ZOTEON PÓ
VIATRIS
tobramicina
Antibiótico. Tratamento da fibrose cística.
Apresentações.
Cápsulas com pó para inalação
ZoteonTM Pó 28 mg - embalagens contendo 224 cápsulas acompanhadas de 5 inaladores.
VIA INALATÓRIA
USO ADULTO E PEDIÁTRICO ACIMA DE 6 ANOS
Composição.
Cada cápsula de ZoteonTM Pó contém 28 mg de tobramicina.
Excipientes: ácido sulfúrico, levoalfafosfatidilcolina distearoila e cloreto de cálcio.
Informações técnicas.
1. INDICAÇÕES
ZoteonTM Pó é indicado para o tratamento da infecção pulmonar por Pseudomonas aeruginosa na fibrose cística (FC) em pacientes com 6 anos de idade ou mais.
A segurança e a eficácia não foram demonstradas em pacientes com idade inferior a 6 anos, em pacientes com VEF1 < 25% ou > 80% do previsto, ou em pacientes infectados por Burkholderia cepacia.
2. RESULTADOS DE EFICÁCIA
A Fase III do programa de desenvolvimento clínico consistiu em dois estudos placebo-controlados, (EVOLVE [20] e EDIT [43]) e um estudo aberto (EAGER [19]) nos quais foram randomizados e dosados 157 e 517 pacientes, respectivamente, com diagnóstico clínico de FC, confirmado pelo teste quantitativo de cloro no suor por iontoforese com pilocarpina, ou pelas mutações bem caracterizadas de cada gene CFTR causadas pela doença, ou pela anormalidade do potencial transepitelial nasal característico da FC. [1]
Nos estudos placebo-controlados, todos os pacientes tinham idade entre 6 e 21 anos e apresentaram na seleção um VEF1(volume expiratório forçado no 1° segundo) ≥ 25% e ≤ 80% dos valores normais para a idade, sexo e altura com base nos critérios de Knudson. Além disso, todos os pacientes eram infectados por P. aeruginosa, como demonstrado por uma cultura positiva do escarro ou garganta (ou lavado broncoalveolar), no prazo de 6 meses antes da seleção, e também em uma cultura de escarro colhida na visita de seleção. Entre os 76 pacientes tratados com ZoteonTM Pó, 37% eram homens e 63% eram mulheres. Trinta e seis pacientes tinham entre 6 e 12 anos de idade e 40 pacientes tinham entre 13 e 21 anos. Os pacientes apresentaram uma média do valor basal de VEF1 de 56% do valor normal previsto.
Em ambos os estudos, > 90% dos pacientes receberam terapias concomitantes com indicação para fibrose cística. Os outros medicamentos antibacterianos usados com mais frequência (qualquer via de
administração) foram azitromicina, ciprofloxacina e ceftazidima. Consistente com a população de pacientes com fibrose cística, os medicamentos concomitantes mais usados incluíram preparações orais de enzimas pancreáticas, mucolíticos (especialmente alfadornase) e agonistas seletivos dos receptores b2-adrenérgicos.
Os seguintes resultados clínicos foram demonstrados:
- Estudo EVOLVE
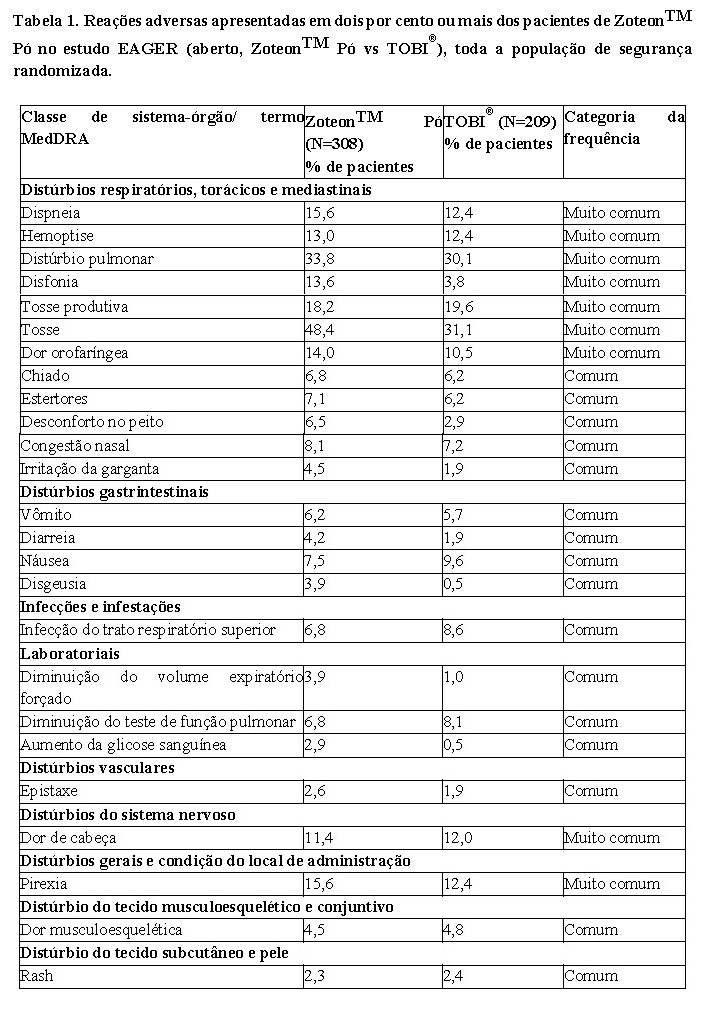
Em um estudo randomizado, duplo-cego, multicêntrico, controlado com placebo (estudo EVOLVE), 112 mg vde ZoteonTM Pó foram administrados duas vezes ao dia (no mesmo horário todas as manhãs e noites), durante três ciclos de 28 dias em tratamento e 28 dias sem tratamento (um período total de tratamento de 24 semanas). Um total de 95 pacientes foram randomizados para o estudo e receberam ZoteonTM Pó (n=46) ou placebo (n=49) no Ciclo 1. Todos os pacientes tinham idade inferior a 22 anos (idade média 13,3 anos) e não tinham recebido antibióticos inalatórios antipseudomonas no prazo de 4 meses antes da seleção, 55,8% eram do sexo feminino e 84,2% eram brancos. Os pacientes que foram randomizados para o grupo de tratamento com placebo, receberam placebo durante o primeiro ciclo de tratamento e ZoteonTM Pó nos dois ciclos subsequentes. [2] ZoteonTM Pó melhorou significativamente a função pulmonar em comparação ao placebo, como demonstrado pelo aumento relativo na porcentagem do VEF1 previsto após 28 dias de tratamento (Figura 1). As melhoras na função pulmonar alcançadas durante o primeiro ciclo de tratamento foram mantidas durante o ciclo subsequente de tratamento com ZoteonTM Pó. Quando os pacientes do grupo de tratamento com placebo foram trocados de placebo para ZoteonTM Pó, no início do segundo ciclo de tratamento, a variação em porcentagem do VEF1 previsto do valor basal foi a mesma que a observada durante o primeiro ciclo de tratamento no grupo tratado com ZoteonTM Pó e as melhoras também foram mantidas ao longo do tempo durante o terceiro ciclo de tratamento. [2,3].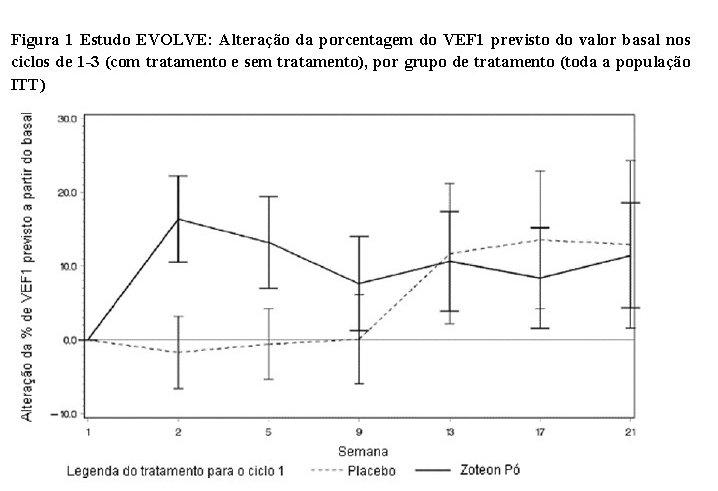
