
BRUKINSA
ADIUM
zanubrutinibe
Antineoplásico.
Apresentações.
Cápsula
BRUKINSA® (zanubrutinibe) é apresentado em embalagem contendo 120 cápsulas.
USO ORAL
USO ADULTO
Composição.
Cada cápsula de Brukinsa® (zanubrutinibe) contém: zanubrutinibe 80mg. Excipientes: celulose microcristalina pH 102, croscarmelose sódica, lauril sulfato de sódio, dióxido de silício coloidal, estearato de magnésio, cápsula (contém tinta preta comestível, gelatina e dióxido de titânio).
Informações técnicas.
1. INDICAÇÕES
BRUKINSA® está indicado no tratamento de pacientes adultos com linfoma de células do manto (LCM) que receberam pelo menos uma terapia anterior.
2. RESULTADOS DE EFICÁCIA
Linfoma de células do manto (LCM)
A eficácia do BRUKINSA® foi avaliada no BGB-3111-206 [NCT03206970], um estudo de fase 2, aberto, multicêntrico, de braço único, com 86 pacientes com LCM previamente tratado, que haviam recebido pelo menos uma terapia anterior. BRUKINSA® foi administrado por via oral na dose de 160 mg duas vezes ao dia até a progressão da doença ou toxicidade inaceitável.
A idade mediana dos pacientes foi de 60,5 anos (variação de 34 a 75) e a maioria era do sexo masculino (78%). O tempo mediano desde o diagnóstico até a entrada no estudo foi de 30 meses (variação: 3 a 102) e o número mediano de terapias anteriores foi de 2 (variação: 1 a 4). Os regimes anteriores mais comuns foram baseados em CHOP (91%), seguidos por aqueles baseados em rituximabe (74%). A maioria dos pacientes apresentou envolvimento extranodal (71%) e doença refratária (52%). A variante blastoide do LCM estava presente em 14% dos pacientes. O escore de risco MIPI foi baixo em 58%, intermediário em 29% e alto em 13% dos pacientes.
A eficácia do BRUKINSA® também foi avaliada no BGB-3111-AU-003 [NCT02343120], um estudo de fase 1/2, aberto, de escalonamento de dose, global, multicêntrico, braço único de doenças malignas de células B, no qual 32 pacientes com LCM previamente tratado receberam BRUKINSA®. BRUKINSA® foi administrado por via oral em doses de 160 mg duas vezes ao dia ou 320 mg uma vez ao dia. A idade mediana dos pacientes com LCM previamente tratado foi de 70 anos (variação de 42 a 86) e 38% dos pacientes tinham idade ≥ 75 anos. A maioria dos pacientes era do sexo masculino (69%) e caucasiana (78%). O escore de risco MIPI foi baixo em 28%, intermediário em 41% e alto risco em 31% dos pacientes.
A resposta tumoral foi avaliada de acordo com a Classificação Lugano de 2014 para ambos os estudos e o desfecho primário de eficácia foi a taxa de resposta global avaliada por um Comitê de Revisão Independente.
Referências Bibliográficas
-Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol. 2014;32:3059-68.
-Song Y, Zhou K, Zou D, et al. Treatment of patients with relapsed or refractory mantle-cell lymphoma with zanubrutinib, a selective inhibitor of Bruton's tyrosine kinase. Clin Cancer Res. 2020;26:4216-24.
-Tam CS, Trotman J, Opat S, et al. Phase 1 study of the selective BTK inhibitor zanubrutinib in B-cell malignancies and safety and efficacy evaluation in CLL. Blood. 2019;134:851-9.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Mecanismo de ação
O zanubrutinibe é uma pequena enzima inibidora da tirosina quinase de Bruton (BTK). O zanubrutinibe forma uma ligação covalente com um resíduo de cisteína no sítio ativo da BTK, levando à inibição da atividade da BTK. BTK é uma molécula sinalizadora das vias do receptor de antígeno das células B (BCR) e do receptor de citocina. Nas células B, a sinalização BTK resulta na ativação de vias necessárias para a proliferação, circulação, quimiotaxia e adesão de células B. Em estudos não clínicos, o zanubrutinibe inibiu a proliferação de células B maligna e reduziu o crescimento do tumor.
