CITALOR®
VIATRIS
atorvastatina
Hipolipidemiante.
Apresentações.
Citalor® 10 mg ou 20 mg em embalagens contendo 30 ou 60 comprimidos revestidos. Citalor® 40 mg ou 80 mg em embalagens contendo 30 comprimidos revestidos.
VIA DE ADMINISTRAÇÃO: USO ORAL
CITALOR® 10 mg e 20 mg -USO ADULTO E PEDIÁTRICO ACIMA DE 10 ANOS DE IDADE
CITALOR® 40 mg e 80 mg - USO ADULTO
Composição.
Cada comprimido revestido de Citalor® 10 mg, 20 mg ou 40 mg contém atorvastatina cálcica equivalente a 10 mg, 20 mg ou 40 mg de atorvastatina base, respectivamente. Excipientes: carbonato de cálcio, celulose microcristalina, lactose monoidratada, croscarmelose sódica, polissorbato 80, hiprolose, estearato de magnésio, corante branco Opadry® (hipromelose, macrogol, dióxido de titânio e talco), emulsão simeticona (simeticona, estearato emulsificante, espessantes, ácido benzóico, ácido sórbico e água) e cera candelila. Cada comprimido revestido de Citalor® 80 mg contém atorvastatina cálcica equivalente a 80 mg de atorvastatina base. Excipientes: carbonato de cálcio, celulose microcristalina, lactose monoidratada, croscarmelose sódica, polissorbato 80, hiprolose, estearato de magnésio, corante branco Opadry® (hipromelose, macrogol, dióxido de titânio e talco) e emulsão simeticona (simeticona, estearato emulsificante, espessantes, ácido benzóico, ácido sórbico e água).
Indicações.
Citalor® (atorvastatina cálcica) comprimidos revestidos é indicado como um adjunto à dieta para o tratamento de pacientes com níveis elevados de colesterol total (CT), lipoproteína de baixa densidade (LDL-C), apolipoproteína B (apo B) e triglicérides (TG), para aumentar os níveis de lipoproteína de alta densidade (HDLC) em pacientes com hipercolesterolemia primária (hipercolesterolemia heterozigótica familiar e não familiar), hiperlipidemia combinada (mista) (Fredrickson tipos IIa e IIb), níveis elevados de triglicérides séricos (Fredrickson tipo IV) e para pacientes com disbetalipoproteinemia (Fredrickson tipo III) que não respondem de forma adequada à dieta. Citalor® também é indicado para a redução do colesterol total e da lipoproteína de baixa densidade em pacientes com hipercolesterolemia familiar homozigótica, quando a resposta à dieta e outras medidas não-farmacológicas forem inadequadas.
Em pacientes com doença cardiovascular e/ou dislipidemia, Citalor® está indicado na síndrome coronária aguda (angina instável e infarto do miocárdio não transmural - sem onda Q) para a prevenção secundária do risco combinado de morte, infarto do miocárdio não fatal, parada cardíaca e re-hospitalização de pacientes com angina do peito.
Prevenção de Complicações Cardiovasculares: Em pacientes sem evidência clínica de doença cardiovascular (DCV) e com ou sem dislipidemia, porém com múltiplos fatores de risco para doença coronariana (DAC) como tabagismo, hipertensão, diabetes, baixo nível de HDL-C ou história familiar de doença coronariana precoce, Citalor® está indicado para redução do risco de doença coronariana fatal e infarto do miocárdio não fatal, acidente vascular cerebral, procedimentos de revascularização e angina do peito. Em pacientes com doença cardíaca coronariana clinicamente evidente, Citalor® é indicado para redução do risco de: infarto do miocárdio não fatal; acidente vascular cerebral fatal e não fatal; procedimentos de revascularização; hospitalização por insuficiência cardíaca congestiva (ICC); angina.
Pacientes Pediátricos (10 a 17 anos): Citalor® também é indicado como um adjuvante à dieta de redução dos níveis de CT, LDL-C e Apo B em meninas pós-menarca e meninos, entre 10 e 17 anos, com hipercolesterolemia familiar heterozigótica se, após a realização de um teste adequado de terapia dietética, os níveis de LDL-C continuarem ≥ 190 mg/dL ou ≥ 160 mg/dL e houver um histórico familiar positivo para doença cardiovascular (DCV) prematura, ou presença de 2 ou mais fatores de risco cardiovascular no paciente pediátrico.
Resultados de eficácia.
Aterosclerose: no estudo de Reversão da Aterosclerose com Terapia Hipolipemiante Intensiva (REVERSAL), o efeito da atorvastatina 80 mg e da pravastatina 40 mg na aterosclerose coronária foi avaliado pelo ultrassom intravascular (USIV), durante a angiografia, em pacientes com doença arterial coronariana. Neste estudo randomizado, duplo-cego, multicêntrico, controlado, o USIV foi realizado em 502 pacientes no período basal e após 18 meses. No grupo tratado com a atorvastatina (n=253), a mudança média percentual observada no volume total do ateroma (critério principal do estudo), quando comparado ao período basal, foi -0,4% (p=0,98) no grupo da atorvastatina e, +2,7% (p=0,001) no grupo da pravastatina (n = 249). Quando comparados aos da pravastatina, os efeitos da atorvastatina foram estatisticamente significantes (p=0,02).
No grupo da atorvastatina, o LDL-C foi reduzido para uma média de 2,04 mmol/L ± 0,8 (78,9 mg/dL ± 30) quando comparado ao período basal de 3,89 mmol/L ± 0,7 (150 mg/dL ± 28), e no grupo da pravastatina o LDLC foi reduzido para uma média de 2,85 mmol/L ± 0,7 (110 mg/dL ± 26) quando comparado ao período basal de 3,89 mmol/L ± 0,7 (150 mg/dL ± 26) (p < 0,0001). A atorvastatina também reduziu significativamente a média do CT em 34,1% (pravastatina: -18,4%, p < 0,0001), os níveis médios de TG em 20% (pravastatina: -6,8%, p < 0,0009) e a média de apolipoproteína B em 39,1% (pravastatina: -22,0%, p < 0,0001). A atorvastatina aumentou o HDL-C em 2,9% (pravastatina: +5,6%, p = NS). Houve uma redução média de 36,4% na proteína reativa C (CRP) no grupo da atorvastatina, comparado com uma redução de 5,2% no grupo da pravastatina (p < 0,0001). O perfil de segurança e tolerabilidade dos 2 grupos de tratamento foi comparável.
