CARVYKTI
JANSSEN-CILAG
ciltacabtageno autoleucel
Antineoplásico.
Apresentações.
Suspensão para infusão de 0,5-1,0 x 106 células T CAR-positivas viáveis/kg com máximo de 1x108 células T CAR-positivas viáveis em 1 bolsa de infusão individual de 30 mL ou 70 mL.
USO INTRAVENOSO
USO ADULTO
Composição.
Uma bolsa contém no máximo 1 x 108 células T CAR-positivas viáveis com 5% de dimetilsulfóxido (DMSO).
Excipientes: Cryostor CS5, dimetilsulfóxido.
CARVYKTI® (ciltacabtageno autoleucel) é uma imunoterapia autóloga de células T geneticamente modificadas direcionada ao antígeno de maturação de células B (BCMA, na sigla em inglês). CARVYKTI® é preparado a partir das células mononucleares do sangue periférico do paciente, que são obtidas através de procedimento padrão de leucaférese. As células mononucleares são enriquecidas com células T e geneticamente modificadas ex-vivo por transdução com um vetor lentiviral incompetente para replicação para expressar um receptor de antígeno quimérico (CAR, na sigla em inglês) contendo um domínio direcionado ao anti-BCMA, que consiste em dois anticorpos de domínio único ligados ao domínio co-estimulador 4-1BB e aos domínios de sinalização CD3-zeta. As células CAR-T anti-BCMA transduzidas são expandidas em cultura celular, lavadas, formuladas em uma suspensão e criopreservadas. O produto deve passar por testes de esterilidade antes da liberação para remessa como uma suspensão congelada em uma bolsa de infusão específica para o paciente. O produto é descongelado e, então, aplicado por infusão intravenosa no paciente, onde as células CAR-T anti-BCMA podem reconhecer e eliminar as células direcionadas que expressam o BCMA. Uma dose única de CARVYKTI® contém 0,5-1,0x106 células T CAR-positivas viáveis por kg de peso corporal até um máximo de 1x 108 células T CAR-positivas viáveis suspensas em uma bolsa de infusão específica para o paciente (veja seção "8. POSOLOGIA E MODO DE USAR", "7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO"). Além das células T, CARVYKTI® pode conter células NK. A fórmula contém 5% de dimetilsulfóxido (DMSO). Para excipientes, veja a seção "COMPOSIÇÃO".
Informações técnicas.
1. INDICAÇÕES
CARVYKTI® é indicado para:
• o tratamento de pacientes com mieloma múltiplo recidivado ou refratário, que receberam anteriormente um inibidor do proteassoma, um agente imunomodulador e um anticorpo anti-CD38.
2. RESULTADOS DE EFICÁCIA
O MMY2001 foi um estudo aberto avaliando o CARVYKTI® no tratamento de pacientes com mieloma múltiplo recidivado ou refratário, que anteriormente receberam um inibidor do proteassoma, um agente imunomodulador e um anticorpo anti-CD38 e que tiveram progressão da doença durante ou após o último regime. No total, 113 pacientes foram submetidos à leucaférese; o CARVYKTI® foi fabricado para todos os pacientes do estudo. A mediana de tempo a partir do dia de recebimento do material de leucaférese na fábrica até a liberação do produto de infusão foi de 29 dias (faixa de 23-64 dias) e a mediana de tempo a partir da leucaférese inicial até a infusão do CARVYKTI® foi de 47 dias (faixa de 41 a 167 dias). Após a leucaférese e antes da administração de CARVYKTI®, 73 (75%) dos 97 pacientes do estudo receberam terapia ponte. Os agentes mais comumente usados como terapia ponte (≥ 20% dos pacientes) incluíram dexametasona: 62 pacientes (64%), bortezomibe: 26 pacientes (27%), ciclofosfamida: 22 pacientes (23%) e pomalidomida: 21 pacientes (22%). O CARVYKTI® foi administrado como uma infusão intravenosa única 5 a 7 dias após o início de uma quimioterapia com depleção linfocitária (300 mg/m2 de ciclofosfamida e 30 mg/m2 de fludarabina, ambos por via intravenosa, diariamente por 3 dias consecutivos. Noventa e sete pacientes receberam CARVYKTI® em uma dose mediana de 0,71×106 células T CARpositivas viáveis/kg (faixa: 0,51 a 0,95×106 células/kg). Todos os pacientes foram hospitalizados para infusão de CARVYKTI® e, posteriormente, por no mínimo 10 dias. Dezesseis pacientes não foram tratados com CARVYKTI® (12 após leucaférese e 4 após terapia de depleção linfocitária), em razão da saída do paciente da pesquisa (n= 5), doença progressiva (n= 2) ou óbito (n= 9). Dos 97 pacientes tratados, 59% eram homens, 71% eram caucasianos e 18% eram afro-americanos. A mediana de idade foi de 61 anos (faixa: 43 a 78 anos). Os pacientes haviam recebido uma mediana de 6 (faixa: 3 a 18) linhas de terapia prévias e 90% dos pacientes haviam recebido Transplante Autólogo de Células-Tronco (TACT). Noventa e nove por cento dos pacientes foram refratários a sua última linha de terapia prévia e 88% foram refratários ao inibidor do proteassoma (IP), agente imunomodulador e anticorpo anti-CD38. Pacientes com histórico anterior ou ativo conhecido de doença do sistema nervoso central (SNC) significativa, incluindo mieloma múltiplo do SNC, transplante alogênico de células-tronco dentro de 6 meses antes da aférese ou sob tratamento com imunossupressor, clearance de creatinina < 40 mL/min, concentração absoluta de linfócitos < 300/mL, transaminases hepáticas > 3 vezes o limite superior da normalidade, fração de ejeção cardíaca < 45% ou com infecção grave ativa foram excluídos do estudo. Os resultados de eficácia tiveram como base uma taxa de resposta global, conforme determinado pela avaliação do Comitê Independente de Revisão usando os critérios da IMWG (veja a Tabela 1).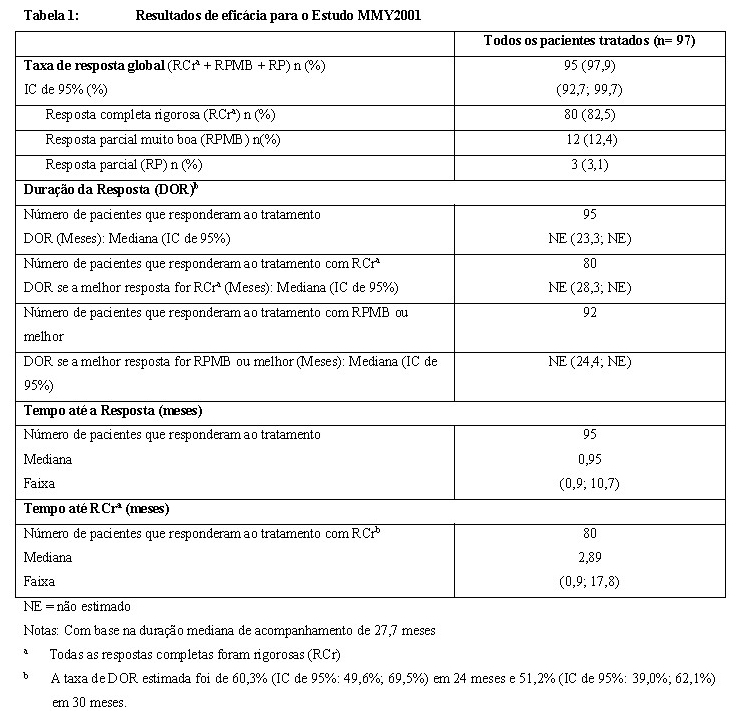

