ALECENSA
ROCHE
alectinibe, cloridrato
Antineoplásico. Inibidor da proteína quinase.
Apresentações.
Alecensa ® 150 mg em embalagens que contém 224 cápsulas duras (4 cartuchos que contém 56 cápsulas duras cada um).
VIA ORAL
USO ADULTO
Composição.
Cada cápsula dura contém:
Princípio ativo: 150 mg de alectinibe (equivalente a 161,3 mg de cloridrato de alectinibe). Excipientes: lactose monoidratada, laurilsulfato de sódio, carmelose cálcica, hiprolose e estearato de magnésio. Cápsula dura: hipromelose, carragenina, cloreto de potássio, dióxido de titânio, amido e cera de carnaúba.
Informações técnicas.
1. INDICAÇÕES
Alecensa® está indicado para o tratamento de primeira linha de pacientes com câncer de pulmão de não pequenas células (CPNPC) positivo para quinase do linfoma anaplásico (ALK) localmente avançado ou metastático.
Alecensa® está indicado para o tratamento de pacientes com CPNPC localmente avançado ou metastático positivo para ALK que tenham progredido durante o uso de crizotinibe, ou que sejam intolerantes a ele.
2. RESULTADOS DE EFICÁCIA
Câncer de pulmão de não pequenas células ALK positivo em pacientes não tratados previamente
A segurança e a eficácia de Alecensa® foram avaliadas em um estudo clínico aberto Fase III randomizado global (BO28984) em pacientes que apresentam CPNPC positivo para ALK, não tratados previamente. Foi exigida positividade no teste central realizado por imuno-histoquímica (IHQ) Ventana anti-ALK (D5F3) para a expressão da proteína ALK de amostras de tecidos de todos os pacientes antes da randomização para o estudo.
No total, 303 pacientes foram incluídos no estudo Fase III, sendo 151 pacientes randomizados para o braço crizotinibe e 152 pacientes randomizados para o braço de Alecensa®, via oral, na dose recomendada de 600 mg duas vezes ao dia.
ECOG (Eastern Cooperative Oncology Group [Grupo Cooperativo Oriental de Oncologia]) PS (Performance Status [Status de performance]) (0/1 versus 2), raça (asiática vs. não asiática) e metástases no sistema nervoso central (SNC) no período basal (sim versus não) foram os fatores de estratificação para randomização. O desfecho primário do estudo foi demonstrar a superioridade de Alecensa® versus crizotinibe com base na sobrevida livre de progressão (SLP) de acordo com avaliação do investigador que utilizou os Critérios de Avaliação de Resposta em Tumores Sólidos (RECIST) versão 1.1. Características demográficas basais e características da doença para Alecensa® foram idade mediana de 58 anos (54 anos para crizotinibe), 55% do sexo feminino (58% para crizotinibe), 55% não asiáticos (54% para crizotinibe), 61% sem histórico de tabagismo (65% para crizotinibe), 93% ECOG PS de 0 ou 1 (93% para crizotinibe), 97% com doença em estágio IV (96% para crizotinibe), 90% de histologia com adenocarcinoma (94% para crizotinibe), 40% com metástases no SNC no período basal (38% para crizotinibe) e 17% que haviam recebido radioterapia prévia para SNC (14% para crizotinibe).
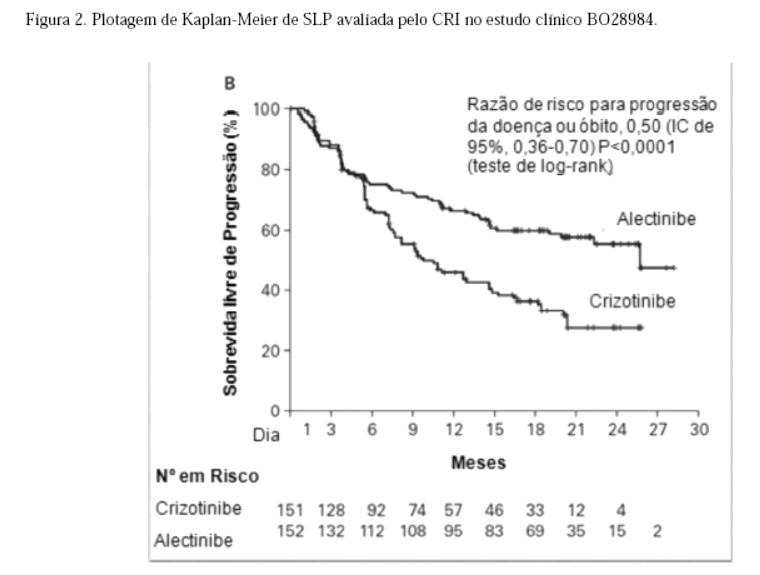
O estudo atingiu seu desfecho primário na análise primária. Dados de eficácia estão resumidos na Tabela 01 e as curvas de Kaplan-Meier para sobrevida livre de progressão (SLP) avaliada pelo investigador (INV) e pelo Comitê de Revisão Independente (CRI) são mostradas nas Figuras 1 e 2.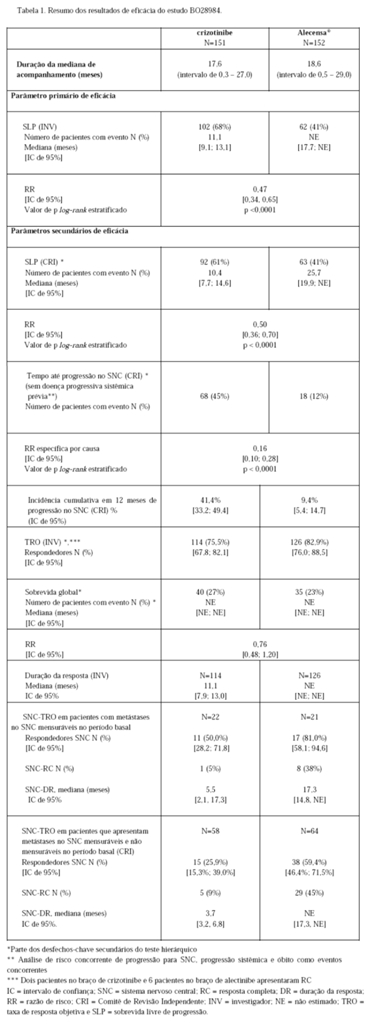