Estudo EDIT:
Este foi um estudo randomizado, duplo-cego, placebo-controlado, com desenho semelhante ao estudo EVOLVE. Os pacientes elegíveis foram randomizados 1:1 para receber ZoteonTM Pó (4 vezes cápsulas de 28 mg duas vezes ao dia) ou placebo por um ciclo (28 dias em tratamento e 28 dias sem tratamento) [43].
Um total de 62 pacientes foram randomizados para este estudo e alocados para o grupo de ZoteonTM Pó (n=32) ou placebo (n=30). Todos os pacientes tinham menos de 22 anos de idade (idade média de 12,9 anos) e não havia recebido antibióticos antipseudomonas inalatórios nos 4 meses anteriores à triagem; 64,5% eram do sexo feminino e 98,4% eram caucasianos [43].
Neste estudo, os resultados não foram estatisticamente significativos para o desfecho da função pulmonar primária ao ajustar para as covariáveis de idade ( < 13 anos, ≥13 anos) e VEF1 % previsto na triagem ( < 50%, ≥50%) e imputação de dados ausentes. Foi avaliada melhoria na função pulmonar para ZoteonTM Pó em comparação com placebo usando a alteração relativa na % FEV1 prevista desde a linha de base até o final da dosagem do Ciclo 1.
O tratamento com ZoteonTM Pó (8,19%) em comparação com o placebo (2,27%) não conseguiu alcançar significância estatística na mudança relativa no VEF1% previsto (diferença média LS = 5,91%; IC 95%: -2,54, 14,37; p=0,167). Análises de mudanças absolutas em % de VEF1 previsto mostrou médias LS de 4,86% para ZoteonTM Pó e 0,48% para placebo com uma diferença de 4,38% (95% CI:-0,17, 8,94) [43].
- Estudo EAGER
Em um segundo estudo aberto, multicêntrico (estudo EAGER), os pacientes receberam tratamento com ZoteonTM Pó (112 mg) ou TOBI® (300 mg) administrados duas vezes ao dia (no mesmo horário todas as manhãs e noites), durante três ciclos de 28 dias de tratamento e 28 dias sem tratamento (um período total de tratamento de 24 semanas).
Um total de 517 pacientes foram randomizados neste estudo e receberam ZoteonTM Pó (n=308) ou TOBI® (n=209). Os pacientes tinham predominantemente 20 anos ou mais (incluindo 4 pacientes com idade superior a 60 anos), sem uso de antibiótico antipseudomonas inalatório no prazo de 28 dias antes do estudo da administração do medicamento, 90% eram brancos e 55% eram do sexo masculino. [1,4]
O tratamento com ambos os medicamentos, TOBI® e ZoteonTM Pó, resultou em um aumento relativo do valor basal do VEF1 de 5,8% e 4,7% do valor previsto, respectivamente, no Dia 28 do terceiro ciclo de tratamento (Figura 2). A melhora no percentual do VEF1 previsto foi numericamente maior no grupo tratado com ZoteonTM Pó e não foi estatisticamente inferior ao TOBI®. Embora a magnitude da melhora da função pulmonar tenha sido menor no presente estudo, isso pode ser explicado pela exposição anterior da população de pacientes ao tratamento com tobramicina inalatória. [3,4]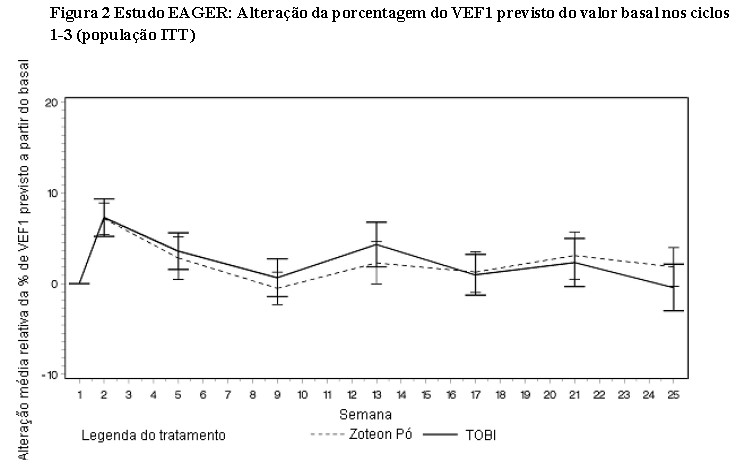
No que diz respeito à supressão da infecção por Pseudomonas aeruginosa (P. aeruginosa), ambos os estudos clínicos demonstraram que o ZoteonTM Pó diminui a densidade da P. aeruginosa no escarro (veja as duas figuras a seguir). O tratamento com ZoteonTM Pó durante 28 dias resultou em uma redução estatisticamente significativa da P. aeruginosa na densidade do escarro (log10 UFC) em comparação ao placebo (diferença média LS = 2,70, 95% IC: -3,60, -1,79, p < 0,001), com a maior diferença observada no 28° dia de tratamento. Após os pacientes terem sido transferidos do tratamento com placebo para o tratamento com ZoteonTM Pó, os resultados foram geralmente semelhantes para os dois grupos de tratamento com uma tendência de recuperação da densidade de P. aeruginosa após 28 dias sem tratamento, que foi revertida após mais 28 dias em tratamento. [3]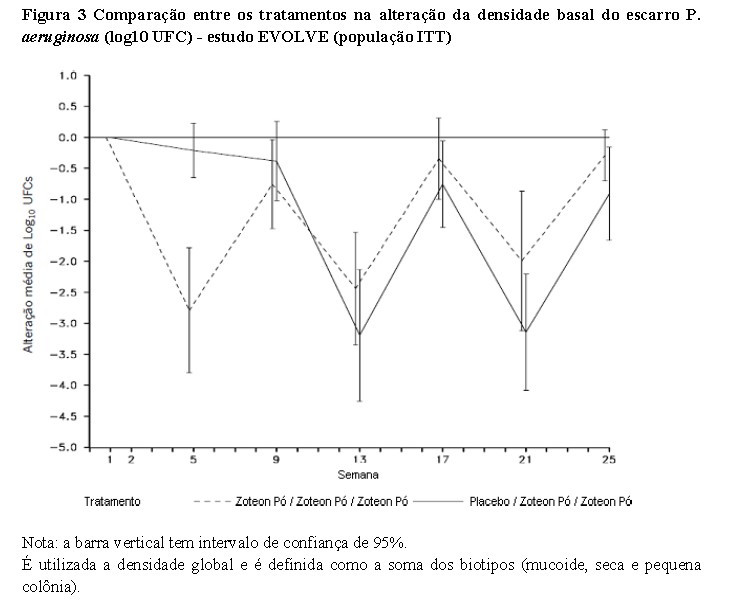