AVC recorrente: no estudo de Prevenção do AVC pela Redução Agressiva nos Níveis de Colesterol (SPARCL - Stroke Prevention by Aggressive Reduction in Cholesterol Levels), os efeitos da atorvastatina 80 mg diários ou placebo sobre o AVC foram avaliados em 4731 pacientes que apresentavam AVC ou ataque isquêmico transitório (AIT) dentro do período de 6 meses e sem histórico de doença arterial coronária (DAC). Os pacientes eram 60% homens, de 21 a 92 anos de idade (idade média de 63 anos) e tinham um baseline médio de 133 mg/dL (3,4 mmol/L). O LDL-C médio foi de 73 mg/dL (1,9 mmol/L) durante o tratamento com atorvastatina e 129 mg/dL (3,3 mmol/L) durante o tratamento com placebo. O acompanhamento médio foi de 4,9 anos.
A atorvastatina 80 mg reduziu o risco de endpoint primário de AVC fatal e não fatal em 15% [hazard ratio (HR)] 0,85; IC 95%, 0,72-1,00; p=0,05 ou 0,84; IC 95%; 0,71-0,99; p=0,03 após ajuste para fatores basais) comparado com o placebo. A atorvastatina 80 mg reduziu significativamente o risco de eventos coronarianos principais (HR 0,67; IC 95%, 0,51-0,89; p=0,006), qualquer evento de DAC (HR 0,60; IC 95%, 0,48-0,74; p < 0,001), e procedimentos de revascularização (HR 0,57; IC 95%, 0,44-0,74; p < 0,001).
Em uma análise post-hoc, a atorvastatina 80 mg reduziu a incidência de AVC isquêmico (218/2.365, 9,2% vs. 274/2.366, 11,6%, p=0,01) e aumentou a incidência de AVC hemorrágico (55/2.365, 2,3% vs. 33/2.366, 1,4%, p=0,02) comparado ao placebo. A incidência de AVC hemorrágico fatal foi similar entre os grupos (17 de atorvastatina vs. 18 de placebo). A redução do risco de eventos cardiovasculares com atorvastatina 80 mg foi demonstrada em todos os grupos de pacientes exceto nos pacientes que entraram no estudo com AVC hemorrágico e apresentaram AVC hemorrágico recorrente (7 de atorvastatina vs. 2 de placebo), onde o número de eventos foi muito pequeno para discernir risco e benefício.
Em pacientes tratados com atorvastatina 80 mg houve poucos casos de AVC de qualquer tipo (265 de atorvastatina vs. 311 de placebo) e poucos eventos de DAC (123 de atorvastatina vs. 204 de placebo). A mortalidade total foi similar nos grupos de tratamento (216 de atorvastatina vs. 211 de placebo). A incidência total de eventos adversos e eventos adversos sérios foi similar entre os grupos de tratamento.
Hipercolesterolemia Familiar Heterozigótica em Pacientes Pediátricos: em um estudo clínico duplo-cego, placebo-controlado, seguido de uma fase aberta, com 187 meninas pósmenarca e meninos, com idades variando entre 10 e 17 anos (média de idade de 14,1 anos), com hipercolesterolemia familiar heterozigótica ou hipercolesterolemia grave, foram randomizados para atorvastatina (n=140) ou placebo (n=47) por 26 semanas e em seguida todos receberam atorvastatina durante 26 semanas. Os critérios para inclusão no estudo foram: um valor basal de LDL-C ³ 190 mg/dL ou; um valor basal de LDL-C ³ 160 mg/dL e histórico familiar positivo de hipercolesterolemia familiar ou doença cardiovascular prematura documentada em parentes de primeiro ou segundo grau. O valor basal médio de LDL-C foi de 218,6 mg/dL (variando entre 138,5 e 385,0 mg/dL) no grupo da atorvastatina comparado a 230,0 mg/dL (variando entre 160,0 e 324,5 mg/dL) no grupo do placebo. A dosagem de atorvastatina (uma vez ao dia) foi de 10 mg para as primeiras 4 semanas e aumentada para 20 mg se o nível de LDL-C fosse > 130 mg/dL. O número de pacientes tratados com atorvastatina que necessitaram de aumento de dose para 20 mg após 4 semanas durante a fase duplo-cega foi de 78 (55,7%). A atorvastatina diminuiu significantemente os níveis plasmáticos de CT, LDL-C, triglicérides e apo B durante as 26 semanas da fase duplo-cega (vide Tabela 1).
TABELA 1
Efeitos da redução de lipídeos da atorvastatina em meninos e meninas adolescentes com hipercolesterolemia familiar heterozigótica ou hipercolesterolemia grave
(Mudança percentual media desde o valor basal ao endpoint na população com intenção de tratamento)
CT = colesterol total, LDL-C = lipoproteína de baixa densidade, HDL-C = lipoproteína de alta densidade, TG = triglicérides.
O valor médio de LDL-C alcançado foi de 130,7 mg/dL (variando entre 70,0 e 242,0 mg/dL) no grupo da atorvastatina em comparação a 228,5 mg/dL (variando entre 152,0 e 385,0 mg/dL) no grupo placebo durante as 26 semanas da fase duplo-cega.
Nesse estudo controlado limitado não foi observado qualquer efeito no crescimento ou maturação sexual em meninos ou alteração na duração do ciclo menstrual em meninas. A atorvastatina não foi avaliada em estudos clínicos controlados envolvendo pacientes pré-púberes ou pacientes menores de 10 anos de idade. A segurança e eficácia das doses superiores a 20 mg não foram avaliadas em estudos controlados realizados com crianças. A eficácia de longo prazo da terapia com atorvastatina durante a infância para a redução da morbidade e mortalidade na idade adulta não foi estabelecida.
Hipercolesterolemia (familiar heterozigótica e não familiar) e Dislipidemia Mista (Fredrickson tipos IIa e IIb): A dose inicial recomendada dos comprimidos de atorvastatina cálcica é de 10 mg ou 20 mg uma vez ao dia. Pacientes que precisam de uma grande redução no LDL-C (mais do que 45%) podem ser iniciados com 40 mg uma vez ao dia. A taxa de dosagem dos comprimidos de atorvastatina de cálcio é de 10 mg a 80 mg uma vez ao dia. Os comprimidos de atorvastatina cálcica podem ser administrados como dose única a qualquer momento do dia, com ou sem alimento. A dose inicial de atorvastatina cálcica deve ser individualizada de acordo com as características do paciente assim como o objetivo da terapia e resposta (vide guias NCEP vigentes). Após iniciação e/ou na titulação dos comprimidos de atorvastatina de cálcio, os níveis de lipídeo devem ser analisados dentro de 2 a 4 semanas e a dosagem ajustada de acordo com a necessidade do paciente.
