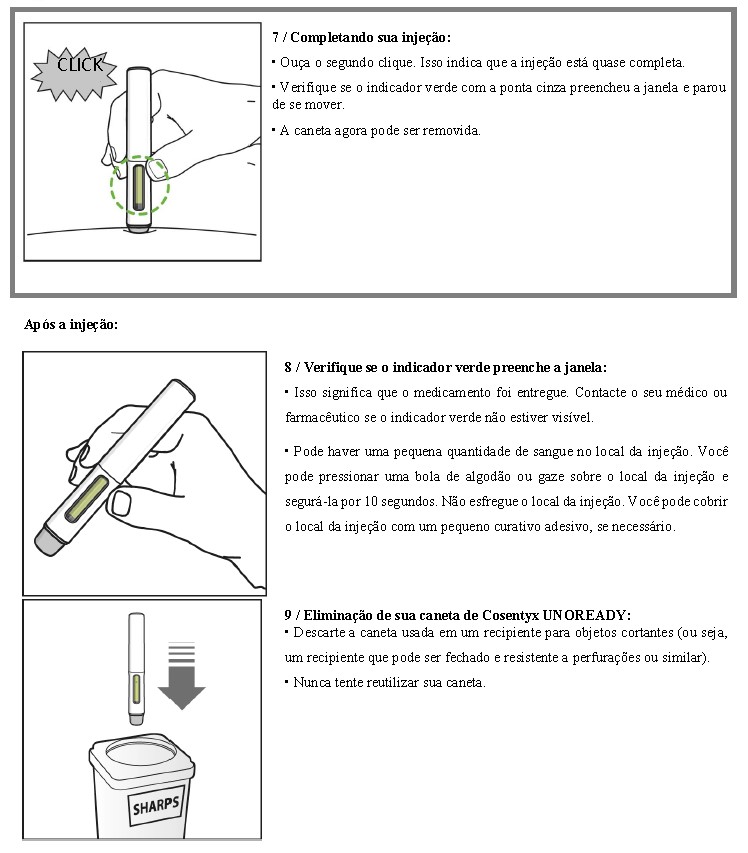
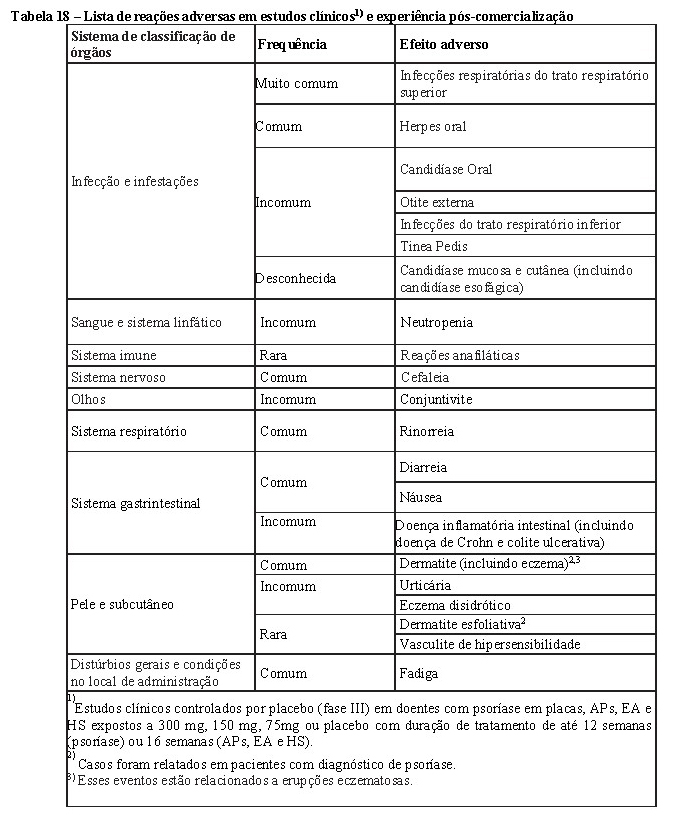
COSENTYX
NOVARTIS
secuquinumabe
Antipsoriásico.
Apresentações.
Cosentyx® 150 mg/mL solução injetável - embalagens contendo 1 ou 2 canetas preenchidas.
Cosentyx® 300 mg/2mL solução injetável - embalagem contendo 1 caneta preenchida.
VIA SUBCUTÂNEA
USO ADULTO E PEDIÁTRICO PARA CRIANÇAS ACIMA DE 6 ANOS DE IDADE (PSORÍASE EM PLACAS)
USO ADULTO (ARTRITE PSORIÁSICA, ESPONDILOARTRITE AXIAL COM OU SEM DANO RADIOGRÁFICO E HIDRADENITE SUPURATIVA)
USO PEDIÁTRICO PARA CRIANÇAS ACIMA DE 2 ANOS DE IDADE (ARTRITE PSORIÁSICA JUVENIL - APJ) e ACIMA DE 4 ANOS DE IDADE (ARTRITE RELACIONADA À ENTESITE - ARE)
Composição.
Cada caneta preenchida de Cosentyx® contém 150 mg de secuquinumabe em 1 mL de solução injetável.
Cada caneta preenchida de Cosentyx® contém 300 mg de secuquinumabe em 2 mL de solução injetável.
Excipientes: trealose di-hidratada, histidina/cloridrato de histidina monoidratado, levometionina, polissorbato 80, água para injetáveis.
Informações técnicas.
1. INDICAÇÕES
Psoríase em placas
Cosentyx® é indicado para o tratamento de psoríase em placas moderada a grave em pacientes com 6 anos de idade ou mais que são candidatos a terapia sistêmica ou fototerapia.
Artrite psoriásica
Cosentyx® é indicado para o tratamento de artrite psoriásica ativa em pacientes adultos, quando a resposta à terapia prévia com medicamentos antirreumáticos modificadores do curso da doença (DMARDs) for inadequada. Cosentyx® pode ser utilizado isoladamente ou em combinação com metotrexato.
Espondiloartrite axial (EpA axial) com ou sem dano radiográfico
• Espondilite anquilosante (EA) / EpA axial com dano radiográfico
Cosentyx® é indicado para o tratamento de espondilite anquilosante ativa em pacientes adultos, que não tenham respondido adequadamente à terapia convencional.
• Espondiloartrite axial não radiográfica (EpAax-nr) / EpA axial sem dano radiográfico
Cosentyx® é indicado para o tratamento de espondiloartrite axial não radiográfica ativa com sinais objetivos de inflamação, como indicado por proteína C reativa (PCR) elevada e/ou evidência na ressonância magnética (RM) em adultos que responderam de maneira inadequada a anti-inflamatórios não esteroidais (AINEs).
Artrite Idiopática Juvenil (AIJ)
• Artrite Relacionada à Entesite (ARE)
Cosentyx® é indicado para o tratamento de artrite relacionada à entesite ativa em pacientes acima de 4 anos de idade.
• Artrite Psoriásica Juvenil (APJ)
Cosentyx® é indicado para o tratamento da artrite psoriásica juvenil ativa em pacientes acima de 2 anos de idade.
Hidradenite Supurativa (HS)
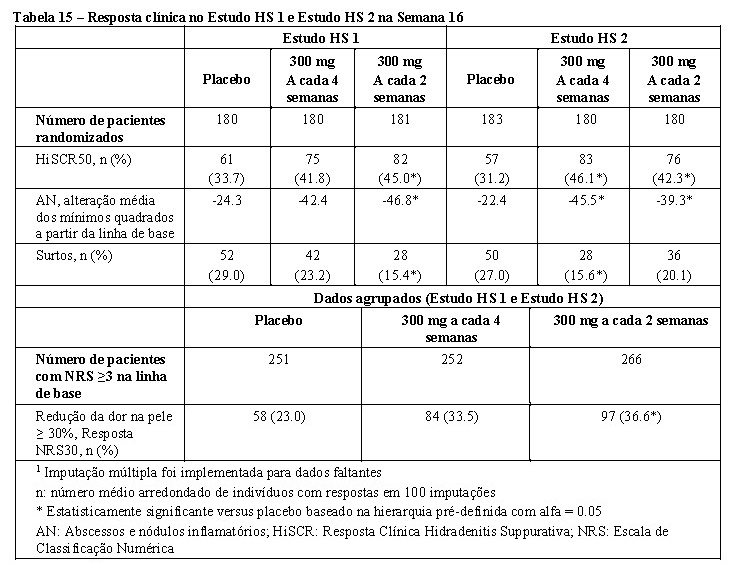
Cosentyx® é indicado para o tratamento de hidradenite supurativa (acne inversa) moderada a grave ativa em pacientes adultos com resposta inadequada à terapia convencional sistêmica.