A magnitude do benefício em SLP foi constante para pacientes que apresentam metástases no SNC no período basal (RR=0,40, IC de 95%: 0,25 -0,64, SLP mediana para Alecensa® = NE, IC de 95% -9,2-NE, SLP mediana para crizotinibe = 7,4 meses, IC de 95%: 6,6 -9,6) e sem metástases no SNC no período basal (RR = 0,51, IC de 95%: 0,33 -0,80, SLP mediana para Alecensa® = NE, IC de 95%: NE, SLP mediana para crizotinibe = 14,8 meses, IC de 95%:10,8-20,3), indicando benefício de Alecensa® em relação a crizotinibe nos dois subgrupos. 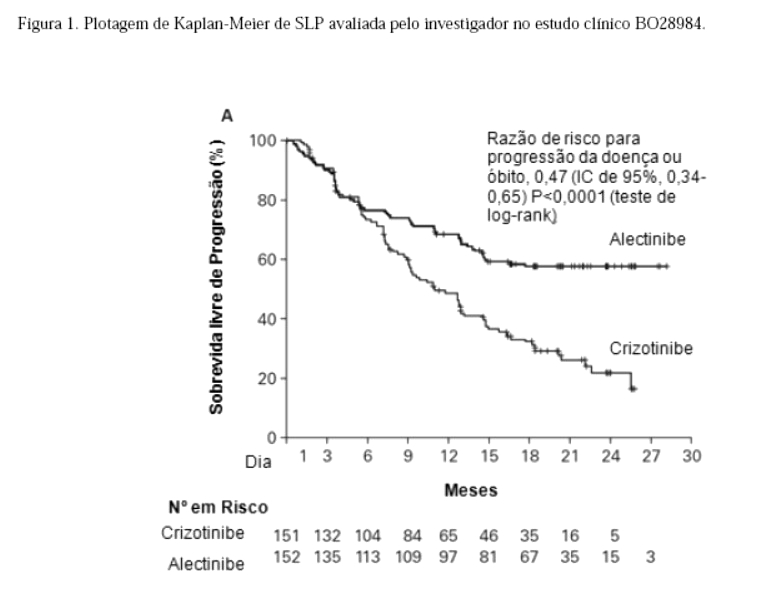
Câncer de pulmão de não pequenas células ALK positivo em pacientes tratados previamente com crizotinibe
A segurança e a eficácia de Alecensa® em pacientes com CPNPC ALK positivo tratados previamente com crizotinibe foram avaliadas em dois estudos clínicos Fase I/II (NP28761 e NP28673).
O estudo NP28761 foi um estudo Fase I/II de braço único, multicêntrico, conduzido em pacientes com CPNPC ALK positivo avançado que haviam progredido previamente durante tratamento com crizotinibe. Adicionalmente ao crizotinibe, os pacientes poderiam ter recebido tratamento prévio com quimioterapia. No total, 87 pacientes foram incluídos na parte da fase II do estudo e receberam Alecensa® via oral na dose recomendada de 600 mg duas vezes ao dia.
O desfecho primário foi avaliar a eficácia de Alecensa® por taxa de resposta objetiva (TRO) de acordo com avaliação utilizando os Critérios de Avaliação de Resposta em Tumores Sólidos (RECIST) versão 1.1.
Os dados demográficos dos pacientes foram compatíveis com os de uma população com CPNPC ALK positivo. As características demográficas da população total do estudo foi 84% de brancos, 8% de asiáticos, 55% do sexo feminino e uma mediana de idade de 54 anos. A maioria dos pacientes não apresentava história de tabagismo (62%). O estado de desempenho ECOG (Eastern Cooperative Oncology Group [Grupo Cooperativo Oriental de Oncologia]) no período basal era 0 ou 1 em 90% dos pacientes e 2 em 10% dos pacientes. No momento da inclusão no estudo, 99% dos pacientes apresentavam doença em estágio IV, 60% apresentavam metástases cerebrais e, em 94% dos pacientes, os tumores foram classificados como adenocarcinoma. Entre os pacientes incluídos no estudo, 26% haviam progredido previamente durante tratamento apenas com crizotinibe e 74% haviam progredido previamente durante tratamento com crizotinibe e quimioterapia.
Como mostrado na plotagem em cascata na Figura 3, a maioria dos pacientes apresentou redução do volume tumoral em suas lesões-alvo definidas, conforme avaliação do CRI de acordo com RECIST 1.1.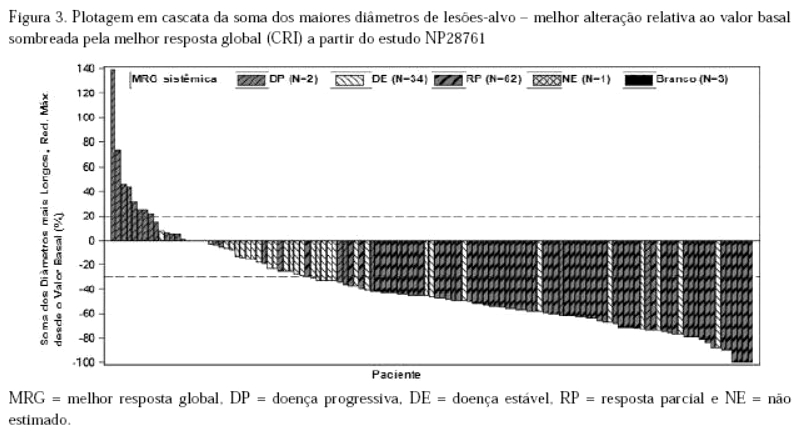
Qualidade de vida (QoL)
Dos itens de QoL analisados (QLQ-C30 e QLQ-LC13), melhoras clinicamente significativas (alteração relativa ao valor basal ≥ 10 pontos) foram observadas nas subescalas de Estado Geral de Saúde, Desempenho Emocional, Desempenho Social, Fadiga e Dor.
O estudo NP28673 foi um estudo Fase I / II de braço único, internacional, multicêntrico, conduzido em pacientes que apresentam CPNPC ALK positivo que haviam progredido anteriormente em tratamento com crizotinibe. Além de crizotinibe, os pacientes poderiam ter recebido tratamento prévio com quimioterapia. No total, 138 pacientes foram incluídos na parte da fase II do estudo e receberam Alecensa® via oral na dose recomendada de 600 mg duas vezes ao dia.