Com uma mediana de duração de acompanhamento de 27,7 meses, a mediana de sobrevida livre de progressão (SLP) não foi alcançada (IC de 95%: 24,5; não estimável). A taxa de SLP em 12 meses (IC de 95%) foi de 76,3% (66,5%; 83,6%). A taxa de SLP em 24 meses (IC de 95%) foi de 62,7% (52,2%; 71,5%). Para pacientes que alcançaram RCr (todas as respostas completas foram rigorosas), a mediana de SLP não foi alcançada (IC de 95%: 30,1%; não estimável) com uma taxa de SLP estimada em 12 meses de 88,8% (IC de 95%: 79,5%; 94,0 %). A taxa de SLP em 24 meses foi de 73,5% (IC 95%: 62,3%, 81,9%). A mediana de sobrevida global (SG) não foi alcançada (IC de 95%: não estimável; não estimável). A taxa de SG em 12 meses foi de 87,6% (IC de 95%: 79,2%; 92,8%). A taxa de SG em 24 meses foi de 76,2% (IC de 95%: 66,5%; 83,5%). A qualidade de vida relacionada à saúde (HRQoL, na sigla em inglês) foi avaliada pelo questionário EORTC QLQ-C30 e concluída no período basal (n= 63) e durante a fase pós-infusão. A alteração mediana ajustada (IC de 95%) a partir do período basal na subescala de dor do EORTC QLQ-C30 foi de -1,9 (-8,5; -4,6) no dia 7, de -9,9 (-16,5; -3,3) no dia 28, de -6,3 (-12,9; 0,4) no dia 56, de -9,4 (-16,3; -2,5) no dia 78 e de -10,5 (-17,3; -3,8) no dia 100, indicando uma redução global na dor após a infusão de CARVYKTI®. Melhorias clinicamente significativas no Dia 100 foram observadas em 72,2% dos pacientes para a subescala de dor, em 53,8% para a subescala de fadiga, em 57,7% para a subescala de funcionamento físico e em 53,7% para subescala da condição global de saúde.
Análise MAMMOTH
Uma análise retrospectiva, agrupada de resultados de pacientes com mieloma múltiplo refratário a anticorpos monoclonais CD38 foi conduzida para fornecer contexto para a interpretação dos resultados de eficácia relatados no Estudo MMY2001. A partir do conjunto de dados MAMMOTH, a análise identificou uma população de pacientes (N = 122). correspondendo à população totalmente tratada do Estudo MMY2001. Para os pacientes do conjunto de dados MAMMOTH, o Dia 1 do estudo foi 47 dias após o início da terapia convencional de tratamento padrão. Para os pacientes do conjunto de dados MAMMOTH, a TRG foi de 38%, a taxa de SLP de 12 meses (IC de 95%) foi de 7% (1%; 13%) e a taxa de SG de 12 meses (IC de 95%) foi de 40% (30 %; 50%). A análise retrospectiva dos resultados do MAMMOTH indicou que os pacientes que receberam CARVYKTI® (Estudo MMY2001) tiveram melhores resultados do que os pacientes que receberam outros tratamentos disponíveis avaliados no estudo MAMMOTH, conforme medido por TRG, SLP e SG.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades farmacodinâmicas
Grupo farmacoterapêutico: Outros agentes antineoplásicos, código ATC: ainda não atribuído
Mecanismo de ação CARVYKTI® é uma imunoterapia autóloga de células T geneticamente modificadas direcionada ao BCMA, que envolve a reprogramação das células T do paciente com uma codificação transgênica de um receptor de antígeno quimérico (CAR) que identifica e elimina as células que expressam o BCMA. BCMA é primariamente expresso na superfície de células da linha B do mieloma múltiplo maligno, bem como em células B de estágio tardio e células plasmáticas. A proteína CAR do CARVYKTI® possui dois anticorpos de domínio único direcionados ao BCMA criada para conferir alta avidez contra o BCMA humano, um domínio coestimulador de 4-1BB e um domínio citoplasmático de sinalização de CD3-zeta (CD3d). Ao ligar-se às células expressando o BCMA, o CAR promove ativação e expansão de células T, e consequente eliminação das células alvo. Experimentos em co-cultura in vitro demonstraram que a citotoxicidade mediada e liberação de citocina pelo ciltacabtageno autoleucel (interferon-gama, [IFN-c], fator alfa de necrose tumoral [TNF-a], interleucina [IL]-2) foram dependentes de BCMA.
Efeitos farmacodinâmicos Após uma infusão única de CARVYKTI®, a expansão de células CAR-T positivas coincidiu com as reduções de BCMA solúvel no soro, proteína M sérica e/ou cadeias leves livres. Dentre todos os pacientes, os níveis do receptor alfa de IL-6, IL-10, IFN-c e IL-2 aumentaram após a infusão e atingiram o pico nos Dias 7-14. Os níveis séricos de todas as citocinas voltaram, de modo geral, aos níveis basais dentro de 2-3 meses após a infusão.