No estudo EAGER, houve uma grande diminuição na variação média do valor basal do log10 UFC no grupo tratado com ZoteonTM Pó comparado ao grupo de tratamento com TOBI®, especialmente durante o terceiro ciclo de tratamento (uma variação média de log10 UFC -1,61 no grupo tratado com ZoteonTM Pó comparado com log10 UFC -0,77 no grupo de tratamento TOBI® (Figura 4)). Assim como no estudo anterior, houve uma recuperação parcial da densidade de P. aeruginosa ao final dos 28 dias da fase sem tratamento em ambos os grupos de tratamento, mas isso foi revertido durante a fase de tratamento de cada ciclo de tratamento. [3,4]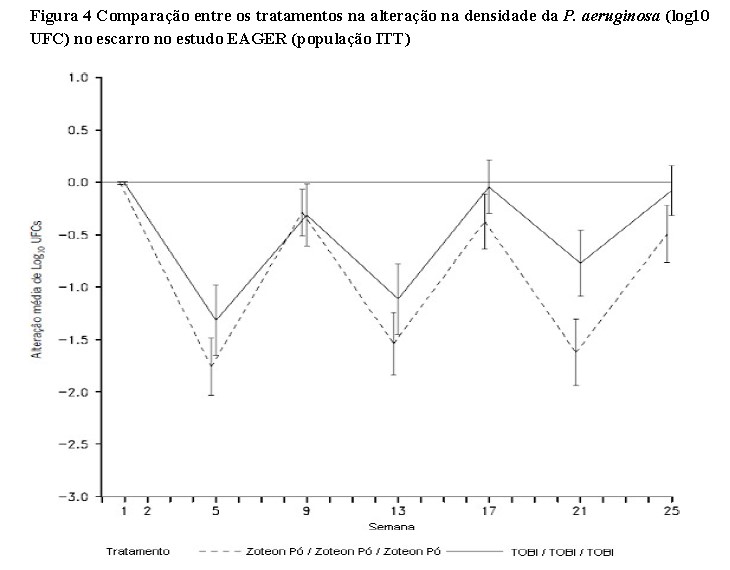
Em ambos os estudos clínicos, mudanças negativas desde o início até o final do período de tratamento foram observadas na CIM (concentração inibitória mínima) máxima de tobramicina para cada biótipo de P. aeruginosa. Em geral, um percentual maior de pacientes tratados com ZoteonTM Pó apresentou aumento na CIM da tobramicina em comparação com diminuição. No entanto, em comparação ao placebo, aproximadamente metade da proporção de pacientes tratados com ZoteonTM Pó tiveram uma diminuição ≥ 2 vezes na CIM da tobramicina na Semana 25 (40,5% e 20,0% em pacientes com o placebo e no grupo tratado com ZoteonTM Pó, respectivamente). [3]
O percentual de pacientes em uso de antibióticos antipseudomonas no Ciclo 1 foi maior no grupo de tratamento com placebo em comparação ao grupo de tratamento com ZoteonTM Pó (20,4% e 13,0%, respectivamente), juntamente com uma maior duração da utilização no grupo de tratamento com placebo (18,2 dias, em comparação com 13,3 dias no grupo de tratamento com ZoteonTM Pó). No ciclo 1, nenhum dos pacientes no grupo de tratamento com ZoteonTM Pó precisou de internações relacionada a problemas respiratórios, em comparação com 6 pacientes (12,2%) no grupo placebo, com duração média de 12,3 dias. [3] No estudo EAGER, mais da metade dos pacientes em ambos os grupos de tratamento ZoteonTM Pó e TOBI®, precisaram de novos antibióticos antipseudomonas ao longo dos três ciclos de tratamento (64,9% e 54,5% respectivamente) e as durações de utilização foram semelhantes em ambos os grupos de tratamento, ZoteonTM Pó e TOBI® (30,9 dias e 33,4 dias, respectivamente). Proporções semelhantes de pacientes em ambos os grupos de tratamento ZoteonTM Pó e TOBI® necessitaram de internação para os eventos respiratórios (24,4% e 22,0% respectivamente) e as durações das internações, também foram semelhantes (15,6 dias e 15,3 dias, respectivamente). [3] Uma das diferenças mais relevantes entre o tratamento com ZoteonTM Pó e TOBI® é o tempo necessário para administrar uma dose. O tempo médio para administrar uma dose nebulizada de TOBI® foi de aproximadamente 20 minutos, comparado com 6 minutos para administrar uma dose de ZoteonTM Pó através do inalador. Desse tempo excluiu-se todo o tempo despendido para preparar e depois guardar o nebulizador usado com TOBI®. [3]
No estudo EAGER, a satisfação dos pacientes ao tratamento foi avaliada através do "Treatment Satisfaction Questionnaire for Medication" (TSQM) modificado. Pacientes relataram consistentemente níveis mais altos de satisfação com o tratamento com ZoteonTM Pó em comparação com TOBI®, particularmente para as avaliações de eficácia, conveniência e satisfação geral. [3]
Medicação concomitante
Em ambos os estudos clínicos mais de 90% dos pacientes receberam terapias concomitantes para indicações relacionadas à FC. Como previsto pelas características basais da doença e em linha com a população de pacientes com FC recrutados neste estudo, os medicamentos mais concomitantemente usados incluíram as preparações de enzimas, especialmente a dornase alfa mucolítica, e os agonistas seletivos dos receptores b2 adrenérgicos. Além disso, a maioria dos pacientes tinha um histórico de uso de antibióticos antipseudomonas e macrolídeos. Os compostos mais usados (qualquer via de administração) foram tobramicina (apenas estudo EAGER), azitromicina, ciprofloxacina e ceftazidima. [1]
Referências bibliográficas
