ZOSTIDE
LIBBS
abiraterona
Citostático.
Apresentações.
Comprimidos de 250 mg de acetato de abiraterona. Embalagem com 120 comprimidos.
USO ORAL
USO ADULTO
Composição.
Cada comprimido contém 250 mg de acetato de abiraterona, que corresponde a 223 mg de abiraterona. Excipientes: celulose microcristalina, croscarmelose sódica, lactose monoidratada, laurilsulfato de sódio, povidona, dióxido de silício e estearato de magnésio.
Informações técnicas.
1. INDICAÇÕES
O medicamento Zostide, em combinação com prednisona ou prednisolona, é indicado para:
- o tratamento de pacientes com câncer de próstata metastático resistente à castração (mCRPC) que são assintomáticos ou levemente sintomáticos, após falha à terapia de privação androgênica.
- o tratamento de pacientes com câncer de próstata avançado metastático resistente à castração (mCRPC) e que receberam quimioterapia prévia com docetaxel.
O medicamento Zostide, em combinação com prednisona e terapia de privação androgênica (agonista de hormônio liberador de gonadotrofina ou castração cirúrgica), é indicado para:
- o tratamento de pacientes com câncer de próstata metastático de alto risco, com diagnóstico recente, não tratados anteriormente com hormônios (mHNPC) ou pacientes que estavam em tratamento hormonal por não mais que três meses e continuam respondendo à terapia hormonal (mHSPC).
2. RESULTADOS DE EFICÁCIA
A eficácia do acetato de abiraterona foi estabelecida em três estudos clínicos Fase 3 (Estudos 3011, 302, e 301), multicêntricos, randomizados, controlados com placebo em pacientes com câncer de próstata metastático de alto risco não tratados anteriormente com hormônio e câncer de próstata metastático e resistente à castração.
O Estudo 3011 incluiu pacientes que foram recentemente diagnosticados (no prazo de 3 meses até a randomização) com câncer de próstata metastático não tratados anteriormente com hormônio e que apresentavam fatores de prognóstico de alto risco. O prognóstico de alto risco foi definido como tendo pelo menos 2 dos 3 seguintes fatores de risco: (1) escore de Gleason ≥ 8; (2) presença de 3 ou mais lesões na varredura óssea; (3) presença de metástases viscerais mensuráveis (excluindo a doença linfonodal). No braço ativo, acetato de abiraterona foi administrado em uma dose de 1000 mg ao dia em combinação com uma dose baixa de prednisona 5 mg uma vez ao dia em adição à ADT (agonista de LHRH ou orquiectomia), que era o tratamento padrão. Os pacientes no braço controle receberam ADT e placebos tanto para acetato de abiraterona como para prednisona.1
O Estudo 302 incluiu pacientes que não haviam recebido quimioterapia prévia e eram assintomáticos ou levemente sintomáticos, enquanto que o Estudo 301 incluiu pacientes que já tinham recebido quimioterapia prévia contendo docetaxel. Em ambos os estudos os pacientes estavam em uso de agonistas de LHRH (hormônio liberador do hormônio luteinizante) ou haviam sido submetidos previamente à orquiectomia. Nos braços experimentais, acetato de abiraterona foi administrado na dose de 1000 mg ao dia, associado a uma dose baixa de prednisona ou prednisolona de 5 mg duas vezes ao dia. Os pacientes do grupo controle receberam placebo e dose baixa de prednisona ou prednisolona de 5 mg duas vezes ao dia.
Devido ao fato de que as alterações na concentração sérica do Antígeno Prostático Específico (PSA) nem sempre podem prever um benefício clínico, em todos os estudos os pacientes foram mantidos em tratamento com acetato de abiraterona até atingirem critério para descontinuação, conforme especificado para cada estudo abaixo.
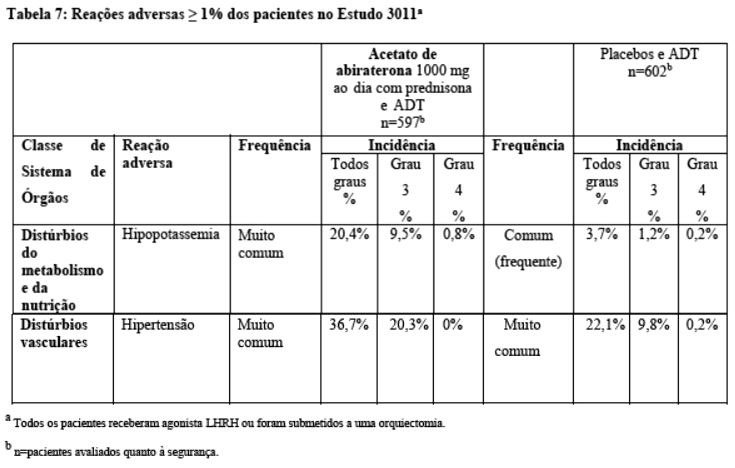
Estudo 3011 [pacientes com câncer de próstata metastático de alto risco não tratados anteriormente com hormônio (mHNPC) recentemente diagnosticado ou câncer de próstata hormônio-sensível metastático de alto risco (mHSPC)]