Imunogenicidade
A imunogenicidade de CARVYKTI® foi avaliada usando um ensaio validado para a detecção de anticorpos de ligação em comparação a pré-dose de CARVYKTI® em vários momentos após a infusão. No Estudo MMY2001, 19 de 97 (19,6%) pacientes foram positivos para anticorpos anti-CAR. Não houve evidência clara para sugerir que os anticorpos anti-CAR observados afetam a cinética de expansão inicial e persistência, eficácia ou segurança de CARVYKTI® .
Propriedades farmacocinéticas
A farmacocinética (PK) de CARVYKTI® foi avaliada em 97 pacientes com mieloma múltiplo que receberam uma infusão única de CARVYKTI® na dose mediana de 0,71×106 células T CAR-positivas viáveis /kg (faixa: 0,51×106 a 0,95×106 células/kg). Após uma infusão única, o CARVYKTI® exibiu uma fase de expansão inicial seguida de um declínio rápido e, então, um declínio mais lento. Contudo, foi observada uma alta variabilidade interindividual.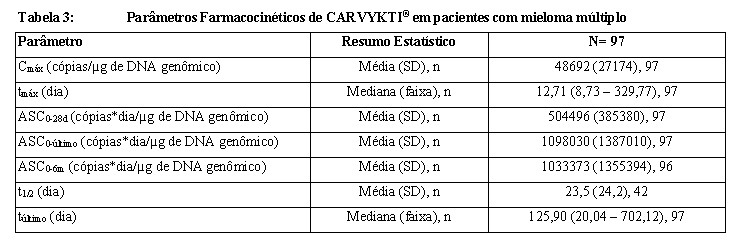
Após a expansão celular, a fase de persistência dos níveis de CARVYKTI® foi observada para todos os pacientes. No momento da análise (n= 65), o tempo mediano para que os níveis transgênicos de CAR no sangue periférico voltassem ao nível basal prédose foi de aproximadamente 100 dias (faixa: 28-365 dias) após a infusão. Exposições detectáveis de CARVYKTI® na medula óssea indicam uma distribuição do CARVYKTI® a partir da circulação sistêmica até a medula óssea. Similar aos níveis transgênicos sanguíneos, os níveis transgênicos na medula óssea diminuíram ao longo do tempo e exibiram alta variabilidade interindividual. Alguns pacientes exigiram tocilizumabe, corticosteroides e anakinra para o tratamento de síndrome de liberação de citocinas (SLC). O CARVYKTI® continua a expandir e persistir após a administração de tocilizumabe. Os pacientes tratados com tocilizumabe (n= 68) tiveram uma Cmáx e ASC0-28d mais alta de 81% e 72%, respectivamente, de CARVYKTI®, conforme comparação com os pacientes (n= 29) que não receberam tocilizumabe. Os pacientes que receberam corticosteroides (n= 28) tiveram uma Cmáx e ASC0-28d mais alta de 75% e 112%, respectivamente, conforme comparação com os pacientes que não receberam corticosteroides (n= 69). Além disso, os pacientes que receberam anakinra (n= 20) tiveram uma Cmáx e ASC0-28d mais alta de 41% e 72%, respectivamente, em comparação com os pacientes que não receberam anakinra (n= 77).
Populações Especiais
A farmacocinética de CARVYKTI® (Cmáx e ASC0-28d) não foi afetada pela idade (intervalo de 43-78 anos), incluindo pacientes < 65 anos de idade [n = 62; 63,9%], 65-75 anos (n = 27; 27,8%) e > 75 anos de idade (n= 8; 8,2%). Da mesma forma, a farmacocinética de CARVYKTI® (Cmáx e ASC0-28d) não foi afetada por gênero, peso corporal e raça.
Insuficiência renal
Não foram realizados estudos com CARVYKTI® em pacientes com insuficiencia renal. Cmáx e ASC0-28d de CARVYKTI® foram semelhantes em pacientes com disfunção renal leve (60 mL/min ≤ depuração da creatinina [CRCL] < 90 mL/min) e pacientes com função renal normal (CRCL ≥ 90 mL/min).
Insuficiência hepática
Não foram realizados estudos com CARVYKTI® em paciente com insuficiencia hepática. A Cmáx e ASC0-28d de CARVYKTI® foram similares nos pacientes com disfunção hepática leve [(bilirrubina total ≤ do limite superior da normalidade (LSN) e aspartato aminotransferase > LSN) ou (LSN < bilirrubina total ≤ 1,5 X LSN)] e pacientes com função hepática normal.
Informação não-clínica A avaliação de segurança não clínica do CARVYKTI® confirmou a especificidade no alvo de CARVYKTI® em relação ao BCMA.
Carcinogenicidade e mutagenicidade
Nenhum estudo de genotoxicidade ou carcinogenicidade foi conduzido. O risco de ocorrer mutagênese insercional durante a fabricação de ciltacabtageno autoleucel após a transdução de células T humanas autólogas com a integração de um vetor lentiviral (VL) foi avaliado ao analisar o padrão de integração do vetor na préinfusão de CARVYKTI® . Essa análise de sítio insercional genômico foi conduzida nos produtos de CARVYKTI® de 7 pacientes e 3 voluntários sadios. Não houve evidência de integração preferencial próxima aos genes em questão. O potencial para proliferação aprimorada de CARVYKTI® foi avaliado em um ensaio de crescimento independente de citocina in vitro. A integração de VL no genoma primário de célula T durante a transdução não levou ao crescimento descontrolado independente de citocina na ausência de IL-2 (a citocina que regula o crescimento de células T e promove a sobrevida de células T) do CARVYKTI® .
Toxicidade reprodutiva e fertilidade Nenhum estudo de toxicidade na reprodução e desenvolvimento em animais foi conduzido com o CARVYKTI® .
4. CONTRAINDICAÇÕES
Nenhuma.
5. ADVERTÊNCIAS E PRECAUÇÕES
Geral
Pacientes com histórico ativo ou anterior de doença do sistema nervoso central (SNC) significativa ou função renal, hepática, pulmonar ou cardíaca inadequada são mais vulneráveis às consequências das reações adversas descritas abaixo e exigem atenção especial.
