CUVITRU
TAKEDA
imunoglobulina humana
Imunização passiva.
Apresentações.
CUVITRU, solução injetável a 20% é um derivado de plasma humano contendo imunoglobulina humana normal.
CUVITRU, solução injetável a 20% é fornecido em embalagem contendo 1 frasco-ampola, nas seguintes apresentações: 5 mL, 10 mL, 20 mL ou 40 mL.
USO ADULTO E PEDIÁTRICO A PARTIR DE 2 ANOS
VIA SUBCUTÂNEA
Composição.
Um mL contém:
Imunoglobulina humana normal 200 mg (pureza mínima de 98% de imunoglobulina G (IgG))
Cada frasco com 5 mL contém: 1 g de imunoglobulina humana normal
Cada frasco com 10 mL contém: 2 g de imunoglobulina humana normal
Cada frasco com 20 mL contém: 4 g de imunoglobulina humana normal
Cada frasco com 40 mL contém: 8 g de imunoglobulina humana normal
Distribuição das subclasses de IgG (valores aproximados): IgG1 ≥ 56,9%
IgG2 ≥ 26,6%
IgG3 ≥ 3,4%
IgG4 ≥ 1,7%
O teor máximo de IgA é de 280 microgramas/mL Excipientes: glicina e água para injetáveis.
Informações técnicas.
1. INDICAÇÕES
Terapia de reposição em adultos, crianças e adolescentes (2 a 18 anos):
• Síndromes de imunodeficiência primária com produção deficiente de anticorpos;
• Hipogamaglobulinemia e infecções bacterianas recorrentes em pacientes com leucemia linfocítica crônica (LLC), nos quais antibióticos profiláticos não tiveram sucesso ou são contraindicados;
• Hipogamaglobulinemia e infecções bacterianas recorrentes em pacientes com mieloma múltiplo (MM);
• Hipogamaglobulinemia em pacientes antes e após transplante alogênico de células tronco hematopoiéticas (HSCT).
2. RESULTADOS DE EFICÁCIA
Estudo Europeu
A eficácia e a segurança durante o tratamento com CUVITRU foram avaliadas em 48 pacientes em um estudo clínico na Europa. CUVITRU foi administrado por uma duração mediana de tratamento de 358 dias (intervalo: 127,0-399 dias) e uma média (± DP) de 347,4 ± 47,9 dias. Tratamento com CUVITRU: 45/48 pacientes tratados com CUVITRU completaram o estudo, incluindo 23/25 indivíduos de 2 a < 18 anos de idade.
Uma infecção bacteriana grave aguda (IBG Aguda) de pneumonia foi relatada em um indivíduo de 12 anos com uma forma mais grave de hipogamaglobulinemia (XLA) enquanto recebia CUVITRU. A estimativa pontual da taxa anualizada de IBG Aguda foi de 0,022 (limite superior de IC de 99%: 0,049) durante o tratamento com CUVITRU. Esta taxa anual de IBG Aguda foi inferior a 1,0 IBG Aguda/ano, (p < 0,0001), o limite especificado como fornecendo evidência substancial de eficácia.
O resumo das infecções e eventos associados para indivíduos no estudo europeu durante o tratamento subcutâneo com CUVITRU está resumido na tabela 1 abaixo: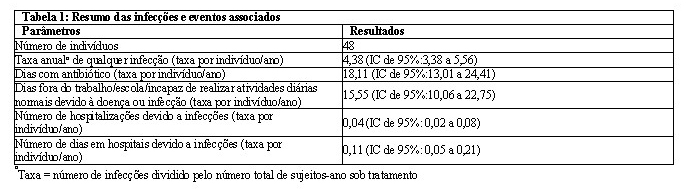
Para resultados de eficácia adicionais, vide item 3. Características Farmacológicas.
Referência:
Borte M, Kriván G, Derfalvi B, et al. Efficacy, safety, tolerability and pharmacokinetics of a novel human immune globulin subcutaneous, 20%: a Phase 2/3 study in Europe in patients with primary immunodeficiencies. Clin Exp Immunol. 2017; 187(1): 146-159.
Estudo norte americano:
Um estudo clínico prospectivo, aberto, não controlado, multicêntrico foi realizado na América do Norte para determinar a eficácia, tolerância e farmacocinética de CUVITRU em 77 indivíduos adultos e pediátricos com imunodeficiência primária. A eficácia foi determinada em 53 adultos a partir de 16 anos, 6 adolescentes entre 12 e < 16 anos, e 15 crianças entre 2 e < 12 anos. CUVITRU foi administrado em 74 indivíduos com uma dose média de 222 mg/kg/semana ± 71 mg/kg/semana por uma duração mediana de tratamento de 380,5 dias (variação: 30 - 629 dias) e uma média (± DP) de 413,1 ± 116,5 dias. A duração mediana de tratamento não variou de forma significativa entre as faixas etárias. A exposição total ao CUVITRU foi de 83,70 indivíduos-anos e 4327 infusões.
