pacientes que receberam fluoruracila, doxorrubicina e ciclofosfamida. Um paciente do grupo TAC faleceu devido a LMA durante o período de acompanhamento (mediana de acompanhamento de 8 anos). Ocorreu Síndrome Mielodisplástica em 2 dos 744 (0,3 %) pacientes que receberam Doceglennu, doxorrubicina e ciclofosfamida e em um dos 736 (0,1 %) pacientes que receberam fluoruracila, doxorrubicina e ciclofosfamida.
Outras reações persistentes
No estudo TAX316, os eventos adversos mais comuns que iniciaram durante o período de tratamento e persistiram até o período de acompanhamento do grupo TAC estão descritos a seguir (mediana de acompanhamento de 8 anos). A maioria dos eventos adversos persistentes foi resolvida durante o período de acompanhamento. O texto a seguir apresenta a frequência das reações persistentes dos pacientes (n = 744) que receberam Doceglennu (75 mg/m2) em combinação com doxorrubicina (50 mg/m2) e ciclofosfamida (500 mg/m2) (TAX 316).
· Muito comuns: (Eventos persistentes do início do tratamento até o período de acompanhamento) alopecia: 687 pacientes (92,3%); asternia: 236 pacientes (31,7%); amenorreia: 202 pacientes (27,2%); edema periférico: 119 pacientes (16,0 %); neuropatia sensorial periférica: 84 pacientes (11,3%). (Eventos em andamento até o fim do período de acompanhamento) amenorreia: 121 pacientes (16,3%).
•Comuns: (Eventos persistentes do início do tratamento até o período de acompanhamento) linfedema: 11 pacientes (1,5%). (Eventos em andamento até o fim do período de acompanhamento) alopecia: 29 pacientes (3,9%); astenia: 29 pacientes (3,9%); edema periférico: 19 pacientes (2,6%); neuropatia sensorial periférica: 10 pacientes (1,3%).
• Incomum: (Eventos em andamento até o fim do período de acompanhamento) linfedema: 6 pacientes (0,8%).
Eventos adversos clinicamente importantes relacionados ao tratamento em pacientes recebendo Doceglennu em associação com doxorrubicina e ciclofosfamida (GEICAM 9805)
O texto a seguir apresenta eventos adversos emergentes relacionados ao tratamento (TEAEs) observado em 532 pacientes com câncer de mama linfonodo-negativo que foram tratados com Doceglennu 75 mg/m2 a cada 3 semanas em associação com doxorrubicina 50 mg/m2 e ciclofosfamida 500 mg/m2 (GEICAM 9805). Tais eventos estão classificados em quaisquer eventos e eventos de Grau 3/4 (G3/4):
• Muito comuns: anemia (94,7%), neutropenia (71,1%, G3/4: 50,8%), pirexia (febre na ausência de infecção) (17,9%), trombocitopenia (12,0%), infecção (15,4%), edema periférico (16,4%), neuropatia sensorial periférica (14,7%), alopecia (95,3%), alterações cutâneas (16,5%), alterações ungueais (19,7%), náusea (70,7%), estomatite (54,5%), vômito (54,3%), diarreia (26,3%), disgeusia (15,8%), constipação (19,7%), anorexia (16,2%), dor abdominal (12,0%), amenorreia (20,3%), fogacho (13,3%), astenia (72,0%), mialgia (19,4%), artralgia (16,4%), conjuntivite (20,1%);
• Comuns: anemia G3/4 (1,3%), trombocitopenia G3/4 (1,1%), infecção G3/4 (1,1%), neutropenia febril (9,6%), infecção neutropênica (6,6%, G3/4: 1,3%), reações de hipersensibilidade (3,6%), ganho de peso (3,4%), neuropatia motora periférica (2,3%), náusea G3/4 (4,9%), estomatite G3/4 (4,5%), vômito G3/4 (4,1%), diarreia G3/4 (3,6%), tosse (2,1%), arritmia (2,1%), flebite (1,1%), astenia G3/4 (8,5%), aumento na lacrimação (5,1%);
• Incomuns: reações de hipersensibilidade G3/4 (0,2%), linfedema (0,8%), perda de peso (0,8%), neuropatia sensorial periférica G3/4 (0,2%), sonolência (0,2%), neurotoxicidade (0,6%), síncope (0,6%), alopecia G3/4 (0,2%), alterações cutâneas G3/4 (0,6%), alterações ungueais G3/4 (0,6%), disgeusia G3/4 (0,6%), constipação G3/4 (0,8%), anorexia G3/4 (0,6%), dor abdominal G3/4 (0,2%), arritmia G3/4 (0,2%), hipotensão (0,8%), mialgia G3/4 (0,6%), conjuntivite G3/4 (0,2%).
Os dados a seguir demonstram que a incidência de neutropenia grau 4, neutropenia febril e infecção neutropênica foi diminuída em pacientes que receberam profilaxia primária com G-CSF após obrigatoriedade desse tratamento no braço TAC.
Complicações neutropênicas em pacientes recebendo TAC com ou sem profilaxia primária com G- CSF (GEICAM 9805)
Com profilaxia primária com G-CSF: N = 421, n(%):
•Muito comum: neutropenia grau 4: 135 (32,1%);
• Comuns: neutropenia febril: 23 (5,5%); infecção neutropênica: 21 (5,0%); infecção neutropênica grau 3/4: 5 (1,2%).
Sem profilaxia primária com G-CSF: N = 111, n(%):
• Muito comuns: neutropenia grau 4: 104 (93,7%); neutropenia febril: 28 (25,2%); infecção neutropênica: 14 (12,6%);
• Comum: infecção neutropênica grau 3/4: 2 (1,8%).
Dos 532 pacientes tratados com TAC, 28,2% apresentaram eventos adversos severos e relacionados ao tratamento. Reduções de dose devido à toxicidade hematológica ocorreram em 1,5% dos ciclos. 4,7% dos pacientes descontinuaram o tratamento devido a eventos adversos; febre na ausência de infecção e neutropenia sendo as razões mais comuns para descontinuação. Não houve óbito no período de 30 dias após o último tratamento do estudo. Nenhum óbito foi considerado como relacionado ao Doceglennu.
Febre e infecção
Não houve óbitos devido à sepse.
Eventos gastrintestinais
Não foram relatados casos de colite/enterite/perfuração ampla do intestino. Outros eventos gastrintestinais estão relatados acima.
Eventos cardiovasculares
Três pacientes (0,6%) desenvolveram insuficiência cardíaca congestiva durante o período de acompanhamento. No final do período de acompanhamento (tempo mediano de acompanhamento de 10 anos e 5 meses), nenhum paciente apresentou insuficiência cardíaca congestiva no grupo TAC e um paciente faleceu por causa da cardiomiopatia.
Leucemia Aguda / Síndrome Mielodisplástica
Durante o período de acompanhamento (tempo mediano de acompanhamento de 10 anos e 5 meses), leucemia aguda ocorreu em 1 de 532 (0,2%) pacientes no braço TAC. Não houve casos relatados de pacientes no braço FAC. Nenhum paciente foi diagnosticado com síndrome mielodisplástica em nenhum dos braços de tratamento.
Reações persistentes
No estudo GEICAM 9805, os eventos adversos mais comuns, que iniciaram durante o período de tratamento e persistiram no período de acompanhamento do grupo TAC, foram descritos a seguir (mediana de tempo de acompanhamento 10 anos e 5 meses). A maioria dos eventos adversos persistentes foram resolvidos durante o período de acompanhamento. O texto a seguir apresenta a frequência das reações persistentes dos pacientes (n = 532) que receberam Doceglennu (75 mg/m2) em combinação com doxorrubicina (50 mg/m2) e ciclofosfamida (500 mg/m2) (GEICAM 9805).
• Comuns: (Eventos persistentes do início do tratamento até o período de acompanhamento) alopecia*: 49 pacientes (9,2%); astenia: 12 pacientes (2,3%); amenorreia: 18 pacientes (3,4%); neuropatia sensorial periférica: 10 pacientes (1,9%). (Andamento durante o período de acompanhamento) amenorreia: 7 pacientes (1,3%).