Síndrome de Liberação de Citocinas (SLC)
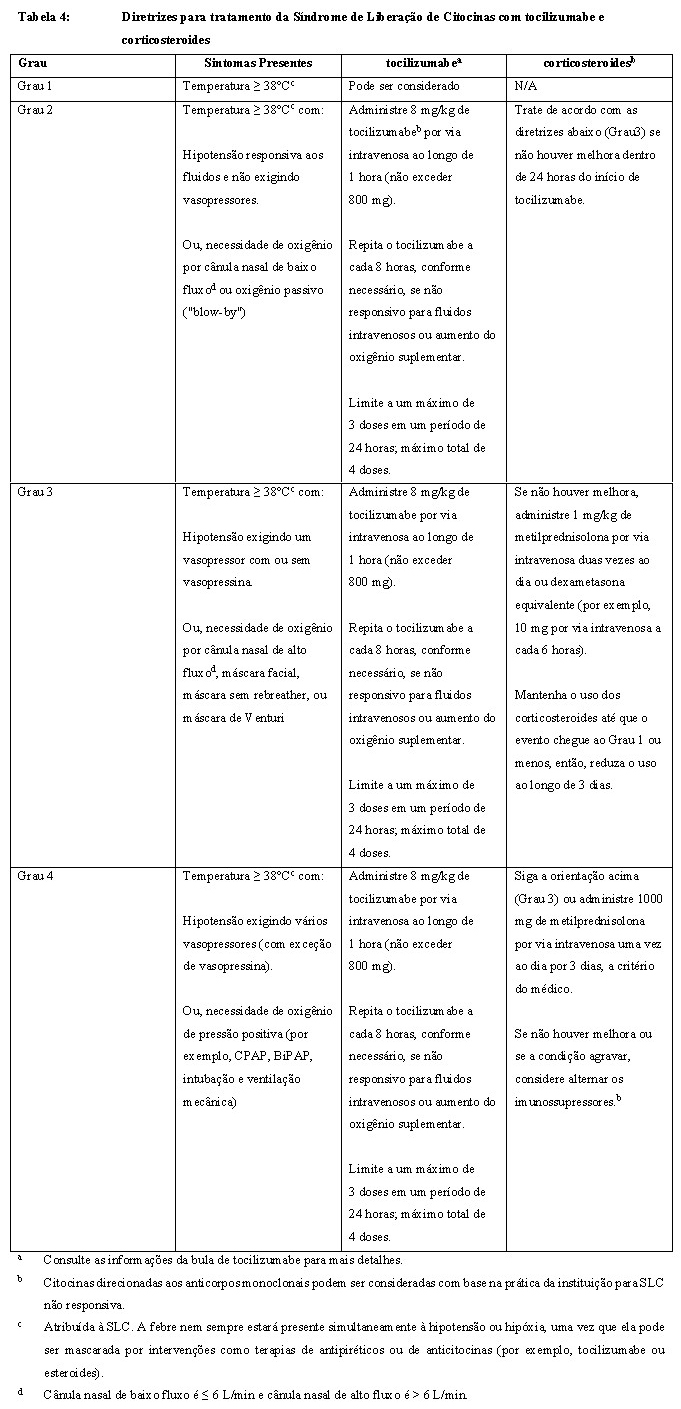
Síndrome de liberação de citocinas, incluindo reações fatais e com risco à vida, podem ocorrer após a infusão de CARVYKTI® . Quase todos os pacientes apresentaram SLC após a infusão de CARVYKTI® com a maioria sendo de Grau 1 ou Grau 2 (veja seção "9. REAÇÕES ADVERSAS"). O tempo mediano entre a infusão de CARVYKTI® (Dia 1) e o início da SLC foi de 7 dias (faixa de 1 a 12 dias). Aproximadamente 90% dos pacientes apresentaram início da SLC após o Dia 3 da administração da infusão de CARVYKTI® . Em quase todos os casos, a duração da SLC variou de 1 a 14 dias (duração mediana de 4 dias) com 88% dos pacientes tendo uma duração da SLC de ≤ 7 dias.
Sinais e sintomas clínicos de SLC podem incluir, entre outros, febre (com ou sem calafrios), calafrios, hipotensão, hipóxia e enzimas hepáticas elevadas. Possíveis complicações com risco à vida da SLC podem incluir disfunção cardíaca, toxicidade neurológica e linfo-histiocitose hemofagocítica (LHH). Pacientes que desenvolvem LHH podem ter um risco aumentado de sangramento grave. Os pacientes devem ser monitorados atentamente para sinais ou sintomas desses eventos, incluindo febre. Os fatores de risco para SLC severa incluem alta carga tumoral pré-infusão, infecção ativa e início precoce de febre ou febre persistente após 24 horas de tratamento sintomático. Adie a infusão de CARVYKTI® se o paciente apresentar reações adversas graves não solucionadas decorrentes da depleção linfocitária ou terapia ponte precedente (incluindo toxicidade cardíaca e toxicidade pulmonar), progressão rápida da doença e infecção ativa clinicamente significativa (veja seção "8. POSOLOGIA E MODO DE USAR"). Tratamentos profiláticos e terapêuticos adequados para infecções devem ser fornecidos e a resolução total de qualquer infecção ativa deve ser garantida antes da infusão de CARVYKTI®. Infecções também podem ocorrer concomitantemente com a SLC e podem aumentar o risco de um evento fatal. Certifique-se de que pelo menos duas doses de tocilizumabe estejam disponíveis antes da infusão de CARVYKTI®. Monitore os pacientes para sinais e sintomas de SLC diariamente por 14 dias após a infusão de CARVYKTI® em uma unidade de cuidados de saúde certificada e, então, periodicamente, por outras duas semanas após a infusão de CARVYKTI® . Aconselhe os pacientes a buscarem atenção médica imediata caso sinais ou sintomas de SLC ocorram a qualquer momento. Aos primeiros sinais de SLC, avalie imediatamente o paciente para hospitalização e institua tratamento com cuidados de suporte, tocilizumabe, ou tocilizumabe e corticosteroides, conforme indicado na Tabela 4 (veja seção "8. POSOLOGIA E MODO DE USAR"). A avaliação para linfohistiocitose hemofagocítica (LHH) deve ser considerada em paciente com SLC severa ou não responsiva. Para os pacientes com alta carga tumoral pré-infusão, início precoce de febre ou febre persistente após 24 horas, deve ser considerado o uso precoce de tocilizumabe. O uso de fatores de crescimento mieloide, principalmente fator estimulador das colônias de granulócitos e macrófagos (GM-CSF, na sigla em inglês), deve ser evitado durante a SLC. Considere reduzir a carga basal da doença com terapia ponte antes da infusão de CARVYKTI® em pacientes com alta carga tumoral.