Os indivíduos inicialmente receberam imunoglobulina 10% via intravenosa (IGIV) a cada 3 ou 4 semanas em uma dose mensal equivalente àquela recebida antes do estudo durante 13 semanas. O objetivo da parte 1 do estudo foi determinar a AUCIV de IgG total após a administração de IGIV. Na parte 2 do estudo, os indivíduos receberam CUVITRU por via subcutânea em uma dose ajustada de 145% da dose de IGIV (o uso de um fator de ajuste de dose na substituição do tratamento de IG via intravenosa por subcutânea é uma exigência da FDA nos EUA). O objetivo da parte 2 foi determinar a AUCSC da IgG total após a administração semanal de CUVITRU e calcular uma dose ajustada para ser usada na parte 3. O fator de ajuste de dose foi avaliado como sendo 145% da dose de IGIV 10%, ao comparar a AUCSC com a AUCIV 0-t (padronizado para 1 semana) da parte 1 para os primeiros 15 indivíduos que concluíram a parte 2. Os indivíduos, que concluíram a parte 1 após essa avaliação ter sido disponibilizada, passaram diretamente para a parte 3. Na parte 3 do estudo, os indivíduos foram tratados semanalmente por 12 semanas na dose ajustada. As proporções de IgG sérica pelos níveis para as partes 1 e 3 foram comparadas ao esperado pelo nível determinado na parte 2 para estabelecer a dose individualmente adaptada para a parte 4 para cada indivíduo. Na parte 4 do estudo, os indivíduos receberam infusão semanalmente com CUVITRU na dose individualmente adaptada por 40 semanas. Durante a parte 4, uma avaliação farmacocinética adicional foi realizada. O acompanhamento com o indivíduo, seja por Sistema de diário ou pelo investigador ocorreu 3-5 dias após cada infusão em cada parte do estudo para documentar eventos adversos. Os eventos adversos foram avaliados usando o diário eletrônico (eDiary) do indivíduo - todos os indivíduos receberam o tablet eDiary para registrar continuamente tratamentos domiciliares, eventos adversos, e informações adicionais conforme elas ocorriam.
Uma infecção bacteriana grave aguda (IBG) de pneumonia foi relatada em um indivíduo de 78 anos de idade que apresentou deficiência de anticorpo específica e aspergilose broncopulmonar alérgica durante o tratamento com CUVITRU. O ponto estimado da taxa anualizada de IBGs foi 0,012 (limite superior de IC de 99%: 0,024) durante o tratamento com CUVITRU. Essa taxa anual de IBGs foi menor do que 1,0 IBGs/ano (p < 0,0001), o limiar especificado como fornecendo evidência substancial de eficácia.
O resumo de infecções e eventos associados para indivíduos durante o tratamento subcutâneo com CUVITRU está descrito na tabela abaixo: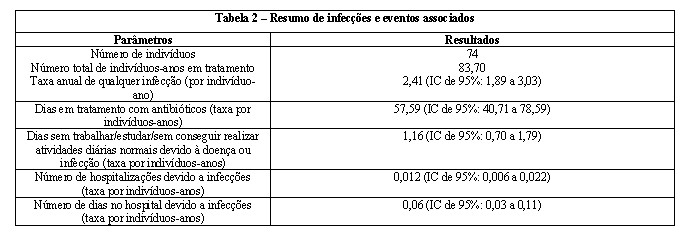
No estudo clínico, entre todas as faixas etárias, a taxa de infusão máxima mediana foi 60 mL/h/local. Essa taxa de infusão foi alcançada em 57,3% (2480/4327) das infusões de CUVITRU concluídas. A taxa de infusão de CUVITRU de 60 mL/h/local foi alcançada em 28,6% (6/21) de indivíduos pediátricos (2 anos a < 16 anos de idade), em 88,7% (47/53) de indivíduos adultos (16 anos de idade ou mais) e em 71,6% (53/74) de todos os indivíduos. Para mais da metade de infusões de CUVITRU (2393/4327), um volume de 30 a 39 mL (1096/4327 infusões) ou de 40 a 49 mL (1297/4327 infusões) foi aplicado por local. Para 320/4327 das infusões de CUVITRU, um volume de 60 mL/local ou mais foi aplicado. Os parâmetros de infusão resultaram em uma mediana de 2 locais de infusão (variação: 1 a 4) por administração de CUVITRU. Durante o tratamento com CUVITRU, 84,9% (3662/4314) de infusões foram administradas usando 1 local de infusão (18,5%; 798/4314) ou 2 locais de infusão (66,4%; 2864/4314) entre todas as idades. A duração mediana de infusões foi menos de 1 hora (0,95 h; variação: 0,2 - 6,4 horas). Durante todos os períodos de tratamento, 99,8% das infusões foram concluídas sem uma redução, interrupção ou descontinuação por razões de tolerância. As características das infusões não diferiram de forma significativa entre os indivíduos adultos e pediátricos.
Durante todo o estudo, a qualidade de vida relacionada à saúde foi avaliada usando o questionário do Inventário Pediátrico de Qualidade de Vida™ (PEDS-QL)1 (indivíduos pediátricos) ou o questionário autoadministrado SF-362 (indivíduos adultos). A qualidade de vida foi analisada separadamente para os grupos de idade 2 a 4 e 5 a 7 anos (PEDS-QL, observador: pais), 8 a 12 e 13 anos (PEDS-QL, observador: indivíduo) e 14 anos ou mais (SF-36, observador: indivíduo). A satisfação do tratamento foi medida usando o questionário de Índice de Qualidade de Vida (LQI) 3,4 e o Questionário de Satisfação de Tratamento para Medicação (TSQM-9)5. O LQI foi avaliado para o grupo de idade 2 anos a 12 anos (observador: pais) e o grupo de idade 13 anos ou mais (observador: indivíduo) em três domínios: Interferência de tratamento, Problemas relacionados à terapia e Ambiente da terapia. O TSQM-9 foi avaliado em indivíduos entre 2 e 12 anos (observador: pais) e 13 anos ou mais (observador: indivíduo) em 3 domínios: Eficiência, Conveniência e Satisfação global. As diferenças entre as pontuações durante a parte intravenosa do estudo e da parte subcutânea do estudo à 20% foram calculadas para domínios selecionados dos instrumentos, vide Tabela 3.
Referências bibliográficas:
1. Varni JW, Seid M, Rode CA. The PedsQL: measurement model for the pediatric quality of life inventory. Med.Care 1999;37:126-139.
2. Ware JE, Jr., Sherbourne CD. The MOS 36-item short-form health survey (SF-36). I. Conceptual framework and item selection. Med.Care 1992;30:473-483.