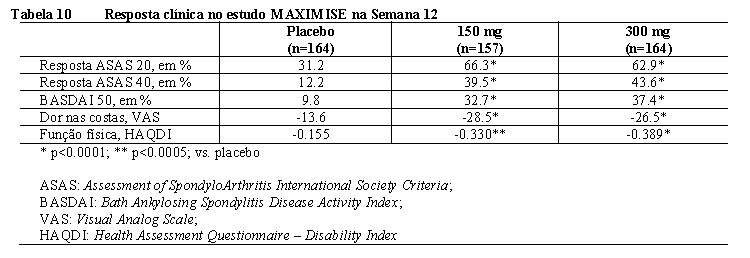
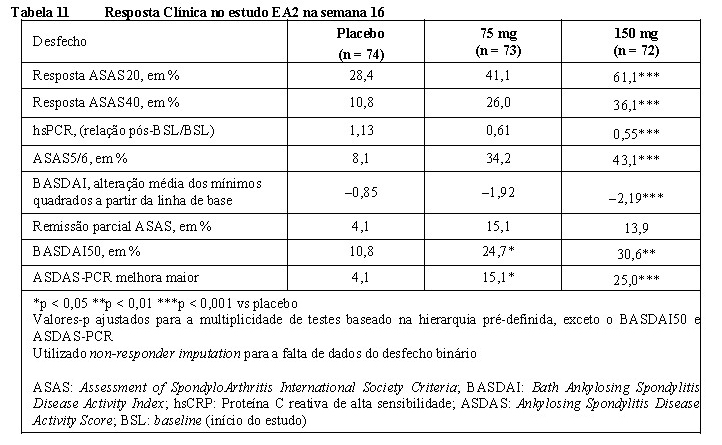
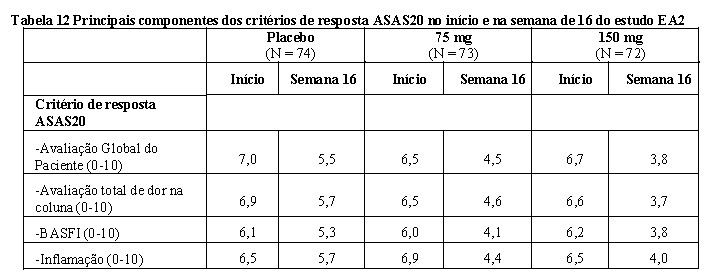
2. RESULTADOS DE EFICÁCIA
Psoríase em Placa 1,2,3,5
Pacientes adultos
A segurança e a eficácia do Cosentyx® foram avaliadas em quatro estudos fase 3 randomizados, duplo-cegos e controlados por placebo em pacientes com psoríase em placas moderada a grave que eram candidatos a fototerapia ou terapia sistêmica [ERASURE, FIXTURE, FEATURE, JUNCTURE]. A eficácia e a segurança de Cosentyx® 150 mg e 300 mg foram avaliadas em comparação ao placebo ou etanercepte. Além disso, um estudo avaliou um regime de tratamento crônico em comparação com um regime de "novo tratamento conforme necessário" [SCULPTURE]. Nestes estudos, cada dose de 300 mg foi administrada em duas injecções subcutâneas de 150 mg.
O Estudo em Psoríase 1 (ERASURE) avaliou 738 pacientes. Os pacientes randomizados para Cosentyx® receberam doses de 150 mg ou 300 mg nas semanas 0, 1, 2, 3 e 4 seguidas pela mesma dose a cada mês. Os pacientes randomizados para receber placebo que eram não responsivos na semana 12 realizaram cruzamento para receber Cosentyx® (150 mg ou 300 mg) nas semanas 12, 13, 14 e 15, seguido pela mesma dose a cada mês, com início na semana 16. Todos os pacientes foram acompanhados por até 52 semanas após a primeira administração do tratamento em estudo.
O Estudo em Psoríase 2 (FIXTURE) avaliou 1.306 pacientes. Os pacientes randomizados para Cosentyx® receberam doses de 150 mg ou 300 mg nas semanas 0, 1, 2, 3 e 4 seguidas pela mesma dose a cada mês. Os pacientes randomizados para etanercepte receberam doses de 50 mg, duas vezes por semana, por 12 semanas, seguido por 50 mg a cada semana. Os pacientes randomizados para receber placebo que não eram responsivos na semana 12 realizaram cruzamento para receber Cosentyx® (150 mg ou 300 mg) nas semanas 12, 13, 14 e 15, seguido pela mesma dose a cada mês, com início na semana 16. Todos os pacientes foram acompanhados por até 52 semanas após a primeira administração do tratamento em estudo.
O Estudo em Psoríase 3 (FEATURE) avaliou 177 pacientes que usaram a seringa preenchida em comparação ao placebo após 12 semanas de tratamento para avaliar a segurança, tolerabilidade e utilização da autoadministração de Cosentyx® por meio da seringa preenchida. Os pacientes randomizados para Cosentyx® receberam doses de 150 mg ou 300 mg nas semanas 0, 1, 2, 3 e 4, seguidas pela mesma dose a cada mês. Os pacientes foram também randomizados para receber placebo nas semanas 0, 1, 2 e 3, seguido pela mesma dose a cada mês, com início na semana 4.
O Estudo em Psoríase 4 (JUNCTURE) avaliou 182 pacientes que usaram a caneta preenchida em comparação ao placebo após 12 semanas de tratamento para avaliar a segurança, tolerabilidade e utilização da autoadministração de Cosentyx® por meio da caneta preenchida. Os pacientes randomizados para Cosentyx® receberam doses de 150 mg ou 300 mg nas semanas 0, 1, 2, 3 e 4, seguidas pela mesma dose a cada mês. Os pacientes foram também randomizados para receber placebo nas semanas 0, 1, 2, 3 e 4, seguido pela mesma dose a cada mês.
O Estudo em Psoríase 5 (SCULPTURE) avaliou 966 pacientes. Todos os pacientes receberam Cosentyx® em doses de 150 mg ou 300 mg nas semanas 0, 1, 2, 3, 4, 8 e 12, e então foram randomizados para receber um regime de manutenção da mesma dose a cada mês, com início na semana 12, ou um regime de "novo tratamento conforme necessário" da mesma dose. Os pacientes randomizados para "novo tratamento conforme necessário" não atingiram uma adequada manutenção da resposta e, portanto, recomenda-se um regime de manutenção mensal fixo.
Desfechos
Os desfechos coprimários nos estudos controlados por ativo e por placebo corresponderam à proporção de pacientes que atingiram uma resposta PASI 75 e uma resposta IGA mod. 2011 "sem lesão" ou "quase sem lesão" em comparação ao placebo na semana 12 (vide Tabelas 1 e 2). A dose de 300 mg forneceu uma melhora no clareamento da pele nos desfechos de eficácia de PASI 75/90/100 e respostas de IGA mod. 2011 "sem lesão" ou "quase sem lesão" em todos os estudos, com efeitos máximos observados na semana 16; portanto, esta dose é recomendada.
Características da linha de base
Dos 2.403 pacientes incluídos nos estudos controlados por placebo, 79% eram virgens de tratamento com medicamentos biológicos, 45% eram falhas de tratamento com agentes não biológicos, 8% eram falhas de tratamento com medicamentos biológicos, 6% eram falhas de tratamento com anti-TNF e 2% eram falhas de tratamento com anti- p40. As características basais da doença eram geralmente compatíveis entre todos os grupos de tratamento, com uma pontuação basal mediana do Índice da Área e da Gravidadeda Psoríase (PASI) entre 19 e 20, uma pontuação do basal do IGA modelo 2011 que variou de "moderada" (62%) a "grave" (38%), uma Superfície de Área Corporal (BSA) basal mediana ≥ 27 e uma pontuação mediana do Índice de Qualidade de Vida em Dermatologia (DLQI) de 10 a 12. Aproximadamente 15 a 25% dos pacientes em estudos de fase III apresentavam artrite psoriásica (AP) no basal.