O desfecho primário foi avaliar a eficácia de Alecensa® por TRO de acordo com a avaliação do CRI central que utilizou RECIST 1.1 na população geral (com e sem exposição prévia a tratamentos quimioterápicos citotóxicos). O desfecho coprimário foi avaliar a TRO de acordo com avaliação do CRI central que utilizou RECIST 1.1 em pacientes que apresentam exposição prévia a tratamentos quimioterápicos.
Os dados demográficos dos pacientes foram compatíveis com os de uma população com CPNPC ALK positivo. As características demográficas da população total do estudo foram: 67% brancos, 26% asiáticos, 56% do sexo feminino e a mediana de idade foi de 52 anos. A maioria dos pacientes não apresentava história de tabagismo (70%). O estado de desempenho ECOG no período basal era 0 ou 1 em 91% dos pacientes e 2 em 9 % dos pacientes. No momento da inclusão no estudo, 99% dos pacientes apresentavam doença em estágio IV, 61% apresentavam metástases cerebrais e, em 96% dos pacientes, os tumores foram classificados como adenocarcinoma. Entre os pacientes incluídos no estudo, 20% haviam progredido previamente durante tratamento apenas com crizotinibe e 80% haviam progredido previamente durante tratamento com crizotinibe e quimioterapia.
Como mostrado na plotagem em cascata na Figura 4, a maioria dos pacientes apresentou redução do volume tumoral de suas lesões-alvo definidas, conforme avaliação do CRI de acordo com o RECIST 1.1.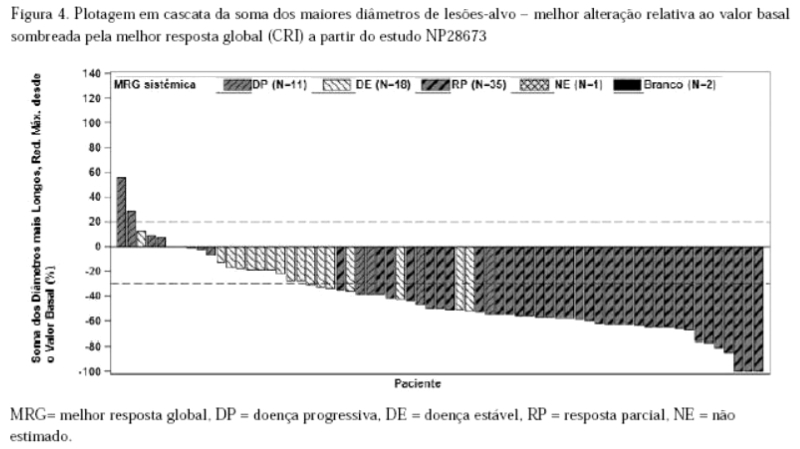
Um resumo das análises agrupadas dos desfechos do sistema nervoso central (SNC) baseadas em RECIST (CRI) realizadas em pacientes que apresentam lesões do SNC mensuráveis no período basal (N=50), incluídos na fase II do NP28761 e do NP28673, é apresentado na tabela a seguir.
Na fase II dos estudos NP28761 e do NP28673, em 136 pacientes incluídos, com lesões mensuráveis e/ou não mensuráveis do SNC no período basal, a taxa de resposta completa do SNC foi de 28,7%. Uma resposta parcial do SNC não pode ser estabelecida em lesões do SNC não mensuráveis de acordo com RECIST. A taxa de controle da doença no SNC foi de 86,0% [IC de 95% (79,1, 91, 4)].
Referências bibliográficas
1. Clinical Study Report NP28761: Primary Clinical Study Report (NP28761/AF002JG) - A Phase I/II Study of the ALK inhibitor alectinib in patients with ALK rearranged non-small cell lung cancer previously treated with crizotinib. Lancet Oncol. 2016 Feb;17(2):234-242. doi: 10.1016/S1470-2045(15)00488-X. Epub 2015 Dec 19.
2. Clinical Study Report NP28673: Primary Clinical Study Report (NP28673) - An open-label, non-randomized, multicenter Phase I/II trial of RO5424802 given orally to non-small cell lung cancer patients who have ALK mutation and who have experienced disease progression on crizotinib. J Clin Oncol. 2016 Mar 1;34(7):661-8. doi: 10.1200/JCO.2015.63.9443. Epub 2015 Nov 23.
3. Clinical Study Report Protocol BO28984 Report Number 1078196 - Global randomized Phase III open label clinical trial (BO28984) in ALK-positive NSCLC patients who were treatment naïve. N Engl J Med 2017; 377:829838August 31, 2017DOI: 10.1056/NEJMoa1704795.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades Farmacodinâmicas
Mecanismo de Ação
Alectinibe é um inibidor altamente seletivo e potente dos receptores tirosina quinase ALK e RET. Em estudos não clínicos, a inibição da atividade de tirosina quinase ALK levou ao bloqueio de vias de sinalização downstream que incluem STAT 3 e PI3K/AKT e induz morte celular tumoral (apoptose).
Alectinibe demonstrou atividade in vitro e in vivo contra formas mutantes de ALK, que incluíram mutações responsáveis pela resistência ao crizotinibe. O principal metabólito de alectinibe (M4) apresentou potência e atividade semelhantes in vitro.
Com base nos dados não clínicos, alectinibe não é um substrato da glicoproteína P (P-gp) ou da Proteína de Resistência ao Câncer de Mama (BCRP), que são transportadores de efluxo na barreira hematoencefálica, e é, portanto, capaz de se distribuir e ser retido dentro do sistema nervoso central. Alectinibe induziu regressão tumoral em modelos não clínicos de xenoenxerto em camundongo, o que inclui atividade antitumoral no cérebro e sobrevida prolongada em modelos de tumor intracraniano em animais.
Propriedades Farmacocinéticas
Os parâmetros farmacocinéticos de alectinibe e de seu principal metabólito ativo (M4) foram caracterizados em pacientes que apresentam CPNPC ALK positivo e em indivíduos saudáveis. As médias geométricas (coeficiente de variação %) de Cmax (concentração máxima), Cmin (concentração mínima) e ASC0-12hr (área sobre a curva de zero a doze horas) do alectinibe em estado de equilíbrio foram aproximadamente 665 ng/mL (44,3%), 572 ng/mL (47,8%) e 7430 ng*h/mL (45,7%), respectivamente. As médias geométricas de Cmax, Cmin e ASC0-12h em estado de equilíbrio do M4 foram aproximadamente 246 ng/mL (45,4 %), 222 ng/mL (46,6 %) e 2810 ng*h/mL (45,9 %), respectivamente.
