ZINPASS EZE
SANOFI MEDLEY
rosuvastatina + ezetimiba
Hipocolesterolemiante.
Apresentações.
Comprimidos revestidos de 10 mg + 10 mg: embalagem com 30 comprimidos. Comprimidos revestidos de 20 mg + 10 mg: embalagem com 30 comprimidos. Comprimidos revestidos de 40 mg + 10 mg: embalagem com 30 comprimidos.
USO ORAL.
USO ADULTO.
Composição.
ZINPASS® EZE 10 mg + 10 mg: cada comprimido revestido contém 10,40 mg de rosuvastatina cálcica equivalente a 10 mg de rosuvastatina base e 10 mg de ezetimiba.
ZINPASS® EZE 20 mg + 10 mg: cada comprimido revestido contém 20,80 mg de rosuvastatina cálcica equivalente a 20 mg de rosuvastatina base e 10 mg de ezetimiba.
ZINPASS® EZE 40 mg + 10 mg: cada comprimido revestido contém 41,60 mg de rosuvastatina cálcica equivalente a 40 mg de rosuvastatina base e 10 mg de ezetimiba.
Excipientes: lactose monoidratada, celulose microcristalina, laurilsulfato de sódio, povidona, croscarmelose sódica, dióxido de silício, estearato de magnésio, hipromelose, macrogol, dióxido de titânio, talco, óxido de ferro amarelo(1), óxido de ferro vermelho(2)
(1) exclusivo do comprimido de 20 mg + 10 mg
(2) exclusivo do comprimido de 40 mg + 10 mg
Informações técnicas.
1. INDICAÇÕES
ZINPASS® EZE é indicado como terapia adjuvante à dieta, em pacientes considerados como de alto ou muito alto risco cardiovascular, quando a resposta à dieta e aos exercícios é inadequada em pacientes adultos com hipercolesterolemia primária (familiar heterozigótica ou não-familiar) ou com dislipidemia mista. Em pacientes adultos com hipercolesterolemia ZINPASS® EZE é indicado para redução do LDL-colesterol, colesterol total e triglicérides elevados, diminuição de ApoB, não HDL-C, das razões LDLC/HDL-C, não HDL-C/HDL-C, ApoB/Apo A-I, C-total/HDL-C e aumento de HDL-C.
2. RESULTADOS DE EFICÁCIA
Estudos utilizando a administração concomitante de rosuvastatina com ezetimiba foram realizados mostrando sua eficácia e segurança na redução dos níveis de LDL C e outros parâmetros relativos ao perfil lipídico.
O estudo EXPLORER (EXamination of Potential Lipidmodifying effects Of Rosuvastatin in combination with Ezetimiba versus Rosuvastatin alone), que avaliou 469 pacientes com hipercolesterolemia e doença cardíaca coronariana, teve como objetivo comparar a eficácia e a segurança da dose mais eficaz de rosuvastatina(40 mg) administrada isoladamente ou em combinação com ezetimiba 10 mg durante 6 semanas em pacientes de alto risco de doença cardíaca coronariana com hipercolesterolemia. Um número significativamente maior de pacientes atingiu sua meta de LDL-C de acordo com o ATP III ( < 100 mg/dl, 94.0% vs 79.1%, p < 0.001) e meta opcional de LDL ( < 70 mg/dl) recebendo rosuvastatina/ezetimiba do que utilizando monoterapia com rosuvastatina para pacientes de alto-risco. A combinação de rosuvastatina/ezetimiba reduziu o LDL-C mais significativamente que a rosuvastatina (69.8% vs 57.1%, p < 0.001). Houve uma melhora significativa (p < 0.001) de outros componentes do perfil lipídico/lipoproteína com a combinação rosuvastatina/ezetimiba.