3. Daly PB, Evans JH, Kobayashi RH et al. Home-based immunoglobulin infusion therapy: quality of life and patient health perceptions. Ann.Allergy 1991;67:504-510.
4. Nicolay U, Haag S, Eichmann F et al. Measuring treatment satisfaction in patients with primary immunodeficiency diseases receiving lifelong immunoglobulin replacement therapy. Qual.Life Res. 2005;14:1683-1691.
5. Bharmal M, Payne K, Atkinson MJ et al. Validation of an abbreviated treatment satisfaction questionnaire for medication (TSQM-9) among patients on antihypertensive medications. Health Qual.Life Outcomes 2009;7:36.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades Farmacodinâmicas
Mecanismo de Ação:
A imunoglobulina humana normal contém principalmente imunoglobulina G (IgG), com um amplo espectro de anticorpos contra agentes infecciosos.
A imunoglobulina humana normal contém anticorpos IgG presentes na população normal. Geralmente são preparados a partir de pools de plasma de não menos que 1000 doadores. Apresentam uma distribuição de subclasses de imunoglobulina G quase proporcional àquela observada no plasma humano nativo. Doses adequadas deste medicamento podem restaurar níveis anormalmente baixos de imunoglobulina G para os valores normais.
População pediátrica
Não há diferenças teóricas ou observadas quanto à ação das imunoglobulinas em crianças em comparação com adultos.
Propriedades Farmacocinéticas
Estudo Europeu
Após a administração subcutânea de CUVITRU, os níveis séricos máximos são atingidos depois de aproximadamente 3 dias.
Em um estudo clínico com CUVITRU (n = 48), os indivíduos atingiram níveis mínimos sustentados de IgG (mediana: 8,26 g/L) em um período de 52 semanas quando receberam doses semanais medianas de 0,125 g/kg.
Dados do estudo clínico de CUVITRU mostram que os níveis séricos mínimos de IgG podem ser mantidos com regimes posológicos de 0,3 a 1,0 g/kg de peso corporal/4 semanas.
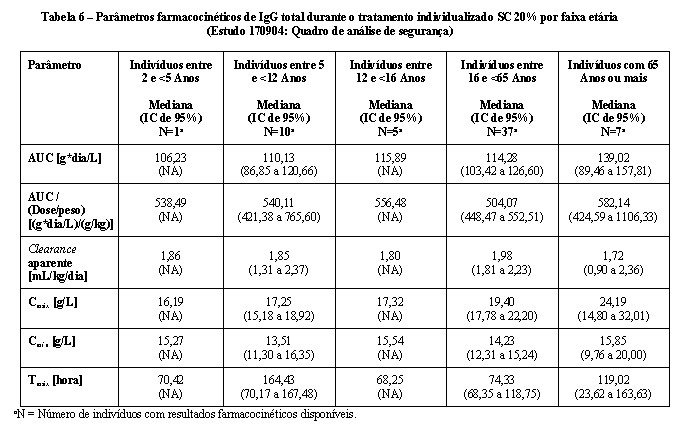
A farmacocinética de CUVITRU foi avaliada no estudo de fase 3 de eficácia e segurança em 31 pacientes com Imunodeficiência primária (IDP) com 12 anos de idade ou mais. Os resultados de farmacocinética são apresentados na tabela abaixo: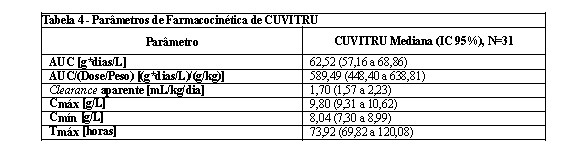
IgG e complexos de IgG são quebrados em células do sistema retículo endotelial.
Administração uma vez por semana, a cada duas semanas ou mais frequente (2-7 vezes por semana)
A caracterização farmacocinética (PK) da administração de CUVITRU a cada duas semanas ou mais frequente foi realizada utilizando simulação e um modelo baseado na PK da população. Os dados de concentração sérica de IgG consistiram em 724 amostras de 32 indivíduos adultos e pediátricos únicos com IDP. Em comparação com a administração uma vez por semana, a simulação e modelo de PK indicou que a administração de CUVITRU a cada duas semanas com o dobro de dose semanal resulta em sobreposição da IgG em todo o intervalo de 2 semanas. Além disso, a simulação e modelo de PK indicou que para a mesma dose semanal total, infusões de CUVITRU administradas 2-7 vezes por semana (administração frequente) resultam também em sobreposição da exposição de IgG durante todo o intervalo de 2 semanas.
População pediátrica
Não há diferenças teóricas ou observadas quanto à farmacocinética das imunoglobulinas em crianças em comparação aos adultos.
Estudo Norte-americano
Os parâmetros farmacocinéticos (PK) de CUVITRU administrado via subcutânea foram avaliados em 60 indivíduos com imunodeficiência primária (IP) durante um estudo clínico na América do Norte. Os indivíduos foram tratados por via intravenosa durante 13 semanas com um produto comparador [GAMMAGARD LIQUID, Imunoglobulina (Humana), 10%] e então passaram para infusões semanais de CUVITRU via subcutânea. Inicialmente, os indivíduos foram tratados por até 12 a 16 semanas na dose subcutânea que foi 145% da dose (o uso de um fator de ajuste de dose na substituição do tratamento de IG via intravenosa por subcutânea é uma exigência da FDA nos EUA). Uma comparação da área sob a curva (AUC) para infusões subcutâneas versus intravenosas foi realizada em 15 indivíduos com 12 anos de idade ou mais. Subsequentemente, todos os indivíduos foram tratados com essa dose durante 12 semanas, após a qual, a dose foi individualizada para todos os indivíduos usando os níveis mínimos de IgG, conforme descrito abaixo. Após aproximadamente 4 meses de tratamento nessa dose subcutânea, uma avaliação farmacocinética foi realizada em todos os indivíduos.