Absorção
Depois da administração oral de 600 mg duas vezes ao dia com alimentos em pacientes que apresentam CPNPC ALK positivo, alectinibe foi rapidamente absorvido, atingiu Tmax depois de aproximadamente 4 (quatro) a 6 (seis) horas.
O estado de equilíbrio de alectinibe foi atingido em torno do Dia 7 com administração contínua da dosagem de 600 mg duas vezes ao dia e permaneceu estável desde então. A razão de acúmulo de médias geométricas estimada por análise da população farmacocinética (PK) para o esquema de 600 mg duas vezes ao dia é 5,6. A análise da população PK apoia a proporcionalidade da dose do alectinibe no intervalo de doses de 300 a 900 mg com alimentos.
A biodisponibilidade absoluta do alectinibe foi de 36,9% (IC de 90%: 33,9%, 40,3%) com alimentos em participantes de pesquisa saudáveis.
Após uma única administração oral de 600 mg com uma refeição rica em gorduras e rica em calorias, a exposição aumentou em 3 vezes em relação às condições de jejum (razão de médias geométricas [IC de 90%] de alectinibe combinado e M4: Cmax: 3,31 [2,79 - 3,93], ASCinf: 3,11 [2,73 - 3,55].
Distribuição
Alectinibe e seu principal metabólito M4 são altamente ligados às proteínas plasmáticas humanas ( > 99%), independentemente da concentração da droga. A média das taxas de concentração no sangue e no plasma humano de alectinibe e M4 in vitro é 2,64 e 2,50, respectivamente, em concentrações clinicamente relevantes.
A média geométrica do volume de distribuição em estado de equilíbrio (Vss) de alectinibe depois de administração IV foi de 475L, que indicou extensa distribuição para os tecidos.
Metabolismo
Estudos de metabolismo in vitro mostraram que a CYP3A4 é a principal isozima CYP mediando o alectinibe e seu principal metabólito, M4. Estima-se que contribua com 40% a 50% do metabolismo de alectinibe nos hepatócitos humanos. Resultados do estudo de balanço de massa humano demonstraram que alectinibe e M4 foram as principais moléculas circulantes no plasma, sendo que alectinibe e M4 juntos constituíram aproximadamente 76% da radioatividade total no plasma. A média geométrica da razão metabólito/droga original em estado de equilíbrio é de 0,399.
Eliminação
Após a administração de uma dose única de alectinibe marcado com 14C, administrado via oral em indivíduos saudáveis, a maior parte da radioatividade foi excretada nas fezes (recuperação média de 97,8%, intervalo de 95,6% a 100%) com excreção mínima na urina (recuperação média de 0,46%, intervalo de 0,30% a 0,60%). Nas fezes, 84% e 5,8% da dose foi excretada como alectinibe inalterado ou M4, respectivamente.
Com base em uma análise de população PK, a depuração (clearance) aparente (CL/F) de alectinibe foi de 81,9 L/hora. A média geométrica da meia-vida de eliminação individual estimada de alectinibe foi de 32,5 horas. Os valores correspondentes para M4 foram 217 L/hora e 30,7 horas, respectivamente.
Farmacocinética em populações especiais
População pediátrica
Não foram conduzidos estudos, a fim de investigar a farmacocinética de Alecensa® nesta população.
População geriátrica
A idade não tem um efeito sobre a exposição a Alecensa® (vide item "8. Posologia e Modo de Usar -Instruções Especiais de Posologia").
Insuficiência renal
Quantidades insignificantes de alectinibe e o metabólito ativo M4 são excretados inalterados na urina ( < 0,2% da dose). Os dados obtidos em pacientes que apresentam insuficiência renal leve e moderada mostram que a farmacocinética de alectinibe não é afetada de forma significativa na insuficiência renal. Nenhum estudo farmacocinético formal foi conduzido e não foi coletado nenhum dado de população PK em pacientes que apresentam insuficiência renal grave; no entanto, como a eliminação de alectinibe através do rim é insignificante, não é necessário nenhum ajuste de dose na insuficiência renal (vide item "8. Posologia e Modo de Usar -Instruções Especiais de Posologia").
Insuficiência hepática
Como a eliminação de alectinibe dá-se predominantemente por meio do metabolismo no fígado, o comprometimento hepático pode aumentar a concentração plasmática de alectinibe e/ou de seu principal metabólito ativo, o M4. Com base em uma análise farmacocinética populacional, as exposições a alectinibe e M4 foram semelhantes em pacientes que apresentam comprometimento hepático leve (bilirrubina total basal menor ou igual a LSN e AST basal maior do que LSN ou bilirrubina total basal maior do que 1,0 a 1,5 vez LSN e qualquer AST basal) e função hepática normal (bilirrubina total menor ou igual a LSN e AST menor ou igual a LSN).
Após a administração de uma dose oral única de 300 mg de alectinibe em indivíduos com insuficiência hepática moderada (Child-Pugh B), a exposição combinada de alectinibe e M4 foi modestamente aumentada em comparação com indivíduos saudáveis (relação da média geométrica [Intervalo de confiança de 90%] para moderado / saudável: Cmax: 1,16 [0,786 -1,72], AUCinf: 1,36 [0,947 -1,96]). A administração de uma dose oral única de 300 mg de alectinibe em indivíduos com insuficiência hepática grave (Child-Pugh C) resultou num aumento maior da exposição de alectinibe e M4 comparados com indivíduos saudáveis (relação da média geométrica [intervalo de confiança de 90%] para grave / saudável: Cmax: 0,981 [0,517 -1,86], AUCinf: 1,76 [0,984 -3,15]).
Nenhum ajuste de dose é requerido para Alecensa® em pacientes com insuficiência hepática subjacente leve ou moderada. Pacientes com insuficiência hepática subjacente grave devem receber uma dose de 450 mg administradas oralmente, duas vezes ao dia (dose diária total de 900 mg).
