ATENTAH
APSEN
atomoxetina
Tratamento do TDHA.
Apresentações.
Cápsulas de 10 mg, 18 mg e 40 mg. Caixas com 10 e 30 cápsulas. Cápsulas de 25 mg, 60 mg e 80 mg. Caixa com 30 cápsulas.
USO ORAL
USO ADULTO E PEDIÁTRICO ACIMA DE 6 ANOS
Composição.
Cloridrato de atomoxetina (equivalente a 10 mg de atomoxetina) 11,428 mg excipientes qsp. 1 cápsula
Excipientes: amido, estearato de magnésio, gelatina e dióxido de titânio.
Cada cápsula de 18 mg contém:
Cloridrato de atomoxetina (equivalente a 18 mg de atomoxetina) 20,570 mg excipientes qsp. 1 cápsula
Excipientes: amido, estearato de magnésio, gelatina, amarelo de quinolina, amarelo crepúsculo e dióxido de titânio.
Cada cápsula de 25 mg contém:
Cloridrato de atomoxetina (equivalente a 25 mg de atomoxetina) 28,569 mg excipientes qsp. 1 cápsula
Excipientes: amido, estearato de magnésio, gelatina, azul brilhante, vermelho allura 129 e dióxido de titânio.
Cada cápsula de 40 mg contém:
Cloridrato de atomoxetina (equivalente a 40 mg de atomoxetina) 45,711 mg excipientes qsp. 1 cápsula
Excipientes: amido, estearato de magnésio, gelatina, azul brilhante, vermelho allura 129 e dióxido de titânio.
Cada cápsula de 60 mg contém:
Cloridrato de atomoxetina (equivalente a 60 mg de atomoxetina) 68,567 mg excipientes qsp. 1 cápsula
Excipientes: amido, estearato de magnésio, gelatina, azul brilhante, vermelho allura 129, amarelo de quinolina, amarelo crepúsculo e dióxido de titânio.
Cada cápsula de 80 mg contém:
Cloridrato de atomoxetina (equivalente a 80 mg de atomoxetina) 91,422 mg excipientes qsp. 1 cápsula
Excipientes: amido, estearato de magnésio, gelatina, azul brilhante, vermelho allura 129, amarelo de quinolina e dióxido de titânio.
Informações técnicas.
1. INDICAÇÕES
ATENTAH (atomoxetina) é indicado para o tratamento do Transtorno de Déficit de Atenção/Hiperatividade (TDAH) em pacientes adultos, adolescentes ou pediátricos com idade superior a 6 anos.
2. RESULTADOS DE EFICÁCIA
A eficácia de atomoxetina no tratamento do TDAH foi estabelecida em sete estudos clínicos randomizados, duplo-cegos e placebo-controlados, realizados em crianças, adolescentes e adultos, que preencheram os critérios da 4ª edição do Manual Estatístico e de Diagnóstico (DSM-IV) para o TDAH.
Estudos de TDAH em Crianças e Adolescentes
A eficácia de atomoxetina no tratamento do TDAH foi estabelecida em 4 estudos randomizados, duplocegos e controlados por placebo de pacientes pediátricos (idades de 6 a 18 anos). Aproximadamente um terço dos pacientes preencheram os critérios do DSM-IV para subtipo desatento e dois terços preencheram os critérios para os subtipos desatento e hiperativo / impulsivo. Os sinais e sintomas de TDAH foram avaliados por uma comparação da mudança média da linha de base até o desfecho de pacientes tratados com atomoxetina e placebo, adotando uma análise por "intenção de tratar" para a Escala para TDAH versão IV para pais (ADHDRS), administrada e pontuada pelo investigador, incluindo a pontuação total da escala e as subescalas: hiperativo/impulsivo e desatento. Cada item na Escala de Avaliação do TDAH avalia um sintoma, de acordo com os critérios para TDAH no DSM-IV. No Estudo 1, um estudo do tratamento agudo de 8 semanas, randomizado, duplo-cego, controlado por placebo, dose-resposta em crianças e adolescentes de 8 a 18 anos (N = 297), os pacientes receberam uma dose fixa de atomoxetina (0,5, 1,2 ou 1,8 mg / kg / dia) ou placebo. A atomoxetina foi administrada como uma dose dividida no início da manhã e no final da tarde / início da noite. Nas 2 doses mais altas, a melhora dos sintomas do TDAH se mostrou estatisticamente superior nos pacientes tratados com atomoxetina, comparado aos pacientes tratados com placebo, conforme medido na escala de ADHDRS. A dose de atomoxetina de 1,8 mg / kg / dia não proporcionou nenhum benefício adicional em relação ao observado com a dose de 1,2 mg / kg / dia. A dose de atomoxetina de 0,5 mg / kg / dia não foi superior ao placebo. No Estudo 2, um estudo de tratamento agudo de 6 semanas, randomizado, duplo-cego, controlado por placebo, com crianças e adolescentes de 6 a 16 anos (N = 171), os pacientes receberam atomoxetina ou placebo. A atomoxetina foi administrada em dose única no início da manhã e sua dose titulada com base no peso e de acordo com a resposta clínica, até uma dose máxima de 1,5 mg / kg / dia. A dose final média de atomoxetina foi de aproximadamente 1,3 mg / kg / dia. Houve uma melhora estatisticamente significativa nos sintomas do TDAH com o uso de atomoxetina comparado ao placebo, conforme escala de avaliação ADHDRS. Este estudo mostra que a atomoxetina é eficaz quando administrado uma vez ao dia pela manhã. Em 2 estudos idênticos, de 9 semanas, agudos, randomizados, duplo-cegos, controlados por placebo com crianças de 7 a 13 anos (Estudo 3, N = 147; Estudo 4, N = 144), atomoxetina e metilfenidato foram comparados com placebo. A atomoxetina foi administrada em uma dose dividida pela manhã e ao final da tarde e titulada com base no peso de acordo com a resposta clínica. A dose máxima recomendada de atomoxetina foi de 2,0 mg / kg / dia. A dose final média de atomoxetina para ambos os estudos foi de aproximadamente 1,6 mg / kg / dia. Em ambos os estudos, os sintomas de TDAH apresentaram uma melhora estatisticamente superior com atomoxetina em comparação ao placebo, conforme medido pela escala de avaliação ADHDRS. O exame dos subconjuntos da população com base no sexo e na idade ( < 12 e 12 a 17) não revelou qualquer resposta diferencial com base nesses subgrupos. Não houve exposição suficiente de grupos étnicos diferentes de caucasianos para permitir a exploração das diferenças nesses subgrupos.