Toxicidades neurológicas
Toxicidades neurológicas frequentemente ocorrem após o tratamento com CARVYKTI® e podem ser fatais (veja seção "9. REAÇÕES ADVERSAS"). Toxicidades neurológicas incluem ICANS, movimento e toxicidade neurocognitiva com sinais e sintomas de parkinsonismo, Síndrome de Guillain-Barré, neuropatias periféricas e paralisia dos nervos cranianos. Continue a monitorar os pacientes para sinais e sintomas de toxicidade neurológica após a recuperação da SLC e/ou ICANS, incluindo mudanças na caligrafia, pois isso pode ser um indicador precoce de possível neurotoxicidade.
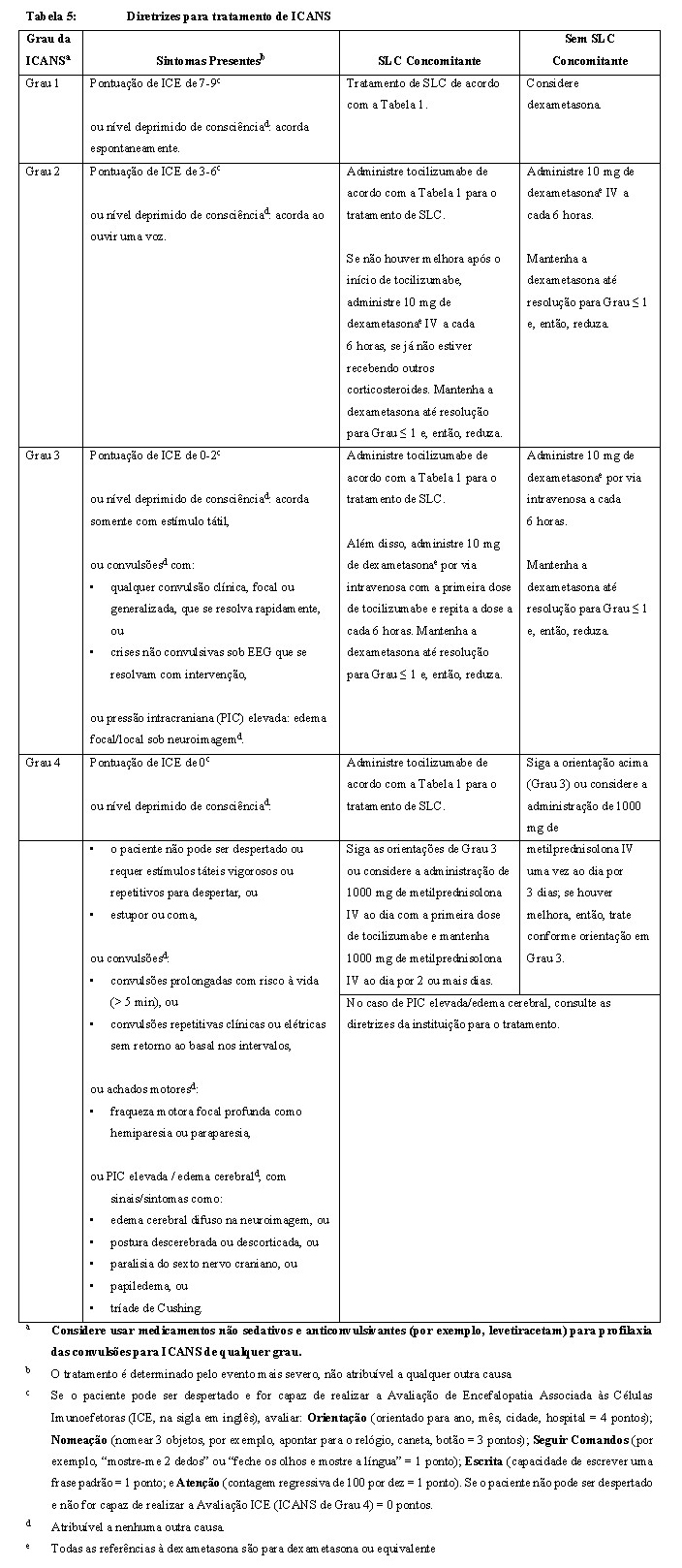
Síndrome de Neurotoxicidade Associada ao Tratamento com Células Imunoefetoras (ICANS):
Pacientes recebendo CARVYKTI® podem experienciar ICANS fatais ou de risco à vida após tratamento com CARVYKTI® , incluindo antes do aparecimento da SLC, concomitante à SLC, após a resolução da SLC, ou na ausência da SLC. Os sintomas incluem afasia, fala lenta, disgrafia, encefalopatia, depressão do nível de consciência e estado de confusão. Considere reduzir a carga basal da doença com terapia ponte antes da infusão de CARVYKTI® em pacientes com alta carga tumoral, o que pode mitigar o risco de desenvolvimento de toxicidade neurológica (veja seção "9. REAÇÕES ADVERSAS"). Monitore os pacientes para sinais ou sintomas de ICANS por 28 dias após a infusão. Aos primeiros sinais de ICANS, avalie imediatamente o paciente para hospitalização e inicie cuidados de suporte conforme indicado na Tabela 5 (veja seção "8. POSOLOGIA E MODO DE USAR"). A detecção precoce e o tratamento agressivo da SLC ou ICANS podem ser importantes para evitar a ocorrência ou agravamento da toxicidade neurológica.