O estudo ACTE foi um ensaio clínico multicêntrico, de 6 semanas, randomizado, duplo-cego, de grupos paralelos que avaliou a segurança e eficácia de ezetimiba (10 mg) adicionado ao tratamento estável de rosuvastatina "versus" o aumento de dose de rosuvastatina de 5 para 10 mg ou de 10 para 20 mg. O estudo incluiu 440 indivíduos com risco moderadamente alto e alto para doença cardíaca coronária e níveis de colesterol LDL (LDL-C) maiores que o recomendado pelo ATP III ( < 100 mg/dl para risco moderadamente alto e risco alto sem doença vascular aterosclerótica ou < 70 mg/dl para indivíduos com alto risco com doença vascular aterosclerótica).. Para avaliação do desfecho primário do estudo foram avaliados os dados agrupados que demonstraram que o ezetimiba adicionado à rosuvastatina estável de 5 mg ou 10 mg reduziu o colesterol LDL em 21%. Em contraste, dobrando a dose da rosuvastatina para 10 mg ou 20 mg reduziu o colesterol LDL em 5,7% (diferença entre os grupos de 15,2%, p < 0,001). Individualmente, ezetimiba com rosuvastatina 5 mg reduziu mais o colesterol LDL do que se aumentar a rosuvastatina para 10 mg (diferença de 12,3%, p < 0,001), e ezetimiba em combinação com rosuvastatina de 10 mg reduziu mais o colesterol LDL do que se aumentar a rosuvastatina para 20 mg (diferença de 17,5%, p < 0,001). Em comparação com o aumento da dose da rosuvastatina, a combinação com ezetimiba obteve índices significativamente maiores de redução do colesterol LDL < 100 mg / dl (59,4% vs 30,9%, p < 0,001) e < 70 mg / dl em todos os indivíduos (43,8% vs 17,5%, p < 0,001); e também produziu uma redução significativamente maior no colesterol total, colesterol não-HDL e apolipoproteína B (p < 0,001) resultando em efeitos semelhantes em outros parâmetros lipídicos.
O estudo GRAVITY (Gauging the lipid effects of Rosuvastatin plus ezetimiba versus simvastatin plus ezetimiba therapy), avaliou a eficácia, segurança e efeito em biomarcadores do uso de rosuvastatina nas doses de 10 e 20 mg adicionadas ao ezetimiba 10 mg com a associação em dose fixa comercialmente disponível de sinvastatina 40 e 80 mg com ezetimiba 10 mg em pacientes com doença arterial coronária ou risco de doença arterial coronária equivalente. Neste estudo multicêntrico de 12 semanas, 833 pacientes foram randomizados a receber as combinações rosuvastatina 10 mg e ezetimiba 10 mg (RSV 10/EZT 10), rosuvastatina 20 mg e ezetimiba 10 mg (RSV 20/EZT 10), sinvastatina 40 mg e ezetimiba 10 mg (SIN 40/EZT 10) ou sinvastatina 80 mg e ezetimiba 10 mg (SIN 80/EZT 10). O desfecho primário foi a mudança no colesterol LDL comparando o valor na entrada no estudo com o valor após 12 semanas. Os pacientes do grupo RSV 20/EZT 10 tiveram seu colesterol LDL reduzido em 63,5% comparado a redução de 55,2% e 57,4% nos pacientes do grupo SIN40/EZE10 e SIN80/EZE10 (p < 0,001). Os pacientes do grupo RSV 10/EZT 10 tiveram redução do colesterol LDL de 59,7% o que foi significativamente maior do que a redução do grupo SIN40/EZE10 que foi de 55,2% (p < 0,002). Uma proporção significativamente (p ≤0,007) maior de pacientes atingiu as metas de LDL-C < 100 mg/dl e < 70 mg/dl com RSV20/EZE10 (95,5% e 77% respectivamente) vs. SIN40/EZE10 (87,4% e 55,3% respectivamente) e SIN80/EZE10 (88,6% e 67,7% respectivamente) e uma proporção significativamente (p =0,03) maior dos pacientes do grupo com RSV10/EZE10 (93,3%) atingiu a meta de colesterol LDL < 100 mg/dl vs. SIN40/EZE10 (87,4%).