Estudo de Manutenção -A eficácia da atomoxetina no tratamento de manutenção do TDAH foi estabelecida em um estudo ambulatorial de crianças e adolescentes (idades de 6 a 15 anos). Pacientes que atenderam aos critérios do DSM-IV para TDAH que mostraram resposta contínua por cerca de 4 semanas durante uma fase inicial de tratamento aberto de 10 semanas com atomoxetina (1,2 a 1,8 mg / kg / dia) foram randomizados para a continuação de sua dose atual de atomoxetina (N = 292) ou placebo (N = 124) em tratamento duplo-cego para observação de recaída. A resposta durante a fase aberta foi definida como pontuação CGI-ADHD-S ≤2 e uma redução de pelo menos 25% em relação ao valor basal em "ADHDRSIV-Parent:Inv total score". Os pacientes que utilizaram atomoxetina e mostraram resposta contínua por aproximadamente 8 meses durante a primeira fase de tratamento duplo-cego foram novamente randomizados para a continuação de sua dose atual de atomoxetina (N = 81) ou para placebo (N = 82) sob tratamento duplo-cego para observação de recaída. A recaída durante a fase duplo-cega foi definida como aumentos de pontuação CGI-ADHD-S de pelo menos 2 desde o final da fase aberta e a pontuação total ADHDRS-IV-Parent:Inv retorna para ≥ 90% da pontuação de entrada no estudo por 2 visitas consecutivas. Em ambas as fases duplo-cegas, os pacientes que receberam tratamento contínuo com atomoxetina tiveram tempos de recaída significativamente mais longos do que aqueles que receberam placebo.
Estudos de TDAH em Adultos
A eficácia da atomoxetina no tratamento do TDAH foi estabelecida em 2 estudos clínicos randomizados, duplo-cegos e controlados por placebo de pacientes adultos, com 18 anos ou mais, que preencheram os critérios do DSM-IV para TDAH. Os sinais e sintomas de TDAH foram avaliados usando a versão de triagem da escala de classificação de TDAH de Conners administrada pelo investigador (CAARS), uma escala de 30 itens. A medida de eficácia primária foi a pontuação total de sintomas de TDAH de 18 itens (a soma das subescalas de desatenção e hiperatividade/impulsividade do CAARS) avaliada pela comparação da alteração média do início ao final do estudo, usando a análise por intenção de tratar.
Em 2 estudos de tratamento agudo idênticos, de 10 semanas, randomizados, duplo-cegos, controlados por placebo (Estudo 5, N = 280; Estudo 6, N = 256), os pacientes receberam atomoxetina ou placebo. A atomoxetina foi administrada em uma dose dividida no início da manhã e no final da tarde / início da noite e titulada de acordo com a resposta clínica em um intervalo de 60 a 120 mg/dia. A dose final média de atomoxetina para ambos os estudos foi de aproximadamente 95 mg/dia. Em ambos os estudos, os sintomas de TDAH melhoraram de forma estatisticamente significativa no grupo que utilizou atomoxetina, conforme medido na pontuação de sintomas de TDAH da escala CAARS. O exame dos subconjuntos da população com base no sexo e na idade ( < 42 e ≥42) não revelou qualquer resposta diferencial com base nesses subgrupos. Não houve exposição suficiente de grupos étnicos diferentes de caucasianos para permitir a exploração das diferenças nesses subgrupos.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Mecanismo de Ação
O mecanismo preciso pelo qual a atomoxetina produz seus efeitos terapêuticos no Transtorno de Déficit de Atenção/Hiperatividade (TDAH) é desconhecido, mas acredita-se que esteja relacionado à inibição seletiva do transportador de norepinefrina pré-sináptica, conforme determinado em estudos ex vivo de captação e depleção de neurotransmissores.
Farmacodinâmica
Uma análise de exposição-resposta abrangendo doses de atomoxetina (0,5, 1,2 ou 1,8 mg/kg/dia) ou placebo demonstrou que a exposição à atomoxetina se correlaciona com a eficácia, conforme medido pela Escala de Classificação de Transtorno de Déficit de Atenção/Hiperatividade-IV-Versão dos Pais: administrado e pontuado pelo investigador. A relação exposição-eficácia foi semelhante à observada entre a dose e a eficácia.