•Incomum: (Eventos persistentes do início do tratamento até o período de acompanhamento) linfedema: 5 pacientes (0,9%); edema periférico: 4 pacientes (0,8%). (Andamento durante o período de acompanhamento) alopecia: 3 pacientes (0,6%); astenia 2 pacientes (0,4%); linfedema 4 pacientes (0,8%) e neuropatia sensorial periférica: 3 pacientes (0,6%)
• Muito raro: (Andamento durante o período de acompanhamento) alopecia: 3 pacientes (0,6%); astenia 2 pacientes (0,4%); linfedema 4 pacientes (0,8%); neuropatia sensorial periférica: 3 pacientes (0,6%).
Obs: Nenhum paciente apresentou edema periférico durante o período de acompanhamento: 0 pacientes (0,0%).
*Relacionada ao fármaco do estudo iniciou ou piorou durante o período de acompanhamento em 42 pacientes (7,9%).
Terapia combinada com Doceglennu e capecitabina para câncer de mama
Para a terapia com associação de Doceglennu e capecitabina, os efeitos indesejáveis mais frequentes relacionados ao tratamento (≥ 5%) relatados no estudo de fase III em pacientes com câncer de mama com falha ao tratamento com antraciclina estão apresentados a seguir:
Resumo de eventos adversos ao menos remotamente relatados em ≥ 5% de pacientes tratados em associação com Doceglennu e capecitabina em estudo com 251 pacientes. Tais eventos estão classificados em qualquer grau de evento e eventos de Grau 3/4 (G3/4):
• Muito comuns: estomatite (67%, G3/4: 18%), diarreia (64%, G3/4: 14%), náusea (43%), vômito (33%), constipação (14%), dor abdominal (14%), dispepsia (12%), síndrome mão-pé (63%, G3: 24%), alopecia (41%), alterações nas unhas (14%), astenia (23%), pirexia (21%), fadiga (21%), fraqueza (13%), alteração do paladar (15%), parestesia (11%), anorexia (12%), diminuição do apetite (10%), lacrimejamento aumentado (12%), mialgia (14%), artralgia (11%), edema do membro inferior (14%), dor de garganta (11%);
• Comuns: náusea (G3/4: 6%), vômito (G3/4: 4%), constipação (G3/4: 1%), dor abdominal (G3/4: 2%), dor abdominal superior (9%), boca seca (5%), alopecia (G3/4: 6%), alterações nas unhas (G3/4: 2%), dermatite (8%), rash eritematoso (8%), descoloração das unhas (6%), onicólise (5%, G3/4: 1%), astenia (G3/4: 3%), pirexia (G3/4: 1%), fadiga (G3/4: 4%), fraqueza (G3/4: 1%), dor no membro (9%), letargia (6%), dor (6%), vertigem (9%), dor de cabeça (7%), neuropatia periférica (5%), anorexia (G3/4: 1%), desidratação (8%, G3/4: 2%), diminuição de peso (6%), mialgia (G3/4: 2%), artralgia (G3/4: 1%), dor nas costas (7%, G3/4: 1%), edema do membro inferior (G3/4: 1%), dor de garganta (G3/4: 2%), dispneia (7%, G3/4: 1%), tosse (6%), epistaxe (5%), candidíase oral (6%);
· Incomuns: rash eritematoso (G3/4: < 1), dor no membro (G3/4: < 1), alteração do paladar (G3/4:
< 1), parestesia (G3/4: < 1), dor de cabeça (G3/4: < 1), tosse (G3/4: < 1), epistaxe (G3/4: < 1), candidíase oral (G3/4: < 1).
As frequentes anormalidades de grau 3 e 4, quando da combinação de Doceglennu e capecitabina, foram:
• Muito comuns: neutropenia (63%), anemia (10%);
• Comuns: trombocitopenia (3%), hiperbilirrubinemia (9%).
Terapia combinada com Doceglennu e trastuzumabe para câncer de mama
Os dados a seguir mostram os eventos adversos (todos os graus), que foram relatados em ≥ 10% de pacientes tratados com Doceglennu e trastuzumabe para câncer de mama metastático, em estudo com 92 pacientes:
• Muito comuns: astenia (45%), fadiga (24%), inflamação na mucosa (23%), pirexia (29%), dor (12%), dor no peito (11%), influenza como doença (12%), calafrios (11%), alopecia (67%), alterações nas unhas (17%), rash (24%), eritema (23%), edema periférico (40%), aumento de peso (15%), linfedema (11%), náusea (43%), diarreia (43%), vômito (29%), constipação (27%), estomatite (20%), dor abdominal (12%), dispepsia (14%), parestesia (32%), dor de cabeça (21%), disgeusia (14%), hipoestesia (11%), neutropenia febril* ou sepse netropênica (23%), nasofaringite (15%), mialgia (27%), artralgia (27%), dor nas extremidades (16%), dor nas costas (10%), dor óssea (14%), tosse (13%), dispneia (14%), dor faringolaríngea (16%), epistaxe (18%), rinorreia (12%), lacrimejamento aumentado (21%), conjuntivite (12%), anorexia (22%), insônia (11%), toxicidade às unhas (11%);
• Comum: letargia (7%).
Esses números incluem pacientes com termos preferidos neutropenia febril, sepse neutropênica ou neutropenia que foi associado com febre (e uso de antibiótico).
Houve uma incidência aumentada de eventos adversos graves (SAEs) (40% versus 31%) e eventos adversos (AEs) de grau 4 (34% versus 23%) no braço associado comparado à monoterapia com Doceglennu.
Toxicidade cardíaca
Foi relatada insuficiência cardíaca sintomática em 2,2% das pacientes que receberam Doceglennu e trastuzumabe comparado a 0% das pacientes tratadas somente com Doceglennu. No braço com Doceglennu e trastuzumabe, 64% receberam anteriormente uma antraciclina como terapia adjuvante, comparada com 55% no braço com Doceglennu isolado.
Toxicidade hematológica
Foi relatada neutropenia grau 3/4 em 32% das pacientes tratadas com Doceglennu e trastuzumabe.
Terapia combinada com Doceglennu para o tratamento adjuvante de pacientes com câncer de mama operável cujos tumores superexpressam HER2 e que receberam ou AC-TH ou TCH - Eventos adversos (AEs) relacionados ao tratamento do estudo, ocorrendo em qualquer período durante o estudo: segurança populacional (incidência ≥ 5% para os AEs não cardíacos; incidência ≥ 1% para os AEs cardíacos)
Pacientes que receberam AC-TH:
• Muito comuns: alopecia (98,0%), hemoglobinab (97,0%), náusea (87,2%), leucócitosb (87,0%, G3/4: 60,2%), neutrófilosb (86,3%, G3/4: 71,3%), fadiga (81,3%), estomatite/faringite (65,0%), vômito (55,3%), TGP (ALT)b (54,2%), retenção hídricab,c (52,2%), mialgia (50,9%), diarreia (45,3%), neuropatia sensorial (44,8%), TGO (AST)b (42,5%), artralgia (39,7%), alterações nas unhas (39,6%), plaquetasb (32,8%), fluxo menstrual irregular (29,1%, G3/4: 19,9%), alteração do paladar (27,2%), constipação (27,1%), rash/descamação (25,9%), fogachos/rubor (21,5%), lacrimejamento (21,3%), fosfatase alcalinab (19,3%), anorexia (19,2%), dispepsia/azia (19,0%), dor de cabeça (16,4%), dispneia (15,5%), aumento de peso (14,9%), infecção sem neutropenia (12,6%), dor abdominal ou cólica (12,4%), insônia (11,1%), neutropenia febril (10,9%, G3/4: 10,9%), febre (sem neutropenia) (10,9%);
· Comuns: hemoglobinab (G3/4: 3,2%), nausea (G3/4: 5,3%), fadiga (G3/4: 6,6%), estomatite/faringite (G3/4: 3,0%), vômito (G3/4: 6,4%), TGP (ALT)b (G3/4: 1,8%), retenção hídricab,c (G3/4: 1,5%), mialgia (G3/4: 4,9%), diarreia (G3/4: 5,1%), neuropatia sensorial (G3/4: 1,9%), artralgia (G3/4: 3,0%), plaquetasb (G3/4: 1,2%), rash/descamação (G3/4: 1,3%), dispneia (G3/4: 1,5%), infecção sem neutropenia (G3/4: 1,9%), reação alérgica/hipersensibilidade (9,8%, G3/4: 1,4%), dor óssea (9,7%), infecção com neutropenia (9,2%, G3/4: 9,2%), dord (8,1%),
conjuntivite (8,1%), vertigem/tonteira (7,3%), creatininab (6,7%), reação mão-pé (6,7%, G3/4:
1,4%), epistaxe (6,7%), perda de peso (6,6%), pele seca (6,5%), tosse (6,2%), rinited (6,0%), tremor/calafrio (5,9%), infecção com contagem absoluta de neutrófilos desconhecida (5,5%, G3/4: 5,5%), neuropatia-motora (5,3%), bilirrubinab (5,1%), reação no local da injeção (4,7%), boca seca (4,0%), função cardíaca ventricular esquerda (3,5%, palpitações (3,4%) taquicardia sinusal (1,8%);
• Incomuns: TGO (AST)b (G3/4: 0,8%) constipação (G3/4: 0,9%), lacrimejamento (G3/4: 0,3%), fosfatase alcalinab (G3/4: 0,3%), anorexia (G3/4: 0,5%), dispepsia/azia (G3/4: 0,3%), dor de cabeça (G3/4: 0,6%), aumento de peso (G3/4: 0,3%), dor abdominal ou cólica (G3/4: 0,4%), insônia (G3/4: 0,1%), febre (sem neutropenia) (G3/4: 0,4%), dor óssea (G3/4: 0,4%), dord (G3/4: 0,4%), vertigem/tonteira (G3/4: 0,7%), creatininab (G3/4: 0,5%), tosse (G3/4: 0,2%), rinited (G3/4: 0,1%), neuropatia-motora (G3/4: 0,4%), bilirrubinab (G3/4: 0,4%), reação no local da injeção (G3/4: 0,1%), função cardíaca ventricular esquerda (G3/4: 0,5%), hipotensão (0,9%).