Resposta Clínica
Os resultados dos estudos 1, 3 e 4 são apresentados na Tabela 1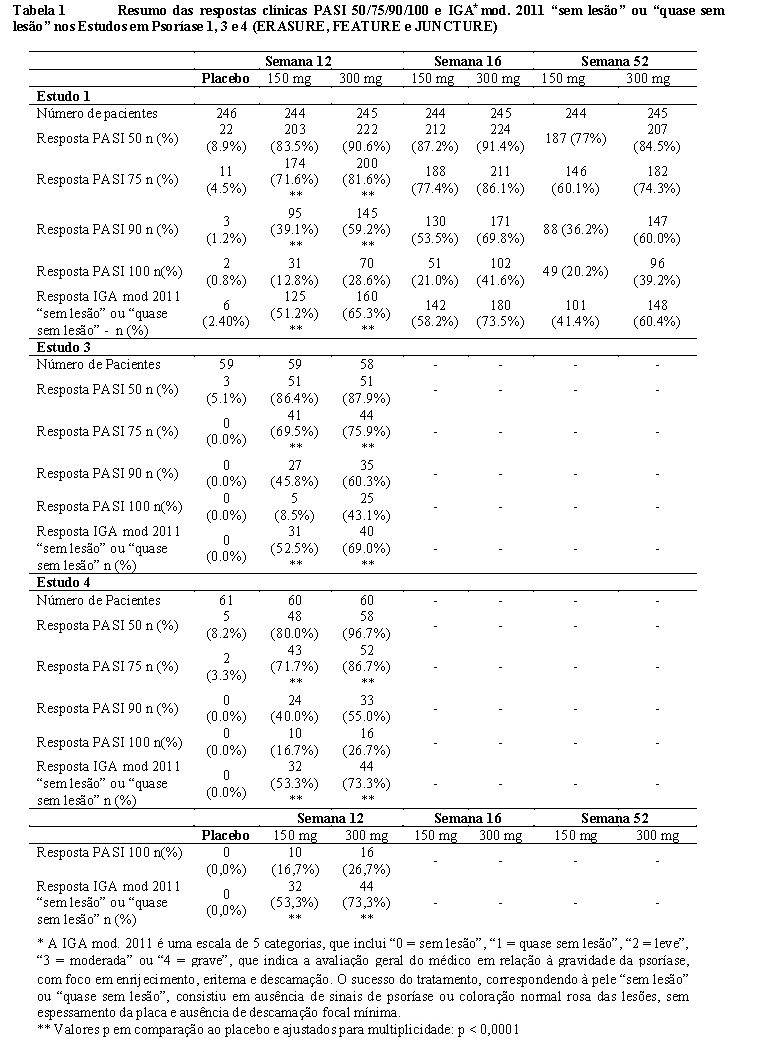
Os resultados dos estudos 2 (Psoríase) estão sendo apresentados na Tabela 2: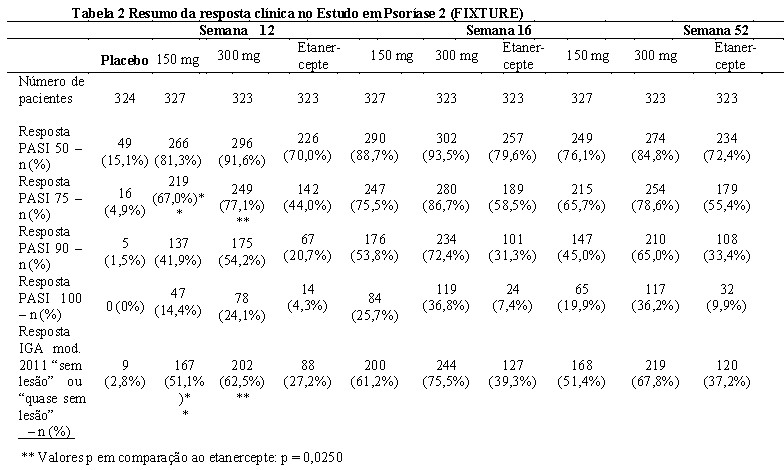
Um estudo adicional em Psoríase (CLEAR) avaliou 676 pacientes. Cosentyx® em dose de 300 mg atingiu os desfechos primário e secundário, exibindo superioridade ao ustequinumabe baseado na resposta do PASI 90 na semana 16 e na velocidade de início da resposta do PASI 75 na semana 4. A eficácia superior de Cosentyx® em comparação ao ustequinumabe em relação aos desfechos PASI 75/90/100 e respostas de IGA mod. 2011 0 ou 1 ("sem lesão" ou "quase sem lesão") foram observadas desde o início do estudo e, continuamente, até a semana 16. Neste estudo, cada dose de 300 mg foi administrada como duas injeções de 150 mg.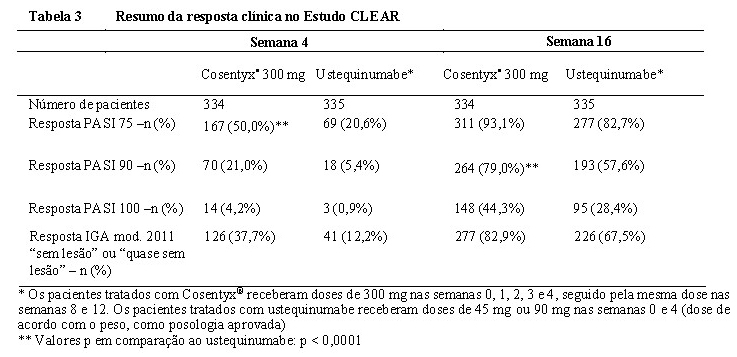
Um estudo adicional de psoríase (CLEAR) avaliou 676 pacientes. Cosentyx® 300 mg atingiu os desfechos primários e secundários principais demonstrando superioridade ao ustequinumabe, com base na resposta PASI 90 na semana 16 (desfecho primário), velocidade de início da resposta (PASI 75 na semana 4) e, a longo prazo, da resposta PASI 90 na semana 52. Foi observado melhor eficácia de Cosentyx® 300 mg comparado ao ustequinumabe desde o início do estudo e até a semana 52 para os desfechos PASI 75/90/100 e resposta IGA mod 2011 0 ou 1 ("sem lesão" ou "quase sem lesão").8 Neste estudo, cada dose de 300 mg foi administrada em duas injeções subcutâneas de 150 mg.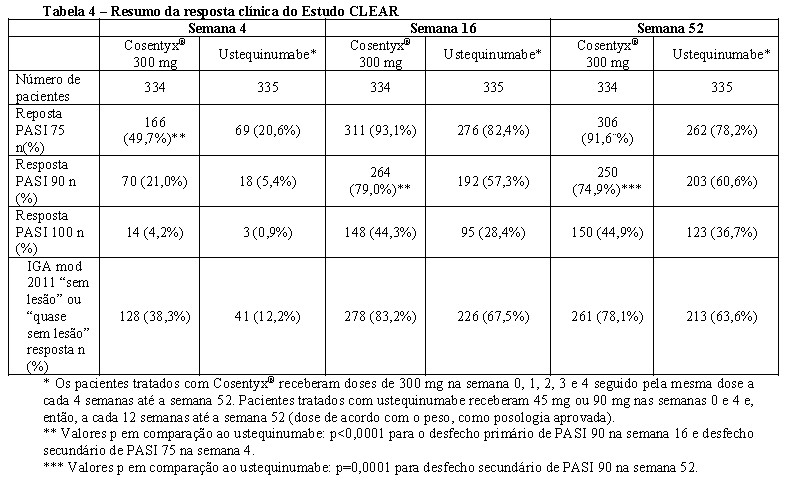
Cosentyx® foi eficaz em pacientes virgens de tratamento com medicamentos biológicos, naqueles expostos a medicamentos biológicos/anti-TNF e nos pacientes com falhas de tratamento com medicamentos biológicos/anti-TNF. Cosentyx® foi associado a uma rápida apresentação de eficácia, conforme demonstrado na figura abaixo, com uma redução de 50% no PASI médio na semana 3 em relação à dose de 300 mg.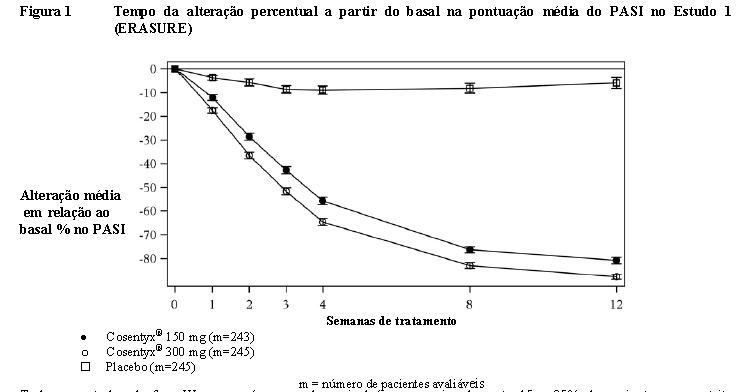
Todos os estudos de fase III em psoríase em placas incluíram aproximadamente 15 a 25% de pacientes com artrite psoriásica concomitante no basal. As melhoras no PASI 75 nesta população de pacientes foram semelhantes àquelas da população geral com psoríase em placas.
Nos estudos 1 e 2 controlados por placebo no subconjunto de pacientes com artrite psoriásica, a função física foi avaliada utilizando o Índice de Incapacidade do HAQ (HAQ-DI). Nestes estudos, os pacientes tratados com 150 mg ou 300 mg de Cosentyx® apresentaram melhora mais favorável a partir do basal na pontuação do HAQ-DI (reduções médias de -27,5% e -50,2% na semana 12) em comparação ao placebo (-8,9%). Essa melhora se manteve até a semana 52.
Localizações específicas/manifestações clínicas da psoríase em placas
Em dois estudos adicionais controlados com placebo, uma melhora foi observada tanto na psoríase ungueal (TRANSFIGURE, 198 pacientes) como na psoríase palmoplantar (GESTURE, 205 pacientes) em pacientes com psoríase em placas moderada a grave. No estudo TRANSFIGURE, Cosentyx® foi superior ao placebo na semana 16 (redução de 46,1% para 300 mg, 38,4% para 150 mg e 11,7% para o placebo), avaliado pela melhora significativa em relação à linha de base no Índice de Gravidade da Psoríase Ungueal (NAPSI%) para pacientes com psoríase em placas moderada a grave com envolvimento ungueal. No estudo GESTURE, Cosentyx® foi superior ao placebo na semana 16 (33,3% para 300 mg, 22,1% para 150 mg, e 1,5% para o placebo), avaliado pela melhora significativa da resposta do IGA palmoplantar 0 ou 1 ("sem lesão" ou "quase sem lesão") para pacientes com psoríase palmoplantar moderada a grave. Nestes estudos, cada dose de 300 mg foi administrada em duas injecções subcutâneas de 150 mg.
Psoríase - Lesões do couro cabeludo
O estudo SCALP, controlado com placebo, avaliou 102 pacientes com psoríase de couro cabeludo moderada a grave, tendo definida pelo PSSI (Índice de Gravidade da Psoríase do couro cabeludo) ≥ 12, uma pontuação na escala IGA mod 2011 somente para o couro cabeludo de 3 ou mais e, pelo menos, 30% da área do couro cabeludo afetada. Neste estudo, 62% dos pacientes apresentaram 50% ou mais de área de superfície do couro cabeludo afetada. Cosentyx® 300 mg foi superior ao placebo na semana 12, demonstrado pela melhora significativa a partir da linha de base tanto na resposta PSSI 90 (52,9% versus 2,0%) quanto na resposta IGA mod 2011 0 ou 1, somente de couro cabeludo (56,9% versus 5,9%). Maior eficácia de Cosentyx® 300 mg em relação ao placebo para ambos os desfechos foi observada na semana 3. A melhora em ambos os parâmetros foi mantida para os pacientes de Cosentyx® que continuaram o tratamento até a semana 24 (resposta PSSI 90 58,8% e IGA mod 2011 0 ou 1 somente resposta de couro cabeludo 62,7%). Nestes estudos, cada dose de 300 mg foi administrada em duas injecções subcutâneas de 150 mg.9
Qualidade de Vida - Resultados relatados pelo paciente
Foram demonstradas melhoras significativas do ponto de vista estatístico na semana 12 (Estudos 1-4) a partir do basal em comparação ao placebo no DLQI (Índice de Qualidade de Vida em Dermatologia), e essas melhoras se mantiveram por 52 semanas (Estudos 1 e 2).