Eletrofisiologia Cardíaca -O efeito de atomoxetina no prolongamento do QTc foi avaliado em um estudo cruzado randomizado, duplo-cego, positivo (moxifloxacina 400 mg) e controlado por placebo em homens saudáveis metabolizadores pobres de CYP2D6. Um total de 120 indivíduos saudáveis receberam atomoxetina (20 mg e 60 mg) duas vezes ao dia durante 7 dias. Não foram observadas grandes alterações no intervalo QTc (ou seja, aumentos > 60 mseg a partir da linha de base, QTc absoluto > 480 mseg) no estudo. No entanto, pequenas alterações no intervalo QTc não podem ser excluídas do estudo atual, porque o estudo falhou em demonstrar a sensibilidade do ensaio. Houve um ligeiro aumento no intervalo QTc com o aumento da concentração de atomoxetina.
Farmacocinética
A atomoxetina é bem absorvida após administração oral e é minimamente afetada pelos alimentos. É eliminado principalmente pelo metabolismo oxidativo através da via enzimática do citocromo P450 2D6 (CYP2D6) e subsequente glucuronidação. A atomoxetina tem meia-vida de cerca de 5 horas. Uma fração da população (cerca de 7% dos caucasianos e 2% dos afro-americanos) são metabolizadores pobres (PMs) de drogas metabolizadas pelo CYP2D6. Esses indivíduos têm atividade reduzida nesta via, resultando em AUCs 10 vezes maiores, pico de concentrações plasmáticas 5 vezes maiores e eliminação mais lenta (meia-vida plasmática de cerca de 24 horas) da atomoxetina em comparação com pessoas com atividade normal [metabolizadores extensivos (EMs)]. Os medicamentos que inibem o CYP2D6, como a fluoxetina, a paroxetina e a quinidina, causam aumentos semelhantes na exposição. A farmacocinética da atomoxetina foi avaliada em mais de 400 crianças e adolescentes em estudos clínicos selecionados, principalmente utilizando estudos farmacocinéticos populacionais. Dados farmacocinéticos individuais em dose única e em estado estacionário também foram obtidos em crianças, adolescentes e adultos. Quando as doses foram normalizadas para uma base de mg/kg, valores semelhantes de meia-vida, Cmax e AUC foram observados em crianças, adolescentes e adultos. A depuração e o volume de distribuição após o ajuste para o peso corporal também foram semelhantes.
Absorção e distribuição: A atomoxetina é rapidamente absorvida após administração oral, com biodisponibilidade absoluta de cerca de 63% em EMs e 94% em PMs. As concentrações plasmáticas máximas (Cmax) são atingidas aproximadamente 1 a 2 horas após a administração. Atomoxetina pode ser administrada com ou sem alimentos. A administração de atomoxetina com uma refeição padrão rica em gordura em adultos não afetou a extensão da absorção oral da atomoxetina (AUC), mas diminuiu a taxa de absorção, resultando em uma Cmax 37% mais baixa e um Tmax retardado em 3 horas. Em ensaios clínicos com crianças e adolescentes, a administração de atomoxetina com alimentos resultou numa Cmax 9% mais baixa.
O volume de distribuição no estado estacionário após a administração intravenosa é de 0,85 L/kg, indicando que a atomoxetina se distribui principalmente na água corporal total. O volume de distribuição é semelhante em toda a faixa de peso do paciente após a normalização pelo peso corporal. Em concentrações terapêuticas, 98% da atomoxetina no plasma liga-se às proteínas, principalmente à albumina.
Metabolismo e eliminação: A atomoxetina é metabolizada principalmente pela via enzimática CYP2D6. Pessoas com atividade reduzida nesta via (PMs) têm maiores concentrações plasmáticas de atomoxetina em comparação com pessoas com atividade normal (EMs). Para PMs, a AUC da atomoxetina é aproximadamente 10 vezes e Css, max é cerca de 5 vezes maior do que EMs. A co-administração de atomoxetina com inibidores potentes do CYP2D6, como fluoxetina, paroxetina ou quinidina, resulta em um aumento substancial na exposição plasmática da atomoxetina e pode ser necessário ajuste da dose. A atomoxetina não inibiu ou induziu a via do CYP2D6. O principal metabólito oxidativo formado, independentemente do status do CYP2D6, é a 4hidroxiatomoxetina, que é glicuronidada. A 4-hidroxatomoxetina é equipotente à atomoxetina como um inibidor do transportador da norepinefrina, mas circula no plasma em concentrações muito mais baixas (1% da concentração de atomoxetina em EMs e 0,1% da concentração de atomoxetina em PMs). A 4hidroxiatomoxetina é formada principalmente pelo CYP2D6, mas nos PMs, a 4-hidroxatomoxetina é formada em uma taxa mais lenta por várias outras enzimas do citocromo P450. A N-desmetilatomoxetina é formada pelo CYP2C19 e outras enzimas do citocromo P450, mas tem substancialmente menos atividade farmacológica em comparação com a atomoxetina e circula no plasma em concentrações mais baixas (5% da concentração de atomoxetina em EMs e 45% da concentração de atomoxetina em PMs). A depuração plasmática aparente média da atomoxetina após administração oral em adultos EMs é de 0,35 L/h/kg e a meia-vida média é de 5,2 horas. Após a administração oral de atomoxetina aos PMs, a depuração plasmática aparente média é de 0,03 L/h/kg e a meia-vida média é de 21,6 horas. Para PMs, a AUC da atomoxetina é aproximadamente 10 vezes e Css, max é cerca de 5 vezes maior do que EMs. A meia-vida de eliminação da 4-hidroxiatomoxetina é semelhante à da N-desmetilatomoxetina (6 a 8 horas) em indivíduos EM, enquanto a meia-vida de N-desmetilatomoxetina é muito maior em indivíduos PM (34 a 40 horas). A atomoxetina é excretada principalmente como 4-hidroxatomoxetina-O-glicuronídeo, principalmente na urina (mais de 80% da dose) e em menor extensão nas fezes (menos de 17% da dose). Apenas uma pequena fração da dose de atomoxetina é excretada como atomoxetina inalterada (menos de 3% da dose), indicando extensa biotransformação.