Nesse ajuste de dose, a proporção média geométrica da AUC para administração de CUVITRU via subcutânea versus intravenosa de imunoglobulina 10% foi 109%. O nível máximo de IgG ocorreu em uma média geométrica de 79 horas após a administração subcutânea de CUVITRU.
Na parte 4 do estudo, os parâmetros farmacocinéticos para CUVITRU foram avaliados para 60 indivíduos com 2 anos de idade ou mais. Os parâmetros farmacocinéticos de CUVITRU administrado via subcutânea são mostrados na Tabela 5. A mediana dos níveis máximos de IgG foram menores (18,09 g/L) durante o tratamento subcutâneo com CUVITRU em comparação à administração de IGIV 10% (26,02 g/L durante intervalos de 3 semanas e 25,21 g/L durante intervalos de 4 semanas), compatível com a dose semanal menor em comparação com a dose administrada a cada 3 ou 4 semanas via intravenosa. Em contraste, os níveis mínimos da média geométrica foram maiores com CUVITRU (14,74 g/L), em comparação àqueles administrados via intravenosa (11,58 g/L para intervalos de 3 semanas e 10,19 g/L para intervalos de 4 semanas), um resultado da dose maior mensal e da administração mais frequente. A administração subcutânea semanal resultou em níveis de IgG séricos relativamente em estado estável em comparação com o IGIV administrado em intervalos de 3 a 4 semanas. Os parâmetros farmacocinéticos para CUVITRU não diferiram de forma significativa entre as faixas etárias. Os parâmetros farmacocinéticos de CUVITRU para os diferentes grupos etários são mostrados na Tabela 6.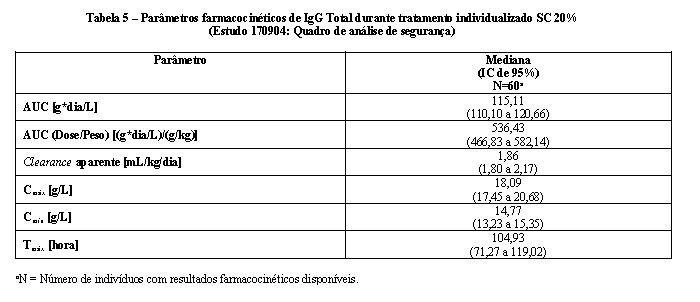
4. CONTRAINDICAÇÕES
Hipersensibilidade ao princípio ativo ou a qualquer um dos componentes do produto.
Deficiência de IgA grave e um histórico de hipersensibilidade ao tratamento com imunoglobulina humana.
CUVITRU não deve ser administrado por via intravascular ou intramuscular.
Este medicamento é contraindicado para menores de 2 anos de idade.
5. ADVERTÊNCIAS E PRECAUÇÕES
Se CUVITRU for acidentalmente administrado em um vaso sanguíneo o paciente pode desenvolver choque.
A taxa de infusão subcutânea recomendada e as instruções de administração devem ser seguidas rigorosamente. Os pacientes devem ser monitorados rigorosamente e observados cuidadosamente quanto a qualquer sintoma durante todo o período de infusão.
Se o produto permanecer em uma seringa siliconizada por mais de duas horas, partículas visíveis podem formar-se.
Certas reações adversas podem ocorrer mais frequentemente em pacientes que recebem imunoglobulina humana normal pela primeira vez ou, em casos raros, quando o produto à base de imunoglobulina humana normal é trocado, ou quando houver um longo intervalo desde a infusão anterior.
Possíveis complicações, geralmente, podem ser evitadas fazendo-se o seguinte:
• injetar inicialmente o produto vagarosamente;
• garantir que os pacientes sejam cuidadosamente monitorados quanto a qualquer sintoma durante todo o período de infusão. Em particular, pacientes que nunca receberam imunoglobulina humana normal, pacientes que recebiam um produto de imunoglobulina alternativo e trocaram de tratamento ou pacientes que ficaram um longo período sem tratamento desde a infusão anterior devem ser monitorados durante a primeira infusão e durante a primeira hora após a primeira infusão, para que se possa detectar possíveis sinais adversos.
Todos os outros pacientes devem ser observados por pelo menos 20 minutos após a administração.
Em caso de reação adversa, a taxa de administração deve ser reduzida ou a infusão deve ser interrompida. Em caso de suspeita de hipersensibilidade grave ou reações anafiláticas é exigida a descontinuação imediata da injeção. O tratamento necessário depende da natureza e da gravidade da reação adversa.
Em caso de choque, o tratamento médico padrão para choque deve ser implementado.
Hipersensibilidade
Reações alérgicas reais são raras. Elas podem ocorrer principalmente em pacientes com anticorpos anti-IgA, que devem ser tratados com cautela especial. Pacientes com anticorpos anti-IgA, cujo tratamento com produtos IgG via subcutânea continua sendo a única opção, devem ser tratados com CUVITRU somente sob rigorosa supervisão médica. CUVITRU contém quantidades residuais de IgA (não mais de 280 microgramas/mL).
Raramente, a imunoglobulina humana normal pode induzir uma queda da pressão arterial com reação anafilática, mesmo em pacientes que haviam tolerado tratamento anterior com imunoglobulina humana normal.