A atorvastatina reduz o CT, LDL-C, VLDL-C, apo B, triglicérides e aumenta o HDL-C em pacientes com hipercolesterolemia e dislipidemia mista. A resposta terapêutica é observada dentro de duas semanas, e a resposta máxima ocorre normalmente em quatro semanas, mantendo-se durante a terapia.
Os estudos em pacientes pediátricos foram limitados a pacientes com hipercolesterolemia familiar homozigótica. Em dois estudos multicêntricos, placebo-controlados, dose-resposta, em pacientes com hipercolesterolemia, a atorvastatina foi administrada uma vez ao dia por 6 semanas, reduzindo significativamente o CT, LDL-C, apo B e triglicérides. A partir de 2 estudos dose-resposta em pacientes com hipercolesterolemia primária foram obtidos os seguintes resultados para placebo (n=21), atorvastatina 10 mg (n=22), 20 mg (n=20), 40 mg (n=21) e 80 mg (n=23) respectivamente: CT = 4%, -29%, -33%, -37%, -45%. LDL-C = 4%, -39%, -43%, -50%, -60%. Apo B = 3%, -32%, -35%, -42%, -50%. Triglicérides = 10%, -19%, -26%, -29%, -37%. HDL-C = -3%, 6%, 9%, 6%, 5%. Não HDL-C/HDL-C = 7%, -34%, -41%, -45%, -53%.
Em pacientes com hiperlipoproteinemia de Fredrickson tipos IIa e IIb, agrupados a partir de 24 estudos clínicos controlados, as mudanças percentuais médias (25° e 75° percentil) do valor basal de HDL-C para atorvastatina 10, 20, 40 e 80 mg foram 6,4 (variando entre -1,4 e 14,0); 8,7 (variando entre 0 e 17); 7,8 (variando entre 0 e 16) e 5,1 (variando entre -2,7 e 15), respectivamente. Adicionalmente, as análises dos dados demonstraram um decréscimo consistente e significativo no CT, LDL-C, triglicérides, razão CT/HDL-C e LDL-C/HDL-C.
Hipertrigliceridemia (Fredrickson tipo IV): a resposta à utilização de atorvastatina em 64 pacientes com hipertrigliceridemia isolada tratados em vários estudos clínicos está na tabela 1. Para os pacientes tratados com atorvastatina, o valor basal médio (mín; máx) de triglicérides foi de 565 (variando entre 267 e 1502).
Tabela 1 - Pacientes com níveis elevados de triglicérides; alterações percentuais médias (mín., máx.) a partir do valor basal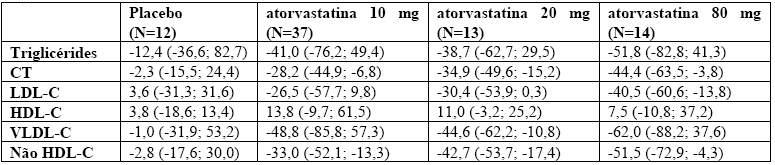
Disbetalipoproteinemia (Fredrickson tipo III): resultados de um estudo cruzado, aberto em 16 pacientes (genótipos 14 apo E2/E2 e 2 apo E3/E2) com disbetalipoproteinemia (Fredrickson tipo III).
Tabela 2 -Estudo aberto, cruzado, em 16 pacientes com disbetalipoproteinemia (Fredrickson tipo III)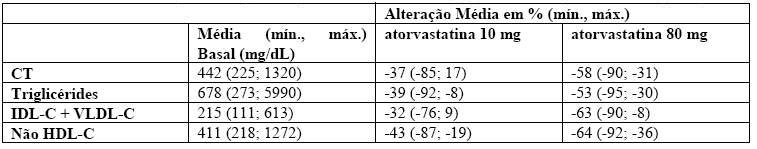
Hipercolesterolemia Familiar Homozigótica: A dose inicial recomendada dos comprimidos de atorvastatina em pacientes com hipercolesterolemia familiar homozigótica é de 10 mg a 80 mg diariamente. Os comprimidos de atorvastatina de cálcio devem ser usados como um complemento a outros tratamentos hipolipemiantes (por exemplo, aferese LDL) nestes pacientes ou se tais tratamentos não estão disponíveis.
Em um estudo sem grupo controle, 29 pacientes com idades variando entre 6 e 37 anos com hipercolesterolemia familiar homozigótica receberam doses diárias de 20 a 80 mg de atorvastatina. A média de redução do LDL-C no estudo foi de 18%. Vinte e cinco pacientes apresentaram uma redução média de LDL-C de 20% (variando entre 7 e 53%, média de 24%); os 4 pacientes restantes tiveram aumentos de 7% a 24% no LDL-C.
Uso em Síndrome Isquêmica Aguda: no estudo clínico "Redução da Isquemia Miocárdica atravès da Redução Intensiva dos Níveis de Colesterol", mais conhecido como estudo MIRACL (Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering), foram estudados os efeitos da terapia com atorvastatina em eventos isquêmicos e sobre a mortalidade total. Nesse estudo multicêntrico, randomizado, duplo-cego, placebocontrolado, foram avaliados 3086 pacientes com síndromes coronárias agudas, angina instável ou infarto do miocárdio não transmural (infarto sem onda Q). Os pacientes foram tratados com procedimentos convencionais, incluindo dieta alimentar mais atorvastatina 80 mg ou placebo, administrado diariamente, por um período médio de tratamento de 16 semanas. Os níveis finais de LDL-C, CT, HDL-C e triglicérides foram 72 mg/dL; 147 mg/dL; 48 mg/dL e 139 mg/dL, respectivamente, no grupo tratado com atorvastatina, e 135 mg/dL; 217 mg/dL; 46 mg/dL e 187 mg/dL, respectivamente, no grupo utilizando placebo. A atorvastatina reduziu significantemente o risco de morte e eventos isquêmicos (Figura 1) em 16%. O risco de re-hospitalização para angina do peito com evidências documentadas de isquemia miocárdica foi reduzido significantemente em 26%. A atorvastatina reduziu o risco de morte e eventos isquêmicos de forma igual e consistente em todos os valores de LDL-C basais. Além disso, reduziu o risco de morte e eventos isquêmicos tanto em pacientes com infarto do miocárdio não transmural (infarto sem onda Q) como em pacientes com angina instável, em homens e em mulheres e em pacientes com idade ≤ 65 anos e > 65 anos.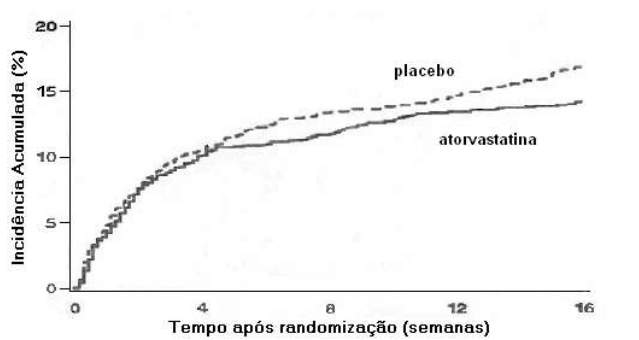
Figura 1. Tempo até o primeiro evento isquêmico ou morte.