Carcinogênese, Mutagênese, Diminuição da Fertilidade
Carcinogênese: O cloridrato de atomoxetina não foi carcinogênico em ratos e camundongos quando administrada na dieta por 2 anos em doses médias ponderadas de até 47 e 458 mg/kg/dia, respectivamente. A dose mais elevada usada em ratos é aproximadamente 8 e 5 vezes a dose humana máxima recomendada (MRHD) em crianças e adultos, respetivamente, numa base de mg /m2. Os níveis plasmáticos (AUC) de atomoxetina nesta dose em ratos são estimados em 1,8 vezes (metabolizadores extensivos) ou 0,2 vezes (metabolizadores pobres) em humanos que recebem a dose humana máxima. A dose mais alta usada em camundongos é aproximadamente 39 e 26 vezes o MRHD em crianças e adultos, respectivamente, em uma base de mg/m2.
Mutagênese: O cloridrato de atomoxetina foi negativo em uma bateria de estudos de genotoxicidade que incluiu um ensaio de mutação de ponto reverso (Teste de Ames), um ensaio de linfoma de camundongo in vitro, um teste de aberração cromossômica em células de ovário de hamster chinês, um teste de síntese de DNA não programado em hepatócitos de rato, e um teste de micronúcleo in vivo em camundongos. No entanto, houve um ligeiro aumento na porcentagem de células de ovário de hamster chinês com diplocromossomos, sugerindo endreduplicação (aberração numérica). O metabólito cloridrato de N-desmetilatomoxetina foi negativo no Teste de Ames, ensaio de linfoma em camundongo e teste de síntese de DNA não programado.
Diminuição da fertilidade: o cloridrato de atomoxetina não prejudicou a fertilidade em ratos quando administrada na dieta em doses de até 57 mg/kg/dia, que é aproximadamente 6 vezes o MRHD em uma base de mg/m2.
4. CONTRAINDICAÇÕES
Hipersensibilidade
ATENTAH é contraindicado em pacientes com hipersensibilidade à atomoxetina ou outros constituintes do produto.
Inibidores da Monoamina Oxidase (IMAO)
ATENTAH não deve ser tomado com um IMAO ou dentro de 2 semanas após a interrupção de uso de um IMAO. O tratamento com um IMAO não deve ser iniciado dentro de 2 semanas após a interrupção do uso de ATENTAH. Com outras drogas que afetam as concentrações de monoaminas cerebrais, houve relatos de reações graves, às vezes fatais (incluindo hipertermia, rigidez, mioclonia, instabilidade autonômica com possíveis flutuações rápidas dos sinais vitais e alterações do estado mental que incluem agitação extrema que progride para delírio e coma) quando tomado em combinação com um IMAO. Alguns casos apresentaram características semelhantes à síndrome neuroléptica maligna. Essas reações podem ocorrer quando esses medicamentos são administrados concomitantemente ou em extrema proximidade.
Glaucoma de Ângulo Estreito
Em estudos clínicos, o uso de atomoxetina foi associado a um risco aumentado de midríase e, portanto, seu uso não é recomendado em pacientes com glaucoma de ângulo estreito.
Feocromocitoma
Reações graves, incluindo pressão arterial elevada e taquiarritmia, foram relatadas em pacientes com feocromocitoma ou história de feocromocitoma que receberam atomoxetina. Portanto, ATENTAH não deve ser administrado por pacientes com feocromocitoma ou história de feocromocitoma.
Doenças Cardiovasculares Graves
ATENTAH não deve ser usado em pacientes com doenças cardíacas ou vasculares graves, cuja condição poderia piorar se experimentarem aumentos na pressão arterial ou frequência cardíaca que poderiam ser clinicamente importantes (por exemplo, 15 a 20 mm Hg na pressão arterial ou 20 batimentos por minuto na frequência cardíaca).
Hipertireoidismo não controlado
Indivíduos com hipertireoidismo apresentam sintomas como hiperatividade, irritabilidade, alterações de humor, insônia, ansiedade etc. Alguns estudos observam que os efeitos adversos da atomoxetina, como alterações de humor, irritabilidade, insônia, arritmia, assemelham-se aos sintomas do hipertireoidismo. Devido às semelhanças entre os sintomas do hipertireoidismo e os efeitos adversos da atomoxetina, as alterações nos níveis de hormônio tireoidiano podem não ser reconhecidas em pacientes em tratamento com atomoxetina, sendo seu uso em pacientes com hipertireoidismo não controlado contraindicado.