Famacodinâmica
Ocupação da BTK em Células Mononucleares do Sangue Periférico (PBMCs) e linfonodos
A ocupação mediana da BTK no estado de equilíbrio nas células mononucleares do sangue periférico foi mantida a 100% ao longo de 24 horas, com uma dose diária total de 320 mg em pacientes com neoplasias das células B. A ocupação mediana da BTK no estado de equilíbrio nos linfonodos foi de 94% a 100% após a dosagem recomendada aprovada.
Eletrofisiologia Cardíaca
Nas doses recomendadas aprovadas (160 mg duas vezes ao dia ou 320 mg uma vez ao dia), não houve efeitos clinicamente relevantes no intervalo QTc. O efeito de BRUKINSA® no intervalo QTc acima da exposição terapêutica não foi avaliado.
Farmacocinética
A concentração plasmática máxima de zanubrutinibe (Cmax) e a área sob a curva de concentração plasmática ao longo do tempo (AUC) aumentam proporcionalmente em uma faixa de dosagem de 40mg a 320mg (0,13 a 1 vez a dose diária total recomendada). Foi observado acúmulo sistêmico limitado de zanubrutinibe após administração repetida.
A média geométrica (% CV) da AUC diária do zanubrutinibe no estado de equilíbrio é de 2,295 (37%) ng·h/mL após 160mg duas vezes ao dia e 2.180 (41%) ng·h/mL após 320mg uma vez ao dia. A média geométrica (% CV) da Cmax do zanubrutinibe no estado estacionário é de 314 (46%) ng/mL após 160mg duas vezes ao dia e 543 (51%) ng/mL após 320mg uma vez ao dia.
Absorção
O tmax mediano do zanubrutinibe é de 2 horas.
Efeito dos Alimentos
Não foram observadas diferenças clinicamente significativas na AUC ou Cmax do zanubrutinibe após a administração de uma refeição rica em gordura (aproximadamente 1.000 calorias com 50% do conteúdo calórico total em gordura) em indivíduos saudáveis.
Distribuição
A média geométrica (% CV) do volume de distribuição aparente no estado de equilíbrio do zanubrutinibe é 881 (95%) L. A ligação do zanubrutinibe às proteínas plasmáticas é de aproximadamente 94% e a razão sangue-plasma é 0,7 a 0,8.
Eliminação
A meia-vida média (t½) de zanubrutinibe é de aproximadamente 2 a 4 horas após uma dose oral única de zanubrutinibe de 160mg ou 320mg. A média geométrica (% CV) da depuração oral aparente (CL / F) de zanubrutinibe é 182 (37%) L / h.
Metabolismo
O zanubrutinibe é metabolizado principalmente pelo citocromo P450 (CYP) 3A.
Excreção
Após uma dose única de 320 mg de zanubrutinibe radiomarcada em indivíduos saudáveis, aproximadamente 87% da dose foi recuperada nas fezes (38% inalteradas) e 8% na urina (menos de 1% inalterada).
Populações especiais
Não foram observadas diferenças clinicamente significativas na farmacocinética do zanubrutinibe com base na idade (19 a 90 anos), sexo, raça (asiática, caucasiana e outras), peso corporal (36 a 140kg) ou insuficiência renal leve ou moderada (depuração de creatinina [CLcr] ≥ 30 mL/min, conforme estimado por Cockcroft-Gault). O efeito de insuficiência renal grave (CLcr < 30 mL / min) e diálise na farmacocinética do zanubrutinibe é desconhecido.
Comprometimento hepático
A AUC total do zanubrutinibe aumentou 11% em indivíduos com comprometimento hepático leve (classe A de Child-Pugh), 21% em indivíduos com comprometimento hepático moderado (classe B de Child-Pugh) e 60% em indivíduos com comprometimento hepático grave (Classe C de Child-Pugh) em relação a indivíduos com função hepática normal. A AUC não ligada do zanubrutinibe aumentou 23% em indivíduos com comprometimento hepático leve (classe A de Child-Pugh), 43% em indivíduos com comprometimento hepático moderado (classe B de Child-Pugh) e 194% em indivíduos com comprometimento hepático grave (Classe C de Child-Pugh) em relação a indivíduos com função hepática normal.