Segurança não clínica
Carcinogenicidade
Não foram realizados estudos de carcinogenicidade para estabelecer o potencial carcinogênico de Alecensa®.
Genotoxicidade
Alectinibe não foi mutagênico in vitro no ensaio de mutação reversa bacteriana (Ames), mas induziu discreto aumento em aberrações numéricas no ensaio citogenético in vitro que utilizou células de Pulmão de Hamster Chinês (CHL) com ativação metabólica e micronúcleos em um teste de micronúcleo em uma medula óssea de rato. O mecanismo da indução de micronúcleo foi de segregação anormal de cromossomos (aneugenicidade), e não um efeito clastogênico sobre os cromossomos.
Comprometimento da fertilidade
Não foram realizados estudos de fertilidade em animais para avaliar o efeito de Alecensa®. Não foram observados efeitos adversos sobre órgãos reprodutivos de machos e fêmeas em estudos de toxicologia geral conduzidos em ratos e macacos em exposições iguais ou maiores do que 2,6 e 0,5 vezes, respectivamente, da exposição humana com base na ASC na dose recomendada, de 600 mg duas vezes ao dia.
Toxicidade reprodutiva
Em estudos com animais, uma dose maternal de alectinibe equivalente a 2,7 vezes a dose humana recomendada, de 600 mg duas vezes ao dia (com base em ASC) causou perda embriofetal (abortamento) em coelhas prenhes. A mesma dose equivalente administrada a ratas prenhes resultou em fetos pequenos com ossificação retardada e discretas anormalidades dos órgãos.
Outros
Alectinibe absorve a luz UV entre 200 e 400 nm e demonstrou potencial fototóxico em um teste de fotossegurança in vitro em cultura de fibroblastos murinos depois de irradiação UVA.
Órgãos-alvo tanto em ratos quanto em macacos com exposições clinicamente relevantes nos estudos de toxicologia de dose repetida incluíam, entre outros, o sistema eritroide, trato gastrintestinal e sistema hepatobiliar.
Foi observada uma morfologia eritrocitária anormal em exposições iguais ou maiores que 10 a 60% da exposição humana por ASC na dose recomendada. A extensão de zona proliferativa em mucosa GI nas duas espécies foi observada em exposições iguais ou maiores do que 20% a 120% da exposição ASC humana na dose recomendada. O aumento da fosfatase alcalina (FA) hepática e da bilirrubina direta, bem como vacuolização/degeneração/necrose do epitélio do ducto biliar e aumento / necrose focal de hepatócitos foi observada em ratos e/ou macacos nas exposições iguais ou maiores do que 20% a 30% da exposição humana por ASC na dose recomendada.
Foi observado um efeito hipotensor discreto em macacos em exposições clinicamente relevantes próximas.
Alectinibe atravessou a barreira hematoencefálica em ratos e foi retido dentro do tecido cerebral, com uma razão de concentração entre SNC e plasma que variou de 0,9 a 1,5, 24 horas depois da administração.
4. CONTRAINDICAÇÕES
Alecensa® é contraindicado a pacientes com hipersensibilidade conhecida a alectinibe ou a quaisquer de seus excipientes.
5. ADVERTÊNCIAS E PRECAUÇÕES
Gerais
Doença pulmonar intersticial (DPI) / pneumonite
Casos de DPI/pneumonite foram reportados em estudos clínicos com Alecensa® (vide item "9. Reações Adversas"). Os pacientes devem ser monitorados em relação a sintomas pulmonares indicativos de pneumonite. Alecensa® deve ser imediatamente interrompido em pacientes diagnosticados com DPI/pneumonite e deve ser definitivamente descontinuado se não for identificada nenhuma outra potencial causa de DPI/pneumonite (vide item "8. Posologia e Modo de Usar").
Hepatotoxicidade
Elevações na alanina aminotransferase (ALT) e aspartato aminotransferase (AST) acima de cinco vezes o limite superior do normal (LSN), bem como elevações de bilirrubina maiores do que três vezes o LSN ocorreram em pacientes em estudos clínicos pivotais com Alecensa® (vide item "9. Reações Adversas"). A maior parte desses eventos ocorreu durante os primeiros três meses de tratamento. Nos estudos clínicos pivotais de Alecensa®, reportou-se que três pacientes que apresentam elevações graus 3-4 de AST/ALT apresentavam lesão hepática induzida por droga. Elevações concomitantes de ALT ou AST maiores ou iguais a três vezes o LSN e de bilirrubina total maiores ou iguais a duas vezes o LSN com fosfatase alcalina normal ocorreram em 1 paciente tratado em estudos clínicos com Alecensa®.
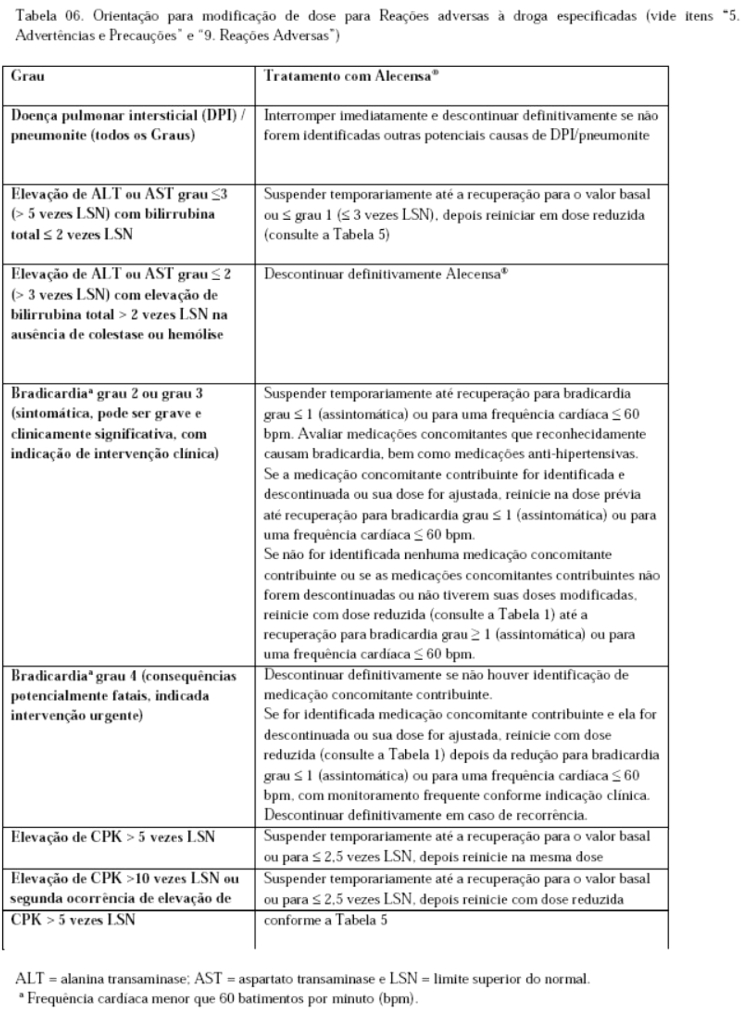
Função hepática, o que inclui ALT, AST e bilirrubina total deve ser monitorada no período basal e depois a cada 2 semanas durante os primeiros 3 meses de tratamento. Depois, o monitoramento deve ocorrer periodicamente, porque os eventos podem ocorrer depois dos três meses, com testes mais frequentes em pacientes que desenvolverem elevações de transaminase e bilirrubina. Com base na severidade da reação adversa à droga, suspenda Alecensa® e reinicie com uma dose reduzida ou descontinue definitivamente Alecensa®, conforme descrito na Tabela 06 (vide item "8. Posologia e Modo de Usar").