No Estudo 3011 (n=1199) a idade mediana dos pacientes incluídos foi de 67 anos.2 O "performance status ECOG" (Eastern Cooperative Oncology Group) foi de 0 ou 1 para 97% dos pacientes.3 Foram excluídos os pacientes com hipertensão não controlada, doença cardíaca significativa ou NYHA Classe II ou maior ou insuficiência cardíaca.4 Os desfechos de eficácia coprimária foram sobrevida global (OS) e sobrevida livre de progressão radiográfica (rPFS).5 A mediana do escore basal de dor, medido pelo Formulário Abreviado da Dor (Brief Pain Inventory Short Form (BPI-SF)) foi 2,0 nos grupos de tratamento e placebo.6 Além dos desfechos de avaliação coprimária, o benefício também foi avaliado usando tempo para eventos esqueléticos relacionados (SRE), tempo para terapia subsequente para câncer de próstata, tempo para início de quimioterapia, tempo para progressão de dor e tempo para progressão do PSA.7 No Estudo 3011, o tratamento continuou até a progressão da doença, a retirada da permissão, a ocorrência de toxicidade inaceitável ou óbito.8
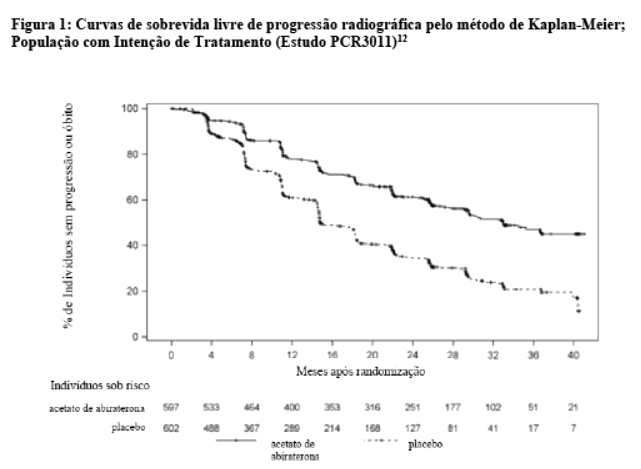
A sobrevida livre de progressão radiográfica foi definida como o tempo desde a randomização até a ocorrência de progressão radiográfica ou óbito por qualquer causa. A progressão radiográfica incluiu progressão por varredura óssea (de acordo com PCWG2 modificado) ou progressão de lesões de tecido mole por TC ou RNM (de acordo com RECIST 1.1).9
Na análise rPFS planejada haviam ocorrido 593 eventos; 239 (40,0%) dos pacientes tratados com acetato de abiraterona e 354 (58,8%) dos pacientes tratados com placebo apresentaram evidência radiográfica de progressão ou evoluíram para óbito. Observou-se uma diferença significativa no rPFS entre os grupos de tratamento (vide Tabela 1 e Figura 1).10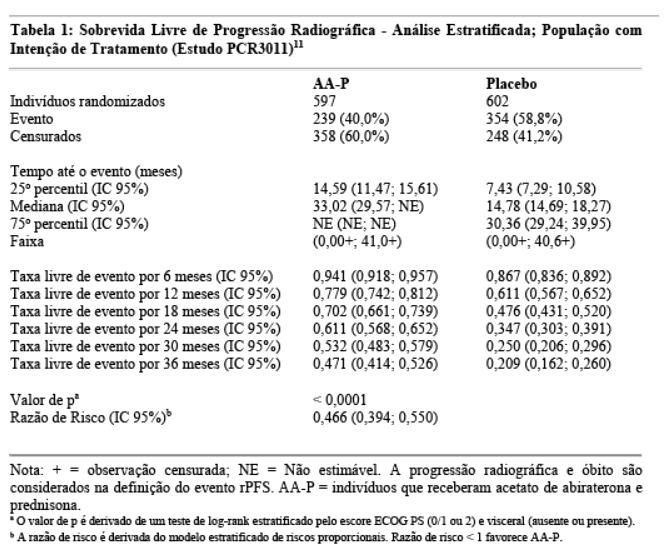
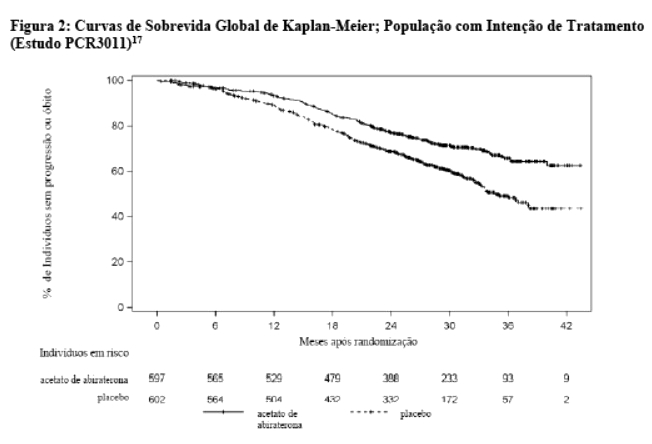
Na primeira Análise Interina (IA-1) de sobrevida global planejada, ocorreram quatrocentos e seis óbitos (406; 47,7% do número total de óbitos requerido para a análise final) (169 indivíduos no grupo AA-P e 237 indivíduos no grupo placebo). Observou-se uma melhora estatisticamente significativa no OS em favor de AA-P mais ADT com uma redução de 38% no risco de morte (HR = 0,621; IC 95%: 0,509; 0,756) comparado ao placebo mais ADT. A sobrevida mediana não foi atingida no grupo AA-P versus 34,7 meses no grupo placebo (p < 0,0001, cruzando o limite pré-especificado para OS na Análise Interina-1 de 0,010) (vide Tabela 2 e Figura 2).13 O estudo foi aberto com base na magnitude do benefício clínico observado e aos pacientes do grupo placebo foram oferecidos tratamento com acetato de abiraterona.14 A sobrevida continuou a ser seguida após esta Análise Interina.15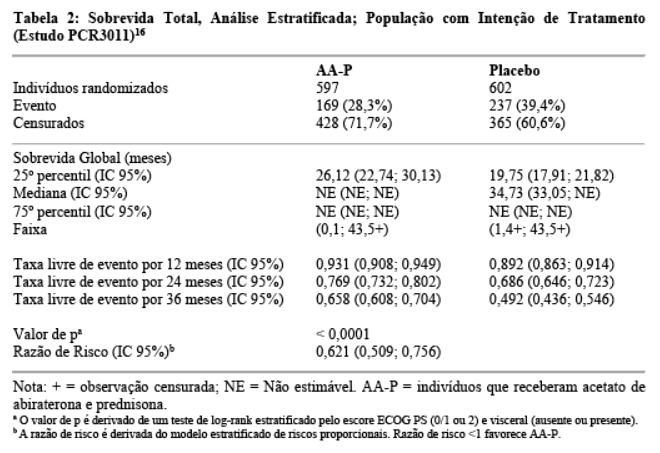
As análises de subgrupos favorecem consistentemente o tratamento com acetato de abiraterona (vide Figura 3).18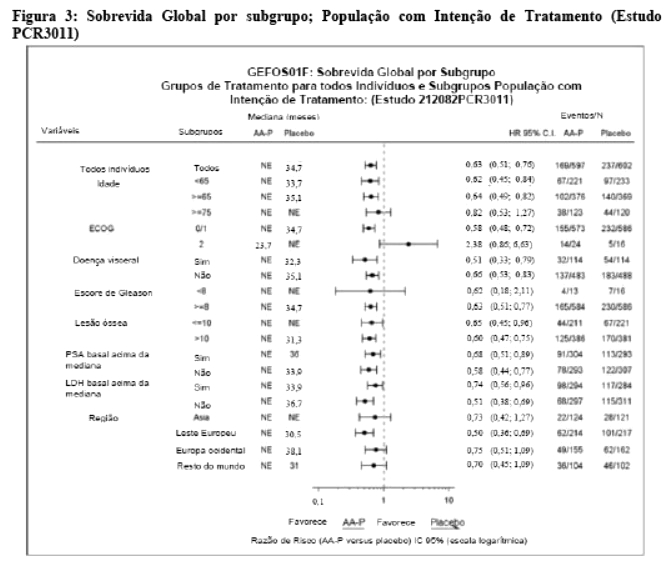
Em adição às melhorias observadas na sobrevida global e na rPFS, foi demonstrado benefício para o tratamento com acetato de abiraterona versus placebo em todas as medidas de desfecho secundário prospectivamente definidas, como segue:
Tempo para eventos relacionados ao esqueleto (SRE): houve uma redução de 30% no risco de eventos relacionados ao esqueleto (HR=0,703; IC 95%: [0,539; 0,916] p < 0,0086). O tempo mediano para o SRE não foi atingido para o braço do estudo acetato de abiraterona ou placebo.19
Tempo para a progressão do PSA com base nos critérios PCWG2: o tempo mediano para a progressão do PSA foi de 33,2 meses para os pacientes que receberam acetato de abiraterona e de 7,4 meses para os pacientes que receberam placebo (HR=0,299; IC 95%: [0,255; 0,352], p < 0,0001).20
Tempo para a terapêutica subsequente: o tempo mediano para a terapêutica subsequente no momento da análise interina não foi atingido para os pacientes que receberam acetato de abiraterona e foi de 21,6 meses para os pacientes que receberam placebo (HR = 0,415; IC 95%: [0,346; 0,497], p < 0,0001).21
Tempo para início da quimioterapia: o tempo mediano para o início da quimioterapia não foi atingido para os pacientes que receberam acetato de abiraterona e foi de 38,9 meses para os pacientes que receberam placebo (HR=0,443, IC 95%: [0,349; 0,561], p < 0,0001).22