Foram demonstradas melhoras significativas do ponto de vista estatístico na semana 12 (Estudos 1 e 2) a partir do basal nos sinais e sintomas relatados pelo paciente de prurido, dor e descamação no Psoriasis Symptom Diary© validado. 4
Pacientes tratados com Cosentyx® apresentaram melhora estatisticamente significativa no DLQI versus ustequinumabe (CLEAR) na semana 4, e essa melhora foi mantida até 52 semanas. Os resultados do questionário de Comprometimento da Produtividade no Trabalho e Atividades em portadores de psoríase (Work Productivity and Activity Impairment-- WPAI-PSO), apresentaram maior melhora nos pacientes tratados com Cosentyx®, quando comparada aos pacientes tratados com ustequinumabe.
Houve melhora estatisticamente significativa do prurido, dor e descamação nos pacientes tratados com Cosentyx® quando comparados aos pacientes tratados com ustequinumabe nas semanas 16 e 52 (CLEAR), relatadas no Psoriasis Symptom Diary© (Diário de Sintomas da Psoríase).
Melhora estatisticamente significativa na semana 12, a partir da linha de base comparado ao placebo (SCALP) foi demonstrada no HRQoL (Índice de Qualidade de Vida Relacionada a Saúde) medido pelo Scalpdex. Essas melhoras foram observadas começando na semana 4 e foram mantidas durante 24 semanas.
Pacientes em uso de Cosentyx® apresentaram melhora estatisticamente significativa do prurido no couro cabeludo (-59,4%), da dor (-45,9%) e descamação (-69,5%) na semana 12 comparado ao início do estudo (SCALP), enquanto que os pacientes tratados com placebo demonstraram piora (aumento) do prurido do couro cabeludo (7,7%) e dor (38,5%) e menor melhora na descamação do couro cabeludo (-4,7%).
Flexibilidade de dose de psoríase em placa
A eficácia, segurança e tolerabilidade de Cosentyx® 300 mg administrado a cada 4 semanas versus Cosentyx® 300 mg administrado a cada 2 semanas em pacientes adultos com peso ≥ 90 kg com psoríase em placas moderada a grave foram avaliadas em um estudo multicêntrico, duplo-cego, randomizado de 331 pacientes. Os pacientes foram randomizados 1: 1 da seguinte forma:
• secucinumabe 300 mg nas semanas 0, 1, 2, 3 e 4 seguido pela mesma dose a cada 2 semanas até a semana 52 (n = 165).
• secucinumabe 300 mg nas semanas 0, 1, 2, 3 e 4 seguido pela mesma dose a cada 4 semanas até a semana 16 (n = 166).
• Pacientes randomizados para receber secucinumabe 300 mg a cada 4 semanas que responderam ao PASI 90 na semana 16 continuaram a receber o mesmo regime de dosagem até a semana 52. Pacientes randomizados para receber Cosentyx® 300 mg a cada 4 semanas que não responderam ao PASI 90 na semana 16 continuaram com o mesmo regime de dosagem ou foram realocados no grupo para receber Cosentyx® 300 mg a cada 2 semanas até a Semana 52.
Os desfechos primários e secundários principais foram a proporção de pacientes que alcançaram uma resposta PASI 90 e IGA mod 2011 'sem lesão' ou 'quase sem lesão' (0 ou 1) resposta na semana 16. Na semana 16, a proporção de pacientes que eram PASI 90 respondentes foi maior no grupo tratado com o regime a cada 2 semanas vs. o regime a cada 4 semanas (73,2% versus 55,5%, respectivamente). A diferença de tratamento foi clinicamente relevante e estatisticamente significativa (valor p unilateral = 0,0003). A proporção de pacientes que alcançaram uma resposta 'sem lesão' ou 'quase sem lesão' do mod IGA 2011 também foi maior no grupo tratado com o regime a cada 2 semanas versus o grupo tratado com o regime a cada 4 semanas (74,2% vs. 65,9%, respectivamente).
Na Semana 52, a proporção de pacientes que responderam ao PASI 75, PASI 90, PASI 100 e IGA 0/1 foi maior em pacientes tratados com o regime a cada 2 semanas vs. pacientes tratados com o regime a cada 4 semanas (88,9% vs. 74,8%, 76,4% vs. 52,4%, 46,7% vs. 27,3%, 75,9% vs. 55,6%, respectivamente). Para todos os desfechos, a diferença de tratamento foi estatisticamente significativa (p-valor < 0,05). A resposta DLQI 0/1 na Semana 52 foi superior no grupo tratado com o regime a cada 2 semanas vs. o grupo tratado com o regime a cada 4 semanas (66,1% vs. 48,8%).
Os que não responderam ao PASI 90 na Semana 16 foram realocados no grupo para receber o regime a cada 2 semanas tiveram uma resposta PASI 90 mais alta na Semana 32 do que o grupo que continuou no regime de tratamento a cada 4 semanas (38,7% vs. 16,5%). A diferença de tratamento foi estatisticamente significativa (valor de p bilateral = 0,0439).
Da mesma forma, a resposta PASI 75 na semana 32 nos pacientes realocados no grupo para receber o regime a cada 2 semanas foi significativamente maior (valor p bilateral = 0,0015) do que para o grupo que continuou no regime de tratamento a cada 4 semanas (90,3% vs. 49,3%). A resposta IGA 0/1 na Semana 52 também foi maior no grupo de pacientes que recebeu o regime a cada 2 semanas (41,9% vs. 29,0%), assim como a resposta DLQI 0/1 (41,9% vs. 30,0%).
Os pacientes no regime a cada 2 semanas versus o regime a cada 4 semanas (Figura 2) mostraram benefício geral consistente e benefício para todos os subgrupos (peso, IGA e tempo desde o diagnóstico). O maior benefício incremental, conforme mostrado pelas diferenças de risco calculadas, foi para pacientes com doença grave (IGA 4), doença de longa duração ( > 25 anos) e maior peso.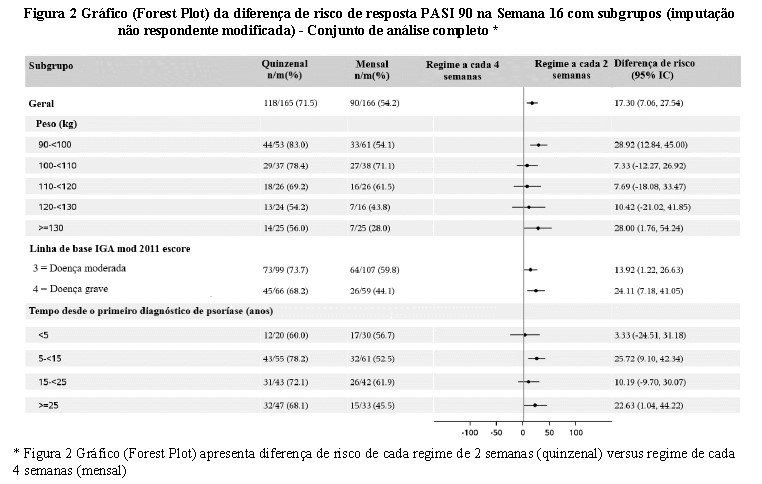
Os perfis de segurança dos dois regimes de dosagem, Cosentyx® 300 mg administrado a cada 4 semanas e Cosentyx® 300 mg administrado a cada 2 semanas, em pacientes com peso ≥ 90 kg foram comparáveis e consistentes com o perfil de segurança relatado em pacientes com psoríase.
Pacientes pediátricos
Psoríase em placa grave15
Um estudo de fase III de 52 semanas, randomizado, duplo-cego, controlado por placebo e etanercepte envolveu 162 pacientes pediátricos de 6 a 18 anos de idade, com psoríase em placas grave (conforme definido por um escore PASI ≥20, um IGA mod 2011 escore de 4 e envolvendo ≥10% da área de superfície corporal) que eram candidatos à terapia sistêmica.
Aproximadamente 43% tiveram exposição prévia à fototerapia, 53% à terapia sistêmica convencional, 3% a produtos biológicos e 9% tiveram artrite psoriática concomitante.
Os pacientes foram randomizados para receber um dos quatro tratamentos a seguir:
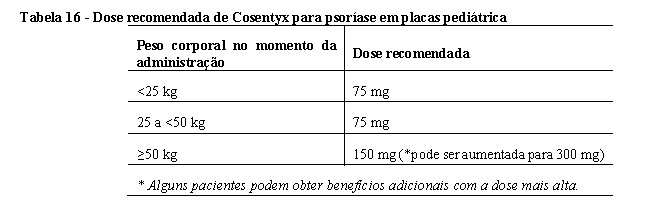
secuquinumabe em dose baixa (75 mg para peso corporal < 50 kg ou 150 mg para peso corporal ≥50 kg) nas semanas 0, 1,
2, 3 e 4 seguido pela mesma dose a cada 4 semanas,
secuquinumabe de alta dose (75 mg para peso corporal < 25 kg, 150 mg para peso corporal ≥25 kg e < 50 kg, ou 300 mg
para peso corporal ≥50 kg) nas semanas 0, 1, 2, 3 e 4 seguido por a mesma dose a cada 4 semanas,
placebo nas semanas 0, 1, 2, 3 e 4, seguido pela mesma dose a cada 4 semanas
etanercepte (0,8 mg / kg) semanalmente (até um máximo de 50 mg)
Os pacientes randomizados para receber placebo que não responderam na semana 12 foram transferidos para o grupo de dose baixa ou alta de secuquinumabe (dose com base no grupo de peso corporal) e receberam o medicamento do estudo nas semanas 12, 13, 14 e 15, seguido pela mesma dose a cada 4 semanas a partir da Semana 16.