1.[Summary of Clinical Safety (2009)] TBM100C (tobramycin inhalation powder) - 2.7.4 Summary of Clinical Safety in Cystic Fibrosis. Novartis. Basel, Switzerland. [5];
2.[Study TBM100C2301 (2008)] A randomized, double-blind, placebo-controlled, multicenter, phase 3 trial to assess the efficacy and safety of tobramycin inhalation powder (TIP) in cystic fibrosis (CF) subjects. Novartis Pharma AG. Basel, Switzerland. [20];
3.[Summary of Clinical Efficacy (2009)] TBM100 (tobramycin inhalation powder) - 2.7.3 Summary of Clinical Efficacy in Cystic Fibrosis. Novartis. Basel, Switzerland. [4]
4.[Study TBM100C2302 (2009)] A randomized, open-label, multicenter, phase 3 trial to assess the safety of tobramycin inhalation powder compared to TOBI® in cystic fibrosis subjects. Novartis Pharma AG. Basel, Switzerland. [19]
5. [Study TBM100C2303 (2011)] A randomized, double-blind, placebo-controlled, multicenter, phase III study in cystic fibrosis (CF) subjections to assess the efficacy, safety and pharmacokinetics of tobramycin inhalation powder from a modified manufacturing process (TIPnew). Novartis Pharma AG. Basel, Switzerland. [43]
3. CARACTERÍSTICAS FARMACOLÓGICAS
Grupo farmacoterapêutico: Antibacterianos Aminoglicosídeos
Código ATC: J01GB01
Mecanismo de ação
A tobramicina é um antibiótico aminoglicosídeo produzido por Streptomyces tenebrarius. Ele age principalmente interrompendo a síntese de proteínas, levando à alterações da permeabilidade da membrana celular, a interrupção progressiva do envelope celular e eventual morte celular. É bactericida em concentrações iguais ou ligeiramente superiores às concentrações inibitórias.
Propriedades farmacodinâmicas
- Microbiologia
Nos estudos clínicos, alguns pacientes em terapia com ZoteonTM Pó mostraram um aumento na concentração inibitória mínima de aminoglicosídeo para isolados de P. aeruginosa testados.
No estudo EAGER, a distribuição da concentração inibitória mínima (CIM) da tobramicina para isolados de Pseudomonas aeruginosa foi caracterizada pelo biotipo: mucoide, seco, variante de pequena colônia e geral. Avaliações de escarro neste estudo mostraram que no início do estudo, 91% dos pacientes tratados com ZoteonTM Pó tinham colônias de P. aeruginosa com CIM de pelo menos 11 vezes menor do que a concentração média de escarro observada dentro de 30 minutos após a administração. No final do 28° dia do terceiro ciclo de dose, 86% dos pacientes tratados com ZoteonTM Pó apresentaram P. aeruginosa com uma CIM pelo menos 21 vezes menor e 89% dos pacientes tratados com ZoteonTM Pó apresentaram P. aeruginosa com uma CIM pelo menos 10 vezes menor que a concentração média observada no escarro 30 minutos após a administração.
O tratamento com ZoteonTM Pó durante 25 semanas em dois estudos clínicos não afetou a susceptibilidade da maioria das colônias de P. aeruginosa testadas. A maioria das amostras apresentou uma CIM da tobramicina entre 0,5 mcg/mL e 8 mcg/mL. Não houve mudança clinicamente relevante na distribuição da CIM para a soma de todos os biotipos de P. aeruginosa com o tratamento com ZoteonTM Pó no estudo EAGER. A distribuição da CIM para colônias mucoides manteve-se praticamente idêntica no início e após 25 semanas de tratamento e para o biótipo seco, houve uma pequena mudança que se manteve abaixo do corte da resistência à exposição sistêmica convencional de 8 mcg/mL. A CIM máxima de todos os biotipos em ambos os grupos de tratamento, ZoteonTM Pó e TOBI® permaneceu relativamente consistente ao longo do estudo, para cada biotipo.
O significado clínico das alterações da CIM para P. aeruginosa não foi claramente estabelecido no tratamento de pacientes com FC. Estudos clínicos demonstraram que um relatório microbiológico indicando resistência ao fármaco in vitro não exclui necessariamente um benefício clínico para o paciente. No estudo EAGER o subgrupo de pacientes com valores de CIM > 8 mcg/mL no início do tratamento teve uma melhora no percentual previsto do VEF1 medida após 3 ciclos de tratamento, quando tratados com ZoteonTM Pó.