Tromboembolismo
Eventos tromboembólicos arteriais e venosos, incluindo infarto do miocárdio, derrame, trombose venosa profunda e embolia pulmonar, foram associados com o uso de imunoglobulinas. Deve-se ter cautela em pacientes com fatores de risco preexistentes para eventos trombóticos (tais como idade avançada, hipertensão, uso de estrógenos, cateteres vasculares permanentes, diabetes mellitus e histórico de doença vascular ou episódios trombóticos, pacientes com distúrbios trombofílicos congênitos ou adquiridos, pacientes com períodos prolongados de imobilização, pacientes com hipovolemia grave, pacientes com doenças que aumentam a viscosidade sanguínea). Os pacientes devem ser informados sobre os primeiros sintomas de eventos tromboembólicos, incluindo falta de ar, dor e inchaço de um membro, déficits neurológicos focais e dor no peito, e devem ser aconselhados a contatarem seus médicos imediatamente diante da ocorrência de sintomas.
Considerar a avaliação basal da viscosidade do sangue em pacientes com risco de hiperviscosidade, incluindo aqueles com crioglobulinas, quilomicronemia em jejum / triacilgliceróis (triglicérides) marcadamente altos ou gamopatias monoclonais. Para pacientes com risco de trombose, administrar CUVITRU na dose mínima e na taxa de infusão praticável. Assegurar uma hidratação adequada em pacientes antes da administração. Monitorar sinais e sintomas de trombose e avaliar a viscosidade do sangue em pacientes com risco de hiperviscosidade.
Complicações renais
Reações adversas renais graves foram relatadas em pacientes recebendo tratamento com imunoglobulina, principalmente produtos que contêm sacarose (CUVITRU não contém sacarose). Essas reações incluem insuficiência renal aguda, necrose tubular aguda, nefropatia tubular proximal e nefrose osmótica. Fatores que influenciam o risco de complicações renais incluem, entre outros, insuficiência renal preexistente, diabetes mellitus, hipovolemia, produtos medicinais nefrotóxicos concomitantes, idade superior a 65 anos, sepse, hiperviscosidade e paraproteinemia.
Garantir que os pacientes não estejam desidratados antes do início da infusão de CUVITRU. Em pacientes que correm o risco de desenvolver disfunção renal devido a insuficiência renal pré-existente ou predisposição à insuficiência renal aguda, monitorar a função renal e considerar uma dosagem mais baixa e mais frequente.
O monitoramento periódico da função renal e da produção de urina é particularmente importante nos pacientes predispostos a estarem em risco aumentado de desenvolver insuficiência renal aguda. Avaliar a função renal, incluindo a medição de nitrogênio uréico no sangue (BUN) e creatinina sérica, antes da infusão inicial de CUVITRU e novamente a intervalos apropriados posteriormente. Se a função renal se deteriorar, considerar a descontinuação de CUVITRU.
Orientar os pacientes para informar imediatamente os seguintes sinais e sintomas ao seu médico: diminuição da produção de urina, aumento súbito de peso, retenção de líquidos / edema e/ou falta de ar.
Síndrome da Meningite Asséptica (SMA)
Síndrome da meningite asséptica (SMA) foi relatada em associação com o tratamento com imunoglobulinas, incluindo Cuvitru (veja seção 9. REAÇÕES ADVERSAS). A SMA pode ocorrer mais frequentemente em pacientes do sexo feminino.
A descontinuação do tratamento com Ig pode resultar na remissão da SMA dentro de vários dias sem sequelas. A síndrome geralmente começa dentro de algumas horas até 2 dias após o tratamento com Ig. Estudos de líquido cefalorraquidiano são frequentemente positivos, com pleocitose de até milhares de células por mm3, predominantemente da série granulocítica, e níveis elevados de proteína de até centenas de mg/dL.
Os pacientes devem ser informados sobre os primeiros sintomas que incluem dor de cabeça grave, rigidez da nuca, sonolência, febre, fotofobia, náusea e vômito.
Hemólise
CUVITRU contém anticorpos de grupo sanguíneo que podem agir como hemolisinas e induzir o revestimento in vivo de eritrócitos (RBC) com imunoglobulina. Isso causa uma reação antiglobulina direta positiva (DAT, teste de Coombs direto) e, raramente, hemólise. Anemia hemolítica tardia pode desenvolver-se após o tratamento com Ig devido ao aumento do sequestro de eritrócitos. Anemia hemolítica aguda, consistente com hemólise intravascular, foi relatada.
Os seguintes fatores de risco podem estar relacionados ao desenvolvimento de hemólise: doses elevadas (por exemplo, ≥2 gramas/kg, administração única ou dividida em vários dias) e grupo sanguíneo O negativo. O estado inflamatório subjacente em um paciente individual pode aumentar o risco de hemólise, mas seu papel é incerto.
Monitorar pacientes com sinais clínicos e sintomas de hemólise, particularmente, pacientes com fatores de risco acima mencionados. Considerar testes laboratoriais adequados em pacientes de alto risco, incluindo medição de hemoglobina ou hematócrito antes da infusão e dentro de aproximadamente 36 a 96 horas após a infusão.
Informar os pacientes para notificar imediatamente os seguintes sinais e sintomas ao seu médico: aumento da frequência cardíaca, fadiga, amarelamento da pele ou dos olhos e urina de cor escura.
Agentes transmissíveis
Medidas padrão para prevenir infecções resultantes do uso de produtos preparados a partir de sangue ou plasma humano incluem a seleção de doadores, triagem de doações individuais e pools de plasma quanto a marcadores específicos de infecção, e a inclusão de etapas eficazes de fabricação para a inativação/remoção de vírus. Apesar disso, quando produtos medicinais preparados a partir de sangue ou plasma humano são administrados, não se pode excluir totalmente a possibilidade de transmissão de agentes infecciosos. Isso também se aplica a vírus desconhecidos ou emergentes e outros patógenos.
As medidas tomadas são consideradas eficazes para vírus envelopados, como o vírus da imunodeficiência humana (HIV), vírus da hepatite B (HBV) e vírus da hepatite C (HCV), e para os vírus não envelopados da hepatite A e parvovírus B19.