Os desfechos co-primários foram a proporção de pacientes que alcançaram uma redução na pontuação PASI de pelo menos 75% (PASI 75) e IGA mod 2011 "sem lesão" ou "quase sem lesão" (0 ou 1) com pelo menos 2 pontos de melhora da linha de base até a semana 12. O principal desfecho secundário foi a proporção de pacientes que alcançaram uma redução na pontuação PASI de pelo menos 90% (PASI 90) da linha de base até a semana 12. Outros desfechos secundários incluíram taxas de resposta PASI 50, 100 na semana 12, Taxas de respondentes PASI 50, 75, 90, 100 e IGA 0/1 na semana 16 e ao longo do tempo até e incluindo a semana 52, alteração na pontuação PASI ao longo do tempo até e incluindo a semana 52 e pontuação IGA ao longo do tempo até e incluindo Semana 52, a proporção de pacientes com uma pontuação de Índice de Qualidade de Vida em Dermatologia Infantil (CDLQI) de 0 ou 1 na Semana 12 e ao longo do tempo até e incluindo a Semana 52, e alteração da linha de base no CDLQI em comparação com o placebo na Semana 12 e ao longo do tempo até a Semana 52, inclusive.
Durante o período de 12 semanas controlado com placebo, a eficácia tanto da dose baixa quanto da dose alta de secuquinumabe foi comparável para os desfechos co-primários. As estimativas de odds ratio a favor de ambas as doses de
secuquinumabe foram clinicamente relevantes e estatisticamente significativas para as respostas PASI 75 e IGA mod 2011 "sem lesão" ou "quase sem lesão" (0 ou 1).
Todos os pacientes foram acompanhados quanto à eficácia e segurança durante as 52 semanas após a primeira dose. A proporção de pacientes que alcançaram PASI 75 e IGA mod 2011 respostas "sem lesão" ou "quase sem lesão" (0 ou 1) mostrou separação entre os grupos de tratamento com secuquinumabe e placebo na primeira visita pós-linha de base na Semana 4, a diferença se tornando mais proeminente na Semana 12. A resposta foi mantida ao longo do período de 52 semanas. A melhora nas taxas de resposta PASI 50, 90, 100 e na pontuação CDLQI 0 ou 1 também foi mantida ao longo do período de 52 semanas.
Além disso, as taxas de resposta PASI 75, IGA 0 ou 1 e PASI 90 nas semanas 12 e 52 para os grupos de dose baixa e alta de secuquinumabe foram maiores do que as taxas de pacientes tratados com etanercepte.
Após a semana 12, a eficácia de ambas as doses baixa e alta de secuquinumabe foi comparável, embora a eficácia da dose
alta tenha sido maior para pacientes ≥50 kg. Os perfis de segurança da dose baixa e da dose alta foram comparáveis.
Os resultados de eficácia nas semanas 12 e 52 são apresentados na Tabela 5.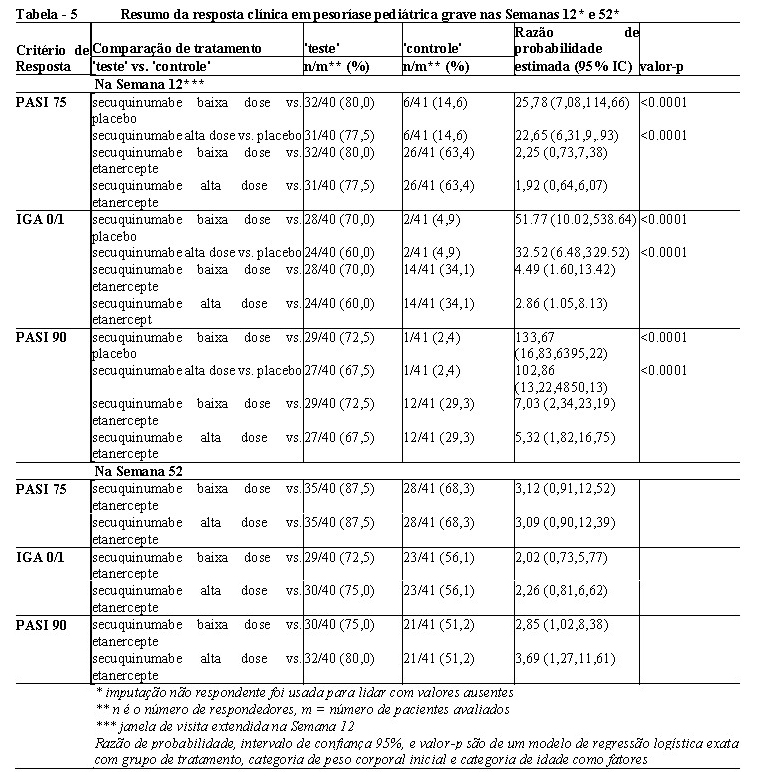
Uma proporção maior de pacientes pediátricos tratados com secuquinumabe relatou melhora na qualidade de vida relacionada à saúde medida por uma pontuação CDLQI de 0 ou 1 em comparação com o placebo na Semana 12 (dose baixa 44,7%, dose alta 50%, placebo 15%). Da Semana 12 à Semana 52, a proporção de pacientes pediátricos em ambos os grupos de dose de secuquinumabe com uma pontuação de CDLQI de 0 ou 1 foi numericamente maior do que para o grupo de etanercepte (dose baixa 60,6%, dose alta 66,7%, etanercepte 44,4%).
Psoríase em placas moderada a grave 16
Um estudo aberto, de dois braços, de grupo paralelo, multicêntrico de fase III inscreveu 84 pacientes pediátricos de 6 a menos de 18 anos de idade com psoríase em placas moderada a grave (conforme definido por um escore PASI ≥12, um escore IGA mod 2011 ≥ 3, e envolvendo ≥10% da área de superfície corporal) que eram candidatos à terapia sistêmica.
Os pacientes foram randomizados para receber secuquinumabe nas semanas 0, 1, 2, 3 e 4, seguido pela mesma dose a cada 4 semanas da seguinte forma:
• secuquinumabe em dose baixa (75 mg para peso corporal < 50 kg ou 150 mg para peso corporal ≥50 kg),
• secuquinumabe em dose elevada (75 mg para peso corporal < 25 kg, 150 mg para peso corporal entre ≥25 kg e < 50 kg, ou 300 mg para peso corporal ≥50 kg).
Os desfechos co-primários foram a proporção de pacientes que alcançaram uma redução na pontuação PASI de pelo menos 75% (PASI 75) e IGA mod 2011 "sem lesão" ou "quase sem lesão" (0 ou 1) com pelo menos uma melhoria de 2 pontos da linha de base até a Semana 12. Os desfechos secundários e adicionais incluíram resposta PASI 90 na Semana 12, PASI 75, 90, 100 e IGA mod 2011 "sem lesão" ou "quase sem lesão" (0 ou 1), e respostas CDLQI ao longo do tempo até fim do tratamento.
A eficácia de ambas as doses baixa e alta de secuquinumabe foi comparável e mostrou melhora estatística e clinicamente significativa em comparação com o placebo histórico para os desfechos co-primários. As estimativas de razão de probabilidade a favor de ambas as doses de secuquinumabe foram clinicamente relevantes e estatisticamente significativas para as respostas PASI 75 e IGA mod 2011 0 ou 1 versus placebo histórico. A probabilidade posterior estimada de um efeito positivo do tratamento foi de 100%.
Todos os pacientes foram acompanhados quanto à eficácia por pelo menos 24 semanas após a primeira administração. A eficácia (definida como resposta PASI 75 e IGA mod 2011 "sem lesão" ou "quase sem lesão" [0 ou 1]) foi observada já na Semana 2 e a proporção de pacientes que alcançaram uma resposta PASI 75 e IGA mod 2011 "sem lesão" ou "quase sem lesão" (0 ou 1) aumentou ao longo do período de 24 semanas. A melhora no PASI 90 e no PASI 100 também foi observada na semana 12 e aumentou ao longo do período de 24 semanas.
Após a semana 12, a eficácia da dose baixa e da dose alta de secuquinumabe foi comparável. Os perfis de segurança da dose baixa e da dose alta foram comparáveis.
Os resultados de eficácia nas semanas 12 e 24 são apresentados na Tabela 6.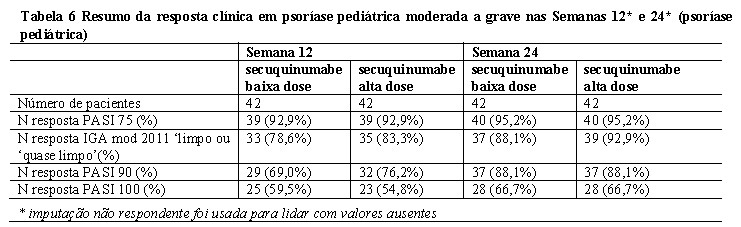
No grupo de dose baixa, 50% e 70,7% dos pacientes alcançaram uma pontuação CDLQI 0 ou 1 nas semanas 12 e 24, respectivamente. No grupo de dose alta, 61,9% e 60,5% alcançaram uma pontuação CDLQI 0 ou 1 nas semanas 12 e 24, respectivamente.
Pacientes Adultos
Psoríase em placa 300 mg/2mL solução injetável em seringa preenchida ou caneta preenchida
Dois estudos randomizados, duplo-cegos, controlados por placebo em pacientes com psoríase em placas foram conduzidos para avaliar a segurança e eficácia de secuquinumabe 300 mg quando administrado por via subcutânea como uma única seringa preenchida de 2 mL (ALLURE, 214 pacientes) ou como uma única caneta preenchida de 2 mL (MATURE, 122 pacientes) em comparação com secuquinumabe 300 mg quando administrado como duas injeções subcutâneas em uma seringa preenchida de 150 mg / 1 mL. Os desfechos co-primários foram a proporção de pacientes que alcançaram uma resposta PASI 75 e resposta IGA mod 2011 'clara' ou 'quase clara' versus placebo na Semana 12.