Tempo para a progressão da dor: o tempo mediano para a progressão da dor não foi atingido para os pacientes que receberam acetato de abiraterona e foi de 16,6 meses para os pacientes que receberam placebo (HR=0,695; IC 95%: [0,583; 0,829], p = < 0,0001).23
A maioria dos pontos de desfechos exploratórios favoreceu o tratamento com acetato de abiraterona e prednisona (AA-P) em relação ao placebo. Observou-se uma melhora estatisticamente significativa na sobrevida global específica do câncer de próstata para o tratamento com AA-P em comparação com o placebo (HR=0,547; p < 0,0001). Observou-se uma resposta confirmada do PSA em 91,0% nos indivíduos do grupo AA-P e 66,8% nos indivíduos do grupo placebo (risco relativo = 1,362; p < 0,0001). A taxa de resposta global (resposta completa e resposta parcial) em indivíduos com doença mensurável no início foi significativamente maior no grupo AA-P comparada com os do grupo placebo (p=0,0002).24
O tempo para deterioração na análise das medidas de resultado relatadas pelo paciente (PRO) demonstrou consistentemente que o tratamento com AA-P retardou a piora e progressão da dor, o estado funcional, a fadiga e a qualidade de vida relacionada à saúde. Com base na variação a partir do valor basal utilizando medidas repetidas de modelo com efeitos mistos, foram observadas diferenças estatisticamente significativas entre AA-P e placebo no início do Ciclo 2 e mantidas ao longo do estudo.25
Estudo 302 (pacientes assintomáticos ou levemente sintomáticos que não receberam quimioterapia prévia)
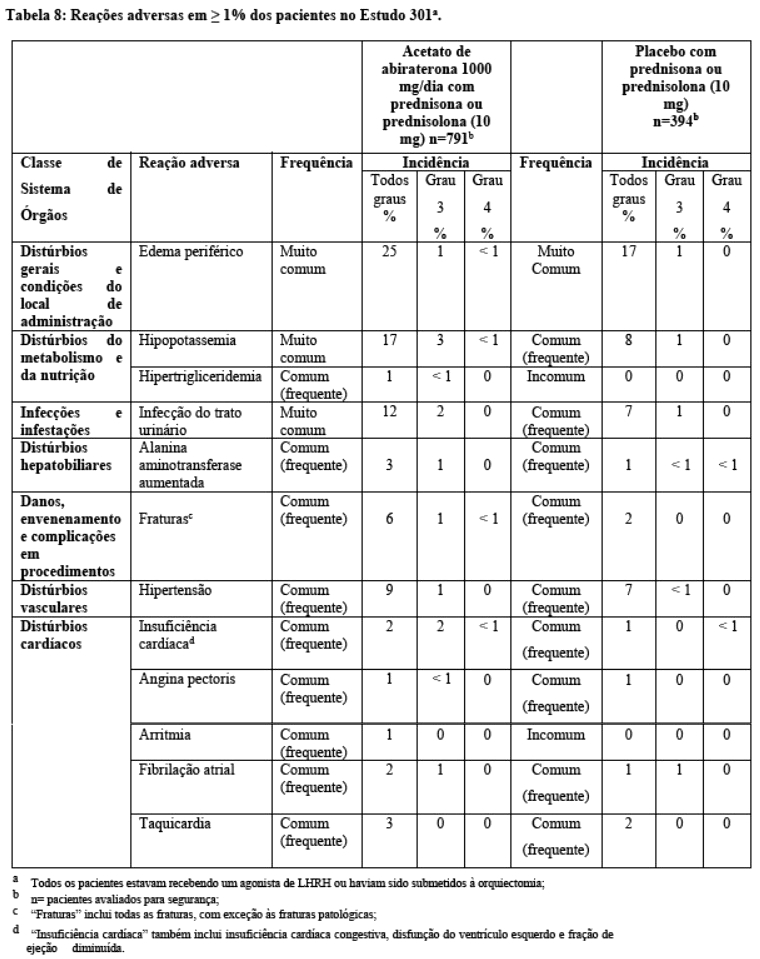
No Estudo 302, (n = 1.088) a mediana de idade dos pacientes incluídos foi de 71 anos para os pacientes tratados com acetato de abiraterona associado à prednisona ou prednisolona e 70 anos para os pacientes tratados com placebo mais prednisona ou prednisolona. O "performance status ECOG" (Eastern Cooperative Oncology Group) era de 0 em 76% dos pacientes e de 1 em 24% dos pacientes em ambos braços. Os pacientes com metástase visceral foram excluídos. Os desfechos de eficácia coprimários foram sobrevida global e sobrevida livre de progressão radiográfica (rPFS). Uma avaliação de dor na linha de base foi de 0-1 (assintomático) em 66% dos pacientes e 2-3 (levemente sintomático) em 26% dos pacientes, conforme definido pelo Formulário Abreviado da Dor (Brief Pain Inventory-Short Form) (pior dor ao longo das últimas 24 horas). Além da avaliação dos desfechos coprimários, a eficácia também foi avaliada observando-se o tempo até o uso de opiáceos para o controle da dor oncológica, o tempo para início de quimioterapia citotóxica, o tempo para queda no escore de "performance status ECOG" em ≥ 1 ponto e o tempo para progressão do PSA conforme os critérios do "Prostate Cancer Working Group-2" (PCWG2).
No Estudo 302, os tratamentos foram descontinuados no momento de progressão clínica inquestionável. Os tratamentos também podiam ser descontinuados no momento de progressão radiográfica confirmada, a critério do investigador.
A sobrevida livre de progressão radiográfica (rPFS) foi avaliada empregando-se exames de imagens sequenciais, definidas pelos critérios de PCWG2 (para lesões ósseas) e "Response Evaluation Criteria In Solid Tumors" (RECIST) modificado (para lesões em partes moles). A análise da rPFS utilizou uma avaliação da progressão radiográfica revisada em laboratório central.
Na análise de rPFS planejada houve 401 eventos; 150 (28%) em pacientes tratados com acetato de abiraterona e 251 (46%) dos pacientes tratados com placebo tiveram evidência radiográfica de progressão
ou vieram a óbito. Uma diferença significativa na rPFS entre os grupos de tratamento foi observada (vide Tabela 3 e Figura 4).
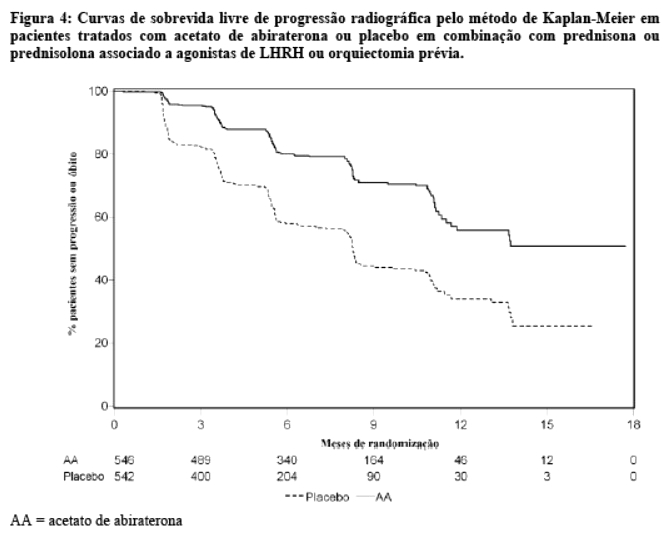
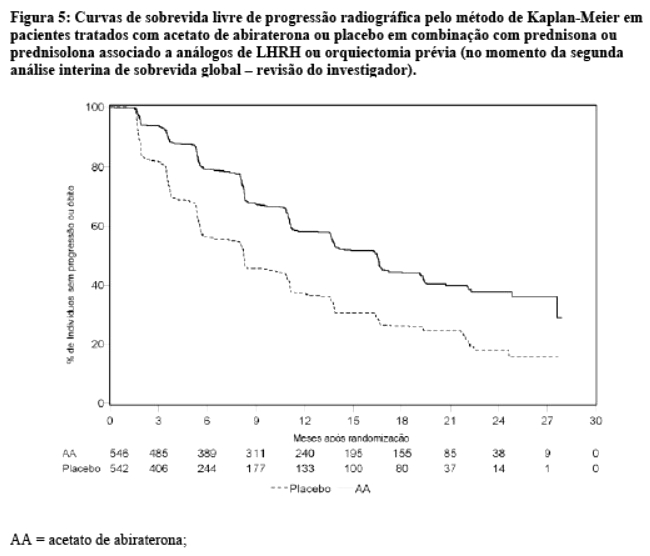
Entretanto, os dados dos indivíduos continuaram a ser coletados até a data da segunda análise interina de sobrevida global (OS). Na Tabela 4 e Figura 5 é apresentada a revisão radiográfica do investigador da rPFS, realizada como uma análise sensitiva de seguimento.