Existe experiência clínica referente à ausência de transmissão do vírus da hepatite A ou do parvovírus B19 com imunoglobulinas, e se supõe que o teor de anticorpos representa uma contribuição importante para a segurança viral.
Recomenda-se fortemente, que toda vez que CUVITRU for administrado a um paciente, o nome e o número de lote do produto sejam registrados para manter uma ligação entre o paciente e o lote do produto.
Lesão pulmonar aguda relacionada à transfusão (TRALI)
Nenhum edema pulmonar não cardiogênico (TRALI) foi reportado em pacientes após o tratamento com produtos de imunoglobulina. TRALI é caracterizada por dificuldade respiratória severa, edema pulmonar, hipoxemia, função ventricular esquerda normal e febre. Os sintomas geralmente ocorrem dentro de 1 a 6 horas após o tratamento.
Monitorar os pacientes para reações adversas pulmonares. Em caso de suspeita de TRALI, realizar testes adequados para a presença de anticorpos anti-neutrófilos e anti-HLA tanto no produto como no soro do paciente. TRALI pode ser gerenciado utilizando oxigenoterapia com suporte ventilatório adequado
Informar aos pacientes para notificar imediatamente os seguintes sinais e sintomas ao seu médico: dificuldade em respirar, dor no peito, lábios ou extremidades azuis ou febre que pode ocorrer 1 a 6 horas após uma infusão de CUVITRU.
Teor de Sódio
CUVITRU é praticamente isento de sódio.
População pediátrica
As advertências e precauções listadas se aplicam a adultos e crianças.
A segurança e a eficácia não foram estabelecidas em crianças abaixo de 2 anos de idade.
Fertilidade, gravidez e lactação
Os médicos devem ponderar os possíveis riscos e só prescrever CUVITRU se for claramente necessário.
Gravidez
A segurança deste medicamento para o uso em gestantes não foi estabelecida em estudos clínicos controlados e, portanto, o medicamento deve ser administrado com cautela em gestantes e mães que estejam amamentando. Os produtos à base de imunoglobulina demonstraram atravessar a placenta, principalmente durante o terceiro trimestre. A experiência clínica com imunoglobulinas sugere que não são esperados efeitos prejudiciais durante a gravidez, no feto ou neonato.
Categoria "C" de risco na gravidez.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Lactação
As imunoglobulinas são excretadas no leite materno e podem contribuir para a proteção do neonato contra patógenos que têm a mucosa como porta de entrada.
Fertilidade
A experiência clínica com imunoglobulinas sugere que não são esperados efeitos prejudiciais na fertilidade.
Condução e utilização de máquinas
A capacidade de dirigir e operar máquinas pode ser comprometida por algumas reações adversas associadas ao CUVITRU. Pacientes que apresentam reações adversas durante o tratamento devem aguardar que estas reações desapareçam antes de dirigir ou operar máquinas.
Incompatibilidades
A administração de CUVITRU com outros medicamentos não é recomendada.
Na ausência de estudos de compatibilidade, este medicamento não deve ser administrado junto com outros produtos. CUVITRU não deve ser diluído.
6. INTERAÇÕES MEDICAMENTOSAS
Interferência no teste sorológico
Após a infusão de imunoglobulina, o aumento temporário dos vários anticorpos transferidos passivamente no sangue do paciente pode resultar em resultados falso-positivos no teste sorológico - por exemplo, Hepatite A, Hepatite B, sarampo e catapora. A transmissão passiva de anticorpos para os antígenos de eritrócitos, por exemplo, A, B, D, pode interferir em alguns testes sorológicos de anticorpos de eritrócitos, como, por exemplo, o teste direto de imunoglobulina (DAT, teste de Coombs direto).
A administração de CUVITRU pode levar a leituras falso-positivas em ensaios que dependem da detecção de beta-D-glucanos para o diagnóstico de infecções fúngicas; isso pode persistir durante semanas após a infusão do produto.
Vacinas com vírus vivo atenuado
A administração de imunoglobulina pode prejudicar, por um período de pelo menos 6 semanas e por até 3 meses, a eficácia de vacinas com vírus vivo atenuado, como de sarampo, caxumba, rubéola e catapora. Após a administração de CUVITRU, um intervalo de 3 meses deve ser respeitado antes da aplicação de vacinas com vírus vivo atenuado. No caso de sarampo, esse comprometimento do efeito pode persistir por até 1 ano. Portanto, pacientes que tiverem vacina de sarampo aplicada devem ter seu status de anticorpos verificado.
População pediátrica
As interações listadas aqui aplicam-se a adultos e crianças.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Conservar sob refrigeração entre 2°C e 8°C. Não congelar. Manter o frasco dentro do cartucho para proteger da luz.
Prazo de validade: CUVITRU, solução injetável, tem validade de 36 meses a partir da data de sua fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Após aberto, utilizar imediatamente.
O produto deverá atingir temperatura ambiente antes do uso.
Os frascos fechados devem ficar em temperatura ambiente por no mínimo 90 minutos antes de serem utilizados e mantidos em temperatura ambiente durante a administração. Não usar dispositivos de aquecimento, incluindo micro-ondas. Soluções turvas ou com depósito não devem ser usadas.
Qualquer medicamento não utilizado ou material residual deve ser descartado de acordo com as exigências locais.
A solução é límpida e incolor ou amarelo pálido ou marrom claro. Não utilizar soluções turvas ou que apresentam depósitos.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
A terapia de reposição deve ser iniciada e monitorada sob a supervisão de um médico com experiência no tratamento de imunodeficiência.
Posologia.
A dose e o regime posológico dependem da indicação.
Terapia de reposição
O produto deve ser administrado por via subcutânea.