4. CONTRAINDICAÇÕES
BRUKINSA® é contraindicado em pacientes com hipersensibilidade conhecida a zanubrutinibe ou qualquer componente da fórmula.
5. ADVERTÊNCIAS E PRECAUÇÕES
Hemorragia
Eventos hemorrágicos fatais e graves ocorreram em pacientes com neoplasias hematológicas tratadas com BRUKINSA® em monoterapia. Foram notificados eventos hemorrágicos de grau 3 ou superior, incluindo hemorragia intracraniana e gastrointestinal, hematúria e hemotórax em 2% dos pacientes tratados com BRUKINSA® em monoterapia. Ocorreram eventos hemorrágicos de qualquer grau, incluindo púrpura e petéquias, em 50% dos pacientes tratados com BRUKINSA® em monoterapia.
Ocorreram eventos hemorrágicos em pacientes com e sem terapia antiplaquetária ou anticoagulação concomitante. A administração concomitante de BRUKINSA® com medicamentos antiplaquetários ou anticoagulantes pode aumentar ainda mais o risco de hemorragia.
Monitorar sinais e sintomas de sangramento. Descontinue BRUKINSA® se ocorrer hemorragia intracraniana de qualquer grau. Considere o risco-benefício de suspender BRUKINSA® por 3-7 dias antes e após a cirurgia, dependendo do tipo de cirurgia e do risco de sangramento.
Infecções
Ocorreram infecções fatais e graves (incluindo bactérias, vírus ou fungos) e infecções oportunistas em pacientes com neoplasias hematológicas tratadas com BRUKINSA® em monoterapia. Infecções de grau 3 ou superior ocorreram em 23% dos pacientes tratados com BRUKINSA® em monoterapia. A infecção mais comum de grau 3 ou superior foi pneumonia. Ocorreram infecções devido à reativação do vírus da hepatite B (HBV).
Considere a profilaxia para o vírus herpes simples, pneumonia por pneumocystis jiroveci e outras infecções, de acordo com o padrão de cuidado em pacientes com risco aumentado de infecções. Monitore e avalie os pacientes em busca de febre ou outros sinais e sintomas de infecção e trate adequadamente.
Citopenias
Citopenias de grau 3 ou 4, incluindo neutropenia (27%), trombocitopenia (10%) e anemia (8%) com base em medições laboratoriais, foram relatadas em pacientes tratados com BRUKINSA® em monoterapia.
Monitore a contagem sanguínea completa durante o tratamento e trate usando fator de crescimento ou transfusões, conforme necessário.
Segunda malignidade primária
As segundas malignidades primárias, incluindo carcinoma não cutâneo, ocorreram em 9% dos pacientes tratados com BRUKINSA® em monoterapia. A segunda malignidade primária mais frequente foi o câncer de pele (carcinoma basocelular e carcinoma espinocelular da pele), relatado em 6% dos pacientes. Aconselhe os pacientes a usarem proteção solar.
Arritmias cardíacas
Ocorreram fibrilação atrial e flutter atrial em 2% dos pacientes tratados com BRUKINSA® em monoterapia. Pacientes com fatores de risco cardíaco, hipertensão e infecções agudas podem ter risco aumentado. Eventos de grau 3 ou superior foram relatados em 0,6% dos pacientes tratados com BRUKINSA® em monoterapia. Monitore sinais e sintomas de fibrilação atrial e flutter atrial e maneje conforme apropriado.
Populações Especiais
Pacientes idosos
Dos 641 pacientes em estudos clínicos com BRUKINSA®, 49% tinham ≥ 65 anos de idade, enquanto 16% tinham ≥ 75 anos de idade. Não foram observadas diferenças gerais em segurança ou eficácia entre pacientes jovens e idosos.
Pacientes pediátricos
Segurança e eficácia em pacientes pediátricos não foram estabelecidas.
Pacientes com insuficiência renal
Nenhuma modificação da dose é recomendada em pacientes com insuficiência renal leve a moderada (CLcr ≥ 30 mL/min, estimado por Cockcroft-Gault). Monitore as reações adversas de BRUKINSA® em pacientes com insuficiência renal grave (CLcr < 30 mL/min) ou em diálise.