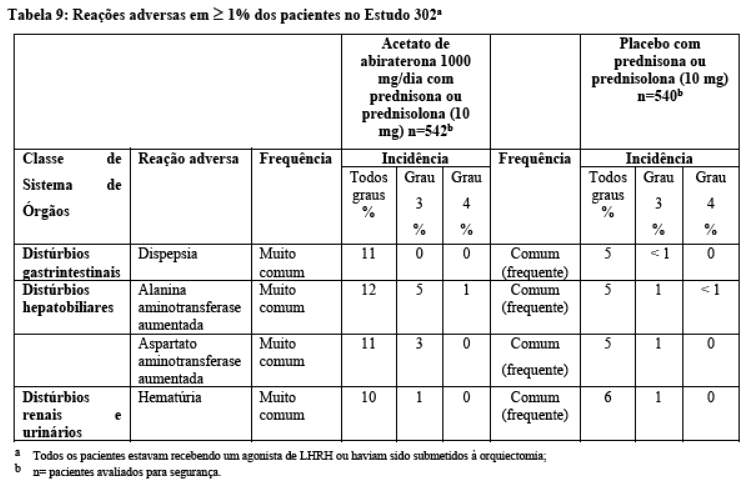
Seiscentos e sete (607) indivíduos tiveram progressão radiográfica ou vieram a óbito: 271 (50%) no grupo do acetato de abiraterona e 336 (62%) no grupo placebo. O tratamento com acetato de abiraterona reduziu o risco de progressão radiográfica ou óbito em 47% comparado com placebo (Razão de Risco = 0,530; IC de 95%: [0,451; 0,623], p < 0,0001). A mediana de rPFS foi de 16,5 meses no grupo acetato de abiraterona e 8,3 meses no grupo placebo.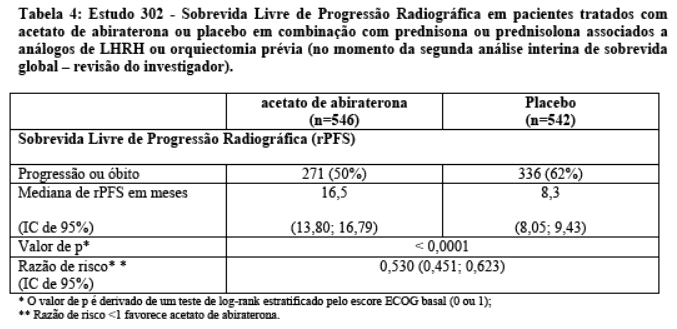
Foi realizada uma análise interina (IA) da sobrevida global planejada após a ocorrência de 333 óbitos. O estudo foi aberto com base na magnitude do benefício clínico observado e aos pacientes no grupo placebo foram oferecidos tratamento com acetato de abiraterona. A sobrevida global foi maior para acetato de abiraterona do que para placebo, com uma redução de 25% no risco de morte (HR = 0,752, IC 95%: [0,606; 0,934], p = 0,0097), mas os resultados interinos da sobrevida global não estavam maduros e não atingiram os limites pré-especificados para significância estatística (vide Tabela 5). A sobrevida continuou a ser seguida após esta IA.
A análise final para OS planejada foi realizada após 741 óbitos observados (mediana de acompanhamento de 49 meses). Sessenta e cinco por cento (354 de 546) dos pacientes tratados com acetato de abiraterona, em comparação com 71% (387 de 542) dos pacientes tratados com placebo, evoluíram para óbito. Foi demonstrado um benefício estatisticamente significativo da OS a favor do grupo tratado com acetato de abiraterona com uma redução de 19,4% no risco de morte (HR = 0,806; IC 95%: [0,697; 0,931], p = 0,0033) e uma melhora na mediana de OS de 4,4 meses (acetato de abiraterona 34,7 meses, placebo 30,3 meses) (vide Tabela 5 e Figura 6). Esta melhora foi demonstrada apesar da terapêutica subsequente ser comum, independentemente se os pacientes receberam inicialmente acetato de abiraterona ou placebo. As terapias subsequentes nos grupos de pacientes de acetato de abiraterona e placebo incluíram acetato de abiraterona, 69 (13%) e 238 (44%); docetaxel, 311 (57%) e 331 (61%); cabazitaxel, 100 (18%) e 105 (19%); e enzalutamida 87 (16%) e 54 (10%) pacientes respectivamente.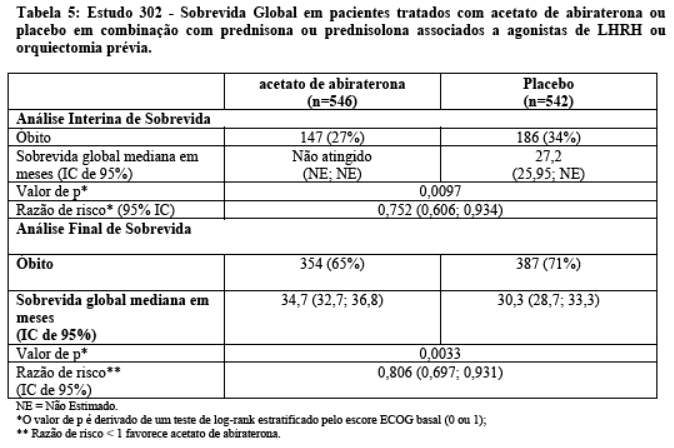
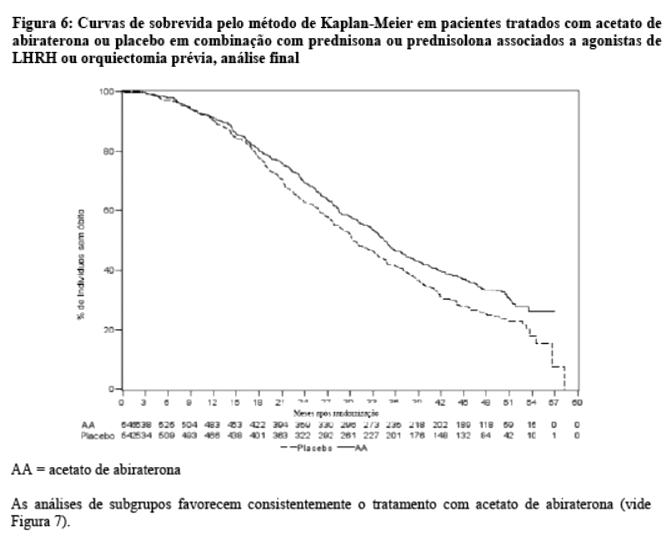
As análises de subgrupos favorecem consistentemente o tratamento com acetato de abiraterona (vide Figura 7).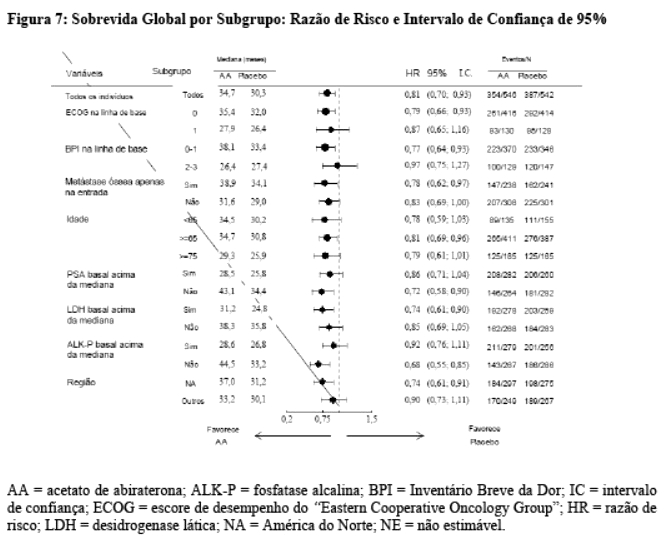
Além das melhoras observadas na sobrevida global e rPFS foram observados benefícios do tratamento com acetato de abiraterona quando comparado ao placebo em todas as medidas de desfechos secundários prospectivamente definidas, como segue:
Tempo para progressão do PSA, de acordo com os critérios PCWG2: o tempo mediano para a progressão do PSA foi de 11,1 meses para pacientes que receberam acetato de abiraterona e 5,6 meses para os pacientes que receberam placebo (Razão de risco = 0,488; IC 95%: [0,420; 0,568], p < 0,0001). O tempo para a progressão do PSA foi aproximadamente o dobro no grupo tratado com acetato de abiraterona (Razão de risco = 0,488). A proporção de indivíduos com resposta confirmada de PSA foi maior no grupo de acetato de abiraterona do que no grupo placebo (62% versus 24%; p < 0,0001).