- Teste de susceptibilidade
Limites de susceptibilidade para a administração parenteral de tobramicina não se aplicam à administração da tobramicina inalada.
Os critérios interpretativos para produtos antibacterianos inalatórios não estão definidos. Os métodos de teste de suscetibilidade antimicrobiana in vitro utilizados para determinar a suscetibilidade para a terapia parenteral com tobramicina podem ser utilizados para monitorar a suscetibilidade de P. aeruginosa isolada de pacientes com fibrose cística. A relação entre os resultados do teste de suscetibilidade in vitro e o resultado clínico da terapia com ZoteonTM Pó é incerta. Uma única amostra de escarro de um paciente com fibrose cística pode conter diversos morfotipos de P. aeruginosa e cada morfotipo pode exigir uma concentração diferente de tobramicina para a inibição de seu crescimento in vitro. Os pacientes devem ser monitorados quanto às alterações na suscetibilidade da tobramicina.
- Desenvolvimento de resistência
Em estudos clínicos, algumas elevações desde o início até o final do período de tratamento foram observadas na CIM (concentração inibitória mínima) de tobramicina para morfotipos de P. aeruginosa. No geral, uma porcentagem mais elevada de pacientes tratados com ZoteonTM Pó apresentou elevações na CIM de tobramicina em comparação ao placebo ou aos pacientes tratados com TOBI® solução inalatória.
- Resistência cruzada
Certa resistência emergente à aztreonam, ceftazidima, ciprofloxacino, imipenem ou meropenem foi observada nos estudos clínicos com ZoteonTM Pó. Como outros antibióticos antipseudomonas foram utilizados concomitantemente em muitos pacientes nos estudos clínicos, a associação com ZoteonTM Pó é incerta.
Farmacocinética
- Absorção
A exposição sistêmica à tobramicina após a inalação de ZoteonTM Pó é esperada como resultado da absorção pulmonar da fração de dose que chega aos pulmões uma vez que a tobramicina não é absorvida em uma extensão considerável quando administrada por via oral.
Concentrações séricas:
após inalação de uma dose única de 112 mg de ZoteonTM Pó (4 cápsulas x 28 mg) em pacientes com fibrose cística, a concentração sérica máxima (Cmáx) da tobramicina foi de 1,02 ± 0,53 mcg/mL (média ± DP) e o tempo médio para alcançar a concentração máxima (tmáx) foi de uma hora. Em comparação, após a inalação de uma única dose de 300 mg de TOBI®, a Cmáx foi de 1,04 ± 0,58 mcg/mL e o tmáx médio foi de uma hora. O grau de exposição sistêmica (AUC) foi também semelhante para a dose de 112 mg de ZoteonTM Pó e para dose de 300 mg de TOBI®. Ao final de 4 semanas de ciclo de tratamento com ZoteonTM Pó (112 mg duas vezes ao dia), a concentração sérica máxima de tobramicina 1 hora após a dose foi de 1,99 ± 0,59 mcg/mL.
Concentrações do escarro: após inalação de uma dose única de 112 mg de ZoteonTM Pó (4 cápsulas x 28 mg) em pacientes com FC, a Cmáx da tobramicina no escarro foi de 1,080 ± 1,048 mcg/g (média ± DP). Em comparação, após a inalação de uma única dose de TOBI® 300 mg, a Cmáx da tobramicina no escarro foi de 737 ± 1.028 mcg/g. A variabilidade nos parâmetros farmacocinéticos no escarro foi maior que em relação ao soro.
- Distribuição
A análise farmacocinética da população para ZoteonTM Pó em pacientes com FC estimou o volume aparente de distribuição da tobramicina no compartimento central para 85,1 L para um paciente típico de FC. Enquanto o volume de distribuição se mostrou variável de acordo com o índice de massa corporal (IMC) e função pulmonar (como o percentual previsto de VEF1), simulações com base no modelo mostraram que as alterações no IMC e na função pulmonar não tiveram impacto clinicamente relevante nas concentrações no pico (Cmáx) e no vale (Cvale).
A ligação da tobramicina às proteínas séricas é desprezível.
- Metabolismo
A tobramicina não é metabolizada e é excretada principalmente inalterada na urina.
- Eliminação
A tobramicina é eliminada da circulação sistêmica principalmente por filtração glomerular do composto inalterado.
A meia-vida terminal aparente da tobramicina no soro após a inalação de uma dose única de 112 mg de ZoteonTM Pó foi de aproximadamente 3 horas em pacientes com FC e está de acordo com a meia-vida da tobramicina após a inalação de TOBI®.
A análise farmacocinética da população para ZoteonTM Pó em pacientes de 6 a 58 anos com fibrose cística estimou o clearance (depuração) sérico aparente de tobramicina a 14,5 L/h. Esta análise não demonstrou diferenças farmacocinéticas relacionadas ao sexo ou à idade.
Dados de segurança pré-clínicos
Estudos de toxicologia de doses inaladas repetidas foram realizados com ZoteonTM Pó em ratos e cachorros. Estes resultados foram avaliados juntamente com aqueles estudos de toxicologia anteriores em TOBI® e em tobramicina administrada por via parenteral (não inalatória). Dados pré-clínicos revelaram que os principais danos aos humanos, baseados nos estudos clínicos de segurança farmacológica, toxicidade de dose repetida, genotoxicidade, ou toxicidade na reprodução, consistiram em toxicidade renal e ototoxicidade. Em geral, a toxicidade é vista em níveis mais elevados de tobramicina sistêmica do que são alcançados por inalação da dose clínica recomendada.
Estudos de carcinogenicidade não foram conduzidos com ZoteonTM Pó. Foi realizado um estudo de toxicologia de inalação de dois anos em ratos para avaliar o potencial carcinogênico de TOBI®. Os ratos foram expostos à TOBI® por até 1,5 horas por dia durante 95 semanas. Os níveis séricos de tobramicina de até 35 mcg/mL foram medidos em ratos, em contraste com o nível máximo de 1,99 ± 0,59 mcg/mL observado em pacientes com fibrose cística nos estudos clínicos. Não houve aumento relacionado ao fármaco na incidência de qualquer variedade de tumor.