Na terapia de reposição, a dose talvez precise ser individualizada para cada paciente, dependendo da resposta clínica e farmacocinética. Os regimes posológicos a seguir são fornecidos como diretriz.
O regime posológico deve atingir um nível mínimo de IgG (medido antes da infusão seguinte) de pelo menos 5-6 g/L e deve estar dentro do intervalo de referência de IgG sérica para a idade. Uma dose de ataque de pelo menos 0,2 a 0,5 g/kg (1 a 2,5 mL/kg) de peso corporal pode ser necessária. Essa dose talvez precise ser dividida em vários dias, com uma dose diária máxima de 0,1 a 0,15 g/kg. Depois que níveis de IgG em estado estável forem atingidos, doses de manutenção serão administradas em intervalos repetidos para se atingir uma dose mensal cumulativa na faixa de 0,3-1,0 g/kg. Cada dose única talvez precise ser injetada em locais anatômicos diferentes.
Os níveis mínimos devem ser medidos e avaliados com a incidência de infecção. Para reduzir a taxa de infecção, talvez seja necessário aumentar a dose e buscar níveis mínimos maiores.
População pediátrica
A posologia em crianças e adolescentes (2-18 anos) não é diferente daquela em adultos, já que a posologia para cada indicação depende do peso corporal e é ajustada para o resultado clínico das indicações mencionadas acima.
Não foram conduzidos estudos clínicos de CUVITRU em crianças com idades entre 0 e < 2 anos.
Método de administração
Somente para uso subcutâneo.
CUVITRU deve ser inspecionado visualmente quanto a material particulado e alteração da cor antes da administração. Não use se for observado material particulado e/ou alteração da cor.
A infusão deve ser iniciada imediatamente após a transferência de CUVITRU para dentro da seringa. A administração está prevista para durar até duas horas. Se uma administração menor que duas horas não for possível devido à dose requerida ou à taxa de administração de CUVITRU, a dose requerida deve ser dividida e administrada em diferentes locais de infusão. Se CUVITRU permanecer nas seringas siliconizadas por mais de duas horas, partículas visíveis podem se formar.
CUVITRU não deve ser diluído.
A infusão subcutânea deve ser iniciada e monitorada no começo por um profissional de saúde com experiência na orientação de pacientes para tratamento domiciliar com acompanhamento regular. Bombas de infusão apropriadas para administração subcutânea de imunoglobulinas podem ser usadas. O paciente ou cuidador deve ser instruído sobre o uso de uma bomba de seringa, técnicas de infusão, manutenção do diário de tratamento, reconhecimento de reações adversas graves e medidas a serem tomadas nesse caso.
CUVITRU pode ser injetado em locais como abdômen, coxa, braço e lateral do quadril.
Ajustes da taxa de infusão e do volume de infusão por local são baseados na tolerância de cada indivíduo.
Recomenda-se o uso de uma velocidade de administração inicial de 10 mL/h/local de infusão. Se bem tolerada, a taxa de infusão pode ser aumentada em intervalos de pelo menos 10 minutos até um máximo de 20 mL/h/local de infusão nas duas infusões iniciais. Mais de uma bomba pode ser usada simultaneamente. A quantidade de produto infundida em determinado local varia. Em crianças, o local de infusão pode ser trocado a cada 5-15 mL. Em adultos, doses acima de 30 mL podem ser divididas de acordo com a preferência do paciente. Não há limite para o número de locais de infusão.
Instruções detalhadas de uso:
Preparar o(s) frasco(s) de CUVITRU:
• Remover o CUVITRU da caixa. Caso o produto esteja armazenado em uma geladeira, permitir que os frascos atinjam a temperatura ambiente. Isso pode levar até 90 minutos.
• Não aplicar calor ou colocar no micro-ondas.
• Não agitar o(s) frasco(s).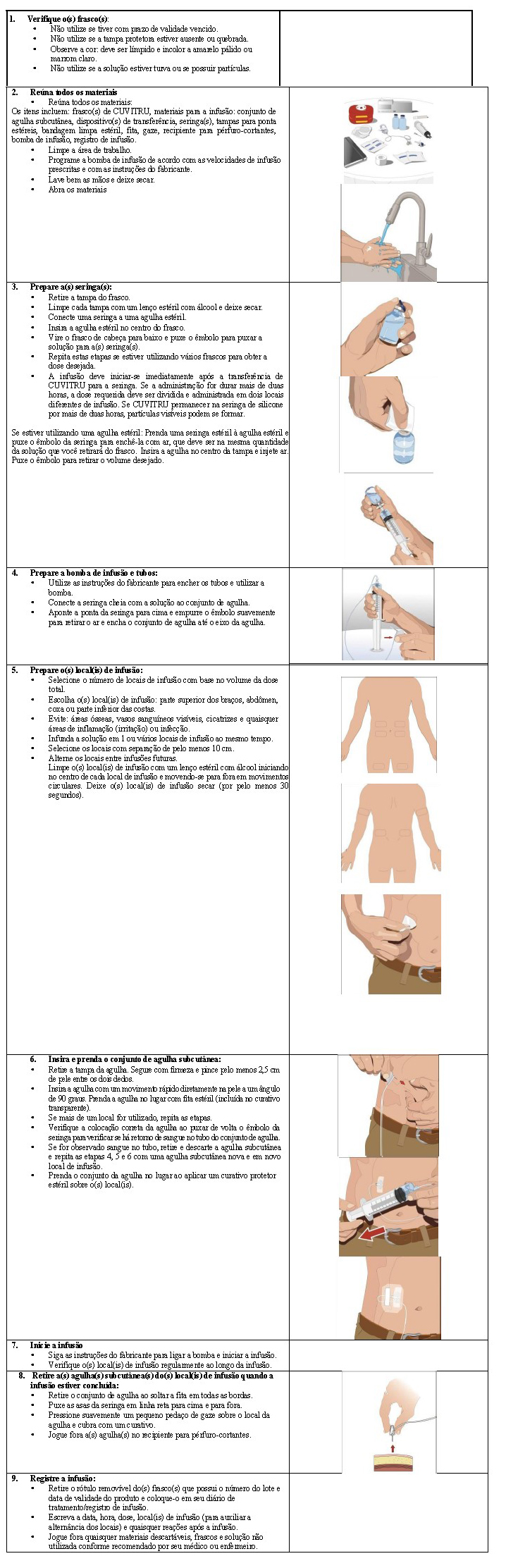
9. REAÇÕES ADVERSAS
Reações adversas (RAs) a CUVITRU como calafrios, cefaleia, tontura, febre, vômito, reações alérgicas, náusea, artralgia, hipotensão e dor lombar moderada, podem ocorrer ocasionalmente.