Pacientes que receberam TCH:
• Muito comuns: alopecia (95,8%), hemoglobinab (96,3%), náusea (80,8%), leucócitosb (83,0%, G3/4: 48,0%), neutrófilosb (81,3%, G3/4: 65,9%), fadiga (80,4%), estomatite/faringite (51,8%), vômito (39,4%), TGP (ALT)b (53,1%), retenção hídricab,c (51,0%), mialgia (33,4%), diarreia (55,8%), neuropatia sensorial (29,9%), TGO (AST)b (38,0%), artralgia (21,8%), alterações nas unhas (23,3%), plaquetasb (63,2%), fluxo menstrual irregular (32,2%, G3/4: 21,4%), alteração do paladar (29,5%), constipação (22,0%), rash/descamação (22,8%), fogachos/rubor (18,2%), lacrimejamento (10,3%), fosfatase alcalinab (20,4%), anorexia (21,0%), dispepsia/azia (20,0%), dor de cabeça (15,2%), dispnéia (14,9%), aumento de peso (14,6%), dor abdominal ou cólica (13,4%), reação alérgica/hipersensibilidade (13,2%);
• Comuns: hemoglobinab (G3/4: 5,8%), náusea (G3/4: 4,6%), fadiga (G3/4: 6,9%), estomatite/faringite (G3/4: 1,4%), vômito (G3/4: 3,0%), TGP (ALT)b (G3/4: 2,4%), retenção hídricab,c (G3/4: 1,4%), mialgia (G3/4: 1,4%), diarreia (G3/4: 4,9%), TGO (AST)b (G3/4: 1,0%), artralgia (G3/4: 1,0%), plaquetasb (G3/4: 5,4%), dispneia (G3/4: 1,7%), infecção sem neutropenia (9,3%, G3/4: 1,5%), insônica (8,8%), neutropenia febril (9,8%, G3/4: 9,8%), febre sem neutropenia (6,6%), reação alérgica/hipersensibilidade (G3/4: 2,5%), dor nos ossos (6,3%), infecção com neutropenia grau 3/4 (7,7%, G3/4: 7,7%), dord (5,4%), conjuntivite (3,3%), vertigem/tonteira (6,6%), creatininab (9,7%), reação mão-pé (2,7%), epistaxe (9,8%), perda de peso (5,3%), pele seca (3,9%), tosse (3,4%), rinited (4,5%), tremor/calafrio (5,1%), infecção com contagem absoluta de neutrófilos desconhecida (3,6%, G3/4: 3,6%), neuropatia-motora (3,6%), bilirrubinab (5,8%), reação no local da injeção (5,8%), boca seca (2,7%), função cardíaca ventricular esquerda (1,4%), palpitações (4,5%) taquicardia sinusal (2,2%), hipotensão (1,2%);
• Incomuns: neuropatia sensorial (G3/4: 0,6%), constipação (G3/4: 0,6%), rash/descamação (G3/4: 0,4%), fosfatase alcalinab (G3/4: 0,3%), anorexia (G3/4: 0,5%), dispepsia/azia (G3/4: 0,4%), dor de cabeça (G3/4: 0,3%), aumento de peso (G3/4: 0,2%), dor abdominal ou cólica (G3/4: 0,5%), febre (sem neutropenia) (G3/4: 0,3%), dor óssea (G3/4: 0,1%), vertigem/tonteira (G3/4: 0,4%), creatininab (G3/4: 0,6%), epistaxe (G3/4: 0,4%), perda de peso (G 3/4: 0,1%), neuropatia-motora (G3/4: 0,3%), bilirrubinab (G3/4: 0,4%), reação no local da injeção (G3/4: 0,2%), função cardíaca ventricular esquerda (G3/4: 0,1%), hipotensão (G3/4: 0,2%).
AC-TH = doxorrubicina e ciclofosfamida, seguida de Doceglennu em associação com trastuzumabe TCH = Doceglennu em associação com trastuzumabe e carboplatina
b Independente de causalidade
c Eventos adversos (AEs) retenção hídrica são definidos como somente edema ou somente aumento de peso ou somente edema pulmonar ou aumento de peso e edema ou edema e edema pulmonar ou edema + aumento de peso + edema pulmonar.
Retenção de líquido corresponde a inchaço no termo NCI-CTC.
d Termo COSTART.
Os 3 anos de incidência cumulativa de todos os eventos cardíacos sintomáticos foi 2,36% e 1,16% nos braços AC-TH e TCH, respectivamente (versus 0,52% no braço controle AC-T). Os 3 anos de incidência cumulativa de eventos ICC (insuficiência cardíaca congestiva) (Grau 3 ou 4) foi 1,9% e 0,4% nos braços AC-TH e TCH, respectivamente (versus 0,3% no braço controle AC-T).
Terapia combinada com Doceglennu em câncer de pulmão de não-pequenas células (NSCLC) - Eventos adversos clinicamente importantes relacionados ao tratamento em pacientes com câncer de pulmão de células não-pequenas recebendo Doceglennu em associação com cisplatina (Cis)
Os eventos adversos clinicamente importantes relacionados ao tratamento estão mostrados abaixo:
Nesta tabela estão incluídos os dados de segurança para um total de 807 pacientes com câncer de pulmão de não-pequenas células não-ressecável estágio IIIB ou IV e sem história de quimioterapia prévia que foram tratados no braço com associação de Doceglennu do estudo controlado de 3 braços, randomizado, aberto. Estas reações são descritas utilizando os Critérios de Toxicidade Comum NCI e são considerados possivelmente ou provavelmente relacionados ao estudo de tratamento, exceto para as toxicidades hematológicas ou como notado de outra maneira.