Movimento e Toxicidade Neurocognitiva com Sinais e Sintomas de Parkinsonismo:
Toxicidade neurológica de movimento e toxicidade neurocognitiva com sinais e sintomas de parkinsonismo foi relatada em estudos clínicos de CARVYKTI®. Foi observado um conjunto de sintomas com início variável abrangendo mais de um domínio de sintomas, incluindo movimento (por exemplo, micrografia, tremor, bradicinesia, rigidez, postura encurvada, marcha arrastada), cognitivo (por exemplo, perda de memória, distúrbio na atenção, confusão), e alteração de personalidade (por exemplo, expressão facial reduzida, embotamento afetivo, hipomimia, apatia), muitas vezes com início sutil (por exemplo, micrografia, embotamento afetivo), que em alguns pacientes evoluiu para incapacidade de trabalhar ou cuidar de si mesmo. Todos esses pacientes apresentaram uma combinação de dois ou mais fatores, como alta carga tumoral (plasmócitos da medula óssea ≥80% ou pico M sèrico ≥ 5 g/dL ou cadeia leve livre sèrica ≥ 5000 mg/L), SLC anterior de Grau 2 ou maior, ICANS anterior, e alta expansão e persistência das células CAR-T. O tratamento com levodopa/carbidopa (n=2) não foi eficaz em melhorar a sintomatologia destes pacientes.
Monitore os pacientes quanto a sinais e sintomas de parkinsonismo que podem ter início tardio e estes devem ser tratados com medidas de suporte.
Síndrome de Guillain-Barré:
Síndrome de Guillain-Barré (SGB) foi relatada após o tratamento com CARVYKTI® . Os sintomas relatados incluem os consistentes com a variação Miller-Fisher da SGB, fraqueza motora, distörbios da fala e polirradiculoneurite (vide "9. REAÇÕES ADVERSAS").
Monitore quanto a SGB. Avalie os pacientes que apresentam neuropatia periférica para SGB. Considerar o tratamento com imunoglobulina intravenosa (IVIG) e encaminhar para plasmaférese, dependendo da gravidade da toxicidade.
Neuropatia periférica:
A ocorrência de neuropatia periférica, incluindo sensorial, motor ou sensório-motora, foi relatada em estudos clínicos com CARVYKTI® . Monitore os pacientes quanto a sinais e sintomas de neuropatias periféricas. Considere o tratamento com corticosteroides sistêmicos de curta duração, dependendo da gravidade e progressão dos sinais e sintomas.
Paralisia dos nervos cranianos:
A ocorrência de paralisia dos 7°, 3°, 5° e 6° nervos cranianos, das quais algumas foram bilaterais, recorrência da paralisia do nervo craniano, e ocorrência de neuropatia periférica em pacientes com paralisia do nervo craniano foram relatadas em estudos clínicos com CARVYKTI® . Monitore os pacientes quanto a sinais e sintomas de paralisia de nervos cranianos. Considere o tratamento com corticosteroides sistêmicos de curta duração, dependendo da gravidade e progressão dos sinais e sintomas.
Citopenias Prolongadas e Recorrentes
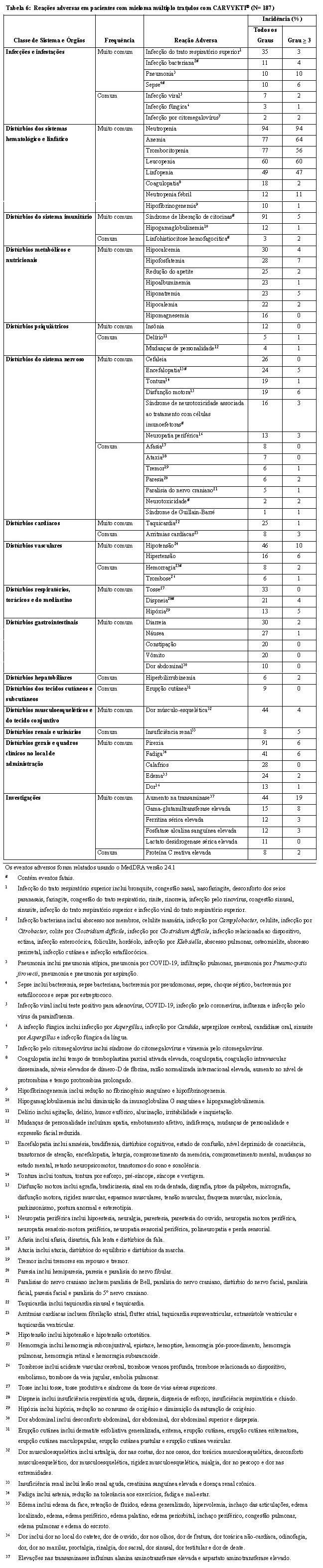
Os pacientes podem exibir citopenias por várias semanas após a quimioterapia com depleção linfocitária e infusão de CARVYKTI® e devem ser tratados de acordo com as diretrizes locais. No Estudo MMY2001, quase todos os pacientes tiveram uma ou mais reações adversas citopênicas de Grau 3 ou 4. A maioria dos pacientes apresentou uma mediana de tempo entre a infusão e o início da citopenia de Grau 3 ou 4 de menos de duas semanas, sendo que a maioria dos pacientes recuperou para≤ Grau 2 até o Dia 30 (veja seção "9. REAÇÕES ADVERSAS"). Monitore as contagens sanguíneas após a infusão de CARVYKTI®. Para trombocitopenia, considere tratamento de suporte com transfusões. Neutropenia prolongada foi associada a um risco elevado de infecção. Fatores de crescimento mieloide, principalmente GM-CSF, têm o potencial de agravar os sintomas de SLC e não são recomendados durante as 3 primeiras semanas após CARVYKTI® ou até a SLC ser solucionada.
Infecções graves e neutropenia febril
Infecções graves, incluindo infecções com risco à vida ou fatais, ocorreram em pacientes após a infusão de CARVYKTI® (veja seção "9. REAÇÕES ADVERSAS"). Monitore os pacientes para sinais e sintomas de infecção, empregue exames de vigilância antes e durante o tratamento com CARVYKTI® e trate os pacientes de forma adequada. É recomendado administrar profilaxia antimicrobiana e antifúngica, de acordo com as diretrizes locais, quando o paciente atinge contagens absolutas de neutrófilos < 500/mL e antiviral, de acordo com as diretrizes locais, no dia 1 da infusão. Infecções são conhecidas por complicar o curso e o tratamento da SLC concomitante. Pacientes com infecção ativa clinicamente significativa não devem iniciar o tratamento com CARVYKTI® até a infecção ser controlada. No evento de neutropenia febril, a infecção deve ser avaliada e tratada de forma adequada com antibióticos de amplo espectro, fluidos e outros cuidados de suporte, conforme clinicamente indicado.