Adicionalmente, a tobramicina foi avaliada para genotoxicidade na bateria de testes in vitro e in vivo. O teste de Ames de reversão bacteriana, conduzido com cinco estirpes para testes, falhou em demonstrar um aumento significativo em revertentes com ou sem ativação metabólica em todas as estirpes. A tobramicina foi negativa no ensaio de mutação avançado de linfoma em camundongos, não induziu aberrações cromossômicas em células ovarianas de hamsters chineses e foi negativa no teste de micronúcleo em camundongos.
Nenhum estudo de reprodução toxicológica foi conduzido com tobramicina administrada por inalação. No entanto, a administração subcutânea de tobramicina em doses de até 100 (ratos) ou 20 (coelhos) mg/kg/dia durante a organogênese, não foi teratogênica. Doses de tobramicina de ≥ 40 mg/kg/dia foram maternalmente severamente tóxicas às coelhas fêmeas (por ex., nefrotoxicidade levando a abortos espontâneos e morte) e impediram a avaliação da teratogenicidade. A ototoxicidade não foi avaliada nas proles durante estudos pré-clínicos de toxicidade na reprodução com tobramicina. Baseado na disponibilidade dos dados em animais, o risco de toxicidade (por ex., ototoxicidade) em níveis de exposição pré-natal não pode ser excluído.
A administração subcutânea de até 100 mg/kg de tobramicina não afetou o comportamento de acasalamento ou causou danos na fertilidade em ratos machos ou fêmeas.
4. CONTRAINDICAÇÕES
ZoteonTM Pó é contraindicado para pacientes com hipersensibilidade conhecida a qualquer aminoglicosídeo.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
5. ADVERTÊNCIAS E PRECAUÇÕES
Ototoxicidade
Ototoxicidade, manifestada como a toxicidade auditiva (surdez) e toxicidade vestibular, foi relatada com aminoglicosídeos parenterais. A toxicidade vestibular pode se manifestar por vertigem, ataxia ou tontura. O zumbido pode ser um sintoma sentinela de ototoxicidade e, portanto o início deste sintoma justifica cautela.
Perda auditiva e zumbido foram relatados por pacientes em estudos clínicos com ZoteonTM Pó (vide "Reações adversas"). Recomenda-se precaução ao prescrever ZoteonTM Pó a pacientes com disfunção auditiva ou vestibular, suspeita ou conhecida. Os médicos deverão considerar um audiograma para pacientes que demonstrem qualquer evidência de disfunção auditiva ou àqueles que tenham um risco aumentado para disfunção auditiva.
- Risco de ototoxicidade devido a variantes do DNA mitocondrial
Casos de ototoxicidade com aminoglicosídeos foram observados em pacientes com certas variantes no gene 12S rRNA codificado mitocondrialmente (MT-RNR1), particularmente a variante m.1555A > G. A ototoxicidade ocorreu em alguns pacientes mesmo quando os seus níveis séricos de aminoglicosídeos estavam dentro da faixa recomendada. As variantes do DNA mitocondrial estão presentes em menos de 1% da população geral dos EUA, e a proporção de portadores de variantes que podem desenvolver ototoxicidade, bem como a gravidade da ototoxicidade, é desconhecida. Em caso de história materna conhecida de ototoxicidade devido ao uso de aminoglicosídeos, ou uma variante de DNA mitocondrial conhecida no paciente, considerar tratamentos alternativos, exceto aminoglicosídeos, a menos que o risco aumentado de perda auditiva permanente seja superado pela gravidade da infecção e falta de terapias alternativas seguras e eficazes.
Se um paciente relatar zumbido ou perda de audição durante o tratamento com ZoteonTM Pó, o médico deve encaminhá-lo para avaliação audiológica.
Vide "Teste Laboratorial" abaixo para o monitoramento das concentrações séricas de tobramicina.
Nefrotoxicidade
Nefrotoxicidade foi relatada com o uso de aminoglicosídeos parenterais.
Nefrotoxicidade não foi observada durante os estudos clínicos com ZoteonTM Pó. Recomenda-se precaução ao se prescrever ZoteonTM Pó para pacientes com disfunção renal suspeita ou conhecida.
Vide "Teste Laboratorial" abaixo para o monitoramento das concentrações séricas de tobramicina. Os testes laboratoriais devem ser realizados para o monitoramento da função renal conforme clinicamente apropriado.
Testes laboratoriais e monitoramento - concentrações séricas
As concentrações séricas de tobramicina devem ser monitoradas em pacientes com disfunções auditiva ou renal conhecidas ou suspeitas. Se ocorrer ototoxicidade ou nefrotoxicidade em um paciente recebendo ZoteonTM Pó, a terapia com tobramicina deve ser descontinuada até que a concentração sérica caia abaixo de 2 mcg/mL.
As concentrações séricas de tobramicina são aproximadamente de 1 a 2 mcg/mL uma hora após a administração de ZoteonTM Pó.
As concentrações séricas de tobramicina devem ser monitoradas em pacientes recebendo terapia com aminoglicosídeos por via parenteral (ou outros medicamentos que possam afetar a excreção renal). Estes pacientes devem ser monitorados conforme clinicamente apropriado.
As concentrações séricas da tobramicina só devem ser monitoradas através de punção venosa e não através de coleta de sangue com picada no dedo. A contaminação da pele dos dedos com tobramicina pode levar a medidas falsamente aumentadas dos níveis séricos do fármaco. Esta contaminação não pode ser totalmente evitada com a lavagem das mãos antes do teste.
Broncoespasmo
Pode ocorrer broncoespasmo com o uso de medicação inalatória e foi relatado com ZoteonTM Pó durante os estudos clínicos. O broncoespasmo deve ser tratado conforme clinicamente apropriado.
Disfunção neuromuscular
Recomenda-se precaução ao se prescrever ZoteonTM Pó a pacientes com doenças neuromusculares conhecidas ou suspeitas, como miastenia grave ou doença de Parkinson. Aminoglicosídeos podem agravar a debilidade muscular devido a um potencial efeito do tipo curare na função neuromuscular.
Tosse
Tosse foi relatada durante os estudos clínicos com ZoteonTM Pó.
Se, com o uso de ZoteonTM Pó, este sintoma se tornar incômodo ou intolerável, será necessário considerar terapias alternativas.