• Muito comuns: neutropeniad,c (91,1%, G3/4: 74,8%), anemia (88,6%), trombocitopeniac (14,9%), infecção (14,3%), febre na ausência de infecção (17,2%), reação de hipersensibilidadea (10,6%), alterações nas unhasb (13,3%), pele (11,1%), retenção hídricab (25,9%), náusea/vômito (73,9%, G3/4: 12,1%), diarreia (41,1%), anorexiab (28,8%), estomatite (23,4%), neurossensorial (40,4%), neuromotor (12,8%), alopecia (73,6%), asteniab (51,5%), mialgiab (13,8%);
• Comuns: anemia (G3/4: 6,9%), trombocitopeniac (G3/4: 2,7%), infecção (G3/4: 5,7%), febre na ausência de infecção (G3/4: 1,2%), neutropenia febrilc (4,9%), reações de hipersensibilidadea (G3/4: 2,5%), diarreia (G3/4: 6,4%), anorexiab (todos os AEs severos) (4,9%), estomatite (G3/4: 2,0%), constipação (9,4%), neurossensorial (G3/4: 3,7%), neuromotor (G3/4: 2,0%), asteniab (todos os AEs severos) (9,9%), reações no local de infusão (6,2%), dor (5,4%);
• Incomuns: alterações nas unhasb (todos os AEs severos) (0,7%), retenção hídricab (todos os AEs severos) (0,7%), alopecia (G3: 0,7%), mialgiab (todos os AEs severos) (0,5%).
a Substitui o termo NCI "alergia"
b Termo COSTART e sistema de graduação
c Incidências são apresentadas independente de relação
d Ciclos onde pacientes receberam G-CSF foram considerados não avaliáveis para neutropenia, a menos que neutropenia fosse equivalente a Grau 4.
Terapia associada com Doceglennu em pacientes com câncer de próstata - Eventos adversos clinicamente importantes relacionados ao tratamento em pacientes com câncer de próstata que receberam Doceglennu em associação com prednisona ou prednisolona (TAX327)
Os seguintes dados estão baseados na experiência de 332 pacientes, que foram tratados com Doceglennu 75mg/m2 a cada 3 semanas em associação com prednisona ou prednisolona 5 mg oral duas vezes ao dia (TAX 327). Tais eventos estão classificados em quaisquer eventos e eventos de Grau 3/4 (G3/4):
• Muito comuns: anemia (66,5%), infecção (12,0%), ne
• Comuns: anemia G3/4 (4,9%), infecção G3/4 (3,3%), trombocitopenia (3,4%), neutropenia febril (2,7%), epistaxe (3,0%), reações alérgicas (6,9%), neuropatia sensorial G3/4 (1,2%), neuropatia motora (3,9%), rash/descamação (3,3%), náusea G3/4 (2,4%), diarreia G3/4 (1,2%), vômito G3/4 (1,2%), tosse (1,2%), dispneia (4,5%), função cardíaca ventricular esquerda (3,9%), fadiga G3/4 (3,9%), mialgia (6,9%), lacrimejamento (9,3%), artralgia (3,0%);
• Incomuns: trombocitopenia G3/4 (0,6%), reações alérgicas G3/4 (0,6%), retenção hídrica G3/4 (0,6%), rash/descamação G3/4 (0,3%), estomatite/faringite G3/4 (0,9%), anorexia G3/4 (0,6%), dispneia G3/4 (0,6%), função cardíaca ventricular esquerda G3/4 (0,3%), mialgia G3/4 (0,3%), lacrimejamento G3/4 (0,6%), artralgia G3/4 (0,3%).
Terapia combinada com Doceglennu no adenocarcinoma gástrico - Eventos adversos clinicamente importantes relacionados ao tratamento em pacientes com adenocarcinoma gástrico recebendo Doceglennu em combinação com cisplatina e fluoruracila (TAX 325)
Os dados a seguir estão baseados na experiência de 221 pacientes com adenocarcinoma gástrico avançado e nenhuma história de quimioterapia prévia para a doença avançada, que foram tratados com Doceglennu 75mg/m2 em associação com cisplatina e fluoruracila (TAX 325). Tais eventos estão classificados em quaisquer eventos e eventos de Grau 3/4 (G3/4):
· Muito comuns: anemia (96,8%; G3/4: 18,2%), neutropenia (95,5%, G3/4: 82,3%), febre na
ausência de infecção (30,8%), trombocitopenia (25,5%), infecção (16,7%, G3/4: 12,7%), neutropenia febril (15,9%), infecção neutropênica (14,1%), retenção hídrica (14,9%), letargia (56,1%, G3/4: 18,6%), neurossensorial (38,0%), alopecia (66,5%), náusea (71,9%, G3/4:
14,5%), vômito (61,1%, G3/4: 14,5%), anorexia (44,8%, G3/4: 10,4%), estomatite (59,3%,
G3/4: 20,8%), diarreia (74,7%, G3/4: 19,5%), constipação (10,0%);
· Comuns: febre na ausência de infecção G3/4 (1,8%), trombocitopenia G3/4 (7,7%), reações alérgicas (9,0%, G3/4: 1,8%), neurossensorial G3/4 (7,7%), neuromotor (relacionado ao movimento) (6,3%, G3/4: 1,8%), tontura (8,1%, G3/4: 2,7%), alopecia (5,0%), rash/coceira (8,1%), alterações nas unhas (8,1%), descamação cutânea (1,8%), esofagite/disfagia/odinofagia (9,0%), dor gastrintestinal/cãibra (7,7%, G3/4: 1,4%), disritmias cardíacas (1,8%), lacrimejamento (8,1%), audição alterada (4,1%);
· Incomuns: rash/coceira G3/4 (0,5%), constipação (0,9%), esofagite/disfagia/odinofagia (0,9%), disritmias cardíacas G3/4 (0,9%).
Neutropenia febril ou infecção neutropênica
• Muito comum: a neutropenia febril e/ou infecção neutropênica ocorreram em 28,6% dos pacientes independente da utilização do G-CSF. O G-CSF foi utilizado para a profilaxia secundária em somente 18,6% dos pacientes (10% dos ciclos) para o braço TCF. A neutropenia febril e/ou infecção neutropênica ocorreram em valores mais baixos, 12,2% quando os pacientes receberam G-CSF profilático e 26,9% sem G-CSF profilático.
Terapia combinada com Doceglennu para câncer de cabeça e pescoço (SCCHN) - Eventos adversos clinicamente importantes relacionados ao tratamento em pacientes com SCCHN recebendo Doceglennu em associação com cisplatina e fluoruracila (TAX 323)
São apresentados, a seguir, os dados de segurança obtidos em 174 pacientes com carcinoma de células escamosas inoperável de cabeça e pescoço localmente avançado (SCCHN), que foram tratados com Doceglennu 75 mg/m2 em associação com cisplatina e fluoruracila.
• Muito comuns: neutropenia (93,1%, G3/4: 76,3%), anemia (89,1%), trombocitopenia (23,6%), infecção (15,5%), febre na ausência de infecção (14,4%), infecção neutropênica (11,0%), retenção hídrica (20,1%), retenção hídrica (somente edema) (12,6%), letargia (37,9%), neurossensorial (16,7%), alopecia (79,9%, G3/4: 10,9%), náusea (43,7%), estomatite (42,0%), diarreia (29,3%), vômito (25,9%), anorexia (15,5%), alteração do paladar e do olfato (10,3%);
• Comuns: anemia (G3/4: 9,2%), trombocitopenia (G3/4: 5,2%), infecção (G3/4: 6,3%), neutropenia febrila (5,2%), alergia (2,9%), retenção hídrica (somente aumento de peso) (5,7%), letargia (G3/4: 3,4%), vertigem (1,1%), rash/coceira (8,6%), pele seca (5,2%), descamação (4,0%), estomatite (G3/4: 4,0%), diarreia (G3/4: 2,9%), constipação (6,9%), esofagite/disfagia/odinofagia (5,7%), dor gastrintestinal/cólica (5,2%), azia (4,0%), sangramento gastrintestinal (1,1%), isquemia do miocárdio (1,7%, G3/4: 1,7%), venoso (1,1%), mialgia (6,3%), dor do câncer (1,1%), lacrimejamento (1,7%), conjuntivite (1,1%), audição alterada (5,7%), perda de peso (9,8%);
· Incomuns: febre na ausência de infecção (G3/4: 0,6%), neurossensorial (G3/4: 0,6%), descamação (G3/4: 0,6%), náusea (G3/4: 0,6%), vômito (G3/4: 0,6%), anorexia (G3/4: 0,6%), esofagite/disfagia/odinofagia (G3/4: 0,6%), sangramento gastrintestinal (G3/4: 0,6%), disritmia cardíaca (0,6%, G3/4: 0,6%), venoso (G3/4: 0,6%), mialgia (G3/4: 0,6%), dor do câncer (G3/4: 0,6%).
a Neutropenia febril: febre grau ≥ 2 concomitante com neutropenia grau 4 requerendo antibióticos IV e/ou hospitalização.