Este medicamento é contraindicado para menores de 6 anos
5. ADVERTÊNCIAS E PRECAUÇÕES
Pensamento Suicida
Atomoxetina aumentou o risco de pensamento suicida em estudos de curto prazo em crianças e adolescentes com Transtorno de Déficit de Atenção/Hiperatividade (TDAH). Análises agrupadas de estudos clínicos de curto prazo controlados por placebo (6 a 18 semanas) de atomoxetina em crianças e adolescentes revelaram um maior risco de pensamento suicida no início do tratamento em pacientes que receberam atomoxetina. Houve um total de 12 estudos (11 em TDAH e 1 em enurese) envolvendo mais de 2.200 pacientes (incluindo 1.357 pacientes recebendo atomoxetina e 851 recebendo placebo). O risco médio de pensamento suicida em pacientes recebendo atomoxetina foi de 0,4% (5/1357 pacientes), em comparação com nenhum em pacientes tratados com placebo. Houve 1 tentativa de suicídio entre esses aproximadamente 2.200 pacientes, ocorrendo em um paciente tratado com atomoxetina. Nenhum suicídio ocorreu nesses estudos. Todas as reações ocorreram em crianças com idade igual ou inferior a 12 anos. Todas as reações ocorreram durante o primeiro mês de tratamento. Não se sabe se o risco de pensamento suicida em pacientes pediátricos se estende ao uso de longo prazo. Uma análise semelhante em pacientes adultos tratados com atomoxetina para TDAH ou transtorno depressivo maior (TDM) não revelou um risco aumentado de pensamento ou comportamento suicida em associação com o uso de atomoxetina.
Todos os pacientes em tratamento com atomoxetina devem ser monitorados adequadamente e observados atentamente quanto a agravamento clínico, tendências suicidas e alterações incomuns no comportamento, especialmente durante os primeiros meses da terapia medicamentosa ou em períodos de mudanças de dose, tanto aumento quanto diminuição.
Os seguintes sintomas foram relatados com atomoxetina: ansiedade, agitação, ataques de pânico, insônia, irritabilidade, hostilidade, agressividade, impulsividade, acatisia (inquietação psicomotora), hipomania e mania. Embora não tenha sido estabelecido um nexo de causalidade entre o surgimento desses sintomas e o surgimento de impulsos suicidas, existe a preocupação de que esses sintomas possam ser precursores do aparecimento de pensamentos suicidas. Portanto, os pacientes em tratamento com atomoxetina devem ser observados quanto ao surgimento de tais sintomas. Deve-se considerar a alteração do regime terapêutico, incluindo a possível descontinuação da medicação, em pacientes que estão apresentando tendências suicidas ou sintomas que podem ser precursores de tendências suicidas, especialmente se esses sintomas forem graves ou de início abrupto, ou se não fizessem parte dos sintomas presentes do paciente. Famílias e cuidadores de pacientes pediátricos em tratamento com atomoxetina devem ser alertados sobre a necessidade de monitorar os pacientes quanto ao surgimento de agitação, irritabilidade, mudanças incomuns no comportamento e outros sintomas descritos acima, bem como o surgimento de tendência suicida e relatar tais sintomas imediatamente aos profissionais de saúde. Esse monitoramento deve incluir observação diária por familiares e cuidadores.
Lesões Hepáticas Graves
Relatos pós-comercialização indicam que atomoxetina pode causar lesões hepáticas graves. Embora nenhuma evidência de lesão hepática tenha sido detectada em estudos clínicos com cerca de 6.000 pacientes, houve casos raros de lesão hepática clinicamente significativa que foram considerados provavelmente ou possivelmente relacionados ao uso de atomoxetina na experiência póscomercialização. Também foram relatados casos raros de insuficiência hepática, incluindo um caso que resultou em um transplante de fígado. Devido à provável subnotificação, é impossível fornecer uma estimativa precisa da verdadeira incidência dessas reações. Casos relatados de lesão hepática ocorreram dentro de 120 dias do início da atomoxetina na maioria dos casos e alguns pacientes apresentaram enzimas hepáticas marcadamente elevadas [ > 20 X limite superior do normal (LSN)] e icterícia com níveis de bilirrubina significativamente elevados ( > 2 X LSN), seguido de recuperação após a descontinuação da atomoxetina. Em um paciente, a lesão hepática, manifestada por enzimas hepáticas elevadas até 40 X LSN e icterícia com bilirrubina até 12 X LSN, reapareceu após a reintrodução e foi seguida por recuperação após a descontinuação do medicamento, fornecendo evidências de que atomoxetina provavelmente causou a lesão hepática. Essas reações podem ocorrer vários meses após o início da terapia, mas as anormalidades laboratoriais podem continuar a piorar por várias semanas após a interrupção do medicamento. O paciente descrito acima se recuperou de sua lesão hepática e não precisou de um transplante de fígado. Atomoxetina deve ser descontinuada em pacientes com icterícia ou evidências laboratoriais de lesão hepática e não deve ser reiniciado. Os exames laboratoriais para determinar os níveis de enzimas hepáticas devem ser realizados após o primeiro sintoma ou sinal de disfunção hepática (por exemplo, prurido, urina escura, icterícia, sensibilidade no quadrante superior direito ou sintomas inexplicáveis de "gripe").
Eventos Cardiovasculares
Graves Morte Súbita e Anormalidades Cardíacas Estruturais Pré-existentes ou Outros Problemas Cardíacos Graves
Crianças e adolescentes - A morte súbita foi relatada em associação com o tratamento com atomoxetina em doses usuais em crianças e adolescentes com anomalias cardíacas estruturais ou outros problemas cardíacos graves. Embora alguns problemas cardíacos graves por si só representem um risco aumentado de morte súbita, a atomoxetina geralmente não deve ser usada em crianças ou adolescentes com anomalias estruturais cardíacas graves conhecidas, cardiomiopatia, anormalidades graves do ritmo cardíaco ou outros problemas cardíacos graves que podem aumentar a vulnerabilidade aos efeitos noradrenérgicos da atomoxetina.