Pacientes com insuficiência hepática
Não são necessárias modificações da dose em pacientes com insuficiência hepática leve ou moderada.
Pacientes com insuficiência hepática leve ou moderada foram tratados em estudos clínicos com zanubrutinibe.
A dose recomendada de zanubrutinibe para pacientes com insuficiência hepática grave é de 80 mg por via oral, duas vezes ao dia. A segurança de zanubrutinibe não foi avaliada em pacientes com insuficiência hepática grave. Recomenda-se monitorar esses pacientes de perto para reações adversas com zanubrutinibe.
Gravidez
Teste de gravidez
O teste de gravidez é recomendado para mulheres com potencial reprodutivo antes de iniciar a terapia com BRUKINSA®.
Com base nos resultados de estudos em animais, BRUKINSA®
pode causar danos fetais quando administrado a mulheres grávidas. Não existem dados disponíveis sobre o uso de BRUKINSA® em mulheres grávidas para avaliar o risco associado ao medicamento de defeitos congênitos graves, aborto espontâneo ou resultados maternos ou fetais adversos. Em estudos de reprodução animal, a administração oral de zanubrutinibe em ratas grávidas durante o período de organogênese foi associada à malformação cardíaca fetal em exposições aproximadamente 5 vezes maiores que a humana. As mulheres devem ser aconselhadas a evitar a gravidez enquanto estiverem tomando BRUKINSA®. Se BRUKINSA® for utilizado durante a gravidez ou se a paciente engravidar enquanto estiver tomando BRUKINSA®, ela deve ser informada do risco potencial para o feto.
O risco estimado de defeitos congênitos importantes e aborto para a população indicada é desconhecido. Todas as gestações têm um risco histórico de defeitos de nascença, aborto ou outros resultados adversos. Na população geral dos EUA, o risco estimado de defeitos congênitos importantes e aborto em gestações clinicamente reconhecidas é de 2% a 4% e 15% a 20%, respectivamente.
Os homens devem ser aconselhados a não conceber um filho durante o tratamento com BRUKINSA® e por pelo menos 1 semana após a última dose de BRUKINSA®.
Categoria C: Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Lactação
Não existem dados sobre a presença de zanubrutinibe ou dos seus metabólitos no leite humano, os efeitos na criança amamentada ou os efeitos na produção de leite. Devido ao potencial de reações adversas graves de BRUKINSA® em uma criança amamentada, as mulheres lactantes devem ser aconselhadas a não amamentarem durante o tratamento com BRUKINSA® e por pelo menos duas semanas após a última dose.
Efeitos sobre a capacidade de dirigir e operar máquinas: fadiga, tontura e astenia foram reportadas em alguns pacientes tomando BRUKINSA® e devem ser considerados quando avaliar a capacidade do paciente para dirigir e operar máquinas.
6. INTERAÇÕES MEDICAMENTOSAS
Efeito de outras drogas:
Estudos clínicos e abordagens informadas por modelo
Inibidores do CYP3A: A coadministração de doses múltiplas de inibidores do CYP3A aumenta a Cmax e a AUC do zanubrutinibe (Tabela 5).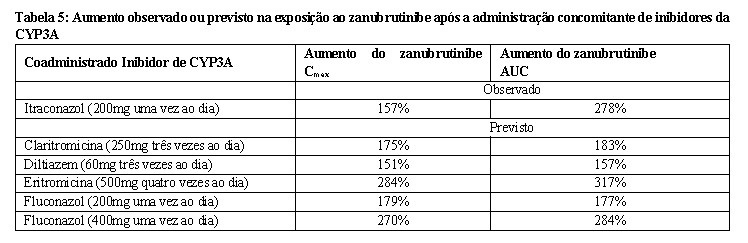
Indutores do CYP3A: A administração concomitante de doses múltiplas de rifampicina (forte indutor do CYP3A) diminuiu a Cmax do zanubrutinibe em 92% e a AUC em 93%.
Prevê-se que a administração concomitante de doses múltiplas de efavirenz (indutor moderado do CYP3A) diminua a Cmax do zanubrutinibe em 58% e a AUC em 60%.
Substratos do CYP3A: A administração concomitante de doses múltiplas de zanubrutinibe diminui a Cmax do midazolam em 30% (substrato do CYP3A) e AUC em 47%.