Raramente, imunoglobulinas humanas normais podem causar uma queda súbita da pressão arterial e, em casos isolados, choque anafilático, mesmo quando o paciente não tiver demonstrado hipersensibilidade na administração anterior.
Reações no local da infusão: inchaço, sensibilidade, vermelhidão, endurecimento, aquecimento do local, dor no local, coceira, hematoma e erupção cutânea, podem ocorrer com frequência.
Lista tabelada de Reações Adversas
A segurança de CUVITRU administrado por via subcutânea foi avaliada em dois estudos prospectivos, abertos, multicêntricos, não controlados, em 122 indivíduos com imunodeficiência primária (IDP).
A maioria (98,8%) das reações adversas (RAs) locais foram de intensidade leve. Um indivíduo descontinuou o tratamento devido a uma RA no local (dor). 112 de 122 indivíduos tratados com CUVITRU completaram o estudo.
A tabela apresentada abaixo está de acordo com a classe de órgãos dos sistemas MedDRA. As frequências foram avaliadas de acordo com o seguinte critério:
• muito comum (≥1/10);
• comum (≥1/100 a < 1/10);
• incomum (≥1/1.000 a < 1/100);
• rara (≥1/10.000 a < 1/1.000);
• muito rara ( < 1/10.000);
• desconhecida (não pode ser estimada com os dados disponíveis).
Dentro de cada grupo de frequência, as reações adversas são apresentadas por ordem decrescente de gravidade.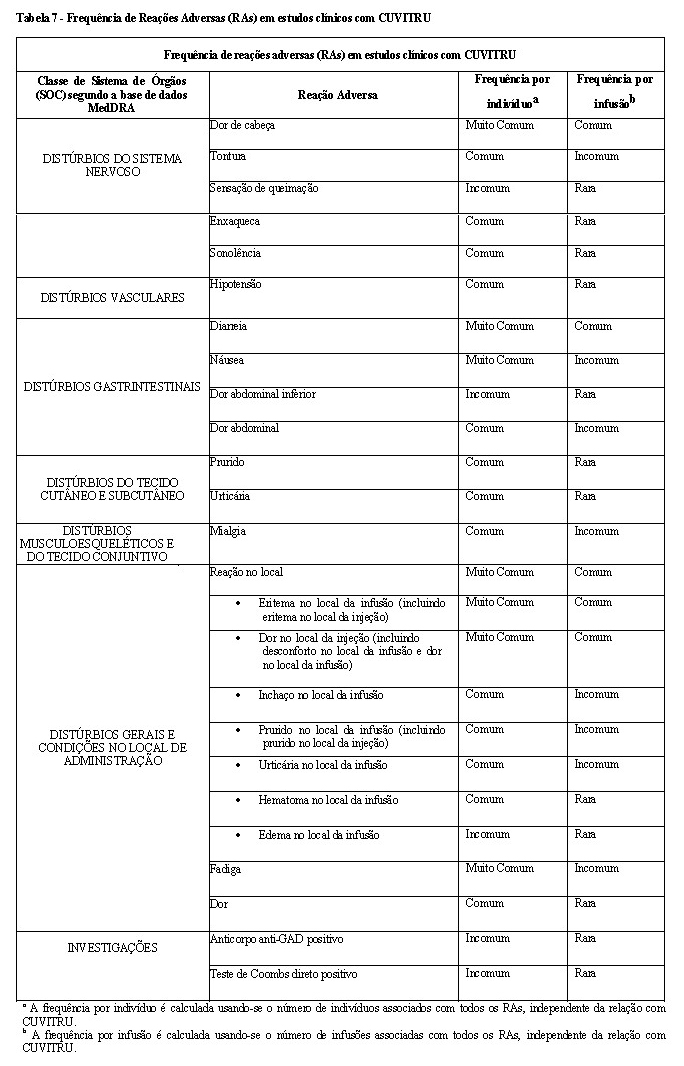

As seguintes RAs adicionais foram identificadas e relatadas durante o uso pós-comercialização de outro produto de imunoglobulina
subcutânea: reação anafilática, parestesia, tremor, taquicardia, dispneia, laringoespasmo, desconforto no tórax, reações no local de injeção (como: endurecimento e calor).
População pediátrica (2 a 16 anos de idade)
O perfil de segurança da população pediátrica foi similar ao dos indivíduos adultos.
Relato de suspeita de reações adversas
O relato de suspeita de reações adversas após a autorização do medicamento é importante. Isto permite o monitoramento contínuo da relação risco/benefício do produto.
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo
que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos pelo Sistema VigiMed, disponível no Portal da Anvisa.
10. SUPERDOSE
As consequências de uma superdosagem de CUVITRU são desconhecidas.
Em caso de intoxicação ligue para 0800 722 6001, se você precisar de mais orientações.
Dizeres legais.
MS 1.0639.0291
USO RESTRITO A HOSPITAIS.
VENDA SOB PRESCRIÇÃO MÉDICA.