Referências Bibliográficas
1. Ballantyne, C. M., et al & EXPLORER Study Investigators. Efficacy and safety of rosuvastatin 40 mg alone or in combination with ezetimibe in patients at high risk of cardiovascular disease (results from the EXPLORER study).. Am J Cardiol.. 2007 Mar 1;99(5):673-80.
2. Bays HE. et al. Safety and efficacy of ezetimibe added on to rosuvastatin 5 or 10 mg versus up-titration of rosuvastatin in patients with hypercholesterolemia (the ACTE Study). Am J Cardiol. 2011 Aug 15;108(4):523-30.
3. Ballantyne CM, Hoogeveen RC, Raya JL, Cain VA, Palmer MK, Karlson BW; GRAVITY Study Investigators. Efficacy, safety and effect on biomarkers related to cholesterol and lipoprotein metabolism of rosuvastatin 10 or 20 mg plus ezetimibe 10 mg vs. simvastatin 40 or 80 mg plus ezetimibe 10 mg in high-risk patients: Results of the GRAVITY randomized study. Atherosclerosis. 2014 Jan;232(1):86-93.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades farmacodinâmicas
Grupo farmacoterapêutico: inibidores de HMG-CoA redutase em combinação com outros agentes modificadores de lipídios
Código ATC: C10BA06 rosuvastatina e ezetimiba
ZINPASS® EZE (rosuvastatina cálcica + ezetimiba) é um produto hipolipemiante que inibe seletivamente a absorção intestinal de colesterol e esteróis vegetais relacionados e inibe a síntese endógena de colesterol.
Mecanismo de ação:
O colesterol plasmático é derivado da absorção intestinal e síntese endógena. ZINPASS® EZE contém ezetimiba e rosuvastatina, dois compostos hipolipemiantes com mecanismos de ação complementares. Este medicamento reduz o colesterol total elevado (total-C), LDL-C, apolipoproteína B (Apo B), triglicerídeos (TG) e colesterol de lipoproteína de densidade não alta (C-não-HDL) e aumenta o colesterol de lipoproteína de alta densidade (HDL-C) através da dupla inibição da absorção e síntese de colesterol.
-ezetimiba
A ezetimiba inibe a absorção intestinal do colesterol. A ezetimiba é oralmente ativa e tem um mecanismo de ação que difere de outras classes de compostos hipocolesterolêmicos (p. ex., estatinas, sequestrantes de ácido biliar [resinas], derivados de ácido fíbrico e estanóis vegetais). O alvo molecular de ezetimiba é o transportador de esteróis, Niemann-Pick C1-Like 1 (NPC1L1), que é responsável pela absorção intestinal de colesterol e fitoesteróis.
A ezetimiba localiza-se na borda em escova do intestino delgado e inibe a absorção de colesterol, levando a uma diminuição no fornecimento de colesterol intestinal ao fígado; as estatinas reduzem a síntese de colesterol no fígado e, em conjunto, esses mecanismos distintos proporcionam redução complementar do colesterol. Em um estudo clínico de 2 semanas em 18 pacientes hipercolesterolêmicos, a ezetimiba inibiu a absorção intestinal de colesterol em 54%, em comparação com o placebo.
Uma série de estudos pré-clínicos foi realizada para determinar a seletividade da ezetimiba para inibir a absorção de colesterol. A ezetimiba inibiu a absorção de [14C]-colesterol sem efeito sobre a absorção de triglicerídeos, ácidos graxos, ácidos biliares, progesterona, etinilestradiol ou vitaminas lipossolúveis A e D.
-rosuvastatina
A rosuvastatina é um inibidor seletivo e competitivo de HMG-CoA redutase, a enzima limitante de taxa que converte a 3-hidroxi-3-metilglutaril coenzima A em mevalonato, um precursor do colesterol. O principal local de ação da rosuvastatina é o fígado, o órgão alvo para a redução do colesterol. A rosuvastatina aumenta o número de receptores hepáticos ao LDL na superfície celular, aumentando a captação e o catabolismo de LDL e inibe a síntese hepática de VLDL, reduzindo assim o número total de partículas de VLDL e LDL.