Pacientes tratados com CARVYKTI® podem estar sob risco aumentado de infecção grave/fatal por COVID-19. Aconselhe os pacientes sobre a importância das medidas de precaução.
Reativação viral
A reativação do VHB, em alguns casos resultando em hepatite fulminante, insuficiência hepática e morte, pode ocorrer em pacientes com hipogamaglobulinemia. Atualmente, não existe experiência de fabricação de CARVYKTI® para pacientes que testaram positivo para HIV, HTLV, VHB ativo ou VHC ativo. A triagem para VHB, VHC, HIV, HTLV e outros agentes infecciosos deve ser conduzida de acordo com as diretrizes clínicas locais antes da coleta das células para a fabricação. Pacientes com qualquer infecção ativa clinicamente significativa, incluindo essas infecções virais, não devem receber o regime de depleção linfocitária até que a infecção seja controlada (veja seção "8. POSOLOGIA E MODO DE USAR"). A avaliação do risco-benefício deve ser realizada pelo médico responsável pelo tratamento dos pacientes com essas infecções antes da aférese para a fabricação de CARVYKTI® e depleção linfocitária.
Hipogamaglobulinemia
Hipogamaglobulinemia pode ocorrer em pacientes que receberem CARVYKTI® . Monitore os níveis de imunoglobulina após o tratamento com CARVYKTI® e administre IVIG caso os níveis de IgG sejam menores que 400 mg/dL. Tratar de acordo com as diretrizes clínicas padrões, incluindo a profilaxia com antibióticos e/ou antivirais e monitoramento de infecções.
Vacinas vivas A segurança da imunização com vacinas virais vivas durante ou após o tratamento com CARVYKTI® não foi estudada. A vacinação com vacinas com vírus vivo não é recomendada por pelo menos 6 semanas antes do início da quimioterapia com depleção linfocitária, durante o tratamento com CARVYKTI® e até a recuperação imunológica após o tratamento com CARVYKTI® .
Malignidades secundárias
Pacientes tratados com CARVYKTI® podem desenvolver malignidades secundárias. Um caso de linfoma de células T CARpositivo foi relatado em um estudo em andamento. Monitore ao longo da vida para o surgimento de malignidades secundárias. Caso ocorram malignidades secundárias, entre em contato com a empresa para relatar e obter instruções sobre coleta de amostra do paciente para testes de malignidade secundária originadas de célula T. Em pacientes com infecção pelo HIV, entre em contato com a empresa para o teste de todos os tipos de malignidade secundária, incluindo aquelas não originadas de célula T.
Hipersensibilidade
Reações alérgicas podem ocorrer com a infusão de CARVYKTI®. Reações de hipersensibilidade graves, incluindo anafilaxia, podem ocorrer em razão do dimetilsulfóxido (DMSO), ou canamicina residual no CARVYKTI® . Os pacientes devem ser monitorados cuidadosamente por 2 horas após a infusão quanto a sinais e sintomas de reações graves. Tratar prontamente e manejar os pacientes adequadamente de acordo com a gravidade da reação de hipersensibilidade.
Doação de sangue, órgãos, tecidos e células Os pacientes tratados com CARVYKTI® não devem doar sangue, órgãos, tecidos nem células para transplante.
Gravidez, Amamentação e Fertilidade
Gravidez (categoria C)
Não existem dados disponíveis quanto ao uso de CARVYKTI® em mulheres grávidas. Nenhum estudo de toxicidade na reprodução e desenvolvimento em animais foi conduzido com o CARVYKTI®. Não se sabe se o CARVYKTI® tem o potencial de ser transferido para o feto e causar toxicidade fetal. Portanto, o CARVYKTI® não é recomendado para mulheres grávidas, ou mulheres com potencial de engravidar que não fazem uso de contraceptivos. Mulheres grávidas devem ser informadas sobre os riscos ao feto. A gravidez após a terapia com CARVYKTI® deve ser discutida com o médico responsável pelo tratamento. Mulheres grávidas que receberem CARVYKTI® podem desenvolver hipogamaglobulinemia. A avaliação dos níveis de imunoglobulina em recém-nascidos de mães que forem tratadas com CARVYKTI® deve ser considerada.
Este medicamento não deve ser usado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Homens e Mulheres férteis
Teste de gravidez O status de gravidez para mulheres em idade com potencial para engravidar deve ser verificado antes do início do tratamento com CARVYKTI® .
Contracepção
Os dados de exposição são insuficientes para fornecer uma recomendação quanto a duração da contracepção após o tratamento com CARVYKTI® . Nos estudos clínicos, as pacientes mulheres com potencial para engravidar foram aconselhadas a fazer uso de métodos contraceptivos altamente eficazes e os pacientes homens com parceiras com potencial para engravidar ou que estavam grávidas, foram instruídos a usar método contraceptivo de barreira até um ano após o paciente ter recebido a infusão com o CARVYKTI® . Veja as informações de prescrição para quimioterapia com depleção linfocitária para informações sobre a necessidade de contraceptivos em pacientes que recebem a quimioterapia com depleção linfocitária.
Amamentação
Não existem informações quanto a presença de CARVYKTI® no leite humano, o efeito nas crianças amamentadas e os efeitos na produção de leite. Os benefícios da amamentação para o desenvolvimento e a saúde da criança devem ser considerados juntamente com a necessidade clínica da mãe para o CARVYKTI® e quaisquer efeitos adversos em potencial do CARVYKTI® ou da condição subjacente materna na criança amamentada.
Fertilidade
Não existem dados sobre o efeito de CARVYKTI® na fertilidade. Os efeitos do CARVYKTI® na fertilidade masculina e feminina não foram avaliados em estudos com animais.
Efeitos sobre a capacidade de dirigir e usar máquinas
Em razão do potencial para eventos neurológicos, os pacientes recebendo CARVYKTI® correm risco de alteração ou redução da consciência ou coordenação nas 8 semanas após a infusão de CARVYKTI®. Aconselhe os pacientes a evitarem dirigir e se envolver em ocupações ou atividades perigosas, como operar máquinas pesadas ou potencialmente perigosas durante esse período inicial e no caso de novo início de quaisquer sintomas neurológicos.