Tempo para uso de opiáceos para dor oncológica: o tempo mediano para o uso de opiáceos para a dor causada pelo câncer de próstata no momento final da análise foi de 33,4 meses para pacientes recebendo acetato de abiraterona e foi de 23,4 meses para pacientes recebendo placebo (Razão de risco = 0,721; IC 95%: [0,614; 0,846], p < 0,0001).
Tempo para início de quimioterapia citotóxica: o tempo mediano para o início de quimioterapia citotóxica foi de 25,2 meses para os pacientes que receberam acetato de abiraterona e 16,8 meses para os pacientes que receberam placebo (Razão de risco = 0,580; IC 95% [0,487; 0,691], p < 0,0001).
Tempo para a piora do escore "performance status (ECOG)" em ≥ 1 ponto: o tempo mediano para piora do escore "performance status (ECOG)" em ≥ 1 ponto foi de 12,3 meses para pacientes que receberam acetato de abiraterona e 10,9 meses para os pacientes que receberam placebo (Razão de risco = 0,821; IC 95%: [0,714; 0,943], p = 0,0053).
Os seguintes desfechos do estudo demonstraram uma vantagem estatisticamente significativa em favor do tratamento com acetato de abiraterona:
Resposta objetiva: a resposta objetiva foi definida como a proporção de indivíduos com doença mensurável que atingiram uma resposta completa ou parcial, conforme critério RECIST (para ser considerado lesão alvo, o linfonodo deveria ter um tamanho ≥ 2 cm no período basal). A proporção de indivíduos com doença mensurável na linha de base que tiveram resposta objetiva foi de 36% no grupo de acetato de abiraterona e de 16% no grupo placebo (p < 0,0001).
Dor: o tratamento com acetato de abiraterona reduziu significativamente o risco de progressão da intensidade média da dor em 18% comparado com placebo (p = 0,0490). O tempo mediano para a progressão foi de 26,7 meses no grupo de acetato de abiraterona e 18,4 meses no grupo placebo.
Tempo para degradação no FACT-P (escore total): o tratamento com acetato de abiraterona diminuiu o risco da degradação no FACT-P (escore total) em 22% comparado com placebo (p = 0,0028). O tempo mediano para a degradação no FACT-P (escore total) foi de 12,7 meses no grupo de acetato de abiraterona e 8,3 meses no grupo placebo.
Estudo 301 (pacientes que receberam quimioterapia prévia)
Onze por cento (11%) dos pacientes incluídos no Estudo 301 tinham "performance status ECOG" (Eastern Cooperative Oncology Group) igual a 2; 70% tinham evidência radiográfica de progressão da doença com ou sem progressão do PSA; 70% haviam recebido um esquema de quimioterapia citotóxica anteriormente e 30% haviam recebido dois. Metástase hepática estava presente em 11% dos pacientes tratados com acetato de abiraterona.
Foi recomendado que os pacientes continuassem recebendo os respectivos medicamentos do estudo até que houvesse progressão do PSA (25% de aumento confirmado em relação ao nível basal/nadir do paciente), em conjunto com a progressão radiográfica definida no protocolo e a progressão sintomática ou clínica. O desfecho primário de eficácia foi a sobrevida global.