Substratos do CYP2C19: A administração concomitante de doses múltiplas de zanubrutinibe diminui a Cmax do omeprazol em 20% (substrato do CYP2C19) e AUC em 36%.
Outros substratos do CYP: Não foram observadas diferenças clinicamente significativas com a farmacocinética da varfarina (substrato CYP2C9) ou previstas com a farmacocinética da rosiglitazona (substrato CYP2C8) quando coadministradas com zanubrutinibe.
Sistemas de Transporte: A coadministração de doses múltiplas de zanubrutinibe aumentou a Cmax da digoxina em 34% (substrato da gp-P) e na AUC em 11%. Não foram observadas diferenças clinicamente significativas na farmacocinética da rosuvastatina (substrato BCRP) quando coadministrado com zanubrutinibe.
Agentes redutores de ácido gástrico: Não foram observadas diferenças clinicamente significativas na farmacocinética do zanubrutinibe quando coadministrado com agentes redutores de ácido gástrico (inibidores da bomba de prótons, antagonistas do receptor H2).
Estudos In Vitro
Enzimas CYP: O zanubrutinibe é um indutor do CYP2B6.
Sistemas de transporte: É provável que o zanubrutinibe seja um substrato da P-gp. O zanubrutinibe não é um substrato ou inibidor de OAT1, OAT3, OCT2, OATP1B1 ou OATP1B3.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
BRUKINSA® (zanubrutinibe) apresenta prazo de validade de 24 meses a partir da data de fabricação, devendo ser armazenado em temperatura ambiente (15°C a 30°C).
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Após aberto, BRUKINSA® é válido por 4 (quatro) meses.
Características físicas e organolépticas:
Cápsula opaca branca a esbranquiçada marcada com "ZANU 80" em tinta preta.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance de crianças.
8. POSOLOGIA E MODO DE USAR
A dose recomendada de BRUKINSA® (zanubrutinibe) é de 160 mg por via oral duas vezes ao dia ou 320 mg por via oral uma vez ao dia até a progressão da doença ou toxicidade inaceitável.
BRUKINSA® pode ser tomado com ou sem alimentos. Oriente os pacientes a engolir as cápsulas inteiras com água. Oriente os pacientes a não abrir, quebrar ou mastigar as cápsulas. Se perder uma dose de BRUKINSA®, esta deve ser tomada o mais rapidamente possível no mesmo dia, com o retorno ao horário normal no dia seguinte.
Este medicamento não deve ser partido, aberto ou mastigado.
Insuficiência hepática:
A dose recomendada de BRUKINSA® para pacientes com insuficiência hepática grave é de 80 mg por via oral duas vezes ao dia [ver item 5. ADVERTÊNCIAS E PRECAUÇÕES e item 3. CARACTERÍSTICAS FARMACOLÓGICAS].
Interações medicamentosas:
As modificações de dose recomendadas de BRUKINSA® para interações medicamentosas são fornecidas na Tabela 6 [ver item 6. INTERAÇÕES MEDICAMENTOSAS].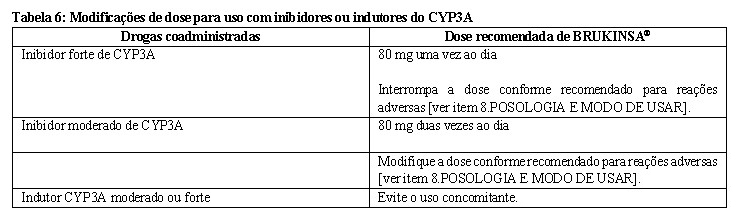
Após a descontinuação de um inibidor da CYP3A, retome a dose anterior de BRUKINSA® [ver item 8. POSOLOGIA E MODO DE USAR e item 6. INTERAÇÕES MEDICAMENTOSAS].