Efeitos farmacodinâmicos
A rosuvastatina reduz o colesterol LDL, o colesterol total e os triglicerídeos elevados, e aumenta o colesterol-HDL. Também reduz ApoB, C-não-HDL, C-VLDL, TG-VLDL e aumenta ApoA-I (veja a Tabela 1). A rosuvastatina também reduz as taxas de LDL-C/HDL-C, C-total/HDL-C e C-não-HDL/HDL-C e as proporções de ApoB/ApoA-I.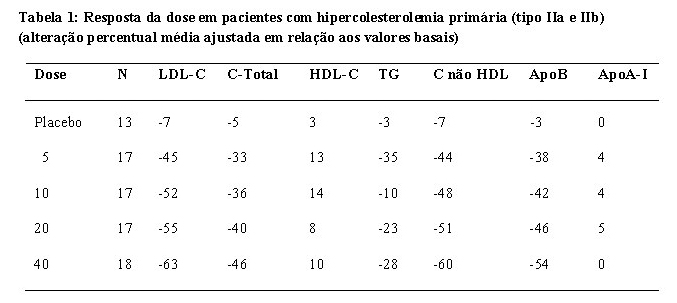
Propriedades farmacocinéticas
Não existe uma interação farmacocinética substancial entre os dois componentes desta preparação de dose fixa.
Os valores médios de ASC e Cmax para ezetimiba e rosuvastatina total não foram diferentes entre os grupos de monoterapia e coadministração de rosuvastatina 10 mg e ezetimiba 10 mg.
Absorção
-ezetimiba
Após a administração oral, a ezetimiba é rapidamente absorvida e amplamente conjugada com um glicuronídeo fenólico farmacologicamente ativo (ezetimiba-glicuronídeo). As concentrações plasmáticas máximas (Cmax) médias ocorrem dentro de 1 a 2 horas para o grupo de ezetimiba-glicuronídeo e de 4 a 12 horas para o grupo de ezetimiba. A biodisponibilidade absoluta da ezetimiba não pode ser determinada porque o composto é praticamente insolúvel em meio aquoso adequado para injeção.
A administração concomitante de alimentos (refeições ricas ou pobres em gordura) não apresentou efeito sobre a biodisponibilidade oral de ezetimiba quando administrada como comprimidos de ezetimiba 10 mg. A ezetimiba pode ser administrada com ou sem alimentos.
-rosuvastatina
As concentrações plasmáticas máximas de rosuvastatina são alcançadas aproximadamente 5 horas após a administração oral. A biodisponibilidade absoluta é de aproximadamente 20%.
Distribuição
-ezetimiba
A ezetimiba e ezetimiba-glicuronídeo estão ligadas 99,7% e 88 a 92% a proteínas plasmáticas humanas, respectivamente.
-rosuvastatina
A rosuvastatina é amplamente absorvida pelo fígado, que é o local primário da síntese de colesterol e depuração de LDL-C. O volume de distribuição da rosuvastatina é de aproximadamente 134 L.
Aproximadamente 90% da rosuvastatina está ligada às proteínas plasmáticas, principalmente à albumina.
Biotransformação
-ezetimiba
A ezetimiba é metabolizada principalmente no intestino delgado e no fígado através da conjugação com glicuronídeo (uma reação de fase II) com excreção biliar subsequente. O metabolismo oxidativo mínimo (uma reação de fase I) foi observado em todas as espécies avaliadas. A ezetimiba e ezetimibaglicuronídeo são os principais compostos derivados de fármacos detectados no plasma, constituindo aproximadamente 10 a 20% e 80 a 90% do fármaco total no plasma, respectivamente. Tanto a ezetimiba como a ezetimiba-glicuronídeo são lentamente eliminadas do plasma com evidência de reciclagem entero-hepática significativa. A meia-vida da ezetimiba e ezetimiba-glicuronídeo é de aproximadamente 22 horas.