Adultos - Morte súbita, acidente vascular cerebral e infarto do miocárdio foram relatados em adultos tomando atomoxetina em doses usuais para TDAH. Embora o papel da atomoxetina nesses casos em adultos também seja desconhecido, os adultos têm maior probabilidade do que as crianças de apresentarem anormalidades cardíacas estruturais graves, cardiomiopatia, anormalidades graves do ritmo cardíaco, doença arterial coronariana ou outros problemas cardíacos graves. Deve-se considerar o não tratamento de adultos com anormalidades cardíacas clinicamente significativas.
Avaliação do Estado Cardiovascular em Pacientes Tratados com Atomoxetina
Crianças, adolescentes ou adultos que estão sendo considerados para tratamento com atomoxetina devem passar por um exame físico e de histórico cuidadoso (incluindo avaliação de histórico familiar de morte súbita ou arritmia ventricular) para avaliar a presença de doença cardíaca, e devem realizar posterior avaliação cardíaca se os achados sugerirem tal doença (por exemplo, eletrocardiograma e ecocardiograma). Os pacientes que desenvolverem sintomas como dor no peito por esforço, síncope inexplicada ou outros sintomas sugestivos de doença cardíaca durante o tratamento com atomoxetina devem ser submetidos a uma avaliação cardíaca imediata.
Efeitos Sobre a Pressão Arterial e Frequência Cardíaca
A atomoxetina deve ser usada com cautela em pacientes cujas condições médicas subjacentes podem ser agravadas por aumentos na pressão arterial ou frequência cardíaca, como certos pacientes com hipertensão, taquicardia ou doença cardiovascular ou cerebrovascular. Não deve ser utilizado em pacientes com doenças cardíacas ou vasculares graves, cuja situação possa ser agravada caso apresentem aumentos clinicamente importantes da pressão arterial ou frequência cardíaca. O pulso e a pressão arterial devem ser medidos no início do tratamento, após aumentos da dose de atomoxetina e periodicamente durante a terapia para detectar possíveis aumentos clinicamente importantes. A tabela a seguir fornece dados de estudos clínicos de curto prazo controlados por placebo para as proporções de pacientes com aumento em: pressão arterial diastólica ≥ 15 mm Hg; pressão arterial sistólica ≥ 20 mm Hg; frequência cardíaca maior ou igual a 20 bpm, tanto na população pediátrica quanto na adulta (ver Tabela 1).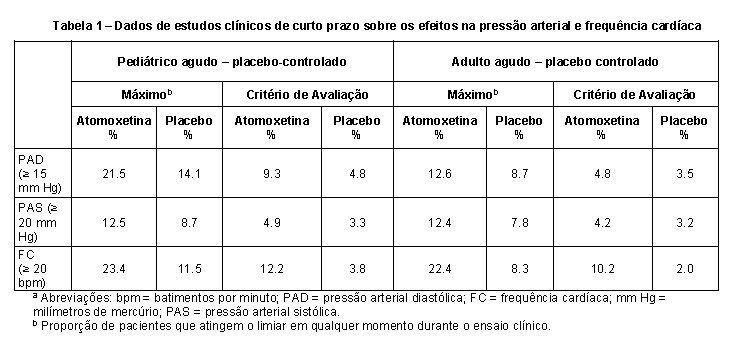
Em estudos para registro controlados por placebo envolvendo pacientes pediátricos, a taquicardia foi identificada como um evento adverso em 0,3% (5/1597) desses pacientes com atomoxetina em comparação com 0% (0/934) dos pacientes com placebo. O aumento médio da frequência cardíaca em pacientes com metabolizador extensivo (EM) foi de 5,0 batimentos/minuto, e em pacientes com metabolizador pobre (PM), 9,4 batimentos/minuto. Em estudos clínicos em adultos onde a condição EM/PM estava disponível, o aumento médio da frequência cardíaca em pacientes com PM foi significativamente maior do que em pacientes com EM (11 batimentos/minuto versus 7,5 batimentos/minuto). Os efeitos da frequência cardíaca podem ser clinicamente importantes em alguns pacientes PM. Em estudos para registro controlados por placebo envolvendo pacientes adultos, a taquicardia foi identificada como um evento adverso em 1,5% (8/540) dos pacientes com atomoxetina em comparação com 0,5% (2/402) dos pacientes com placebo.
Em estudos clínicos em adultos onde a condição EM/PM estava disponível, a mudança média da linha de base na pressão arterial diastólica em pacientes com PM foi maior do que em pacientes com EM (4,21 versus 2,13 mm Hg) como foi a mudança média da linha de base na pressão arterial sistólica (PM: 2,75 versus EM: 2,40 mm Hg).Os efeitos da pressão arterial podem ser clinicamente importantes em alguns pacientes PM. Hipotensão ortostática e síncope foram relatadas em pacientes utilizando atomoxetina. Em estudos para registro de crianças e adolescentes, 0,2% (12/5596) dos pacientes tratados com atomoxetina apresentaram hipotensão ortostática e 0,8% (46/5596) apresentaram síncope. Em estudos para registro de crianças e adolescentes de curto prazo, 1,8% (6/340) dos pacientes tratados com atomoxetina apresentaram hipotensão ortostática em comparação com 0,5% (1/207) dos pacientes tratados com placebo. A síncope não foi relatada durante estudos de curto prazo para registro, placebo controlados, em crianças e adolescentes com TDAH. A atomoxetina deve ser usada com cuidado em qualquer condição que possa predispor os pacientes à hipotensão ou condições associadas a alterações abruptas da frequência cardíaca ou da pressão arterial.