Em uma análise planejada conduzida após a ocorrência de 552 óbitos, 42% (333 de 797) dos pacientes tratados com acetato de abiraterona morreram em comparação com 55% (219 de 398) dos pacientes tratados com placebo. Uma melhora estatisticamente significativa na mediana da sobrevida global foi observada em pacientes tratados com acetato de abiraterona (vide Tabela 6 e Figura 8). Uma análise de sobrevida atualizada foi conduzida quando 775 óbitos foram observados (97% do número planejado de óbitos para a análise final). Os resultados desta análise de sobrevida atualizada foram consistentes com a primeira análise de sobrevida (vide Tabela 6).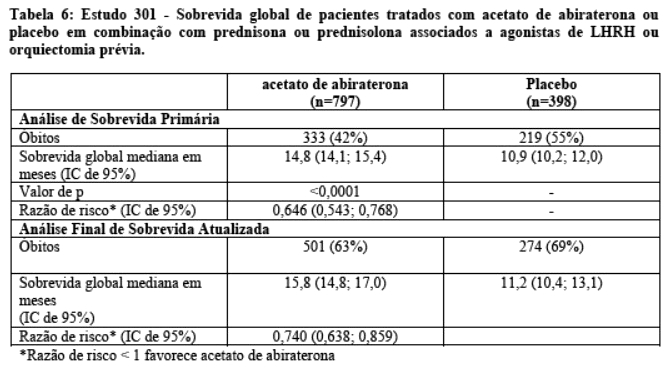
Em todos os pontos de avaliação após os primeiros meses de tratamento, uma proporção maior de pacientes tratados com acetato de abiraterona continuava viva em comparação com a proporção de pacientes tratados com placebo (vide Figura 8).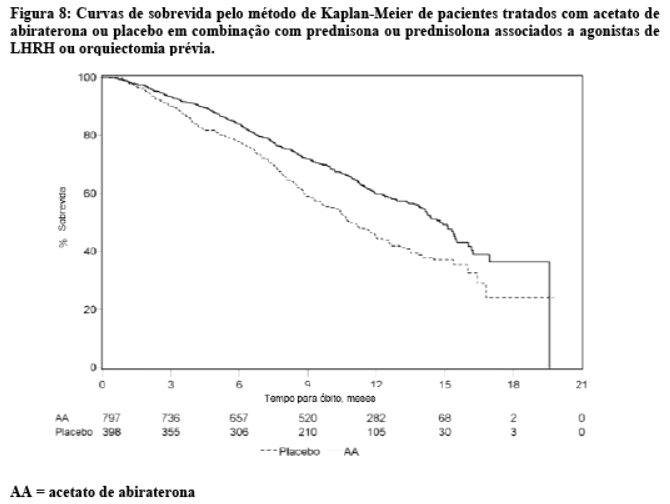
As análises de sobrevida por subgrupo mostraram um benefício consistente de sobrevida para o tratamento com acetato de abiraterona (vide Figura 9).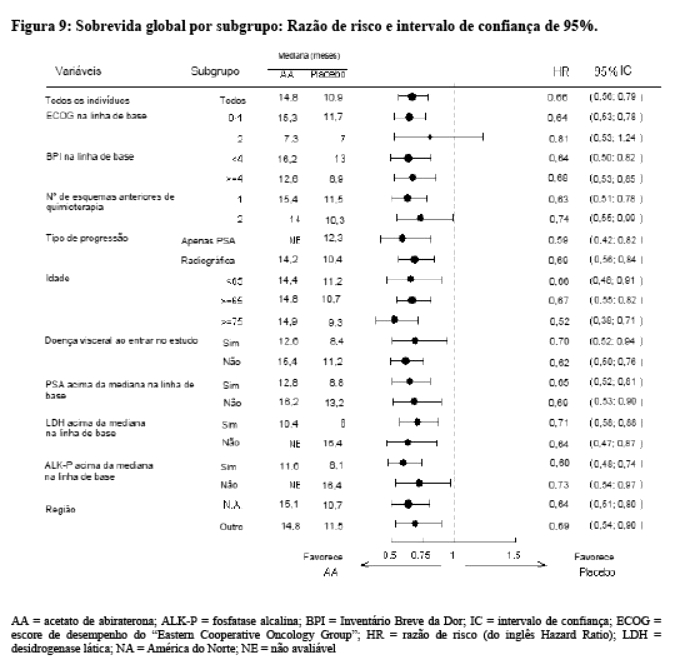
Além da melhora observada na sobrevida global, todos os desfechos secundários do estudo favoreceram acetato de abiraterona e foram estatisticamente significativos depois do ajuste para multiplicidade como segue:
Os pacientes que receberam acetato de abiraterona demonstraram uma taxa de resposta do PSA total significativamente maior (definida como redução ≥ 50% em relação à linha de base) em comparação com os pacientes que receberam o placebo: 38% versus 10%, p < 0,0001.
O tempo mediano para progressão do PSA foi 10,2 meses para os pacientes tratados com acetato de abiraterona e 6,6 meses para os pacientes tratados com placebo (Razão de risco = 0,580; IC 95%: [0,462; 0,728], p < 0,0001).
A mediana de sobrevida livre de progressão radiográfica foi 5,6 meses para os pacientes tratados com acetato de abiraterona e 3,6 meses para os pacientes que receberam placebo (Razão de risco = 0,673; IC 95%: [0,585; 0,776], p < 0,0001).
Dor
A proporção de pacientes com alívio da dor foi maior e estatisticamente significativo no grupo acetato de abiraterona comparado ao grupo placebo (44% versus 27%, p=0,0002). Um paciente com resposta para alívio da dor foi definido como aquele paciente que apresentou redução de pelo menos 30% no escore de intensidade da pior dor durante as últimas 24 horas pelo Inventário Breve da Dor (BPI-SF) em relação à linha de base sem aumento do escore de uso de analgésicos observado em duas avaliações consecutivas com 4 semanas de intervalo. Apenas pacientes com escore de dor ≥ 4 na linha de base e pelo menos um escore de dor depois da linha de base foram analisados (n=512) quanto ao alívio da dor.
Uma proporção menor de pacientes tratados com acetato de abiraterona apresentou progressão da dor em comparação aos pacientes tomando placebo em 6 (22% versus 28%), 12 (30% versus 38%) e 18 meses
(35% versus 46%). Progressão da dor foi definida como um aumento ≥ 30% no escore de intensidade da pior dor nas últimas 24 horas pelo BPI-SF da linha de base, sem diminuição do escore de uso de analgésicos observado em duas visitas consecutivas ou um aumento ≥ 30% no escore de uso de analgésicos em duas visitas consecutivas. O tempo para progressão da doença no 25° percentil foi 7,4 meses no grupo acetato de abiraterona versus 4,7 meses no grupo placebo.
Eventos relacionados ao esqueleto
Uma proporção menor de pacientes no grupo acetato de abiraterona teve eventos relacionados ao esqueleto em comparação ao grupo placebo em 6 meses (18% versus 28%), 12 meses (30% versus 40%) e 18 meses (35% versus 40%). O tempo para o primeiro evento relacionado ao esqueleto no 25° percentil no grupo acetato de abiraterona foi duas vezes maior em relação ao grupo controle no tempo 9,9 meses versus 4,9 meses. Um evento relacionado ao esqueleto foi definido como uma fratura patológica, compressão da medula espinhal, radiação paliativa no osso ou cirurgia óssea.
Referências
1. H. I. Scher, C. Logothetis, A. Molina, et al. Improved survival outcomes in clinically relevant patient subgroups from COU-AA-301, a phase III study of abiraterone acetate (AA) plus prednisone (P) in patients with metastatic castration-resistant prostate cancer (mCRPC) progressing after docetaxel-based chemotherapy. J Clin Oncol 29: 2011 (Suppl 7; Abstr 4).
2. J.S. de Bono, C.J. Logothetis, K. Fizazi, et al. Abiraterone Acetate (AA) Plus Low Dose Prednisone (P) Improves Overall Survival (Os) In Patients (Pts) With Metastatic Castration Resistant Prostate Cancer (Mcrpc) Who Have Progressed After Docetaxel-Based Chemotherapy (Chemo): Results Of COU-AA-301, A Randomized Double-Blind Placebo-Controlled Phase III. Study Annals of Oncology 21 (Suppl 8): viii1-viii12, LBA5, 2010.
3. Adolfsson J, Steineck G, Hedlund PO. Deferred treatment of locally advanced non-metastatic prostate cancer: a long-term follow-up. J Urol. 1999;161:505-508.
4. Amit O, Mannino F, Stone AM, et al. Blinded independent central review of progression in cancer. EU J Cancer. 2011;47:1772-1778.
5. Basch E, Jia X, Heller G, et al. Adverse symptom event reporting by patients versus clinicians: Relationship with clinical outcomes. J Natl Cancer Inst 2009;101:1624-1632.
6. de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995-2005.
7. Fitzpatrick JM. Management of localized prostate cancer in senior adults: the crucial role of comorbidity. BJU Int. 2008;101 Suppl 2:16-22.
8. Flamand V, Zini L, Salleron J, Fantoni J-C, Biserte J, Villers A. Observational survey on variations of prostate cancer incidence by stage in the Nord-Pas-de-Calais region between 1998 and 2004. Prog Urol. 2008;18:53-59.