-rosuvastatina
A rosuvastatina sofre um metabolismo limitado (aproximadamente 10%). Estudos de metabolismo "in vitro" usando hepatócitos humanos indicam que a rosuvastatina é um substrato fraco para o metabolismo com base no citocromo P450. O CYP2C9 foi a principal isoenzima envolvida, com 2C19, 3A4 e 2D6 envolvidos em menor grau. Os principais metabolitos identificados são os metabólitos N-desmetil e lactona. O metabólito N-desmetil é aproximadamente 50% menos ativo do que a rosuvastatina, enquanto que a forma de lactona é considerada clinicamente inativa. A rosuvastatina representa mais de 90% da atividade circulante do inibidor de HMG-CoA redutase.
Eliminação
-ezetimiba
Após a administração oral de 14C-ezetimiba (20 mg) a sujeitos humanos, a ezetimiba total respondeu por aproximadamente 93% da radioatividade total no plasma. Aproximadamente 78% e 11% da radioatividade administrada foram recuperados nas fezes e urina, respectivamente, durante um período de coleta de 10 dias. Após 48 horas, não houve níveis detectáveis de radioatividade no plasma.
-rosuvastatina
Aproximadamente 90% da dose de rosuvastatina é excretada inalterada nas fezes (consistindo do princípio ativo absorvido e não absorvido) e a parte restante é excretada na urina. Aproximadamente 5% é excretada inalterada na urina. A meia-vida de eliminação plasmática é de aproximadamente 19 horas. A meia-vida de eliminação não aumenta em doses mais elevadas. A média geométrica de depuração plasmática é de aproximadamente 50 litros/hora (coeficiente de variação de 21,7%). Tal como acontece com outros inibidores de HMG-CoA redutase, a captação hepática de rosuvastatina envolve o transportador de membrana OATP-C. Este transportador é importante na eliminação hepática da rosuvastatina.
Linearidade: a exposição sistêmica da rosuvastatina aumenta de forma proporcional à dose. Não há alterações nos parâmetros farmacocinéticos após múltiplas doses diárias.
Populações especiais:
Insuficiência hepática
-ezetimiba
Após uma única dose de 10 mg de ezetimiba, a ASC média da ezetimiba total aumentou aproximadamente 1,7 vezes em pacientes com insuficiência hepática leve (pontuação de Child Pugh 5 ou 6) em comparação com sujeitos saudáveis. Em um estudo de doses múltiplas de 14 dias (10 mg por dia) em pacientes com insuficiência hepática moderada (pontuação de Child Pugh 7 a 9), a ASC média para ezetimiba total foi aumentada aproximadamente 4 vezes no Dia 1 e Dia 14 em comparação com os sujeitos saudáveis. Não é necessário ajuste de dose para pacientes com insuficiência hepática leve. Devido aos efeitos desconhecidos do aumento da exposição à ezetimiba em pacientes com insuficiência hepática moderada ou grave (pontuação de Child Pugh > 9), a ezetimiba não é recomendada nestes pacientes (veja a seção "Advertências e precauções").
-rosuvastatina
Em um estudo com sujeitos com diferentes graus de insuficiência hepática, não houve evidência de exposição aumentada à rosuvastatina em sujeitos com pontuações de Child-Pugh 7 ou inferior. No entanto, dois sujeitos com pontuação Child-Pugh 8 e 9 mostraram um aumento na exposição sistêmica de pelo menos 2 vezes em comparação com sujeitos com pontuações menores de Child-Pugh. Não há experiência em sujeitos com pontuação de Child-Pugh acima de 9.
Insuficiência renal
-ezetimiba
Após uma única dose de 10 mg de ezetimiba em pacientes com doença renal grave (n=8, CrCl média de ≤30 mL/min/1,73m2), a ASC média para a ezetimiba total foi aumentada em aproximadamente 1,5 vezes, em comparação com sujeitos saudáveis (n=9). Este resultado não é considerado clinicamente significativo. Não é necessário ajuste de dose para pacientes com problemas renais.