Eventos adversos clinicamente importantes relacionados ao tratamento em pacientes com SCCHN recebendo Doceglennu em associação com cisplatina e fluoruracila (TAX 324)
São apresentados, a seguir, os dados de segurança obtidos em 251 pacientes com tumor maligno de células escamosas de cabeça e pescoço localmente avançado que foram tratados com Doceglennu 75 mg/m2 em associação com cisplatina e fluoruracila.
• Muito comuns: neutropenia (94,8%, G3/4: 83,5%), anemia (90,0%, G3/4:12,4%), trombocitopenia (27,5%), infecção (13,1%), febre na ausência de infecção (26,3%), neutropenia febrila (12,1%), retenção hídrica (13,1%), retenção hídrica (somente edema) (12,0%), letargia (58,6%), neurossensorial (11,6%), alopecia (67,7%), rash/coceira (12,7%), náusea (75,7%, G3/4:
13,9%), estomatite (64,5%, G3/4: 20,7%), diarreia (42,2%), vômito (56,2%), anorexia (37,8%, G3/4: 12,0%), constipação (13,9%), esofagite/disfagia/odinofagia (21,9%, G3/4: 12,0%), alteração do paladar e do olfato (19,5%), audição alterada (11,2%), perda de peso (11,2%);
• Comuns: trombocitopenia (G3/4: 4,0%), infecção (G3/4: 3,6%), febre na ausência de infecção (G3/4: 3,6%), infecção neutropênica (6,5%), retenção hídrica (G3/4: 1,2%), retenção hídrica (somente edema) (G3/4: 1,2%), letargia (G3/4: 4,0%), neurossensorial (G3/4: 1,2%), neuromotor (7,2%), vertigem (9,6%, G3/4: 2,0%), alopecia (G3/4: 4,0%), pele seca (2,8%), descamação (2,0%), diarreia (G3/4: 6,8%), vômito (G3/4: 8,4%), dor gastrintestinal, cólica (6,0%, G3/4: 1,2%), azia (8,8%), sangramento gastrintestinal (2,0%), disritmia cardíaca (3,2%), mialgia (5,2%), dor do câncer (3,2%, G3/4: 1,2%), lacrimejamento (1,6%), audição alterada (G3/4: 1,2%);
• Incomuns: alergia (0,4%), retenção hídrica (somente ganho de peso) (0,4%), neuromotor (G3/4: 0,4%), pele seca (G3/4: 0,4%), constipação (G3/4: 0,4%), azia (G3/4: 0,8%), sangramento gastrintestinal (G3/4: 0,4%), alteração do paladar e do olfato (G3/4: 0,4%), disritmia cardíaca (G3/4: 0,2%), isquemia do miocárdio (0,8%, G3/4: 0,8%), distúrbio venoso (0,8%, G3/4: 0,4%), mialgia (G3/4: 0,4%), conjuntivite (0,8%).
Os TEAEs clinicamente importantes foram determinados baseados na frequência, severidade e impacto clínico do evento adverso.
a Neutropenia febril: febre grau ≥ 2 concomitante com neutropenia grau 4 requerendo antibióticos IV e/ou hospitalização.
Experiência pós-comercialização Reações de Hipersensibilidade
Foram relatados raros casos de choque anafilático. Estes casos, muito raramente, resultaram em um desfecho fatal em pacientes que receberam pré-medicação.
Reações de hipersensibilidade com desfecho potencialmente fatal (frequência desconhecida) foram reportadas com docetaxel em pacientes que previamente apresentaram reações de hipersensibilidade ao paclitaxel.
Reações Cutâneas
Casos muito raros de lúpus eritematoso cutâneo e erupções bolhosas como eritema multiforme, síndrome de Stevens-Johnson, necrólise epidérmica tóxica e alterações parecidas com esclerodermia usualmente precedidas por linfedema periférico têm sido relatados com Doceglennu. Em alguns casos vários fatores como infecções simultâneas, uso concomitante de medicamentos e doenças pré-existentes pode ter contribuído para o desenvolvimento destas reações.
Foram reportados casos de alopecia permanente (frequência desconhecida).
Retenção hídrica
Desidratação e edema pulmonar têm sido raramente relatados.
Reações gastrintestinais
Enterocolite (frequência desconhecida), incluindo colite, colite isquêmica e enterocolite neutropênica, foram reportadas com desfecho potencialmente fatal (frequência desconhecida).
Foram relatados raros casos de desidratação resultante de eventos gastrintestinais, incluindo enterocolite e perfuração gastrintestinal.
Casos raros de obstrução intestinal e do íleo foram reportados.
Reações neurológicas
Observou-se raramente casos de convulsão ou perda transitória da consciência com a administração de Doceglennu. Algumas vezes estas reações aparecem durante a infusão do medicamento.
Reações cardiovasculares
Foram relatados raros episódios de tromboembolismo venoso e infarto do miocárdio.
Arritmia ventricular, incluindo taquicardia ventricular (frequência desconhecida) algumas vezes fatal, foi reportada em pacientes tratados com Doceglennu em combinação com tratamentos incluindo doxorrubicina, fluoruracila e/ou ciclofosfamida.
Reações hepáticas
Foram relatados casos muito raros de hepatite, algumas vezes fatal principalmente em pacientes com distúrbios hepáticos pré-existentes.
Distúrbios auditivos e do labirinto
Foram relatados raros casos de ototoxicidade, distúrbios auditivos e/ou perda na audição, incluindo casos associados com outras drogas ototóxicas.
Distúrbios oculares
Foram relatados raros casos de lacrimejamento com ou sem conjuntivite e muito raramente casos de obstrução do ducto lacrimal resultando no lacrimejamento excessivo, principalmente em pacientes recebendo terapia combinada com outros agentes antitumorais.
Foram relatados raros casos de distúrbios visuais transitórios (flashes, feixes de luz e escotomas), ocorrendo tipicamente durante a infusão do medicamento e em associação com reações de hipersensibilidade. Estes raros casos foram reversíveis com a interrupção da infusão.
Casos de Edema Macular Cistoide (EMC) têm sido reportados em pacientes tratados com docetaxel, bem como com outros taxanos.
Distúrbios respiratório, torácico e mediastinal
Casos de síndrome de dificuldade respiratória aguda, pneumonia intersticial/ pneumonite, doença intersticial pulmonar, fibrose pulmonar, insuficiência respiratória e fenômenos de reaparecimento dos efeitos da radiação foram relatados raramente podendo ser fatais. Foram relatados raros casos de pneumonite actínica em pacientes recebendo radioterapia concomitante.
Distúrbios gerais e condições dos locais de administração
Reação recorrente no local de injeção (recorrência da reação cutânea em um local de extravasamento anterior após administração de docetaxel em um local diferente) foi observada no local de extravasamento prévio (frequência desconhecida).
Distúrbios sanguíneo e linfático
Foram relatados casos muito raros de leucemia mieloide aguda e síndrome mielodisplástica em associação com docetaxel quando utilizado em combinação com outros agentes quimioterápicos e/ou radioterapia.
Foi relatada coagulação intravascular disseminada (CID), geralmente em associação com sepse ou insuficiência de múltiplos órgãos.
Desordens renal e urinária
Foram relatadas insuficiência renal e falência renal. A maioria desses casos foi associada com drogas nefrotóxicas concomitantes.
Distúrbios do metabolismo e nutrição
Casos de desequilíbrio eletrolítico foram reportados. Casos de hiponatremia têm sido reportados, em sua maioria associados com desidratação, vômitos e pneumonia. Hipocalemia, hipomagnesemia e hipocalcemia foram observados, usualmente em associação com distúrbios gastrintestinais, em particular diarreia.
Em casos de eventos adversos, notifique ao Sistema de Notificações em Vigilância Sanitária NOTIVISA, disponível em ou para a Vigilância Sanitária Estadual ou Municipal.