9. Gennari JF. Hypokalemia. NJEM. 1998. 339 (7); 451-458.
10. Gravis G, Fiazzi, F, Joly F, et al. Androgen-deprivation therapy alone or with docetaxel in non-castrate metastatic prostate cancer (GETUG-AFU 15): a randomised, open-label, phase 3 trial. Lancet Oncol 2013;14:149-158.
11. Guidelines for Preparing Core Clinical-Safety Information on Drugs. Second edition (Including New Proposals for Investigator's Brochures). Report of CIOMS Working Groups III (Revised) and V (New). CIOMS 1999.
12. Thomson CS, Howard GCW, Stroner PL, Goodman CM, Windsor PM, Brewster DH. Patterns of referral, management and survival of patients diagnosed with prostate cancer in Scotland during 1988 and 1993: results of a national retrospective population-based audit. BJUI. 2001;87:339-347.
13. Jack R, Davies EA, Moller H. Prostate cancer incidence, stage at diagnosis, treatment and survival in ethnic groups in South-East England. BJUI. 2009;105:1226-1230.
14. James ND, Spears MR, Clarke NW et al. Survival with Newly Diagnosed Metastatic Prostate Cancer in the "Docetaxel Era": Data from 917 Patients in the Control Arm of the STAMPEDE Trial (MRC PR08, CRUK/06/019). Eur Urol. 2015 Jun;67(6):1028-38.
15. James ND, Sydes MR, Clarke NW et al. Addition of docetaxel, zolendronic acid, or both to first-line longterm hormone therapy in prostate cancer (STAMPEDE): survival results from an adaptive, multiarm, multistage, platform randomized controlled trial. Lancet. 2016; 387:1163-1177.
16. Jonsson E, Sigbjarnarson H, Tomasson J, Benediktsdottir KR. Adenocarcinoma of the prostate in Iceland: a population-based study of stage, Gleason grade, treatment and long-term survival in males diagnosed between 1983 and 1987. Scandinavian Journal of Urology and Nephrology. 2006;40:265-271
17. Immediate versus deferred treatment for advanced prostatic cancer: initial results of the Medical Research Council Trial. Medical Research Council (MRC) Prostate Cancer Working Party Investigators Group. Br J Urol. 1997;79:234-246.
18. Merseburger AS, Alcaraz A, von Klot CA. Androgen deprivation therapy as backbone therapy in the management of prostate cancer. Onco Targets and Therapy. 2016;9:7263-7274.
19. Norgaard M, Jensen AO, Jacobsen JB, Cetin K, Fryzek JP, Sorensen HT. Skeletal related events, bone metastasis and survival of prostate cancer: a population based cohort study in Denmark. J Urol. 2010;184:162-167.
20. Parker C, Gillessen S, Heidenreich A, Horwich A, on behalf of the ESMO Guidelines Committee. Cancer of the prostate: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Annals Oncol. 2015;26(suppl 5): v69-v77.
21. Quaglia A, Vercelli M, Puppo A. Prostate cancer in Italy before and during the 'PSA era': survival trend and prognostic determinants. Eur J Cancer Prev. 2003 Apr;12(2):145-52.
22. Smith MR. Osteoporosis in men with prostate cancer: now for the fracture data. J Clin Oncol. 2008;26 (27):4371- 4372.
23. Sweeney CJ, Chen Y-H, Carducci M, et al. Chemohormonal therapy in metastatic hormone-sensitive prostate cancer. N Engl J Med 2015; 373:737-746.
24. Watkins PB, Seligman PJ, Pears JS, Avigan MI, Senior JR. Using controlled clinical trials to learn more about acute drug-induced liver injury. Hepatology 2008;48:1680-1689.
25. Wilt T, Nair B, MacDonald R, Rutks I. Early vs. deferred androgen suppression in the treatment of advanced prostate cancer. Cochrane Database System Rev 2002;(1):CD 3506.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades Farmacodinâmicas
Mecanismo de ação
In vivo, o acetato de abiraterona é convertido em abiraterona, um inibidor da biossíntese de androgênios. Especificamente, abiraterona inibe seletivamente a enzima 17alfa-hidroxilase/C17,20-liase (CYP17). Esta enzima é expressa nos tecidos testicular, suprarrenal e do tumor prostático e é necessária para a biossíntese de androgênios nestes tecidos. Ela catalisa a conversão de pregnenolona e progesterona em precursores da testosterona, DHEA e androstenediona, respectivamente, pela 17 alfa-hidroxilação e clivagem da ligação C17, 20. A inibição da CYP17 também resulta em um aumento da produção de mineralocorticoides pelas suprarrenais (vide "Advertências e Precauções - Hipertensão, hipopotassemia e retenção hídrica devido ao excesso de mineralocorticoides").
O carcinoma prostático sensível aos androgênios responde ao tratamento que diminui os níveis de androgênios. Os tratamentos de privação de androgênios, tais como utilização de agonistas de LHRH ou orquiectomia, diminuem a produção de androgênio nos testículos, mas não afetam a produção de androgênios pelas suprarrenais ou no tumor. O tratamento com acetato de abiraterona diminui a testosterona sérica para níveis não detectáveis (utilizando análises comerciais) quando administrado com agonistas de LHRH (ou orquiectomia).
Efeitos farmacodinâmicos
O acetato de abiraterona diminui a testosterona sérica e outros androgênios a níveis menores que aqueles alcançados com o uso de agonistas de LHRH isolados ou pela orquiectomia. Isto é o resultado da inibição seletiva da enzima CYP17 necessária para a biossíntese de androgênios. O antígeno prostático específico (PSA) serve como um biomarcador em pacientes com câncer de próstata. Em um estudo clínico Fase 3 em pacientes que falharam à quimioterapia anterior com taxanos, 38% dos pacientes tratados com acetato de abiraterona tiveram declínio de pelo menos 50% nos níveis de PSA em relação à linha de base comparado a 10% dos pacientes tratados com placebo.
A concentração sérica de testosterona é reduzida dentro de 12 horas após a administração da primeira dose do medicamento.
Uso de Espironolactona
Nos estudos clínicos pivotais com acetato de abiraterona não se permitiu que os pacientes recebessem espironolactona uma vez que a mesma se liga ao receptor de androgênio e pode aumentar os níveis de PSA.
Propriedades Farmacocinéticas
Geral
Após a administração do acetato de abiraterona, a farmacocinética da abiraterona e do acetato de abiraterona foram estudadas em indivíduos saudáveis, pacientes com câncer de próstata avançado metastático e indivíduos sem câncer com insuficiência renal ou hepática. In vivo, o acetato de abiraterona é rapidamente convertido em abiraterona, um inibidor da biossíntese de androgênios (vide "Características Farmacológicas - Mecanismo de Ação").
Absorção
Depois da administração oral do acetato de abiraterona em jejum, o tempo para alcançar a concentração plasmática máxima da abiraterona é de aproximadamente 2 horas.
A administração do acetato de abiraterona com alimento, comparada com a administração em jejum, resulta em aumento de até 17 vezes na exposição sistêmica média da abiraterona, dependendo do conteúdo de gordura da refeição. Em função da variação comum do conteúdo e da composição das refeições, o uso do acetato de abiraterona com estas pode resultar em exposições altamente variáveis. Portanto, acetato de abiraterona não deve ser tomado com alimentos. O acetato de abiraterona deve ser tomado com o estômago vazio, pelo menos uma hora antes ou pelo menos duas horas após uma refeição. Os comprimidos devem ser deglutidos inteiros, com água (vide "Posologia e Modo de usar").