No estudo ALLURE, a proporção de indivíduos que obtiveram respostas PASI 75 e IGA mod 2011 0 ou 1 na Semana 12 foi de 88,9% e 76,4% para o grupo de seringa preenchida de secuquinumabe 300 mg/ 2mL em comparação com 1,7% e 1,4% no grupo do placebo. No estudo MATURE, a proporção de indivíduos que obtiveram respostas PASI 75 e IGA mod 2011 0 ou 1 na Semana 12 foi de 95,1% e 75,6% para o grupo de caneta preenchida de secuquinumabe 300 mg/ 2mL em comparação com 10% e 7,6% no grupo do placebo. A resposta PASI 90 na semana 12 foi alcançada com secuquinumabe 300 mg/ 2mL seringa preenchida (estudo ALLURE) e secuquinumabe 300 mg/ 2mL caneta preenchida (estudo MATURE) em comparação com o placebo em 66,7% versus 1,6% dos indivíduos, respectivamente (estudo ALLURE) e 75,6% versus 5% dos indivíduos, respectivamente (estudo MATURE).
A experiência geral do paciente com a autoinjeção subcutânea com a seringa preenchida de 300 mg/ 2 mL e a caneta preenchida de 300 mg/ 2 mL foi medida usando o Questionário de Avaliação de Auto-injeção (SIAQ). No estudo ALLURE, a proporção de pacientes "muito satisfeitos" e "satisfeitos" atingiu 89,5% na semana 28. No estudo MATURE, a proporção de pacientes "muito satisfeitos" e "satisfeitos" atingiu 91,8% na semana 12.
Com a continuação do tratamento durante 52 semanas, a proporção de respondentes PASI 75/90/100 e IGA mod 2011 0 ou 1 no estudo ALLURE aumentou até a semana 28 e as respostas foram mantidas até a semana 52. Na semana 52, o PASI 75/90/100 e IGA mod 2011 0 ou 1 taxas de resposta para o grupo de seringa preenchida de secuquinumabe 300 mg/2 mL foram 88,2%, 75,6%, 55,2% e 76,5%, respectivamente, e 87,2%, 81,7%, 52,5 % e 76,8%, respectivamente, para o grupo da seringa preenchida de secuquinumabe 2 × 150 mg / 1 mL 23,24.
Artrite psoriásica (AP) 6
Nos estudos clínicos, os pacientes adultos com artrite psoriásica ativa tratados com Cosentyx® apresentaram melhora nos sinais e sintomas da doença, na função física e na qualidade de vida. A inibição da progressão radiográfica (dano estrutural) nos pacientes com AP foi demonstrada no estudo clínico AP1 (FUTURE 1) com doses iniciais intravenosas de Cosentyx® na fase de indução.
A eficácia e segurança de Cosentyx® em AP foram avaliadas em 1.999 pacientes, de três estudos de fase III, randomizados, duplo-cegos, controlados por placebo. Os pacientes apresentavam AP ativa (≥ 3 articulações edemaciadas e ≥ 3 articulações dolorosas), apesar da terapia com anti-inflamatórios não esteroidais (AINE), corticosteroides ou medicamentos antirreumáticos modificadores da doença (DMARDs). Os pacientes nesses estudos tinham diagnóstico de AP há pelo menos cinco anos. A maioria dos pacientes também apresentava lesões de psoríase cutânea ativa ou histórico de psoríase. Mais de 61% e 42% dos pacientes com AP apresentavam, respectivamente, entesite ou dactilite no momento inicial do estudo.
A eficácia e a segurança de Cosentyx® 75 mg, 150 mg e/ou 300 mg foram comparadas ao placebo com doses de indução
intravenosa (i.v.) ou subcutânea (s.c.). No estudo de artrite psoriásica 1 (estudo AP1), no estudo de artrite psoriásica 2 (estudo AP2) e no estudo de artrite psoriásica 3 (AP3), 29%, 35% e 30% dos pacientes, respectivamente, foram tratados previamente com um agente anti-TNF-alfa (pacientes anti-TNF-alfa-IR) e interromperam o uso do agente anti-TNF-alfa por falta de eficácia ou intolerância.
O estudo AP1 (FUTURE 1) avaliou 606 pacientes, dos quais 60,7% receberam metotrexato (MTX) concomitantemente. Pacientes com todos os subtipos de AP foram recrutados, incluindo artrite poliarticular sem evidências de nódulos reumatoides (76,7%), espondilite com artrite periférica (18,5%), artrite periférica assimétrica (60,2%), acometimento predominante das interfalangianas distais (59,6%) e artrite mutilante (7,9%). Os pacientes randomizados para Cosentyx® receberam a dose de 10 mg/kg i.v. nas semanas 0, 2 e 4, seguido de doses mensais de 75 mg s.c. (grupo secuquinumabe 10 mg/kg i.v. - 75 mg s.c.) ou 150 mg s.c. (grupo secuquinumabe 10 mg/kg i.v. - 150 mg s.c.), a partir da semana 8. Na semana 16, os pacientes que foram randomizados para os grupos tratados com Cosentyx® foram caracterizados como respondedores, ou não?respondedores e continuaram com o mesmo tratamento. Os pacientes randomizados para receber placebo que não responderam na semana 16, passaram a receber Cosentyx® (75 mg ou 150 mg, s.c.) em doses mensais, a partir da semana 16. Os pacientes randomizados para receber placebo que responderam na semana 16, passaram a receber Cosentyx® (75 mg ou 150 mg, s.c.) mensalmente, a partir da semana 24. O desfecho primário foi a resposta clínica pelo American College of Rheumatology (ACR) 20 na semana 24.
O estudo AP2 (FUTURE 2) avaliou 397 pacientes, dos quais 46,6% receberam metotrexato concomitantemente. Pacientes com todos os subtipos de AP foram recrutados, incluindo artrite poliarticular sem evidência de nódulos reumatoides (85,9%), espondilite com artrite periférica (21,7%), artrite periférica assimétrica (64,0%), acometimento predominante das interfalangianas distais (57,9%) e artrite mutilante (6,3%). Os pacientes randomizados para Cosentyx® receberam doses de 75 mg, 150 mg ou 300 mg s.c. nas semanas 0, 1, 2, 3 e 4, seguido da mesma dose, mensalmente. Na semana 16, os pacientes que foram randomizados para os grupos tratados com Cosentyx® foram caracterizados como respondedores, ou não?respondedores e continuaram com o mesmo tratamento. Os pacientes randomizados para receber placebo que não responderam na semana 16 passaram a receber Cosentyx® (150 mg ou 300 mg, s.c.) mensalmente, a partir da semana 16. Os pacientes randomizados para receber placebo que responderam na semana 16 passaram a receber Cosentyx® (150 mg ou 300 mg, s.c.) mensalmente, a partir da semana 24. O desfecho primário foi a resposta ACR20 na semana 24.
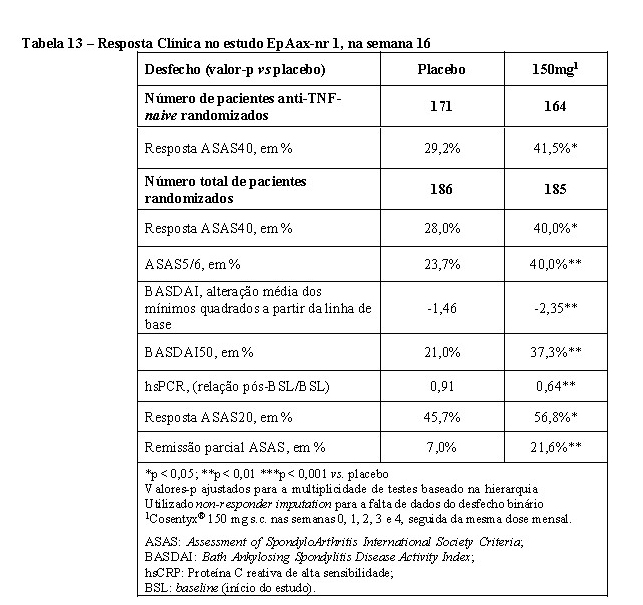
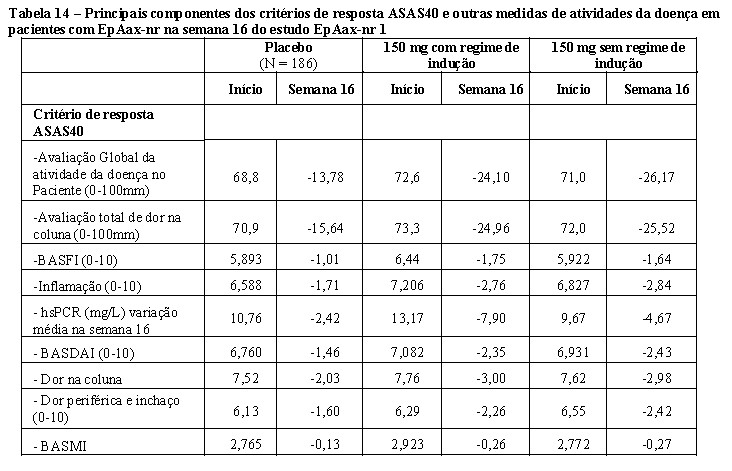
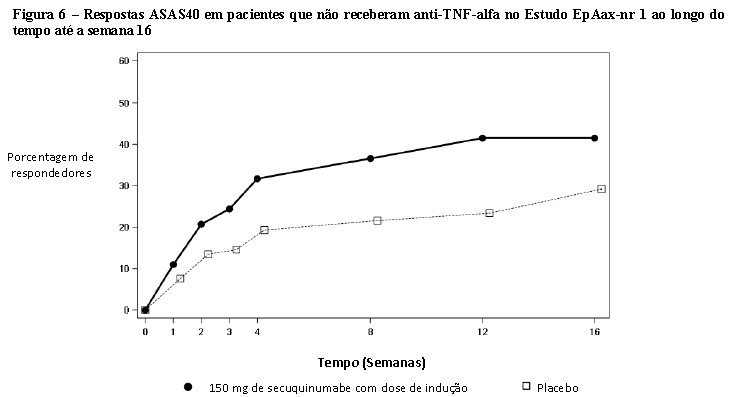
O estudo AP3 (FUTURE 5) avaliou 996 pacientes, dos quais 50,1% tiveram tratamento MTX concomitante. Pacientes com todos os subtipos de AP foram recrutados incluindo artrite poliarticular sem evidência de nódulos reumatoides (78,7%), espondilite com artrite periférica (19,8%), artrite periférica assimétrica (65%), acometimento predominante das interfalangianas distais (56,7%) e artrite mutilante (6,8%). Os pacientes randomizados receberam Cosentyx® 150 mg, 300 mg ou placebo nas semanas 0, 1, 2, 3 e 4 seguido da mesma dose mensalmente, ou Cosentyx® 150 mg solução injetável mensalmente (sem as doses de indução). Na semana 16, os pacientes que foram randomizados para os grupos tratados com Cosentyx® foram caracterizados como respondedores, ou não?respondedores e continuaram com o mesmo tratamento. Já os pacientes tratados com placebo, que foram classificados como não?respondedores na semana 16 foram re? aleatorizados para receber Cosentyx® (150mg ou 300 mg) à semana 16, seguido da mesma dose mensalmente. Na semana 24, os pacientes tratados com placebo, que foram classificados como respondedores na semana 16 foram re?aleatorizados para receber Cosentyx® (150mg ou 300 mg) à semana 24, seguido da mesma dose mensalmente. O objetivo primário foi demonstrar que a eficácia do Cosentyx® 150 mg SC (com ou sem regime de indução), ou 300 mg sc com regime indução, na 16ª semana foi superior ao placebo, com base na proporção de pacientes com AP ativa que atingiram a resposta ACR20, com o desfecho secundário chave de alteração da linha de base na pontuação total Sharp (mTSS) modificada na semana 24.