Um paciente adicional neste estudo (pós-transplante renal e recebendo vários medicamentos, incluindo ciclosporina) apresentou uma exposição 12 vezes maior em relação à ezetimiba total.
-rosuvastatina
Em um estudo em sujeitos com diferentes graus de insuficiência renal, a doença renal leve a moderada não teve influência sobre a concentração plasmática de rosuvastatina ou o metabólito N-desmetil. Os sujeitos com insuficiência grave (CrCl < 30 mL/min) tiveram um aumento de 3 vezes na concentração plasmática e um aumento de 9 vezes na concentração do metabólito N-desmetil em comparação com voluntários saudáveis. As concentrações plasmáticas no estado estacionário de rosuvastatina em sujeitos submetidos à hemodiálise foram aproximadamente 50% maiores em comparação com voluntários saudáveis.
Idade e sexo
-ezetimiba
As concentrações plasmáticas para ezetimiba total são aproximadamente 2 vezes maiores nos idosos (≥ 65 anos) do que nos jovens (18 a 45 anos). A redução do LDL-C e o perfil de segurança são comparáveis entre indivíduos idosos e jovens tratados com ezetimiba. Portanto, não é necessário ajuste de dose nos idosos.
As concentrações plasmáticas para ezetimiba total são ligeiramente maiores (aproximadamente 20%) em mulheres do que em homens. A redução do LDL-C e o perfil de segurança são comparáveis entre homens e mulheres tratados com ezetimiba. Portanto, não é necessário ajuste de dose com base no sexo.
-rosuvastatina
Não houve efeito clinicamente relevante de idade ou sexo na farmacocinética da rosuvastatina em adultos.
Raça
-rosuvastatina
Estudos farmacocinéticos mostram uma elevação aproximada de 2 vezes na ASC média e Cmax em sujeitos asiáticos (japonês, chinês, filipino, vietnamita e coreano) em comparação com os caucasianos. Os indo-asiáticos mostram uma elevação aproximada de 1,3 vezes na ASC mediana e Cmax. Uma análise farmacocinética populacional não revelou diferenças clinicamente relevantes na farmacocinética entre os grupos caucasianos e negros.
Polimorfismos genéticos
-rosuvastatina
A disposição dos inibidores de HMG-CoA redutase, incluindo a rosuvastatina, envolve proteínas transportadoras OATP1B1 e BCRP. Em pacientes com polimorfismos genéticos SLCO1B1 (OATP1B1) e/ou ABCG2 (BCRP) existe o risco de aumento da exposição à rosuvastatina. Os polimorfismos individuais de SLCO1B1 c.521CC e ABCG2 c.421AA estão associados a uma maior exposição à rosuvastatina (ASC) em comparação com os genótipos SLCO1B1 c.521TT ou ABCG2 c.421CC. Esta genotipagem específica não está estabelecida na prática clínica, mas para pacientes que são conhecidos por esses tipos de polimorfismos, recomenda-se uma dose diária menor de rosuvastatina.
Dados de segurança pré-clínica
Em estudos de coadministração com ezetimiba e estatinas, os efeitos tóxicos observados foram essencialmente aqueles tipicamente associados com as estatinas. Alguns dos efeitos tóxicos foram mais pronunciados do que os observados durante o tratamento com estatinas isoladas. Isto é atribuído às interações farmacocinéticas e farmacodinâmicas na terapia de coadministração. Nenhuma dessas interações ocorreu nos estudos clínicos. As miopatias ocorreram em ratos somente após a exposição às doses que foram várias vezes superiores à dose terapêutica humana (aproximadamente 20 vezes o nível de ASC para estatinas e 500 a 2.000 vezes o nível de ASC para os metabólitos ativos).
A coadministração de ezetimiba e de estatinas não foi teratogênica em ratos. Em coelhas prenhas, observou-se um pequeno número de deformidades esqueléticas (vértebra torácica e caudal fundida, número reduzido de vértebras caudais).
Em uma série de ensaios "in vivo" e "in vitro", a ezetimiba, administrada isoladamente ou coadministrada com estatinas, não apresentou potencial genotóxico.