Prevenção de Complicações Cardiovasculares: no estudo Anglo-Scandinavian Cardiac Outcomes Trial Lipid Lowering Arm (ASCOT-LLA), o efeito da atorvastatina na doença coronária fatal e não fatal foi avaliada em 10.305 pacientes hipertensos de 40 a 80 anos de idade (média de idade de 63 anos), sem histórico de infarto do miocárdio e com níveis de triglicérides < 6,5 mmol/L (251 mg/dL). Além disso, todos os pacientes apresentavam pelo menos 3 outros fatores de risco cardiovasculares (CV): sexo masculino, idade > 55 anos, tabagismo, diabetes, história de cardiopatia congênita em um parente de primeiro grau, CT:HDL > 6, doença vascular periférica, hipertrofia ventricular esquerda, acidente vascular cerebral prévio, anormalidade específica em ECG, proteinúria/albuminúria. Neste estudo duplo-cego, placebo-controlado, os pacientes foram tratados com medicação anti-hipertensiva (meta de PA < 140/90 mmHg para não diabéticos e < 130/80 mmHg para diabéticos) e alocados para receber atorvastatina 10 mg/dia (n = 5168) ou placebo (n = 5137). Considerando que o resultado do tratamento com a atorvastatina em comparação ao placebo excedeu o limiar de significância, em uma análise interina dos dados, o braço de redução lipídica foi encerrado precocemente (ASCOT-LLA) com 3,3 anos de seguimento ao invés de 5 anos, como originalmente planejado. Além disso, a pressão arterial foi bem controlada e semelhante em pacientes designados para atorvastatina e placebo. Essas alterações persistiram durante todo o período de tratamento.
A atorvastatina reduziu os índices relacionados aos seguintes eventos: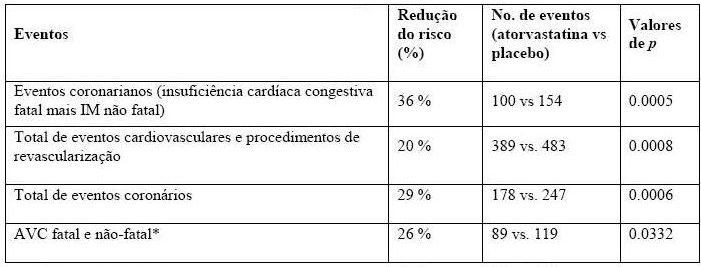
A mortalidade total e a mortalidade cardiovascular não foram reduzidas de forma significativa, apesar de ter sido observada uma tendência favorável.
o Estudo Colaborativo Atorvastatina Diabetes (CARDS), o efeito da atorvastatina na doença cardiovascular fatal ou não fatal foi avaliada em 2.838 pacientes com diabetes tipo 2, com idade entre 40 a 75 anos, sem história prèvia de doença cardiovascular e com LDL ≤ 4,14 mmol/L (160 mg/dL) e triglicèrides ≤ 6,78 mmol/L (600 mg/dL). Além disso, todos os pacientes tinham pelo menos mais um fator dos seguintes fatores de risco cardiovascular: hipertensão, tabagismo, retinoplastia, microalbuminúria ou macroalbuminúria.
Neste estudo randomizado, duplo-cego, multicêntrico, placebo-controlado, os pacientes foram tratados com atorvastatina 10 mg uma vez ao dia (n = 1428) ou com placebo (n = 1410) e acompanhados, em média, por 3,9 anos.
Uma vez que o efeito do tratamento com atorvastatina sobre o endpoint primário preencheu as regras pré-definidas de eficácia para a interrupção do estudo, o CARDS foi terminado 2 anos antes do esperado. O efeito da atorvastatina na redução dos riscos absoluto e relativo foi o seguinte: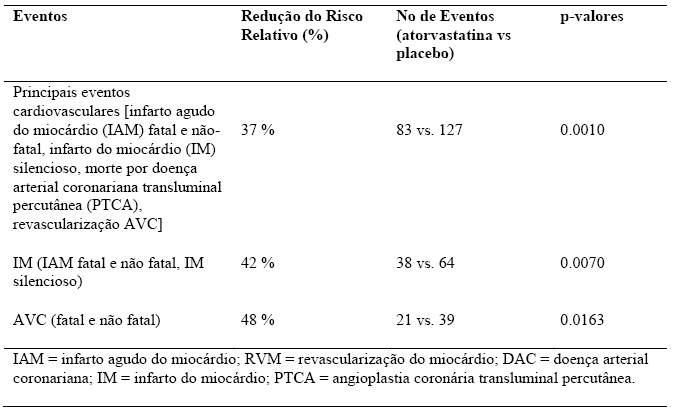
IAM = infarto agudo do miocárdio; RVM = revascularização do miocárdio; DAC = doença arterial coronariana; IM = infarto do miocárdio; PTCA = angioplastia coronária transluminal percutânea.
Não houve evidência de uma diferença no efeito do tratamento do paciente por sexo, idade ou nível de LDL-C da linha de base. Uma redução no risco de morte de 27% (82 mortes no grupo placebo comparado a 61 mortes no braço tratado) foi observada com uma significância estatística limítrofe (p = 0,0592). A incidência geral dos eventos adversos ou eventos adversos sérios foi similar entre os grupos sob tratamento.