Mulheres em idade fértil, gravidez e lactação
- Gravidez
Existe uma quantidade limitada de dados sobre o uso de tobramicina inalada em mulheres grávidas.
Aminoglicosídeos podem causar dano fetal (por exemplo, surdez congênita), quando são atingidas altas concentrações sistêmicas em uma mulher grávida.
Estudos de toxicologia reprodutiva não foram conduzidos com tobramicina administrada por inalação. Estudos com tobramicina em animais utilizando administração subcutânea demonstraram nefrotoxicidade materna (vide "Dados de segurança pré-clínicos").
O tratamento com ZoteonTM Pó durante a gravidez deverá ser administrado somente se os benefícios esperados para a mãe superarem os potenciais riscos para o feto ou bebê. Pacientes que usam ZoteonTM Pó durante a gravidez, ou que engravidaram durante o tratamento com ZoteonTM Pó, devem ser informadas sobre o potencial perigo para o feto.
Este medicamento pertence à categoria de risco na gravidez D, portanto, este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
- Lactação
A quantidade de tobramicina excretada no leite materno após administração por inalação não é conhecida. Devido ao potencial de ototoxicidade e nefrotoxicidade em lactentes, deve-se decidir entre parar a amamentação ou descontinuar o tratamento com o ZoteonTM Pó, levando-se em consideração a importância do medicamento para a mãe.
- Fertilidade
Dados sobre a administração subcutânea de tobramicina em animais não revelaram um problema ou um potencial problema em relação à fertilidade em machos ou fêmeas (vide "Dados de segurança pré-clínicos").
Efeitos na habilidade de dirigir ou operar máquinas
ZoteonTM Pó não tem ou tem influência insignificante na capacidade de dirigir e operar máquinas.
6. INTERAÇÕES MEDICAMENTOSAS
Não foram realizados estudos clínicos de interação medicamentosa com ZoteonTM Pó. Alguns diuréticos podem aumentar a toxicidade dos aminoglicosídeos através da alteração das concentrações do antibiótico no soro e nos tecidos. ZoteonTM Pó não deve ser administrado concomitantemente com ácido etacrínico, furosemida, ureia ou manitol intravenoso. O uso concomitante e/ou sequencial de ZoteonTM Pó com outros medicamentos com potencial neurotóxico, nefrotóxicos ou ototóxicos devem ser evitados.
Ausência de interações
Durante o período de tratamento do estudo EAGER, proporções similares de pacientes que receberam ZoteonTM Pó e TOBI® continuaram a tomar dornase alfa, broncodilatadores, corticoides inalatórios e macrolídeos; nenhuma evidência de interação medicamentosa com estes medicamentos foi identificada.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Conservar em temperatura ambiente (de 15 a 30 °C). Proteger da luz e umidade.
Guardar o inalador no estojo quando não estiver em uso.
O prazo de validade é de 36 meses a partir da data de fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Aspecto físico
Pó branco ou quase branco contido em uma cápsula incolor.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Antes de usar, observe o aspecto do medicamento.
TODO MEDICAMENTO DEVE SER MANTIDO FORA DO ALCANCE DAS CRIANÇAS.
8. POSOLOGIA E MODO DE USAR
ZoteonTM Pó é apenas para inalação por via oral e não deve ser administrado por qualquer outra via.
Modo de administração
ZoteonTM Pó deve ser administrado apenas por via inalatória oral e apenas usando o inalador de ZoteonTM Pó. Não deve ser administrado por qualquer outra via ou usando qualquer outro inalador. As cápsulas de ZoteonTM Pó não devem ser ingeridas.
Quando os pacientes estão recebendo vários tipos diferentes de medicamentos inalatórios e fazendo fisioterapia do tórax, é recomendável que o tratamento com ZoteonTM Pó seja administrado por último.
Para demais instruções sobre a administração do medicamento, vide seção "Instruções de uso e manuseio".
Incompatibilidades
O inalador de ZoteonTM Pó é o único inalador para ser usado com as cápsulas de ZoteonTM Pó, este inalador não deve ser utilizado para qualquer outro medicamento que não seja o ZoteonTM Pó.
Posologia
A dose de ZoteonTM Pó é a mesma para todos pacientes (adultos e crianças com 6 anos de idade ou mais), independentemente da idade ou peso. A dose recomendada é de quatro cápsulas (4 x 28 mg = 112 mg de tobramicina), administradas duas vezes ao dia durante 28 dias. ZoteonTM Pó é administrado em ciclos alternados de 28 dias de tratamento, seguidos por 28 dias sem uso do medicamento. Cada dose de quatro cápsulas deve ser inalada o mais próximo possível de 12 horas e não em menos de 6 horas.
Dosagem em populações especiais
- Pacientes pediátricos ( < 6 anos)
Não há dose recomendada para pacientes pediátricos com idade inferior a 6 anos, uma vez que ZoteonTM Pó não é indicado para esta população.
- Pacientes idosos (≥ 65 anos)
Não existem dados suficientes nessa população que suportam uma recomendação a favor ou contra o ajuste da dose. A função renal em pacientes idosos deve ser levada em consideração durante o uso de ZoteonTM Pó (vide "Advertências e precauções").
- Pacientes com insuficiência renal
A tobramicina é principalmente excretada inalterada na urina e a função renal afeta a exposição à tobramicina. Os pacientes com creatinina sérica de 2 mg/dL ou mais, nitrogênio-urêico no sangue de 40 mg/dL ou mais, não foram incluídos nos estudos clínicos e não há dados nesta população que suportem uma recomendação a favor ou contra o ajuste da dose para ZoteonTMPó. Vide "Advertências e precauções - Nefrotoxicidade".
- Pacientes com insuficiência hepática
Não foram realizados estudos em pacientes com insuficiência hepática. Como a tobramicina não é metabolizada, não é esperado efeito da insuficiência hepática sobre a exposição à tobramicina.
- Pacientes após transplante de órgãos
Não existem dados adequados sobre o uso de ZoteonTM Pó em pacientes após o transplante de órgãos.
Este medicamento não deve ser partido, aberto ou mastigado.
Instruções de uso e manuseio
Cada caixa semanal contém sete blisters (correspondentes aos sete dias da semana) e cada blister contém oito cápsulas (que correspondem a uma dose diária: 4 cápsulas a serem inaladas pela manhã e 4 cápsulas a serem inaladas à noite).