Emergência de Novos Sintomas Psicóticos ou Maníacos
O tratamento de sintomas psicóticos ou maníacos emergentes, como por exemplo, alucinações, pensamento delirante ou mania em crianças e adolescentes sem histórico prévio de doença psicótica ou mania, podem ser causados pela atomoxetina em doses usuais. Se tais sintomas ocorrerem, deve-se considerar um possível papel causal da atomoxetina, e a descontinuação do tratamento deve ser considerada. Em uma análise conjunta de vários estudos de curto prazo controlados por placebo, tais sintomas ocorreram em cerca de 0,2% (4 pacientes com reações dentre 1939 expostos à atomoxetina por várias semanas em doses usuais) de pacientes tratados com atomoxetina em comparação com 0 de 1.056 pacientes tratados com placebo.
Triagem de Pacientes para Transtorno Bipolar
Em geral, deve-se ter cuidado especial no tratamento de TDAH em pacientes com transtorno bipolar comórbido devido à preocupação com a possível indução de um episódio misto/maníaco em pacientes com risco de transtorno bipolar. Não se sabe se algum dos sintomas descritos acima representa tal conversão. No entanto, antes de iniciar o tratamento com atomoxetina, os pacientes com sintomas depressivos comórbidos devem ser examinados adequadamente para determinar se estão em risco de transtorno bipolar; esse rastreamento deve incluir um histórico psiquiátrico detalhado, incluindo histórico familiar de suicídio, transtorno bipolar e depressão.
Comportamento Agressivo ou Hostilidade
Pacientes que iniciam o tratamento para TDAH devem ser monitorados quanto ao aparecimento ou agravamento de comportamento agressivo ou hostilidade. O comportamento agressivo ou hostilidade é frequentemente observado em crianças e adolescentes com TDAH. Em estudos clínicos pediátricos de curto prazo controlados, 21/1308 (1,6%) dos pacientes tratados com atomoxetina versus 9/806 (1,1%) dos pacientes tratados com placebo relataram espontaneamente eventos adversos relacionados à hostilidade emergentes do tratamento (razão de
risco geral de 1,33 [95% C.I. 0,67-2,64 -não estatisticamente significativo]). Em ensaios clínicos controlados com placebo em adultos, 6/1697 (0,35%) dos pacientes tratados com atomoxetina versus 4/1560 (0,26%) dos pacientes tratados com placebo relataram espontaneamente eventos adversos relacionados à hostilidade emergente do tratamento (razão de risco geral de 1,38 [C.I. 95% 0,39-4,88 -não estatisticamente significativo]). Embora não seja uma evidência conclusiva de que atomoxetina cause comportamento agressivo ou hostilidade, esses comportamentos foram observados com mais frequência em estudos clínicos entre crianças, adolescentes e adultos tratados com atomoxetina em comparação com placebo.
Eventos alérgicos
Embora incomuns, reações alérgicas, incluindo reações anafiláticas, edema angioneurótico, urticária e erupção cutânea, foram relatadas em pacientes tomando atomoxetina.
Atenção:
Atentah 18 mg: Contém os corantes amarelo de quinolina e amarelo crepúsculo que podem, eventualmente, causar reações alérgicas.
Atentah 25 mg: Contém os corantes azul brilhante e vermelho allura 129 que podem, eventualmente, causar reações alérgicas.
Atentah 40 mg: Contém os corantes azul brilhante e vermelho allura 129 que podem, eventualmente, causar reações alérgicas.
Atentah 60 mg: Contém os corantes azul brilhante, vermelho allura 129, amarelo de quinolina e amarelo crepúsculo que podem, eventualmente, causar reações alérgicas.
Atentah 80 mg: Contém os corantes azul brilhante, vermelho allura 129 e amarelo de quinolina que podem, eventualmente, causar reações alérgicas.
Efeitos Sobre o Fluxo Urinário da Bexiga
Em estudos controlados em adultos com TDAH, as taxas de retenção urinária (1,7%, 9/540) e hesitação urinária (5,6%, 30/540) aumentaram entre os indivíduos que utilizaram atomoxetina em comparação com os indivíduos que receberam placebo (0%, 0/402; 0,5%, 2/402, respectivamente). Dois sujeitos adultos que receberam atomoxetina e nenhum sujeito que recebeu placebo descontinuaram os estudos clínicos controlados por causa da retenção urinária. Uma queixa de retenção urinária ou hesitação urinária deve ser considerada potencialmente relacionada à atomoxetina.
Priapismo
Casos raros de priapismo pós-comercialização, definidos como ereção peniana dolorosa e não dolorosa com duração superior a 4 horas, foram relatados por pacientes pediátricos e adultos tratados com atomoxetina. As ereções foram resolvidas nos casos em que havia informações de acompanhamento disponíveis, alguns após a descontinuação de atomoxetina. É necessária atenção médica imediata em caso de suspeita de priapismo.