Prevenção Secundária de Eventos Cardiovasculares: no estudo Tratamento até Novas Metas mais conhecido como Treating to New Targets (TNT), o efeito de atorvastatina cálcica 80 mg/dia vs. atorvastatina cálcica 10 mg/dia na redução de eventos cardiovasculares foi avaliado em 10.001 indivíduos (94% de raça branca, 81% de sexo masculino, 38% ≥ 65 anos de idade) com doença cardíaca coronariana clinicamente evidente que tinham atingido a meta de LDL-C < 130 mg/dL após completarem o período de introdução de 8 semanas com atorvastatina cálcica 10 mg/dia, em regime aberto. Os indivíduos foram randomizados para receber 10 mg/dia ou 80 mg/dia de atorvastatina cálcica e acompanhados por uma duração mediana de 4,9 anos. O endpoint primário foi o tempo até a primeira ocorrência de qualquer um dos seguintes eventos cardiovasculares importantes: óbito em decorrência de insuficiência cardíaca congestiva, infarto do miocárdio não fatal, parada cardíaca ressuscitada, e acidente vascular cerebral fatal e não fatal. Os níveis médios de LDL-C, colesterol total, triglicérides e colesterol não-HDL e HDL na semana 12 foram de 73 mg/dL, 145 mg/dL, 128 mg/dL, 98 mg/dL e 47 mg/dL, respectivamente, durante o tratamento com 80 mg de atorvastatina cálcica e 99 mg/dL, 177 mg/dL, 152 mg/dL, 129 mg/dL e 48 mg/dL, respectivamente, durante o tratamento com 10 mg de atorvastatina cálcica. O tratamento com atorvastatina cálcica 80 mg/dia reduziu significativamente a taxa de eventos cardiovasculares maiores (MCVE) (434 eventos no grupo recebendo 80 mg/dia vs. 548 eventos no grupo recebendo 10 mg/dia) com uma redução do risco absoluto de 2,2% e do risco relativo de 22%, razão de risco de 0,78, IC de 95% (0,69-0,89), p=0,0002 (vide Figura 2 e Tabela 3).A redução global do risco foi consistente independentemente da idade ( < 65, ≥ 65) ou sexo.
Figura 2. Efeito da atorvastatina cálcica 80 mg/dia vs. 10 mg/dia no Tempo até a Ocorrência de Eventos Cardíacos Importantes (TNT)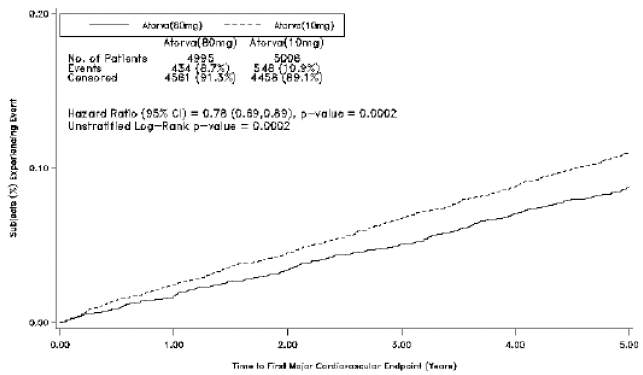
A atorvastatina 80mg reduz significantemente o risco dos seguintes:
Tabela 3 - Visão Geral dos Resultados de Eficácia no Estudo TNT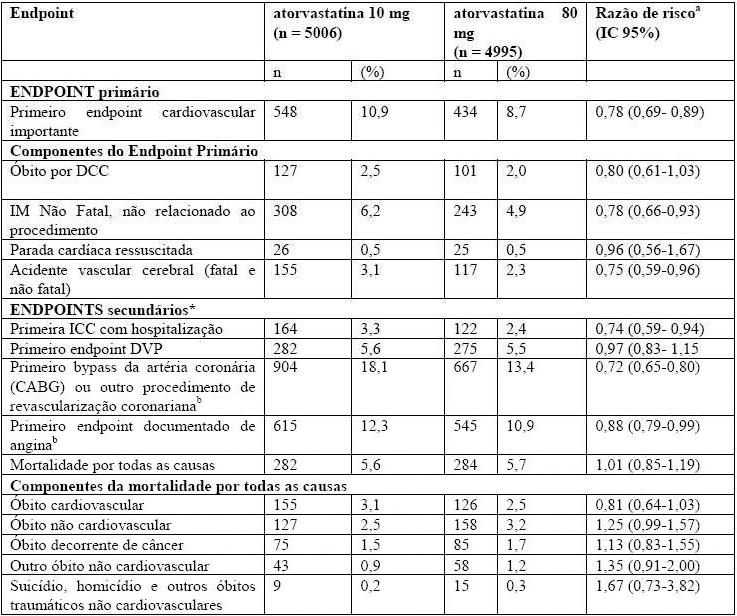
a atorvastatina 80 mg: atorvastatina 10mg.
b componente de outros endpoints secundários.
* principal endpoint cardiovascular (MCVE) = morte devido doença cardíaca coronariana, infarto do miocárdio não-fatal, parada cardíaca ressuscitada e AVC fatal e não-fatal.
** endpoints secundários não incluídos no endpoint primário.
HR = hazard ratio; IC=intervalo de confiança; IM=infarto do miocárdio; ICC=insuficiência cardíaca congestiva; CABG - bypass da artéria coronária.
Os intervalos de confiança dos endpoints secundários não foram ajustados para comparações múltiplas.
Dentre os eventos incluídos no endpoint primário de eficácia, o tratamento com atorvastatina cálcica 80 mg/dia reduziu significativamente a taxa de infarto do miocárdio não fatal, e não relacionado ao procedimento e a taxa de acidente vascular cerebral fatal e não fatal, porém não reduziu a taxa de óbito decorrente de doença cardíaca coronariana ou a taxa de parada cardíaca ressuscitada (Tabela 3). Dentre os endpoints secundários pré-definidos, o tratamento com atorvastatina cálcica 80 mg/dia reduziu significativamente a taxa de revascularização coronariana, angina e hospitalização por insuficiência cardíaca, porém não reduziu a taxa de doença vascular periférica. A redução da taxa de ICC com hospitalização foi observada em apenas 8% dos pacientes com história anterior de ICC.