Mialgia grave e elevação de creatina fosfoquinase (CPK)
Foi reportada mialgia ou dor musculoesquelética em pacientes em estudos pivotais com Alecensa®, incluindo eventos Grau 3.
Ocorreram elevações de CPK em estudos pivotais com Alecensa®, incluindo eventos grau 3. O tempo mediano até a elevação Grau 3 de CPK foi de 14 dias nos estudos pivotais fase II (NP28761, NP28673). O tempo mediano até a elevação Grau 3 de CPK foi de 27,5 dias no estudo clínico pivotal fase III (BO28984) (vide item "9. Reações Adversas").
Oriente os pacientes a reportar qualquer dor, sensibilidade ou fraqueza muscular inexplicada. Avalie os níveis de CPK a cada duas semanas no primeiro mês de tratamento e de acordo com a indicação clínica em pacientes reportando sintomas. Com base na severidade da elevação de CPK, suspenda Alecensa® e depois reinicie ou reduza a dose (vide item "8. Posologia e Modo de Usar").
Bradicardia
Pode ocorrer bradicardia sintomática com Alecensa® (vide item "9. Reações Adversas"). A frequência cardíaca e a pressão arterial devem ser monitoradas conforme indicação clínica. A modificação da dose não é necessária em caso de bradicardia assintomática (vide item "8. Posologia e Modo de Usar"). Se os pacientes apresentarem bradicardia sintomática ou eventos potencialmente fatais, as medicações concomitantes que reconhecidamente provocam bradicardia, bem como as medicações anti-hipertensivas devem ser avaliadas e o tratamento com Alecensa® deve ser ajustado conforme descrito na Tabela 06 (vide itens "8. Posologia e Modo de Usar" e "6. Interações Medicamentosas -Substratos P-gp e BCRP").
Fotossensibilidade
Foi reportada fotossensibilidade à luz solar durante administração de Alecensa® (vide item "9.Reações Adversas"). Os pacientes devem ser orientados a evitar exposição prolongada ao sol enquanto estiverem tomando Alecensa® e durante pelo menos 7 dias depois da descontinuação do tratamento. Os pacientes também devem ser orientados a usar filtro solar de amplo espectro para ultravioleta A (UVA) / ultravioleta B (UVB) e protetor labial (FPS ≤ 50) para ajudar na proteção contra potencial queimadura solar.
Toxicidade embriofetal
Alecensa® pode provocar dano fetal quando administrado a mulheres grávidas. Quando administrado em ratas e coelhas prenhes, alectinibe causou toxicidade embriofetal. Pacientes do sexo feminino férteis ou mulheres férteis que sejam parceiras de pacientes do sexo masculino que recebem Alecensa® precisam utilizar métodos contraceptivos altamente efetivos durante o tratamento e durante pelo menos três meses depois da última dose de Alecensa® (vide item "5. Advertências e Precauções -Uso em Populações Especiais").
Abuso e dependência de drogas
Não se aplica.
Capacidade para dirigir e operar máquinas
Não foram realizados estudos sobre os efeitos na capacidade de dirigir veículos e de operar máquinas.
Intolerância à lactose
Este medicamento contém lactose. Pacientes com problemas hereditários raros de intolerância à galactose, deficiência congênita de lactase ou má absorção de glicose e galactose não devem tomar este medicamento.
Teor de sódio
A dose diária de 1200 mg de Alecensa® contém 48 mg de sódio, equivalente a 2,4% da dose máxima diária recomendada pela Organização Mundial da Saúde (OMS) de 2 g de sódio para um adulto.
Uso em populações especiais
Mulheres e homens com potencial reprodutivo
Contracepção
Pacientes do sexo feminino férteis ou mulheres férteis que sejam parceiras de pacientes do sexo masculino recebendo Alecensa® precisam usar métodos contraceptivos altamente efetivos durante o tratamento e pelo menos 3 meses depois da última dose de Alecensa®.
Gravidez
Categoria de risco na gravidez: D.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
Mulheres férteis precisam ser orientadas a evitar a gravidez enquanto estiverem recebendo Alecensa®. Não foram realizados estudos clínicos de Alecensa® em gestantes. Com base em seu mecanismo de ação, Alecensa® pode causar dano fetal quando administrado a uma gestante.
Pacientes do sexo feminino ou mulheres que sejam parceiras de pacientes do sexo masculino que recebem Alecensa® que ficarem grávidas enquanto recebem Alecensa® ou durante os 3 (três) meses depois da última dose de Alecensa® precisam entrar em contato com seu médico e devem ser orientadas sobre o potencial dano ao feto.
Dados em animais
Em estudos feitos em animais, alectinibe causou toxicidade embriofetal (vide item "3. Características Farmacológicas -Segurança Não Clínica").
Trabalho de parto e parto
A segurança de Alecensa® durante o trabalho de parto e o parto não foi estabelecido.