Modificações de dosagem para reações adversas
As modificações recomendadas da dose de BRUKINSA® para reações adversas de Grau 3 ou superiores são fornecidas na Tabela 7: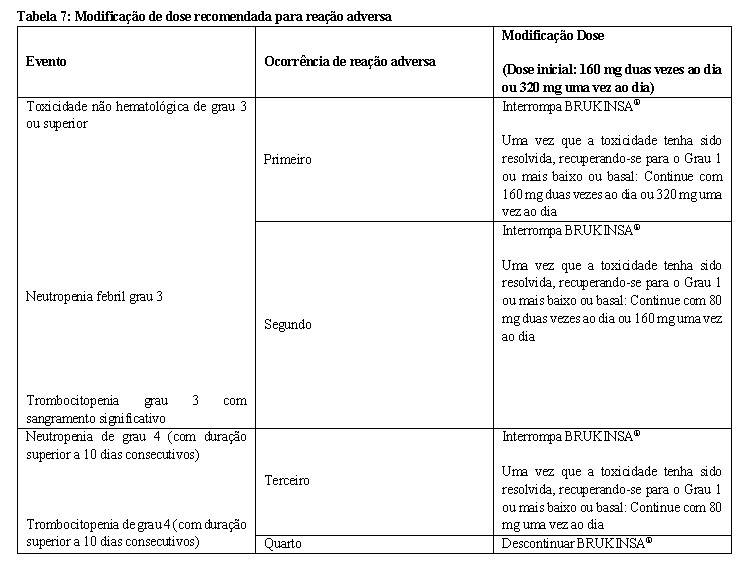
A linfocitose assintomática não deve ser considerada uma reação adversa, e esses pacientes devem continuar tomando BRUKINSA®.
9. REAÇÕES ADVERSAS
Os dados descritos refletem a exposição a BRUKINSA® como agente único, 160 mg duas vezes ao dia em 524 pacientes nos ensaios clínicos BGB-3111-AU-003, BGB-3111-206, BGB-3111-205, BGB-3111- 210 e BGB-3111-1002 e para BRUKINSA® 320 mg uma vez ao dia em 105 pacientes nos ensaios BGB-3111-AU-003 e BGB-3111-1002. Entre os 629 pacientes que receberam BRUKINSA®, 79% foram expostos por 6 meses ou mais e 61% foram expostos por mais de um ano.
A segurança de BRUKINSA® foi avaliada em 118 pacientes com LCM que receberam pelo menos uma terapia anterior em dois ensaios clínicos de braço único, BGB-3111-206 [NCT03206970] e BGB-3111AU-003 [NCT02343120]. A idade mediana dos pacientes que receberam BRUKINSA® nos estudos BGB-3111-206 e BGB-3111-AU-003 foi de 62 anos (intervalo: 34 a 86), 75% eram do sexo masculino, 75% eram asiáticos, 21% eram brancos e 94% tinham um status de desempenho de ECOG de 0 a 1. Os pacientes tinham uma mediana de 2 linhas de terapia anteriores (intervalo: 1 a 4). O estudo BGB-3111-206 exigiu uma contagem de plaquetas ≥ 75 x 109/L e uma contagem absoluta de neutrófilos ≥ 1 x 109/L independente do suporte de fator de crescimento, enzimas hepáticas ≤ 2,5 x limite superior da normalidade, bilirrubina total ≤ 1,5 x limite superior da normalidade. O estudo BGB-3111-AU-003 exigiu uma contagem de plaquetas ≥ 50 x 109/L e uma contagem absoluta de neutrófilos ≥ 1 x 109/L, independentemente do suporte ao fator de crescimento, enzimas hepáticas ≤ 3 x limite superior da bilirrubina total normal ≤ 1,5 x limite superior da normalidade. Ambos os ensaios necessários em CLcr ≥ 30 mL/min. Ambos os ensaios excluíram pacientes com transplante alogênico de células-tronco hematopoiéticas prévio, exposição a um inibidor da BTK, infecção conhecida pelo HIV e evidência sorológica de infecção ativa pela hepatite B ou hepatite C e pacientes que necessitassem de fortes inibidores da CYP3A ou fortes indutores da CYP3A. Os pacientes receberam BRUKINSA® 160 mg duas vezes ao dia ou 320 mg uma vez ao dia. Entre os pacientes que receberam BRUKINSA®, 79% foram expostos por 6 meses ou mais e 68% foram expostos por mais de um ano.
Eventos fatais dentro de 30 dias da última dose de BRUKINSA® ocorreram em 8 (7%) dos 118 pacientes com LCM. Os casos fatais incluíram pneumonia em 2 pacientes e hemorragia cerebral em um paciente.