Não foi observada diferença significativa entre os grupos de tratamento com relação a todas as causas de mortalidade: 282 (5,6%) no grupo atorvastatina 10 mg/dia vs. 284 (5,7%) no grupo atorvastatina 80 mg/dia por todas as causas (Tabela 3). A proporção de pacientes com óbito cardiovascular, incluindo os componentes de óbito decorrente de ICC e acidente vascular cerebral fatal foi numericamente menor no grupo recebendo tratamento com atorvastatina cálcica 80 mg que no grupo recebendo atorvastatina cálcica 10 mg. A proporção de indivíduos com óbito não cardiovascular foi numericamente maior no grupo recebendo tratamento com atorvastatina cálcica 80 mg que no grupo recebendo atorvastatina cálcica 10 mg.
No estudo Redução Incremental nos Endpoints Através da Redução Agressiva dos Lipídeos, mais conhecido como Incremental Decrease in Endpoints Through Aggressive Lipid Lowering Study (IDEAL), o tratamento com atorvastatina cálcica 80 mg/dia foi comparado ao tratamento com sinvastatina 20 mg/dia a 40 mg/dia em 8.888 indivíduos com até 80 anos de idade e com histórico de DCC, para avaliar se poderia ser atingida uma redução no risco cardiovascular. Os pacientes eram na maioria de sexo masculino (81%), de raça branca (99%) e com idade média de 61,7 anos, apresentando níveis médios de LDL-C de 121,5 mg/dL por ocasião da randomização; 76% estavam recebendo terapia com estatina. Neste estudo prospectivo, randomizado, aberto, com endpoint cego (PROBE) sem período de introdução, os indivíduos foram acompanhados por uma duração mediana de 4,8 anos. Os níveis médios de LDL-C, colesterol total, triglicérides e colesterol HDL e não-HDL na Semana 12 eram de 78 mg/dL, 145 mg/dL, 115 mg/dL, 45 mg/dL e 100 mg/dL, respectivamente, durante o tratamento com 80 mg de atorvastatina cálcica e 105 mg/dL, 179 mg/dL, 142 mg/dL, 47 mg/dL e 132 mg/dL, respectivamente, durante o tratamento com 20 mg a 40 mg de sinvastatina.
Não foi observada diferença significativa entre os grupos de tratamento com relação ao endpoint primário, a taxa de primeiro evento coronariano importante (doença cardíaca coronariana fatal, infarto do miocárdio não fatal e parada cardíaca ressuscitada): 411 (9,3%) no grupo recebendo atorvastatina cálcica 80 mg/dia vs. 463 (10,4%) no grupo recebendo sinvastatina 20 mg a 40 mg/dia, razão de risco 0,89, IC de 95% (0,78-1,01), p=0,07.
Não foram observadas diferenças significativas entre os grupos de tratamento com relação à mortalidade por todas as causas: 366 (8,2%) no grupo recebendo atorvastatina cálcica 80 mg/dia vs. 374 (8,4%) no grupo recebendo sinvastatina 20 mg/dia a 40 mg/dia. A proporção de pacientes com óbito cardiovascular ou não cardiovascular foi similar aos grupos recebendo atorvastatina cálcica 80 mg e sinvastatina 20 mg a 40 mg.
Referências Bibliográficas:
McCrindle BW1, Ose L, Marais AD. Efficacy and safety of atorvastatin in children and adolescents with familial hypercholesterolemia or severe hyperlipidemia: a multicenter, randomized, placebo-controlled trial. J Pediatr. 2003 Jul;143(1):74-80.
Sever PS et al. for the ASCOT investigators. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial - Lipid Lowering Arm (ASCOT-LLA): a multicentre, randomized, controlled trial. Lancet 2003;361:1149-58.
Sever PS et al. Different time course for prevention of coronary and stroke events by atorvastatin in the Anglo-Scandinavian Cardiac Outcomes Trial-Lipid-Lowering Arm (ASCOT-LLA). Am J Cardiol 2005;96:39F-44F.
Law MR et al. Quantifying effect of statins on low density lipoprotein cholesterol, ischaemic heart disease, and stroke: systematic review and meta-analysis. BMJ 2003;326:1423.
Colhoun HM et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre, randomized, placebo controlled trial. Lancet 2004;364:685-96.
Steven E. Nissen, Md, et al. Effect of Intensive Compared With Moderate Lipid-Lowering Therapy on Progression of Coronary Atherosclerosis. A Randomized Controlled Trial. JAMA. 2004;291(9):10711080. doi:10.1001/jama.291.9.1071.
The Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) Investigators. High-dose atorvastatin after stroke or transient ischemic attack. N Engl J Med 2006;355:549-59.
LaRosa JC et al. for the Treating to New Targets (TNT) Investigators. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med 2005;352:1425-35.
Pedersen TR et al. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction. The IDEAL study: a randomized controlled trial. JAMA 2005;294:2437-45.
Caract. farmacológicas.
Propriedades Farmacodinâmicas
A atorvastatina cálcica é um agente de redução de lípides sintéticos, que é um inibidor da HMG-CoA redutase. Esta enzima catalisa a conversão de HMG-CoA em mevalonato, uma etapa inicial e limitante da velocidade na biossíntese do colesterol.
A fórmula empírica de atorvastatina cálcica é (C33H34FN2O5)2Ca •3H2O e o seu peso molecular é 1209,42. A sua fórmula estrutural é: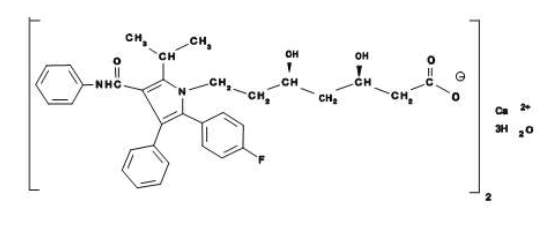
A atorvastatina cálcica é um pó cristalino branco a esbranquiçado, praticamente insolúvel em soluções aquosas de pH 4 e abaixo. Ele é muito pouco solúvel em água destilada, pH 7,4 tampão de fosfato e acetonitrilo; ligeiramente solúvel em etanol; e livremente solúvel em metanol.