10. SUPERDOSE
Poucos casos de superdose com docetaxel foram relatados. Em caso de superdose, o paciente deve ser mantido em unidade especializada com monitorização cuidadosa das funções vitais. Não existe antídoto que possa ser utilizado em caso de superdose com docetaxel. As complicações primárias antecipadas da superdose consistem de supressão da medula óssea, neurotoxicidade periférica e mucosite. Os pacientes devem receber tratamento com G-CSF o mais precocemente possível após o diagnóstico de superdose. Se necessário, devem ser empregadas outras medidas sintomáticas apropriadas.
Em caso de intoxicação ligue para 0800 722 6001, se você precisar de mais orientações.
DIZERES LEGAIS
VENDA SOB PRESCRIÇÃO MÉDICA
USO RESTRITO A HOSPITAIS
MS 1.1013.0282
INSTRUÇÕES DE PREPARO PARA UTILIZAR DOCEGLENNU 20 mg/1,0 mL ou 80 mg/4,0 mL CONCENTRADO PARA INFUSÃO
NÃO utilize a formulação contendo dois frascos-ampola (concentrado para infusão e diluente) com a formulação contendo apenas um frasco-ampola (concentrado para infusão).
É IMPORTANTE QUE SE LEIA ATENTAMENTE ESTA INSTRUÇÃO NA SUA TOTALIDADE ANTES DO PREPARO DA SOLUÇÃO PARA INFUSÃO DE DOCEGLENNU.
1. FÓRMULA
Doceglennu concentrado para infusão é uma solução amarela clara a amarela-acastanhada, contendo 20 mg/mL de docetaxel em polissorbato 80 e álcool etílico (anidro).
2. APRESENTAÇÃO
Doceglennu é apresentado em frascos-ampola.
Cada embalagem contém um frasco-ampola de Doceglennu 20 mg/1,0 mL ou Doceglennu 80 mg/4,0 mL solução concentrada para infusão.
Doceglennu deve ser conservado em temperatura abaixo de 25°C, protegido da luz. Nestas condições, o prazo de validade é de 24 meses tanto para Doceglennu 20 mg/1,0 mL quanto para Doceglennu 80 mg/4,0 mL.
2.1. Frasco-ampola de 20 mg/1,0 mL
• O frasco-ampola de Doceglennu 20 mg/1,0 mL é de vidro incolor, fechado com lacre de alumínio.
• O frasco-ampola de Doceglennu contém 20 mg de docetaxel (anidro) em 1,0 mL de solução de polissorbato 80 e álcool etílico (anidro).
2.2. Frasco-ampola de 80 mg/4,0 mL:
• O frasco-ampola de Doceglennu 80 mg/4,0 mL é de vidro incolor, fechado com lacre de alumínio.
• O frasco-ampola de Doceglennu contém 80 mg de docetaxel (anidro) em 4,0 mL de solução contendo polissorbato 80 e álcool etílico (anidro).
3. RECOMENDAÇÕES PARA O MANUSEIO SEGURO
Doceglennu é um agente antineoplásico e, assim como com outros compostos potencialmente tóxicos, deve-se ter cautela na manipulação e no preparo das soluções de Doceglennu. É recomendado o uso de luvas.
Caso a solução de Doceglennu concentrado ou solução para infusão entre em contato com a pele, lave a região imediata e completamente com água e sabão. Caso a solução de Doceglennu concentrado ou solução para infusão entre em contato com membranas mucosas, lave-as imediata e completamente com água.
4. PREPARO DA SOLUÇÃO PARA ADMINISTRAÇÃO INTRAVENOSA
4.1. Preparo da solução para infusão
4.1.1. Pode ser necessário mais do que um frasco de Doceglennu concentrado para infusão para se obter a dose necessária ao paciente. Retire assepticamente a quantidade necessária de solução concentrada (20 mg/mL) utilizando uma seringa calibrada com agulha de 21G.
4.1.2. Transfira este volume, através de uma injeção única, para uma bolsa ou frasco de infusão com 250 mL de solução glicosilada a 5% ou solução de cloreto de sódio 0,9%. Caso seja necessária uma dose maior que 200 mg de docetaxel, utilize um volume superior de veículo de infusão, visando não exceder a concentração de 0,74 mg/ml de docetaxel.
4.1.3. Misture o conteúdo da bolsa ou frasco de infusão manualmente, utilizando movimento oscilante.

4.1.4. Uma vez adicionada, conforme recomendações, à bolsa de infusão, a solução para infusão de Doceglennu, se armazenada em temperatura abaixo de 25°C e luminosidade normal, é estável por 6 horas. A solução deve ser utilizada dentro de 6 horas (incluindo 1 hora de infusão intravenosa).
A estabilidade física e química da solução para infusão em uso, preparada conforme recomendado, foi demonstrada em bolsas que não contêm PVC por até 48 horas quando armazenada entre 2°C e 8°C e luminosidade normal.
4.1.5. A solução para infusão de Doceglennu é supersaturada, portanto, pode cristalizar com o tempo. Se aparecerem cristais, a solução não deve mais ser utilizada, devendo ser descartada.
5. INUTILIZAÇÃO DE MATERIAIS
Todos os materiais utilizados na diluição e administração de Doceglennu devem ser descartados seguindo procedimento padrão.
2. RESULTADOS DE EFICÁCIA
Câncer de mama(Martin M, et al. N Engl J Med. 2005 Jun;352(22):2302-13)
Estudo randomizado, aberto, multicêntrico suporta o uso de docetaxel para o tratamento adjuvante nas pacientes com câncer de mama operável com linfonodo positivo. 1491 pacientes foram randomizadas para receber tanto docetaxel 75 mg/m2, administrado 1 hora após doxorrubicina 50 mg/m2 e ciclofosfamida 500 mg/m2 (braço TAC) ou doxorrubicina 50 mg/m2 seguida de fluoruracila 500 mg/m2 e ciclofosfamida 500 mg/m2 (braço FAC). Foi demonstrada uma sobrevida livre de doença significativamente mais longa para o braço TAC comparado com o braço FAC. A sobrevida global também foi significativamente maior no braço TAC com as pacientes tratadas com TAC que apresentaram uma redução de 30% no risco de morte comparado ao FAC (hazard ratio=0,70; 95% CI (0,53-0,91), p=0,008).
(GEICAM 9805) (N Engl J Med. 2010 Dec 2;363(23):2200-10)
Estudo randomizado, aberto, multicêntrico (GEICAM 9805) suportam o uso de docetaxel para o tratamento adjuvante de pacientes com câncer de mama operável linfonodo-negativo com um ou mais fatores de alto risco. (tamanho do tumor > 2 cm, idade < 35 anos, status negativo do receptor de hormônio, tumor grau 2 ou 3). 1060 pacientes foram randomizadas para receber ou docetaxel 75 mg/m2administrado 1 hora após doxorrubicina 50 mg/m2 e ciclofosfamida 500 mg/m2 (539 pacientes no braço TAC), ou doxorrubicina 50 mg/m2 seguida de 5-fluorouracil 500 mg/m2 e ciclofosfamida 500 mg/m2 (521 pacientes no braço FAC). Foi demonstrada sobrevida livre de doença significativamente mais longa para o braço TAC comparado ao braço FAC. Sobrevida global mediana também foi mais longa no braço TAC com pacientes tratadas com TAC apresentando uma redução de 24% no risco de morte em comparado ao FAC, porém sem significância estatística, até o momento do acompanhamento de follow- up do estudo (hazard ratio=0,76, 95% IC (0,46-1,26), p=0,28).
(Estudo BCIRG 006 - SABCS 2006, Abstract: 52 e Slamon D, et al. SABCS 2009. Abstract 62)
A eficácia e segurança de docetaxel em associação com trastuzumabe foram avaliadas no tratamento adjuvante para pacientes com câncer de mama operável cujos tumores superexpressam HER2 (com linfonodo positivo e linfonodo negativo de alto risco). Um total de 3.222 mulheres foram randomizadas no estudo e 3.174 foram tratadas com AC-T, AC-TH ou TCH. Sobrevida Livre de Doença (DFS) foi o endpoint primário e a Sobrevida Global (OS) foi o endpoint secundário. Docetaxel e trastuzumabe administrados simultaneamente como parte dos regimes de tratamento adjuvante baseado em antraciclina (AC-TH) ou sem antraciclina (TCH) prolongou significativamente e estatisticamente ambos DFS e OS comparado com o braço controle (AC-T). A redução relativa no risco de morte foi 42% (p=0,0024) e 34% (p=0,0182) para os braços AC-TH e TCH, respectivamente, comparados com o braço AC-T.