6. INTERAÇÕES MEDICAMENTOSAS
Nenhum estudo de interação foi conduzido com o CARVYKTI® . HIV e o lentivírus usados na fabricação do CARVYKTI® possuem extensão curta e limitada de material genético idêntico (RNA). Portanto, alguns testes comerciais de ácido nucleico (NATs, na sigla em inglês) para HIV podem produzir resultados falso-positivos em pacientes que haviam recebido CARVYKTI® .
Incompatibilidades
Na ausência de estudos de compatibilidade, este medicamento não deve ser misturado com outros medicamentos.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Armazene e transporte em temperatura ≤ -120 °C em tanque criogênico contendo nitrogênio líquido em fase de vapor. Armazene na embalagem original contendo o cassete de proteção da bolsa de infusão. Assim que estiver descongelado, o produto deve ser administrado imediatamente e a infusão deve ser concluída dentro de 2,5 horas em temperatura de 20°C a 25°C. O produto descongelado não deve ser agitado, recongelado ou refrigerado novamente.
Número de lote e datas de fabricação e validade: vide embalagem. Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Natureza e Teor do Recipiente
Bolsa de infusão em etileno-acetato de vinila (EVA) com tubo de adição selado e duas portas de perfuração disponíveis contendo 30 mL ou 70 mL de dispersão celular. Cada bolsa de infusão é acondicionada individualmente em um cassete de criogenia de alumínio.
Aspecto físico
Suspensão incolor a branco, incluindo tons de branco, amarelo e rosa. A bolsa deve estar sem defeitos visíveis e vazamentos.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
Instruções de Uso, Manuseio e Descarte
Não irradiar, uma vez que isso pode levar à inativação do produto. O CARVYKTI® deve ser transportado dentro de um recipiente fechado, lacrado e à prova de vazamento. O CARVYKTI® contém células sanguíneas humanas que foram geneticamente modificados com a replicação de vetor lentiviral incompetente. Siga as precauções universais e diretrizes locais para manuseio e descarte do medicamento não usado ou todos os materiais que entraram em contato com o CARVYKTI® (dejetos sólido e líquido) para evitar uma possível transmissão de doenças infecciosas.
8. POSOLOGIA E MODO DE USAR
Somente para uso autólogo. Somente para uso intravenoso.
Dose - Adultos (≥ 18 anos)
CARVYKTI® é fornecido como uma dose única para infusão contendo uma suspensão de receptor de antígeno quimérico de células T CAR-positivas viáveis. A dose contém 0,5-1,0×106 células T CAR-positivas viáveis por kg de peso corporal, com uma dose alvo de 0,75×106 células T CAR-positivas viáveis por kg de peso corporal e uma dose máxima de 1×108 células T CAR-positivas viáveis por infusão única. Devido às singularidades de fabricação do produto, a administração da dose dentro do intervalo mencionado anteriormente é aceitável.
Populações Especiais
População pediátrica (17 anos ou menos) A segurança e a eficácia de CARVYKTI® em crianças com menos de 18 anos de idade ainda não foram estabelecidas.
Não existem dados disponíveis.
Idosos (65 anos ou mais)
Nenhum ajuste de dose è necessário para pacientes ≥ 65 anos de idade.
Administração
Preparando o Paciente para a infusão de CARVYKTI®
Confirme a disponibilidade de CARVYKTI® antes de iniciar o regime de depleção linfocitária.
Regime de depleção linfocitária
Administre um regime para depleção linfocitária de 300 mg/m2 de ciclofosfamida e 30 mg/m2 de fludarabina. Ambos medicamentos devem ser administrados por via intravenosa, diariamente por 3 dias consecutivos. Administre CARVYKTI® de 5 a 7 dias após o início do regime de depleção linfocitária. Se a resolução de toxicidade de Grau 1 ou menor decorrente do regime de depleção linfocitária levar mais de 14 dias, resultando em atraso na administração de CARVYKTI®, o regime de depleção linfocitária deve ser novamente administrado após um mínimo de 21 dias da primeira dose do primeiro regime de depleção linfocitária. Para modificações da dose, vide as informações de prescrição dos fabricantes correspondentes. O regime de depleção linfocitária deve ser adiado se um paciente tiver reações adversas graves após a terapia ponte precedente (incluindo infecção ativa clinicamente significante, toxicidade cardíaca e toxicidade pulmonar).
Avaliação clínica antes da infusão de CARVYKTI®
A infusão de CARVYKTI® deve ser adiada se um paciente apresentar quaisquer das condições a seguir:
• infecção ativa clinicamente significante ou distúrbios inflamatórios.
• toxicidades não-hematológicas de Grau ≥ 3 do condicionamento com ciclofosfamida e fludarabina, exceto náusea, vômito, diarreia ou constipação de Grau 3. A infusão de CARVYKTI® deve ser adiada até que esses eventos sejam solucionados para Grau ≤ 1.
• doença do enxerto contra o hospedeiro ativa.
Pré-medicamentos
Administre os seguintes medicamentos pré-infusão em todos os pacientes (30 a 60 minutos) antes da infusão de CARVYKTI®:
• Antipiréticos (650 a 1000 mg de paracetamol/acetaminofeno oral ou intravenoso).
• Anti-histamínico (25 a 50 mg de difenidramina oral ou intravenosa, ou equivalente).
Evite o uso de corticosteroides sistêmicos profiláticos, uma vez que eles podem interferir na atividade de CARVYKTI® .
Preparação de CARVYKTI® para infusão
Não descongele o produto até que esteja pronto para ser usado. Coordene o tempo entre o descongelamento e a infusão de CARVYKTI® . Confirme primeiro o horário de infusão e ajuste o tempo de início do descongelamento de modo que o CARVYKTI® esteja disponível para infusão quando o paciente estiver preparado para receber o produto.
• Confirme a identificação do paciente: Antes de preparar o CARVYKTI®, verifique se a identificação do paciente no cassete do CARVYKTI® corresponde ao paciente em questão. Não remova a bolsa do produto CARVYKTI® do cassete se a informação no rótulo específico ao paciente não for a mesma do paciente em questão.
• Assim que a identificação do paciente for confirmada, remova a bolsa do produto CARVYKTI® do cassete.
• Inspecione a bolsa do produto quanto a qualquer violaçã