-ezetimiba
Estudos em animais sobre a toxicidade crônica de ezetimiba identificaram órgãos alvo para efeitos tóxicos. Em cães tratados durante quatro semanas com ezetimiba (0,03 mg/kg/dia), a concentração de colesterol na bile cística foi aumentada em um fator de 2,5 a 3,5. No entanto, em um estudo de um ano em cães com doses de até 300 mg/kg/dia, não foi observada incidência aumentada de colelitíase ou outros efeitos hepatobiliares. O significado desses dados para humanos não é conhecido. Um risco litogênico associado ao uso terapêutico de ezetimiba não pode ser descartado.
Os testes de carcinogenicidade em longo prazo sobre a ezetimiba foram negativos.
A ezetimiba não apresentou efeito sobre a fertilidade de ratos machos ou fêmeas, nem foi considerado teratogênico em ratos ou coelhos, nem afetou o desenvolvimento pré-natal ou pós-natal. A ezetimiba atravessou a barreira placentária em ratas e coelhas prenhas que receberam doses múltiplas de 1.000 mg/kg/dia.
-rosuvastatina
Os dados pré-clínicos não revelam riscos especiais para os seres humanos com base nos estudos convencionais de farmacologia de segurança, genotoxicidade e potencial de carcinogenicidade. Testes específicos para os efeitos sobre o hERG não foram avaliados. As reações adversas não observadas em estudos clínicos, mas observadas em animais em níveis de exposição semelhantes aos níveis de exposição clínica foram as seguintes: nos estudos de toxicidade de doses repetidas, foram observadas alterações hepáticas histopatológicas provavelmente devido à ação farmacológica da rosuvastatina em camundongos, ratos e em menor grau com efeitos sobre a vesícula biliar em cães, mas não em macacos. Além disso, a toxicidade testicular foi observada em macacos e cães com doses mais elevadas. A toxicidade reprodutiva foi evidente em ratos, com reduções dos tamanhos da ninhada, peso da ninhada e sobrevida dos filhotes observados em doses tóxicas para a mãe, onde as exposições sistêmicas estavam várias vezes acima do nível de exposição terapêutica.
4. CONTRAINDICAÇÕES
Este medicamento é contraindicado em casos de:
• Hipersensibilidade às substâncias ativas ou a qualquer um dos excipientes listados na seção "Composição".
• Gravidez e amamentação e em mulheres em idade fértil que não utilizam medidas de contracepção adequadas (veja a seção "Advertências e precauções").
• Doença hepática ativa ou elevações persistentes sem explicação nas transaminases séricas e qualquer elevação de transaminases sérica superior a 3x o limite superior de normalidade (LSN) (veja a seção "Advertências e precauções").
• Em pacientes com insuficiência renal grave ("clearance" de creatinina < 30 mL/min).
• Em pacientes com miopatia (veja a seção "Advertências e precauções").
• Em pacientes que recebem ciclosporina concomitante (veja a seção "Interações medicamentosas").
A dose de 40 mg/10 mg é contraindicada em pacientes com fatores de predisposição para miopatia/rabdomiólise. Tais fatores incluem:
• Insuficiência renal moderada ("clearance" de creatinina < 60 mL/min).
• Hipotiroidismo.
• Histórico pessoal ou familiar de distúrbios musculares hereditários.
• Histórico anterior de toxicidade muscular com outro inibidor de HMG-CoA redutase ou fibrato.
• Abuso de álcool.
• Situações em que possa ocorrer um aumento nos níveis plasmáticos de rosuvastatina (uso concomitante com medicamentos que são inibidores das proteínas hepáticas OATP1B1 e BCRP como por exemplo inibidores de protease incluindo combinações de ritonavir com atazanavir, lopinavir e/ou tipranavir e pacientes com polimorfismos genéticos em SLCO1B1 (OATP1B1) e/ou ABCG2 (BCRP) e genótipos c.521CC ou c.421AA.)
• Pacientes asiáticos.
• Uso concomitante de fibratos.