Resposta clínica
- Sinais e Sintomas
Os pacientes do grupo Cosentyx® apresentaram melhora significativa nas medidas de atividade da doença em comparação com o grupo placebo, nas semanas 16, 24 e 52. Estas medidas incluíram a resposta ACR20, ACR50, ACR70, resposta Índice de Área e Gravidade da Psoríase, PASI 75, PASI 90, Escore de Atividade da Doença de 28 articulaçõespela Proteína C reativa (DAS28-PCR), Short Form Health Survey - Physical Component Summary (SF36 - PCS), Health Assessment Questionnaire - Disability Index (HAQ-DI), todas comparadas ao placebo nas semanas 16, 24 e 52 (vide Tabela 7).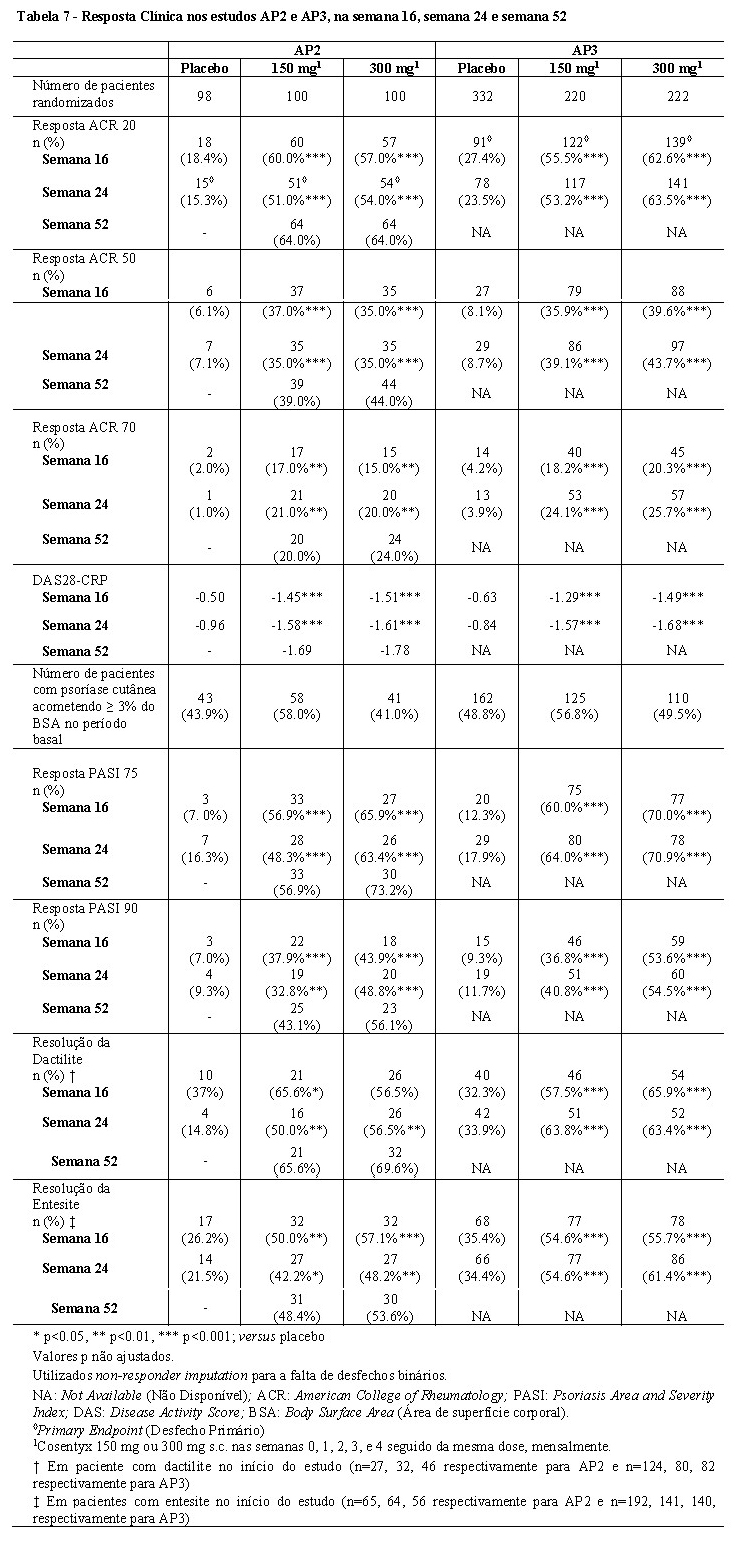
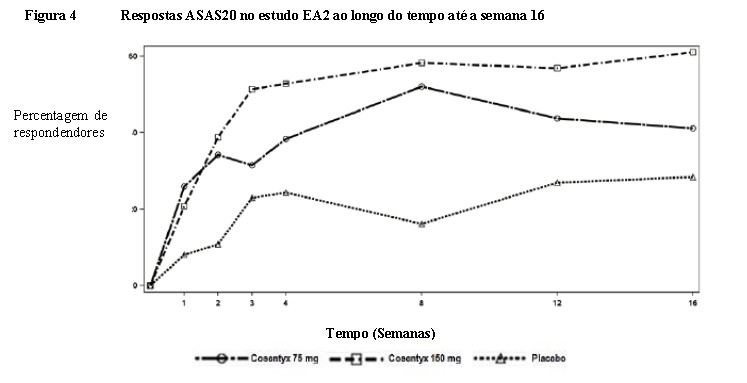
O início de ação de Cosentyx® ocorreu na semana 2. A diferença estatisticamente significante na resposta ACR20
versus o placebo foi alcançada na semana 3. Em AP2 as respostas de eficácia foram mantidas até a semana 104.
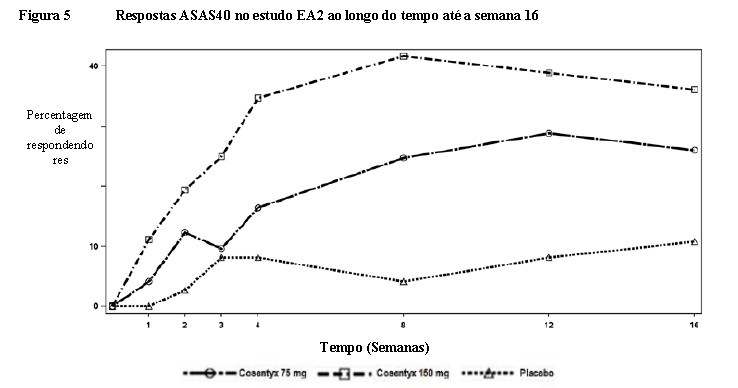
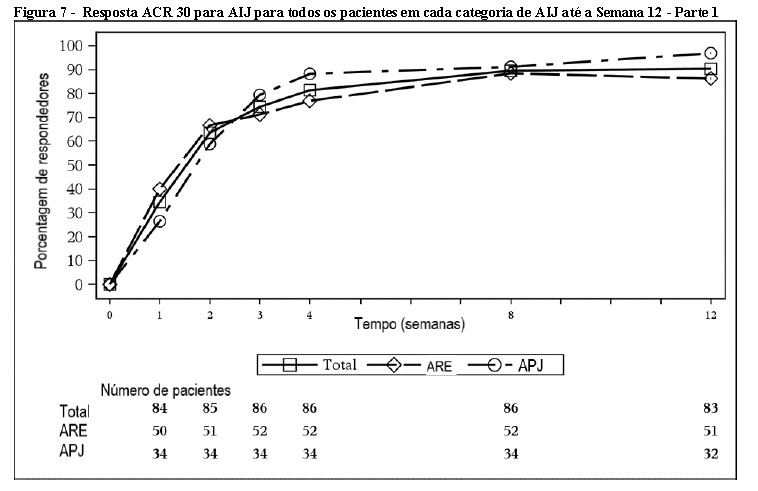
A percentagem de pacientes que atingiu resposta ACR20 por visita é mostrada na Figura 3.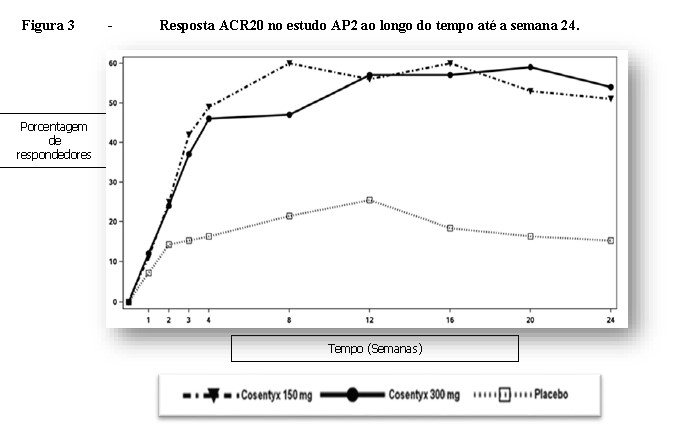
Foram observadas respostas semelhantes ao desfecho primário para os principais desfechos secundários em pacientes com AP, independentemente se eles estavam ou não em tratamento concomitante com metotrexato. Embora os objetivos primários e secundários fossem relacionados à comparação entre as posologias de Cosentyx® com o placebo, a comparação entre as diferentes doses de Cosentyx® fizeram parte dos objetivos exploratórios do estudo.