Mecanismo de Ação: a atorvastatina é um inibidor seletivo e competitivo da HMG-CoA redutase, a enzima limitante da velocidade que converte HMG-Co-A a mevalonato, um precursor de esteróis, incluindo o colesterol. Em pacientes com hipercolesterolemia familiar homozigótica e heterozigótica, formas não familiares de hipercolesterolemia e dislipidemia mista, a atorvastatina reduz o CT (colesterol total), LDL-C (lipoproteína de baixa densidade) e apo B (apolipoproteína B). A atorvastatina também reduz o VLDL-C (lipoproteínas de densidade muito baixa) e os TG (triglicérides) e produz aumentos variáveis no HDL-C (lipoproteínas de alta densidade).
A atorvastatina diminui os níveis plasmáticos de colesterol e lipoproteínas através da inibição da HMG-CoA redutase e da síntese de colesterol no fígado, e aumenta o número de receptores de LDL hepáticos na superfície da célula, aumentando a absorção e o catabolismo do LDL.
A atorvastatina reduz a produção e o número de partículas de LDL. Produz um aumento marcante e prolongado na atividade do receptor de LDL, além de promover uma alteração benéfica na qualidade das partículas de LDL circulantes. O fármaco é eficaz na redução de LDL em pacientes com hipercolesterolemia familiar homozigótica, uma população que não responde normalmente à medicação de redução lipídica.
A atorvastatina e alguns de seus metabólitos são farmacologicamente ativos em humanos. O principal sítio de ação da atorvastatina é o fígado, que é o principal local de síntese de colesterol e clearance de LDL. A redução no LDL-C está mais relacionada à dose do medicamento do que à concentração sistêmica do fármaco. A individualização da dose do medicamento deve ser baseada na resposta terapêutica (vide item 8. Posologia e Modo de Usar).
Em um estudo dose-resposta, a atorvastatina (10 mg - 80 mg) demonstrou reduzir as concentrações de CT (30% - 46%), LDL-C (41% - 61%), apo B (34% - 50%) e triglicérides (14% - 33%). Estes resultados são compatíveis em pacientes com hipercolesterolemia familiar heterozigótica, formas não familiares de hipercolesterolemia e hiperlipidemia mista, incluindo pacientes com diabetes melito não insulino-dependentes.
Em pacientes com hipertrigliceridemia isolada, a atorvastatina reduz o CT, o LDL-C, o VLDL-C, a apo B, triglicérides e não-HDL-C, e aumenta o HDL-C. Em pacientes com disbetalipoproteinemia, reduz a lipoproteína de densidade intermediária-colesterol (IDL-C). Em pacientes com hiperlipoproteinemia de Fredrickson tipos IIa e IIb, reunidos em 24 estudos controlados, o aumento percentual médio a partir do valor basal no HDL-C para atorvastatina (10 mg - 80 mg) foi de 5,1 - 8,7% de maneira não relacionada à dose. Além disso, a análise destes dados demonstrou uma redução significativa relacionada à dose nas proporções de CT/HDL-C e LDL-C/HDL-C, variando de -29% para -44% e 37% para -55%, respectivamente.
Propriedades Farmacocinéticas
Farmacocinética e Metabolismo
Absorção: a atorvastatina é rapidamente absorvida após administração oral e concentrações plasmáticas máximas são atingidas dentro de 1 a 2 horas. A extensão da absorção e as concentrações plasmáticas aumentam em proporção à sua dose. A atorvastatina em comprimidos apresenta biodisponibilidade entre 95% e 99% em comparação à solução. A biodisponibilidade absoluta é de aproximadamente 14% e a disponibilidade sistêmica da atividade inibitória sobre a HMG-CoA redutase é de aproximadamente 30%. A baixa disponibilidade sistêmica é atribuída ao clearance pré-sistêmico na mucosa gastrintestinal e/ou ao metabolismo hepático de primeira passagem. Embora o alimento diminua a taxa e a extensão da absorção do fármaco em aproximadamente 25% e 9%, respectivamente, como observado através da Cmáx e da AUC, a redução no LDLC é semelhante se a atorvastatina for administrada com ou sem alimentos. As concentrações plasmáticas de atorvastatina são mais baixas (aproximadamente 30% para Cmáx e AUC) após a administração do medicamento à noite, quando comparada à administração pela manhã. Entretanto, a redução no LDL-C é a mesma independente da hora em que o fármaco é administrado (vide item 8. Posologia e Modo de Usar).
Distribuição: o volume médio de distribuição da atorvastatina é de aproximadamente 381 litros. A atorvastatina apresenta uma taxa de ligação às proteínas plasmáticas igual ou superior a 98%. Uma proporção glóbulo vermelho do sangue/plasma de aproximadamente 0,25 indica baixa penetração do fármaco nos glóbulos vermelhos do sangue (eritrócitos).
Metabolismo: a atorvastatina é amplamente metabolizada a derivados orto e para-hidroxilados e a vários produtos de beta-oxidação. A inibição da HMG-CoA redutase pelos metabólitos orto e para-hidroxilados in vitro é equivalente àquela observada com a atorvastatina. Aproximadamente 70% da atividade inibitória circulante sobre a HMG-CoA redutase é atribuída aos metabólitos ativos. Estudos in vitro sugerem a importância do metabolismo da atorvastatina pelo citocromo hepático P450 3A4 conforme o aumento das concentrações plasmáticas de atorvastatina em humanos após coadministração de eritromicina, um inibidor conhecido desta isoenzima. Estudos in vitro também indicaram que a atorvastatina é um inibidor fraco do citocromo P450 3A4. A coadministração de atorvastatina e terfenadina não produziu um efeito clinicamente significante nas concentrações plasmáticas da terfenadina, um composto predominantemente metabolizado pelo citocromo P450 3A4. Portanto, é improvável que a atorvastatina altere significantemente a farmacocinética de outros substratos do citocromo P450 3A4 (vide item 6. Interações Medicamentosas). Em animais, o metabólito orto-hidroxilado sofre posteriormente glicuronidação.
Excreção: a atorvastatina e seus metabólitos são eliminados principalmente na bile após metabolismo hepático e/ou extra-hepático; entretanto, o fármaco parece não sofrer recirculação entero-hepática. A meia-vida de eliminação plasmática média em humanos é de aproximadamente 14 horas, mas a meia-vida da atividade inibitória para a HMG-CoA redutase é de 20 a 30 horas, devido à contribuição dos metabólitos ativos. Menos de 2% de uma dose de atorvastatina é recuperada na urina após administração oral.