Lactação
Não se sabe se Alecensa® é excretado no leite materno. Não foram conduzidos estudos para avaliar o impacto de Alecensa® sobre a produção do leite ou sua presença no leite materno. Como muitas drogas são excretadas no leite materno e por causa do potencial dano à criança, as mães devem ser orientadas a não realizar o aleitamento materno enquanto estiverem recebendo Alecensa®.
Uso pediátrico
A segurança e a eficácia em pacientes pediátricos com idade abaixo de 18 anos não foram estabelecidas.
Uso geriátrico
Vide itens "8. Posologia e Modo de Usar -Instruções Especiais de Posologia" e "3. Características Farmacológicas -Farmacocinética em Populações Especiais".
Insuficiência renal
Vide itens "8. Posologia e Modo de Usar -Instruções Especiais de Posologia" e "3. Características Farmacológicas -Farmacocinética em Populações Especiais".
Comprometimento hepático
Vide itens "8. Posologia e Modo de Usar -Instruções Especiais de Posologia" e "3. Características Farmacológicas -Farmacocinética em Populações Especiais".
6. INTERAÇÕESMEDICAMENTOSAS
Efeitos de alectinibe sobre outras drogas
Substratos CYP
Estudos in vitro indicam que nem alectinibe nem seu principal metabólito ativo (M4) inibe CYP1A2, CYP2B6, CYP2C9, CYP2C19 ou CYP2D6 em concentrações clinicamente relevantes. Alectinibe e M4 apresentam uma fraca inibição dependente do tempo de CYP3A4. In vitro, alectinibe apresenta um fraco potencial indutor de CYP3A4 e CYP2B6 em concentrações clínicas.
Resultados de um estudo clínico de interação medicamentosa em pacientes que apresentam CPNPC positivo para ALK demonstraram que múltiplas doses de alectinibe não tiveram nenhuma influência sobre a exposição de midazolam, um substrato CYP3A sensível. Portanto, não é necessário nenhum ajuste da dose para substratos CYP3A administrados concomitantemente.
Embora estudos in vitro indiquem que alectinibe é um inibidor de CYP2C8, o modelo farmacocinético de base fisiológica (PBPK) corrobora que, em concentrações clinicamente relevantes, alectinibe não tem o potencial para aumentar as concentrações plasmáticas de substratos de CYP2C8 administrados concomitantemente.
Substratos P-gp e BCRP
In vitro, alectinibe e M4 são inibidores dos transportadores de efluxo da P-glicoproteína (P-gp) e da Proteína de Resistência do Câncer de Mama (BCRP). Portanto, alectinibe pode ter o potencial para aumentar as concentrações plasmáticas de substratos administrados concomitantemente de transportadores P-gp ou BCRP (não é esperado que o aumento da exposição seja maior do que duas vezes). Quando alectinibe é administrado concomitantemente com substratos P-gp ou BCRP com índice terapêutico estreito (por exemplo: digoxina, dabigatrana e metotrexato), recomenda-se o monitoramento apropriado.
Efeitos de outras drogas sobre alectinibe
Com base em dados in vitro, CYP3A4 é a enzima primária na mediação do metabolismo de alectinibe e de seu principal metabólito ativo, M4, e a CYP3A contribui para 40% - 50% do metabolismo hepático total. Demonstrou-se que M4 tem potência e atividade in vitro semelhantes às do alectinibe contra o ALK.
Indutores CYP3A
A administração concomitante de múltiplas doses orais de 600 mg de rifampicina uma vez ao dia, um potente indutor de CYP3A, com uma única dose oral de 600 mg de alectinibe apresentou efeito mínimo sobre a exposição combinada de alectinibe e M4 (razão de médias geométricas com/sem rifampicina [intervalo de confiança 90%]: Cmax: 0,96 [0,88 - 1,05], ASCinf: 0,82 [0,74 - 0,90]). Portanto, não são necessários ajustes da dose quando Alecensa® é administrado concomitantemente com indutores CYP3A.
Inibidores de CYP3A
A administração concomitante de múltiplas doses orais de 400 mg de posaconazol duas vezes ao dia, um potente inibidor de CYP3A, com uma única dose oral de 300 mg de alectinibe teve um efeito mínimo sobre a exposição combinada de alectinibe e M4 (razão de médias geométricas com/sem posaconazol [intervalo de confiança 90%]: Cmax: 0,93 [0,81 - 1,08], ASCinf: 1,36 [1,24 - 1,49]). Portanto, não é necessário nenhum ajuste de dose quando Alecensa® é administrado concomitantemente com inibidores da CYP3A.
Medicamentos que aumentam o pH gástrico
Embora a solubilidade aquosa de alectinibe in vitro seja dependente do pH, um estudo clínico de interação medicamentosa realizado com 40 mg de esomeprazol, um inibidor de bomba de prótons, uma vez ao dia, não demonstrou efeito clinicamente relevante sobre a exposição combinada de alectinibe e M4. Portanto, não é necessário nenhum ajuste de dose quando Alecensa® é administrado concomitantemente com inibidores de bomba de prótons ou outras drogas que elevem o pH gástrico (por exemplo: antagonistas de receptor H2 ou antiácidos).
Efeito dos transportadores sobre a distribuição de alectinibe
Com base em dados in vitro, alectinibe não é um substrato de P-gp. Alectinibe e M4 não são substratos de BCRP ou do polipeptídeo transportador de ânion orgânico (OATP) 1B1/B3. Por outro lado, M4 é um substrato de P-gp. Alectinibe inibe P-gp e, portanto, não se espera que a medicação concomitante com inibidores P-gp tenha um efeito relevante sobre a exposição a M4.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Alecensa® cápsulas duras deve ser conservado em temperatura ambiente (entre 15 e 30°C) e deve ser mantido dentro do cartucho para proteger da luz e umidade.
Prazo de validade
Este medicamento possui prazo de validade de 36 meses a partir da data de fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Antes de usar, observe o aspecto do medicamento.
As cápsulas duras de Alecensa® apresentam coloração branca com as inscrições "ALE" e "150 mg" em preto.
Descarte de medicamentos não utilizados e/ou com data de validade vencida
O descarte de medicamentos no meio ambiente deve ser minimizado. Os medicamentos não devem ser descartados no esgoto, e o descarte em lixo doméstico deve ser evitado. Utilize o sistema de coleta local estabelecido, se disponível.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
Gerais
É necessário um ensaio ALK validado para seleção de pacientes que apresentam CPNPC ALK positivo. A situação de CPNPC ALK positivo deve ser estabelecida antes do início da terapia de primeira linha com Alecensa®.