Tanto os pacientes não tratados anteriormente com anti-TNF-alfa (anti-TNF-alfa-naive) quanto os anti-TNF-alfa-IR que foram tratados com Cosentyx®, obtiveram respostas ACR20 significativamente maiores em comparação ao placebo nas semanas 16 e 24. Os pacientes anti-TNF-alfa-naive apresentaram percentuais de resposta ACR20 ligeiramente superiores (anti-TNF-alfa-naive: 64% e 58% para 150 mg e 300 mg, respectivamente, em comparação a 15,9% no grupo placebo; anti-TNF-alfa-IR: 30% e 46% para 150 mg e 300 mg, respectivamente, em comparação a 14,3% do grupo placebo). Em pacientes anti-TNF-alfa-IR, o grupo tratado com Cosentyx® 300 mg apresentou resposta ACR20 superior ao grupo placebo (p < 0,05) e demonstrou benefícios clinicamente relevantes em relação aos pacientes tratados com 150 mg nos diversos desfechos secundários. Foi observada melhora na resposta PASI75 independentemente do uso prévio de anti-TNF-alfa.
Em AP2, a proporção de pacientes que atingiu o Critérios de Resposta da Artrite Psoriásica (Psoriatic Arthritis Response Criteria -PsARC) foi maior naqueles tratados com Cosentyx® (59,0% e 61,0% para 150 mg e 300 mg, respectivamente) em comparação ao grupo placebo (26,5%) na semana 24.
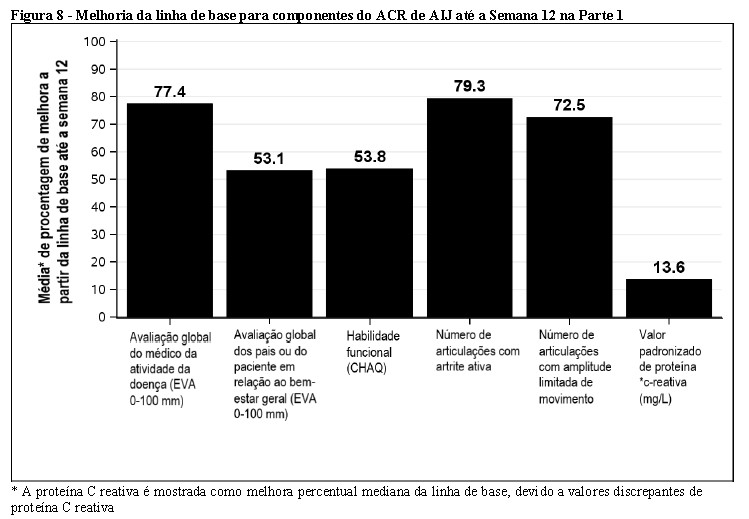
Nas semanas 16 e 24, foi observada melhora nos parâmetros de atividade periférica da artrite psoriásica (por exemplo, número de articulações dolorosas, dactilite, entesite e o índice modificado de gravidade da unha na psoríase (mNAPSI)) em pacientes tratados com Cosentyx® (valor-p nominal p < 0,01).
Os resultados dos componentes do critério de resposta do ACR são apresentados na Tabela 8.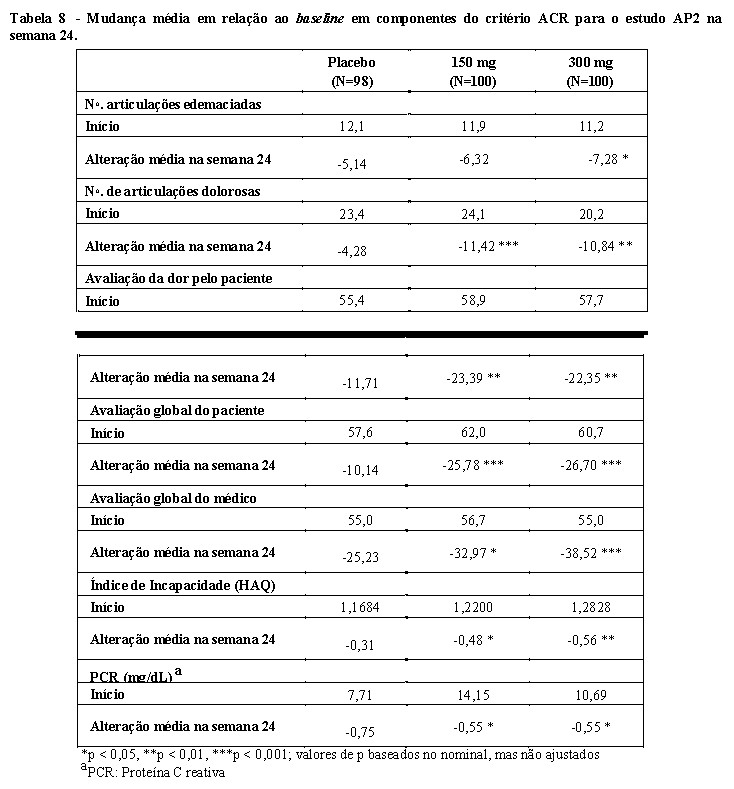
No estudo AP1, os pacientes tratados com Cosentyx® demonstraram melhora significativa nos sinais e sintomas da AP na semana 24 com magnitude similar de resposta ao estudo AP2. A eficácia foi mantida até a semana 104.
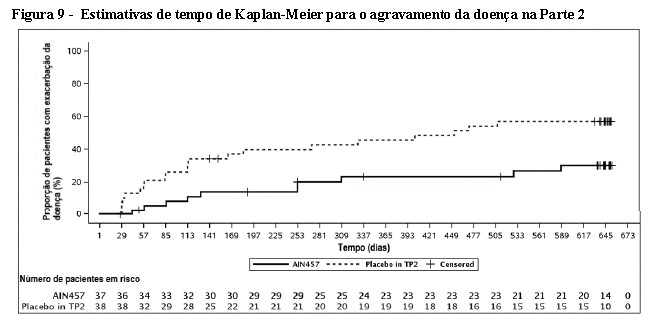
- Resposta na inibição da progressão radiográfica
No estudo AP3, o dano estrutural foi avaliado radiograficamente e expresso pelo Escore Total de Sharp modificado (modified Total Sharp Score - mTSS) e seus componentes, o Escore de Erosão (Erosion Score - ES) e o Escore de Estreitamento do Espaço Articular (Joint Space Narrowing score - JSN). As radiografias das mãos, punhos e pés foram obtidas no início da semana 16 e/ou semana 24 e foram marcadas de forma independente por pelo menos dois leitores cegos para o grupo de tratamento e número de visitas.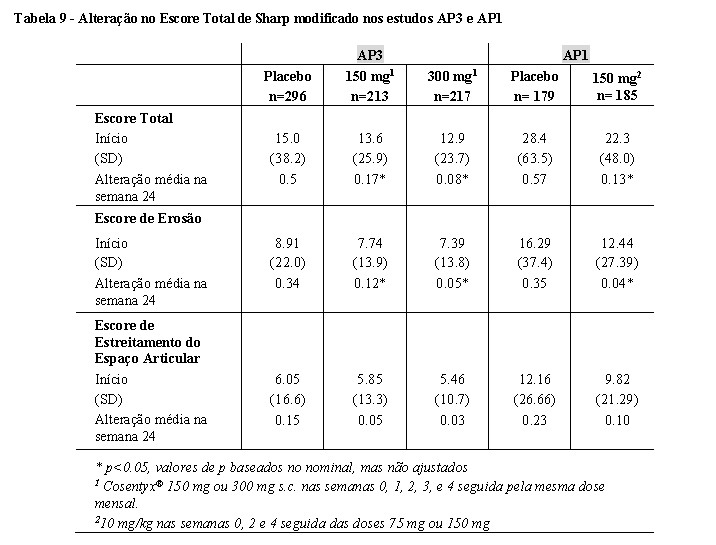
O tratamento com Cosentyx® 150 mg e 300 mg inibiu significativamente a taxa