(Nabholtz JM, et al. J Clin Oncol. 2003 May;21(6):968-75)
Estudo fase III randomizado, envolvendo 429 pacientes com câncer de mama metastático não tratados previamente foi realizado com doxorrubicina (50 mg/m2) em associação com docetaxel (75 mg/m2) (grupo AT) versus doxorrubicina (60 mg/m2) em associação com ciclofosfamida (600 mg/m2) (grupo AC). O tempo para progressão foi significativamente mais prolongado no grupo do docetaxel comparado ao grupo controle, p = 0,0138. A taxa de resposta global foi significativamente maior no grupo do docetaxel (59,3%) comparado ao grupo controle (46,5%), p=0,009.
(Jones SE, et al. J Clin Oncol. 2005 Aug;23(24):5542-51)
Dois estudos comparativos, fase III randomizados, envolvendo 326 pacientes com câncer de mama metastático que falharam aos agentes alquilantes ou 392 que falharam às antraciclinas. Em pacientes que falharam a agentes alquilantes, docetaxel foi comparado à doxorrubicina (75 mg/m2 a cada 3 semanas). O tempo de sobrevida global foi: docetaxel 15 meses versus 14 meses, p =0,38 / A taxa de resposta foi: docetaxel 52% versus 37%, p=0,01 / O tempo de resposta foi: docetaxel 12 semanas versus 23 semanas, p=0,007.
Em pacientes que falharam à antraciclina, docetaxel foi comparado à associação de mitomicina C e vinblastina (12 mg/m2 a cada 6 semanas e 6 mg/ m2 a cada 3 semanas): O tempo de sobrevida global foi: docetaxel 11 meses versus 9 meses, p =0,01 / A taxa de resposta foi: docetaxel 33% versus 12%, p=0,0001.
Um estudo de fase III aberto, multicêntrico, randomizado foi conduzido para comparar docetaxel e paclitaxel no tratamento do câncer de mama avançado nas pacientes cuja terapia prévia deveria ter incluído uma antraciclina. Um total de 449 pacientes foram randomizadas para receberem ou docetaxel 100 mg/m2 ou paclitaxel 175 mg/m2.
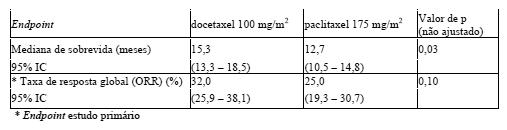
(Shaughnessy J, et al. J Clin Oncol. 2002; 20(12):2812-23)
Estudo clínico fase III controlado, randomizado, multicêntrico suportam o uso de docetaxel em associação com capecitabina para o tratamento de pacientes com câncer de mama localmente avançado ou metastático após falha de quimioterapia citotóxica, incluindo uma antraciclina. Neste estudo, 255 pacientes foram randomizados para tratamento com docetaxel e capecitabina). Foram randomizados 256 pacientes para o tratamento com docetaxel em monoterapia. A sobrevida foi superior no grupo da associação docetaxel + capecitabina (p=0,0126). Sobrevida mediana foi de 442 dias (docetaxel + capecitabina) comparada à 352 dias (docetaxel em monoterapia). A taxa de resposta objetiva global em toda população randomizada (avaliação do investigador) foi de 41,6% (docetaxel + capecitabina) comparado à 29,7% (docetaxel em monoterapia); p= 0,0058. Tempo para progressão da doença ou morte foi superior na associação docetaxel + capecitabina (p < 0,0001). O tempo mediano para progressão foi 186 dias (docetaxel + capecitabina) comparado à 128 dias (docetaxel em monoterapia).
(Marty M, et al. J Clin Oncol. 2005 Jul;23(19):4265-74)
Foi estudada a associação de docetaxel com trastuzumabe para o tratamento de pacientes com câncer de mama metastático cujos tumores superexpressam HER2 e que previamente não receberam quimioterapia para doença metastática. 186 pacientes receberam docetaxel com ou sem trastuzumabe; 60% das pacientes receberam anteriormente quimioterapia adjuvante baseada em antraciclina. Os resultados de eficácia estão resumidos na seguinte tabela:

Câncer de pulmão de não-pequenas células
(Roszkowski K et al. Lung Cancer. 2000 Mar;27(3):145-57)
Estudo fase III comparando docetaxel adicionado a melhor tratamento de suporte (BSC) versus melhor tratamento de suporte, à pacientes com doença localmente avançada (estágio IIIb) e metastática (IV). A sobrevida global foi significativamente mais prolongada em pacientes no grupo do docetaxel (p=0,026) comparada aos pacientes do grupo que recebeu apenas BSC. A taxa de sobrevida de 1 ano foi de 25 % para docetaxel comparado a 16% para BSC. A taxa de resposta global nos pacientes avaliáveis foi de 19,6% com uma duração mediana de resposta de 37,1 semanas.
(Fossella F, et al. J Clin Oncol. 2003 Aug;21(16):3016-24)
Estudo fase III, 1218 pacientes com câncer de pulmão de não-pequenas células no estágio IIIB não ressecado ou IV e sem quimioterapia prévia foram randomizados para receber tanto docetaxel 75 mg/m2 por infusão de 1 hora seguido imediatamente por cisplatina (Cis) 75 mg/m2 por 30-60 minutos a cada 3 semanas, quanto docetaxel 75 mg/m2 por infusão de 1 hora seguido imediatamente por carboplatina (Cb) (AUC 6 mg/mL.min) por 30-60 minutos a cada 3 semanas ou vinorelbina (V) 25 mg/m2 administrado por 6-10 minutos nos dias 1, 8, 15, 22 seguido por Cis 100 mg/m2 administrado no dia 1 dos ciclos repetidos a cada 4 semanas. A sobrevida mediana no grupo docetaxel + Cis foi de 11,3 meses comparado com 10,1 meses no grupo V + Cis, a taxa de sobrevida em 2 anos foi 21% e 14% respectivamente. Hazard ratio foi 1,183 a favor de docetaxel + Cis (95% IC = 1,008 - 1,388). A taxa de resposta global foi mais elevada no grupo docetaxel + Cis comparada ao grupo vinorelbina + cisplatina (31,6% vs. 24,5%). A duração mediana de resposta foi comparável entre os 2 grupos (32 semanas vs. 34 semanas), assim como o tempo para progressão mediano (22,0 semanas vs. 23,0 semanas).
(Fossella F, et al. J Clin Oncol. 2000 Jun;18(12):2354-62)
Em um estudo multicêntrico fase III, 373 pacientes foram randomizados em três grupos de tratamento: A) docetaxel 100 mg/m2 (D/100) [n=125] por infusão IV de 1 hora a cada 3 semanas ou, B) docetaxel 75 mg/m2 (D/75) [n=125] por infusão IV de 1 hora a cada 3 semanas ou, C) de acordo com escolha do médico, tanto vinorelbina (30 mg/m2 [n=89] por infusão IV nos dias 1, 8 e 15 repetidos a 3 semanas), quanto ifosfamida (2 g/m2 [n=34] nos dias 1, 2 e 3 repetidos a cada 3 semanas). A taxa de sobrevida em 1 ano é maior em cada grupo do docetaxel (32%) comparado à 10% no grupo controle de vinorelbina (V) ou isofosfamida (I). Entre os pacientes que foram acompanhados por no mínimo 1 ano antes de quimioterapia subsequente, a sobrevida em 1 ano foi significativamente diferente a favor do grupo do docetaxel (16% com vida) comparado ao grupo V ou I (5% com vida) [p=0,023]. Taxa de resposta para o grupo D/100 foi significativa e estatisticamente maior que o grupo controle V/I na análise dos pacientes avaliáveis (11,9% versus 1,0%; p=0,001). No grupo D/75, a taxa de resposta foi também significativa e estatisticamente maior que o grupo controle V/I (7,5% versus 1,0%; p=0,036).