As cápsulas duras de Alecensa® devem ser ingeridas com alimento, devem ser engolidas inteiras e não devem ser abertas ou dissolvidas.
A dose recomendada de Alecensa® é de 600 mg (quatro cápsulas de 150 mg) administradas por via oral duas vezes ao dia (dose diária total de 1200 mg) (vide item "3. Características Farmacológicas").
Pacientes com insuficiência hepática grave subjacente devem receber a dose de 450 mg, administrada oralmente, duas vezes ao dia (dose diária total de 900 mg) (vide itens "8. Posologia e modo de usar -Instruções especiais de posologia" e "3. Características Farmacológicas -Farmacocinética em populações especiais").
Este medicamento não deve ser partido, aberto ou mastigado.
Duração do tratamento
Recomenda-se que os pacientes sejam tratados com Alecensa® até a progressão da doença ou o aparecimento de toxicidade não manejável (vide item "2.Resultados de Eficácia").
Conduta em caso de esquecimento
Se uma dose programada de Alecensa® for esquecida, os pacientes podem tomar aquela dose, desde que a dose seguinte seja dentro das próximas 6 (seis) horas. Se ocorrerem vômitos depois de tomar uma dose de Alecensa®, os pacientes devem tomar a dose seguinte no horário agendado.
Modificações da dose
O tratamento dos eventos adversos pode exigir interrupção temporária, redução da dose ou descontinuação do tratamento com Alecensa®. A dose de Alecensa® deve ser reduzida em passos de 150 mg duas vezes ao dia, com base na tolerabilidade. O tratamento com Alecensa® deve ser definitivamente descontinuado se os pacientes não conseguirem tolerar a dose de 300 mg duas vezes ao dia.
Instruções Especiais de Posologia
Uso pediátrico
A segurança e a eficácia de Alecensa® em crianças e adolescentes ( < 18 anos) não foram estudadas.
Uso geriátrico
Não é necessário ajuste de dose de Alecensa® em pacientes ≤ 65 anos de idade.
Insuficiência renal
Não é necessário ajuste de dose em pacientes que apresentam insuficiência renal leve ou moderada. Alecensa® não foi estudado em pacientes que apresentam insuficiência renal grave; no entanto, como a eliminação renal de alectinibe é desprezível, nenhum ajuste de dose é necessário em pacientes que apresentam insuficiência renal grave (vide itens "5. Advertências e Precauções -Uso em Populações Especiais" e "3. Características Farmacológicas -Farmacocinética em Populações Especiais").
Insuficiência hepática
Não é necessário ajuste da dose em pacientes que apresentam insuficiência hepática subjacente leve ou moderada. Pacientes com insuficiência hepática subjacente grave devem receber uma dose de 450 mg administrada oralmente, duas vezes ao dia (dose diária total de 900 mg) (vide item "3. Características Farmacológicas -Farmacocinética em Populações Especiais").
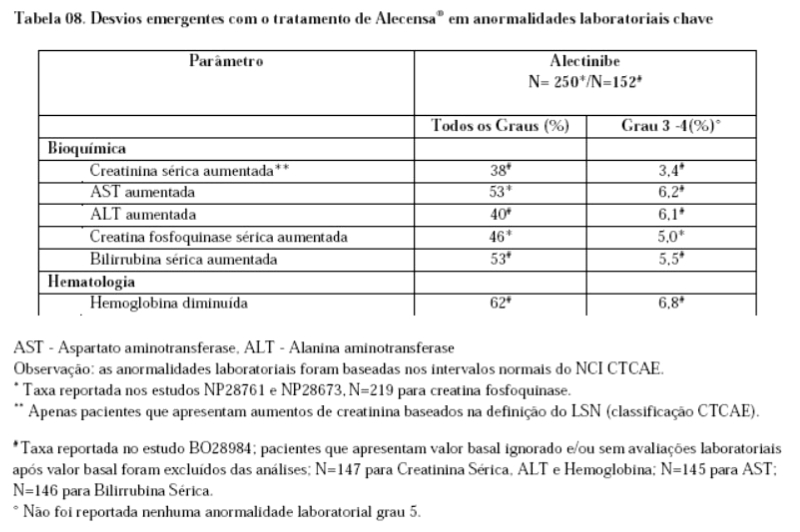
9. REAÇÕES ADVERSAS
Estudos clínicos
Para o programa de desenvolvimento clínico de Alecensa®, estima-se que um total de 928 pacientes tenha recebido Alecensa® e 203 pacientes tenham recebido Alecensa® cego. A segurança de Alecensa® foi avaliada em 253 pacientes nos estudos clínicos pivotais fase II (NP28761, NP28673) que apresentam câncer de pulmão de não pequenas células (CPNPC) positivo para ALK tratados com a dose recomendada de 600 mg duas vezes ao dia. A duração mediana de exposição a Alecensa® foi de 11 meses (intervalo de 0 a 35 meses). A segurança de Alecensa® foi também avaliada em 152 pacientes que apresentam CPNPC positivo para ALK tratados com uma dose de 600 mg duas vezes ao dia no estudo clínico fase III BO28984. A duração mediana de exposição a Alecensa® foi de 17,9 meses.
As reações adversas à droga mais comuns (≤ 20%) foram obstipação (36%), edema (34%, que inclui edema periférico generalizado, de pálpebras e periorbitário), mialgia (31%, que inclui mialgia e dor musculoesquelética), náuseas (22%), bilirrubina aumentada (21%, que inclui bilirrubina sérica aumentada, hiperbilirrubinemia e bilirrubina conjugada aumentada), anemia (20%, que inclui anemia e hemoglobina diminuída) e erupção (20%, que inclui erupção, erupção maculopapular, dermatite acneiforme, eritema, erupção generalizada, erupção papular, erupção pruriginosa e erupção macular).
A Tabela 7 apresenta as reações adversas à droga (RADs) que ocorreram em pacientes que receberam Alecensa® em estudos clínicos (NP28761, NP28673) e no estudo clínico fase III BO28984. Reações adversas à droga a partir de estudos clínicos são listadas por classe de grupo sistêmico do MedDRA. A categoria de frequência correspondent