Efeitos Sobre o Crescimento
Os dados sobre os efeitos a longo prazo de atomoxetina no crescimento vêm de estudos abertos e as alterações de peso e altura são comparadas com dados populacionais normativos. Em geral, o ganho de peso e altura de pacientes pediátricos tratados com atomoxetina fica aquém do previsto por dados populacionais normativos por cerca dos primeiros 9-12 meses de tratamento. Posteriormente, o ganho de peso voltou e, em cerca de 3 anos de tratamento, os pacientes tratados com atomoxetina ganharam 17,9 kg em média, 0,5 kg a mais do que o previsto por seus dados base. Após cerca de 12 meses, o ganho de altura se estabiliza e, em 3 anos, os pacientes tratados com atomoxetina ganharam 19,4 cm em média, 0,4 cm a menos do que o previsto por seus dados de linha de base (ver Figura 1 abaixo).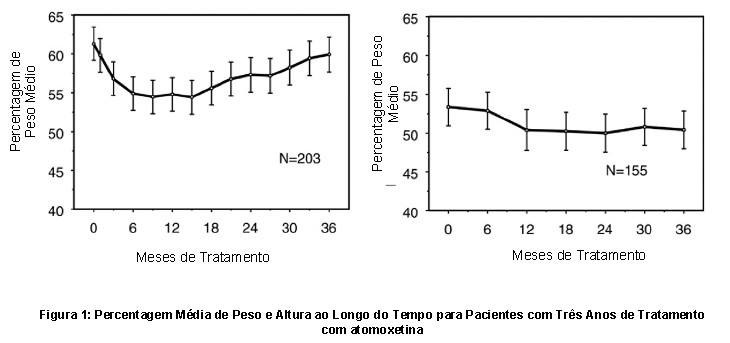
Este padrão de crescimento foi geralmente semelhante, independentemente do estado puberal no momento do início do tratamento. Pacientes que estavam na pré-puberdade no início do tratamento (meninas ≤8 anos, meninos ≤9 anos) ganharam em média 2,1 kg e 1,2 cm menos do que o previsto após três anos. Pacientes que estavam na puberdade (meninas > 8 a ≤13 anos, meninos > 9 a ≤14 anos) ou na puberdade tardia (meninas > 13 anos, meninos > 14 anos) tiveram ganhos médios de peso e altura próximos do previsto ou ultrapassou os previstos após três anos de tratamento. O crescimento seguiu um padrão semelhante tanto em metabolizadores extensivos quanto em metabolizadores pobres (EMs, PMs). PMs tratados por pelo menos dois anos ganharam uma média de 2,4 kg e 1,1 cm menos do que o previsto, enquanto os EMs ganharam uma média de 0,2 kg e 0,4 cm menos do que o previsto. Em estudos controlados de curto prazo (até 9 semanas), os pacientes tratados com atomoxetina perderam em média 0,4 kg e ganharam em média 0,9 cm, em comparação com um ganho de 1,5 kg e 1,1 cm nos pacientes tratados com placebo. Em um estudo clínico controlado com dose fixa, 1,3%, 7,1%, 19,3% e 29,1% dos pacientes perderam pelo menos 3,5% do peso corporal nos grupos de dose com placebo, 0,5, 1,2 e 1,8 mg/kg/dia. O crescimento deve ser monitorizado durante o tratamento com atomoxetina.
Testes de Laboratório
Não são necessários testes laboratoriais de rotina.
Metabolismo de CYP2D6 - Metabolizadores pobres (PMs) de CYP2D6 têm uma AUC 10 vezes maior e um pico de concentração 5 vezes maior para uma determinada dose de atomoxetina em comparação com metabolizadores extensivos (EMs). Aproximadamente 7% da população caucasiana são PMs. Os testes de laboratório estão disponíveis para identificar PMs CYP2D6. Os níveis sanguíneos dos PMs são semelhantes aos obtidos com o uso de inibidores fortes do CYP2D6. Os níveis sanguíneos mais elevados em PMs levam a uma taxa mais alta de alguns efeitos adversos da atomoxetina.
Uso Concomitante de Inibidores Potentes do CYP2D6 ou Uso em Pacientes que são conhecidos como PMs do CYP2D6
A atomoxetina é metabolizada principalmente pela via CYP2D6 em 4-hidroxiatomoxetina. O ajuste posológico de atomoxetina pode ser necessário quando coadministrado com inibidores potentes do CYP2D6 (por exemplo, paroxetina, fluoxetina e quinidina) ou quando administrado aos PMs do CYP2D6.
USO EM POPULAÇÕES ESPECÍFICAS
Gravidez (Categoria C): Não foram realizados estudos apropriados e bem controlados com atomoxetina em mulheres grávidas. ATENTAH (atomoxetina) não deve ser usado durante a gravidez, a menos que o benefício esperado justifique o risco potencial para o feto.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião dentista.
Os estudos disponíveis publicados com uso de atomoxetina em mulheres grávidas são insuficientes para estabelecer um risco de grandes defeitos de nascença, aborto espontâneo ou resultados maternais ou fetais adversos associado à droga.
Alguns estudos de reprodução animal com atomoxetina tiveram resultados adversos no desenvolvimento. Um dos 3 estudos em coelhas grávidas que receberam atomoxetina durante a organogênese resultou na diminuição de fetos vivos e um aumento nas reabsorções precoces, bem como ligeiros aumentos nas incidências na origem da artéria carótida atípica e artéria subclávia ausente. Estes efeitos foram observados em níveis plasmáticos (AUC) 3 vezes e 0,4 vezes os níveis plasmáticos humanos em metabolizadores pobres e extensivos receb