(Shepherd F, et al. J Clin Oncol. 2000 May;18(10):2095-103)
Em um segundo estudo multicêntrico fase III, 204 pacientes foram randomizados dentro de dois grupos de tratamento: - docetaxel 100 [n=49] ou 75 [n=55] mg/m2 de infusão intravenosa de 1 hora a cada 3 semanas comparado a melhor tratamento de suporte (BSC) [n=100]. A sobrevida mediana foi de 7,2 meses para os pacientes tratados nos grupos com docetaxel, comparado à 4,6 meses para os pacientes que receberam tratamento de suporte (p=0,14). No entanto, nos pacientes tratados com docetaxel a 75 mg/m2, a sobrevida global foi significativamente mais prolongada (p=0,016) em favor do grupo do docetaxel, comparado ao grupo BSC, com sobrevida mediana de 9 meses versus 4,6 meses, respectivamente. A sobrevida em 1 ano foi também significativamente mais prolongada (p=0,016) com docetaxel (40%) comparada ao grupo BSC (16%). A taxa de resposta global foi 7,6% nos pacientes avaliáveis, e a mediana da duração de resposta foi de 26,1 semanas.
Câncer de ovário
(Aapro MS, et al. Ann Oncol. 1994; 5(5):508 (abstract) / Francis P, et al. J Clin Oncol. 1994 Nov;12(11):2301-8 /; Piccart MJ, et al. Clin Cancer Res. 1995 May; 87(9):676-81 / Kavanagh JJ, et al. Clin Cancer Res. 1996 May;2(5):837-42)
A segurança e eficácia de docetaxel foi avaliada em quatro estudos Fase II em mulheres com câncer de ovário avançado refratário à platina. No total, 340 pacientes foram incluídas, todas tendo sido tratadas previamente com cisplatina ou carboplatina e portadoras de doença recorrente ou progressiva. As taxas de resposta global entre os quatro estudos clínicos individuais variaram de 26 a 40%.Quando os dados de resposta dos quatro estudos foram compilados, houve 14 respostas completas e 79 respostas parciais entre os 315 pacientes avaliáveis, resultando em uma taxa de resposta global de 30% (IC 95%: 19-36%) A duração mediana da resposta e a sobrevida mediana nos quatro estudos individuais variou de 4,5 a 6,7 meses e de 8 a 10,4 meses, respectivamente.
Câncer de próstata
(Tannock IF, et al. N Engl J Med. 2004 Oct;351(15):1502-12)
Estudo fase III multicêntrico randomizado de docetaxel em associação com prednisona ou prednisolona em pacientes com câncer de próstata metastático androgênio independente (refratário a hormônio). Um total de 1006 pacientes com KPS≥60 foram randomizados nos seguintes grupos de tratamento: A) docetaxel 75 mg/m2 a cada 3 semanas por 10 ciclos ou B) docetaxel 30 mg/m2 administrado semanalmente nas 5 primeiras semanas em um ciclo de 6 semanas por 5 ciclos ou C) Mitoxantrona 12 mg/m2 a cada 3 semanas por 10 ciclos. Pacientes que receberam docetaxel a cada três semanas demonstraram significativo prolongamento da sobrevida global comparada àqueles tratados com mitoxantrona. O aumento na sobrevida observado no grupo semanal de docetaxel não foi estatisticamente significativo comparado ao grupo controle mitoxantrona. Endpoints de eficácia para o grupo do docetaxel comparados ao grupo controle estão resumidos na tabela abaixo:

Adenocarcinoma gástrico
(Van Cutsem E, et al. J Clin Oncol. 2006 Nov;24(31):4991-7)
Um estudo randomizado, aberto, multicêntrico foi conduzido para avaliação de segurança e de eficácia de docetaxel para o tratamento de pacientes com adenocarcinoma gástrico avançado, incluindo adenocarcinoma da junção gastroesofágica, que não receberam quimioterapia prévia para a doença avançada. Um total de 445 pacientes com KPS > 70 foram tratados com docetaxel (T) (75 mg/m2 no dia 1) em combinação com cisplatina (C) (75 mg/m2 no dia 1) e 5-fluorouracil (F) (750 mg/m2 por dia por 5 dias) ou cisplatina (100 mg/m2 no dia 1) e fluoruracila (1000 mg/m2 por dia por 5 dias). A sobrevida global foi significativamente mais longa (p=0,0201) a favor do braço TCF com um risco de redução de mortalidade de 22,7% (sobrevida global mediana de 9,2 meses no braço TCF versus 8,6 meses no braço CF). As taxas de resposta global (resposta completa + resposta parcial) foram 36,7% no braço tratado com TCF e 25,4% no braço tratado com CF, com uma diferença estatisticamente significante (p=0,0106).
Câncer de cabeça e pescoço
(Vermoken J, et al. N Engl J Med. 2007 Oct;357(17):1695-704).
A segurança e eficácia de docetaxel no tratamento de indução de pacientes com carcinoma de células escamosas de cabeça e pescoço (SCCHN) foi avaliada no estudo de fase III, randomizado, aberto, multicêntrico (TAX 323). Neste estudo, 358 pacientes com SCCHN inoperável localmente avançado e com estado de desempenho WHO 0 ou 1, foram randomizados para um dos dois braços de tratamento. Os pacientes no braço docetaxel receberam docetaxel (T) 75 mg/m2 seguido de cisplatina (P) 75 mg/m2 no dia 1, seguido de 5-fluorouracil (F) 750 mg/m2 por dia em infusão contínua nos dias 1-5. Os pacientes no braço comparador receberam cisplatina (P) 100 mg/m2 no dia 1, seguido por 5-fluorouracil (F) 1000 mg/m2 em infusão contínua nos dias 1-5. O endpoint primário neste estudo, a sobrevida livre de progressão (PFS) foi significativamente maior no braço TPF comparado com o braço PF, p=0,0042 (PFS mediano: 11,4 vs. 8,3 meses, respectivamente) com um tempo de acompanhamento mediano global de 33,7 meses. A sobrevida mediana global foi também, significativamente maior a favor do braço TPF comparado ao braço PF (OS mediana: 18,6 vs. 14,5 meses, respectivamente) com uma redução no risco de mortalidade de 28%, p=0,0128.
(Posner M, et al. N Engl J Med. 2007 Oct;357(17):1695-704)
A segurança e eficácia de docetaxel no tratamento de indução de pacientes com SCCHN localmente avançado (não ressecável, cura cirúrgica baixa ou preservação do órgão) foi avaliado num estudo de fase III, randomizado, multicêntrico, aberto (TAX 324). Neste estudo, 501 pacientes, com SCCHN localmente avançado e um estado de desempenho WHO de 0 ou 1, foi randomizado para 1 dos 2 braços. Os pacientes do braço docetaxel receberam docetaxel (T) 75 mg/m2 por infusão IV no dia 1 seguido de cisplatina (P) 100 mg/m2 administrada por infusão IV de 30 minutos a três horas, seguido por infusão contínua IV de fluoruracila (F) 1000 mg/m2/dia do dia 1 ao dia 4. Os pacientes no braço comparador receberam cisplatina
(P) 100 mg/m2 por infusão IV de 30 minutos a 3 horas no dia 1 seguido por infusão contínua IV de fluoruracila (F) 1000 mg/m2/dia do dia 1 ao dia 5. O endpoint de eficácia primário neste estudo, sobrevida global (OS) foi significativamente mais longo (teste long-rank, p=0,0058) com o regime contendo docetaxel comparado ao PF (mediana OS: 70,6 versus 30,1 meses respectivamente), com uma redução do risco de 30% na mortalidade comparada ao PF (hazard ratio (HR) = 0,70, 95% intervalo de confiança (CI) = 0,54 - 0,90). O endpoint secundário, PFS, demonstrou uma redução do risco de 29% de progressão ou morte e uma melhora de 22 meses no PFS mediano (35,5 meses para TPF e 13,1 para PF). Isto também foi estatisticamente significante com um HR de 0,71; 95% CI 0,56 - 0,90; teste de log-rank p=0,004.


 Trocar de país
Trocar de país