Reações adversas graves foram relatadas em 36 pacientes (31%). As reações adversas graves mais frequentes que ocorreram foram pneumonia (11%) e hemorragia (5%).
Dos 118 pacientes com LCM tratados com BRUKINSA®, 8 (7%) pacientes descontinuaram o tratamento devido a reações adversas nos estudos. A reação adversa mais frequente que levou à descontinuação do tratamento foi pneumonia (3,4%). Um (0,8%) paciente apresentou reação adversa, levando à redução da dose (hepatite B).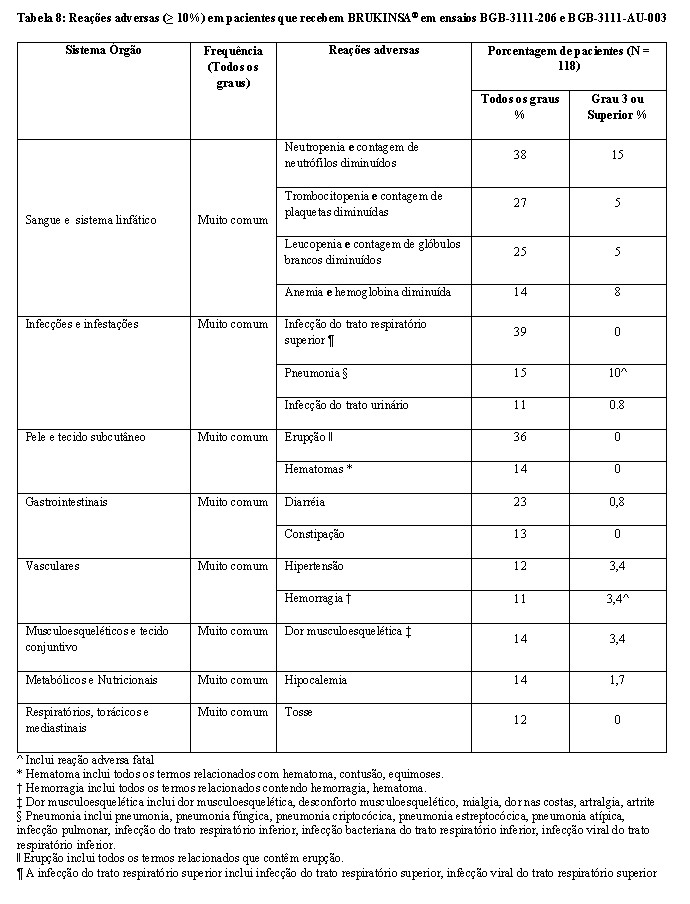
Outras reações adversas clinicamente significativas que ocorreram em < 10% dos pacientes com linfoma de células do manto incluem hemorragia grave (definida como hemorragia ≥ Grau 3 ou hemorragia do SNC de qualquer grau) (5%), hiperuricemia (6%) e dor de cabeça (4,2%).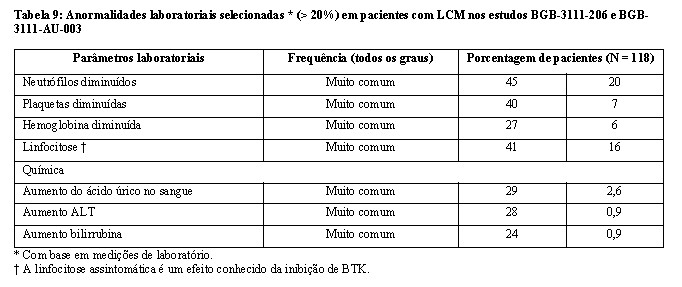
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos pelo Sistema VigiMed, disponível no Portal da Anvisa.
10. SUPERDOSE
Não há dados disponíveis referentes à superdosagem com BRUKINSA®.
Em caso de intoxicação ligue para 0800 722 6001, se você precisar de mais orientações.
Dizeres legais.
MS n°: 1.2214.0116
VENDA SOB PRESCRIÇÃO MÉDICA
Esta bula foi aprovada pela Anvisa em 08/09/2021.


 Trocar de país
Trocar de país