Distribuição e ligação às proteínas
No plasma humano, a ligação da 14C-abiraterona às proteínas plasmáticas é de 99,8%. O volume aparente de distribuição é de aproximadamente 5.630L, sugerindo que a abiraterona é extensivamente distribuída para os tecidos periféricos.
Estudos in vitro demonstraram que o acetato de abiraterona é um inibidor da glicoproteína-P (P-gp). O acetato de abiraterona pode aumentar a exposição a medicamentos administrados concomitantemente os quais são substratos da P-gp, entretanto como o acetato de abiraterona é rapidamente convertido em abiraterona, não se espera inibição sistêmica da P-gp.
Metabolismo
Após a administração oral de 14C-acetato de abiraterona na forma de cápsulas, o acetato de abiraterona é hidrolisado para abiraterona, a qual sofre metabolismo incluindo sulfatação, hidroxilação e oxidação, primariamente no fígado. A maior parte da radioatividade circulante (aproximadamente 92%) é encontrada na forma de metabólitos de abiraterona. Entre os 15 metabólitos detectáveis, dois metabólitos principais, sulfato de abiraterona e sulfato de N-óxido abiraterona, representam aproximadamente 43% da radioatividade total cada um.
Eliminação
A meia-vida média da abiraterona no plasma é de aproximadamente 15 horas, com base em dados de sujeitos sadios. Após a administração oral de 14C-acetato de abiraterona, aproximadamente 88% da dose radioativa é recuperada nas fezes e aproximadamente 5% na urina. Os principais compostos presentes nas fezes são o acetato de abiraterona inalterado e a abiraterona (aproximadamente 55% e 22% da dose administrada, respectivamente).
Populações especiais
Insuficiência hepática
A farmacocinética da abiraterona foi avaliada em indivíduos com insuficiência hepática leve ou moderada pré-existente (Classe A e B de Child-Pugh, respectivamente) e em indivíduos sadios controle. A exposição sistêmica à abiraterona depois de uma dose oral de 1000 mg aumentou em aproximadamente 11% e 260% em indivíduos com insuficiência hepática leve e moderada pré-existente, respectivamente. A meia-vida média da abiraterona é prolongada para aproximadamente 18 horas em indivíduos com insuficiência hepática leve e aproximadamente 19 horas em indivíduos com insuficiência hepática moderada. Nenhum ajuste de dose é necessário para pacientes com insuficiência hepática leve pré-existente. Não existem dados sobre a segurança e a eficácia de doses múltiplas de acetato de abiraterona quando administrados a pacientes com insuficiência hepática moderada ou grave (Child Pugh Classe B ou C). Não é possível prever o ajuste da dose. Zostide deve ser utilizado com precaução em pacientes com insuficiência hepática moderada e somente se o benefício compensar claramente o possível risco (vide "Posologia e Modo de Usar - Insuficiência hepática" e "Advertências e Precauções - Hepatotoxicidade e Insuficiência hepática"). Zostide não deve ser usado em pacientes com insuficiência hepática grave. Para pacientes que desenvolvem hepatotoxicidade durante o tratamento com Zostide, pode ser necessário suspender o tratamento e ajustar a dose (vide "Posologia e Modo de Usar - Insuficiência hepática" e "Advertências e Precauções - Hepatotoxicidade e Insuficiência hepática").
Pacientes com insuficiência renal
A farmacocinética da abiraterona foi comparada entre pacientes com doença renal terminal que estavam em um cronograma de hemodiálise estável e em indivíduos controle correspondentes, com função renal normal. A exposição sistêmica à abiraterona depois de uma dose oral única de 1000 mg não aumentou em pacientes com doença renal terminal em diálise.
A administração de acetato de abiraterona em pacientes com insuficiência renal, incluindo insuficiência renal grave, não requer redução da dose (vide "Posologia e Modo de Usar - Insuficiência renal").
Efeitos no intervalo QT
Em um estudo de segurança cardiovascular em pacientes com câncer de próstata avançado metastático não houve efeitos significativos do acetato de abiraterona sobre o intervalo QT/QTc.
Dados de segurança pré-clínicos
- Toxicidade reprodutiva
Em estudos de fertilidade em ratos machos e fêmeas, o acetato de abiraterona reduziu a fertilidade, o que foi completamente reversível em 4 a 16 semanas após a interrupção da administração do acetato de abiraterona.
Em um estudo de toxicidade do desenvolvimento nos ratos, o acetato de abiraterona afetou a gravidez, incluindo peso fetal reduzido e sobrevivência. Efeitos na genitália externa foram observados apesar do acetato de abiraterona não ser teratogênico.
Nestes estudos de fertilidade e toxicidade do desenvolvimento realizados em ratos, todos os efeitos foram relacionados à atividade farmacológica da abiraterona.
Zostide é contraindicado na gravidez (vide "Contraindicações").
- Carcinogênese e mutagenicidade
O acetato de abiraterona não foi carcinogênico em um estudo de 6 meses, em camundongo transgênico (Tg.rasH2). Em um estudo de carcinogenicidade de 24 meses em ratos, o acetato de abiraterona aumentou a incidência de neoplasias de células intersticiais dos testículos. Este resultado é considerado relacionado à ação farmacológica da abiraterona e a especificidade do rato. O acetato de abiraterona não foi carcinogênico em ratas.
O acetato de abiraterona e a abiraterona foram desprovidos de potencial genotóxico no painel padrão de testes de genotoxicidade, incluindo ensaio de mutação reversa bacteriana in vitro (teste de Ames), teste de aberração cromossômica de mamíferos in vitro (usando linfócitos humanos) e ensaio de micronúcleo de rato in vivo.
- Toxicologia animal
Em todos os estudos de toxicidade em animais, os níveis circulantes de testosterona foram reduzidos de forma significativa. Como resultado, foram observadas reduções no peso de órgãos e alterações morfológicas e/ou histopatológicas nos órgãos reprodutivos e nas glândulas suprarrenal, hipófise e mamária. Todas as alterações foram completas ou parcialmente reversíveis. As alterações nos órgãos reprodutivos e órgãos sensíveis aos androgênios são consistentes com a farmacologia da abiraterona. Todas as alterações hormonais relacionadas ao tratamento foram revertidas ou pareceram estar se resolvendo após um período de recuperação de 4 semanas.
4. CONTRAINDICAÇÕES
Este medicamento é contraindicado em mulheres grávidas ou que potencialmente possam estar grávidas (vide "Advertências e Precauções - Uso durante a gravidez").
Este medicamento é contraindicado em pacientes com hipersensibilidade à substância ativa ou qualquer excipiente presente na formulação.
Este medicamento é contraindicado em pacientes com insuficiência hepática grave.
Categoria X de gravidez. Este medicamento não deve ser utilizado por mulheres grávidas ou que possam ficar grávidas durante o tratamento.
5. ADVERTÊNCIAS E PRECAUÇÕES
Hipertensão, hipopotassemia e retenção hídrica devido ao excesso de mineralocorticoides
Zostide pode causar hipertensão, hipopotassemia e retenção hídrica (vide "Reações Adversas") como consequência dos níveis aumentados de mineralocorticoides resultantes da inibição da CYP17 (vide "Características Farmacológicas - Mecanismos de Ação"). A administração concomitante de um corticosteroide suprime o estímulo do hormônio adrenocorticotrópico (ACTH), resultando em redução da incidência e da gravidade destas reações adversas. É necessário ter cautela ao tratar paci