Populações Especiais
Idosos: as concentrações plasmáticas da atorvastatina se apresentam mais elevadas (aproximadamente 40% para Cmáx e 30% para AUC) em indivíduos idosos sadios (65 anos de idade ou mais) do que em adultos jovens. O estudo ACCESS avaliou especificamente pacientes idosos com relação ao alcance da meta de tratamento segundo o Programa Nacional de Colesterol dos EUA (NCEP - National Cholesterol Education Program). O estudo incluiu 1.087 pacientes com menos de 65 anos, 815 pacientes com mais de 65 anos e 185 pacientes com mais de 75 anos de idade. Não foram observadas diferenças entre pacientes idosos e a população em geral com relação à segurança, eficácia ou alcance do objetivo do tratamento de lípides.
Crianças: não foram conduzidos estudos de farmacocinética na população pediátrica.
Sexo: as concentrações plasmáticas de atorvastatina em mulheres são diferentes das observadas nos homens (aproximadamente 20% mais altas para Cmáx e 10% mais baixas para AUC). Entretanto, não houve diferenças clinicamente significativas do efeito nos lípides entre homens e mulheres.
Insuficiência renal: disfunção renal não apresenta influência nas concentrações plasmáticas ou no efeito da atorvastatina como hipolipemiante. Portanto, não é necessário o ajuste de dose em pacientes com disfunção renal (vide item 8. Posologia e Modo de Usar).
Hemodiálise: apesar de não terem sido realizados estudos em pacientes com insuficiência renal em estágio terminal, não se espera que a hemodiálise aumente significativamente o clearance da atorvastatina, uma vez que este fármaco se liga amplamente às proteínas plasmáticas.
Insuficiência hepática: as concentrações plasmáticas de atorvastatina aumentam acentuadamente (aproximadamente 16 vezes na Cmáx e 11 vezes na AUC) em pacientes com hepatopatia alcoólica crônica (classe B de Child-Pugh) (vide item 4. Contraindicações).
Interações medicamentosas: o efeito de medicamentos coadministrados sobre a farmacocinética de atorvastatina, bem como o efeito da atorvastatina sobre a farmacocinética de medicamentos coadministrados é resumido nas Tabelas 5 e 6 (vide item 5. Advertências e Precauções e item 6. Interações Medicamentosas).
Tabela 4 - Efeitos na farmacocinética da atorvastatina devido coadministração de medicamentos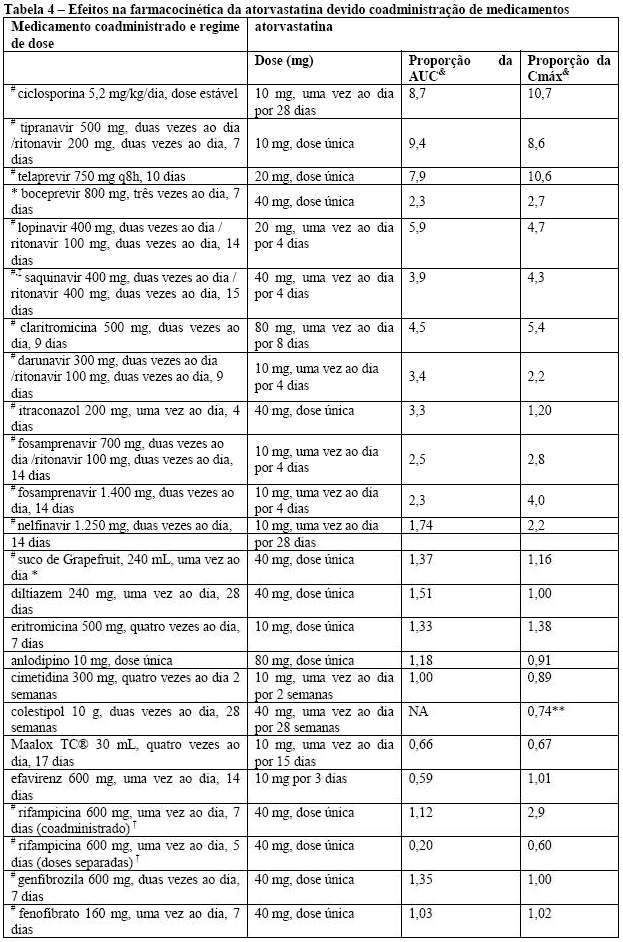
Tabela 5 - Efeito da atorvastatina na farmacocinética de medicamentos coadministrados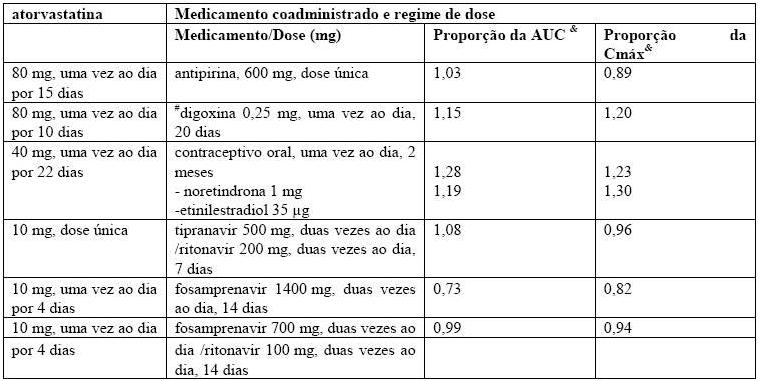
Dados de Segurança Pré-Clínicos
Carcinogênese, Mutagênese e Distúrbios da Fertilidade: a atorvastatina não se mostrou carcinogênica em ratos. A dose máxima utilizada foi 63 vezes maior, em mg/kg de peso corpóreo, do que a dose máxima recomendada para humanos (80 mg/dia) e de 8 a 16 vezes maior baseada nos valores de AUC(0-24). Em um estudo de 2 anos realizado com camundongos, as incidências de adenomas hepatocelulares em machos e de carcinomas hepatocelulares em fêmeas se mostraram aumentadas na dose máxima utilizada, que foi 250 vezes superior, em mg/kg de peso corpóreo, do que a dose máxima recomendada para humanos. A exposição sistêmica foi de 6 a 11 vezes superior, baseada na AUC (0-24). Todos os outros fármacos quimicamente semelhantes desta classe induziram tumores em ratos e camundongos com doses de 12 a 125 vezes superiores à dose clínica máxima recomendada, baseada em mg/kg de peso corpóreo.
A atorvastatina não demonstrou potencial mutagênico ou clastogênico em 4 testes in vitro, com ou sem ativação metabólica, nem em 1 estudo in vi