As cápsulas de ZoteonTM Pó devem ser sempre armazenadas no blister e somente serem retiradas imediatamente antes do uso. Cada inalador é utilizado por sete dias e depois descartado e substituído.
As instruções de uso básicas estão descritas abaixo: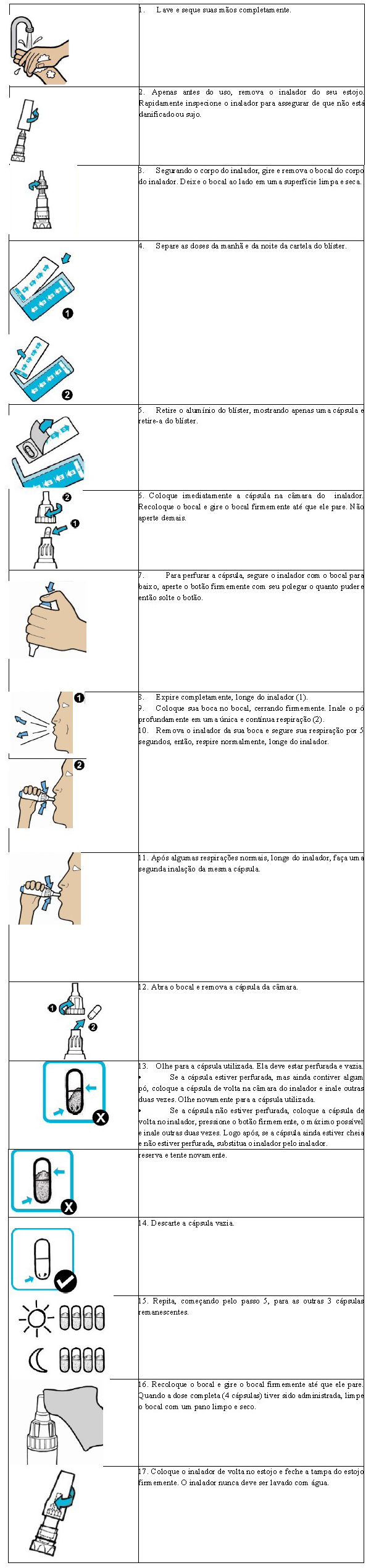
ZoteonTM Pó deve ser mantido fora do alcance e da vista de outras crianças e administrado somente sob a supervisão de um adulto.
Cuidadores devem fornecer assistência as crianças que iniciaram o tratamento com o ZoteonTM Pó, particularmente aquelas com 10 anos ou mais novas, e também devem supervisioná-las até que elas sejam capazes de administrar o medicamento sozinhas.
Para prevenir o desenvolvimento de bactérias resistentes, este medicamento deverá ser usado somente para o tratamento ou prevenção de infecções causadas ou fortemente suspeitas de serem causadas por microrganismos sensíveis a este medicamento.
9. REAÇÕES ADVERSAS
Resumo do perfil de segurança
A segurança de ZoteonTM Pó foi avaliada em 425 pacientes com fibrose cística expostos a pelo menos uma dose de ZoteonTM Pó, incluindo 273 que foram expostos em três ciclos (seis meses) de tratamento. Cada ciclo consistiu 28 dias de tratamento (com 112 mg administrados duas vezes ao dia) e 28 dias sem tratamento.
A população para segurança primária, randomizada em uma proporção de 3:2, consistiu em 308 pacientes tratados com ZoteonTM Pó e 209 pacientes tratados com TOBI® (solução nebulizada de tobramicina 300 mg/5 mL) no estudo EAGER, um estudo aberto comparando ZoteonTM Pó com TOBI® durante três ciclos de tratamento. Para ambos os grupos ZoteonTM Pó e TOBI®, a exposição média ao medicamento em cada ciclo foi de 28-29 dias. A população de segurança suporte reflete pacientes de dois estudos: um adicional de 87 pacientes tratados com ZoteonTM Pó e 49 tratados com placebo no estudo EVOLVE, um desenho duplo-cego, placebo-controlado para o primeiro ciclo de tratamento, seguido por todos os pacientes recebendo ZoteonTM Pó (placebo substituído) por dois ciclos adicionais, e 30 pacientes adicionais tratados com ZoteonTM Pó e 32 tratados com placebo no Estudo EDIT, um estudo duplo-cego, placebo-controlado, para apenas um ciclo de tratamento. O placebo nesses estudos foi o pó inalado sem o ingrediente ativo, tobramicina. A população de pacientes para esses estudos era muito mais jovem do que no estudo EAGER (idade média de 13 anos).
Durante essas exposições, ZoteonTM Pó foi geralmente bem tolerado. No estudo EAGER, as reações adversas mais frequentes que ocorreram estavam relacionadas aos sistemas respiratório, torácico e mediastinal. As reações adversas mais comuns (por termo preferido) foram tosse e doença pulmonar em ambos os grupos de tratamento, ZoteonTM Pó e TOBI®.
Durante o ciclo controlado por placebo do estudo EVOLVER, a incidência global de reações adversas a medicamentos foi menor no grupo de tratamento com ZoteonTM Pó do que no grupo placebo, com exceção da dor orofaríngea, disfonia e disgeusia.
No estudo EDIT, tosse e hipoacusia foram relatados com mais frequência no grupo de ZoteonTM Pó do que no grupo placebo.
- Avaliação Audiométrica
No estudo EAGER, problemas auditivos como zumbido foram relatados em aproximadamente 2% dos pacientes em geral. De um subconjunto dos pacientes no estudo EAGER que receberam a série de testes de audiologia, 25,6% (ZoteonTM Pó) e 15,6% (TOBI®) apresentaram diminuição do valor basal em qualquer consulta (80% do subgrupo tiveram uma avaliação de audição normal no valor basal). No entanto, a maioria dessas alterações foram transitórias e resolvidas até o final do estudo. Quatro pacientes no grupo de tratamento com ZoteonTM Pó apresentaram diminuições significativas na audição, a qual foi transitória em três pacientes e persistentes em um caso. Menos de 3% dos pacientes em ambos os grupos mostraram evidência de perda auditiva significativa. Utilizando o critério de perda para qualquer um dos ouvido