Este medicamento é contraindicado para menores de 18 anos.
Categoria de risco na gravidez: X.
Este medicamento não deve ser utilizado por mulheres grávidas ou que possam ficar grávidas durante o tratamento.
5. ADVERTÊNCIAS E PRECAUÇÕES
Efeitos no músculo esquelético
Na experiência pós-comercialização com ezetimiba, foram relatados casos de miopatia e rabdomiólise. A maioria dos pacientes que desenvolveram rabdomiólise estava tomando estatina concomitantemente com ezetimiba. No entanto, a rabdomiólise tem sido relatada muito raramente com a monoterapia com ezetimiba e muito raramente com a adição de ezetimiba a outros agentes conhecidos por estarem associados ao aumento do risco de rabdomiólise.
Efeitos sobre o músculo esquelético, p. ex., mialgia, miopatia e, raramente, rabdomiólise foram relatados em pacientes tratados com rosuvastatina com todas as doses e, em particular, com doses > 20 mg. Foram notificados casos muito raros de rabdomiólise com o uso de ezetimiba em combinação com inibidores de HMG-CoA redutase. Uma interação farmacodinâmica não pode ser excluída (veja a seção "Interações medicamentosas") e deve-se ter cuidado com o seu uso combinado.
Tal como acontece com outros inibidores de HMG-CoA redutase, a taxa de notificação de rabdomiólise associada à rosuvastatina no uso pós-comercialização é maior na dose de 40 mg.
Se houver suspeita de miopatia com base nos sintomas musculares, ou for confirmada por um nível de creatina fosfoquinase (CPK) > 10 vezes o LSN, ZINPASS® EZE e qualquer um desses outros agentes que o paciente estiver tomando concomitantemente devem ser imediatamente descontinuados. Todos os pacientes que iniciam a terapia com ZINPASS® EZE devem ser informados sobre o risco de miopatia e devem informar imediatamente qualquer dor muscular inexplicável, sensibilidade ou fraqueza (veja a seção "Reações adversas").
Exames Laboratoriais:
Medição da creatina quinase
A creatina quinase (CK) não deve ser medida após exercício extenuante ou na presença de uma causa alternativa plausível de aumento de CK que possa confundir a interpretação do resultado. Se os níveis de CK estiverem significativamente elevados no período basal ( > 5xLSN), um teste de confirmação deve ser realizado dentro de 5-7 dias. Se o teste de repetição confirmar uma CK basal > 5xLSN, o tratamento não deve ser iniciado.
Enzimas hepáticas
Nos estudos clínicos controlados de coadministração em pacientes que receberam ezetimiba com estatina, observaram-se elevações consecutivas de transaminases (≥3 × o limite superior de normalidade [LSN]).
Quando a ezetimiba é administrada concomitantemente com rosuvastatina, os testes de função hepática devem ser realizados no início da terapia e, posteriormente, quando indicado clinicamente (veja a seção "Reações adversas").
Tal como acontece com outros inibidores de HMG-CoA redutase, a rosuvastatina deve ser utilizada com precaução em pacientes que consomem quantidades excessivas de álcool e/ou tenham histórico de doença hepática. Recomenda-se que os testes de função hepática sejam realizados antes e 3 meses após o início do tratamento. A rosuvastatina deve ser interrompida ou a dose reduzida se o nível de transaminases séricas for superior a 3 vezes o limite superior de normalidade. A taxa de notificação para eventos hepáticos graves (consistindo principalmente de transaminases hepáticas aumentadas) no uso pós-comercialização é maior na dose de 40 mg.
Em pacientes com hipercolesterolemia secundária causada por hipotireoidismo ou síndrome nefrótica, a doença subjacente deve ser tratada antes de iniciar a terapia com rosuvastatina.
Antes do tratamento
Deve-se ter cuidado em pacientes com fatores de predisposição para miopatia/rabdomiólise. Tais fatores incluem:
- insuficiência renal,
- hipotireoidismo,
- histórico pessoal ou familiar de distúrbios musculares hereditários,
- histórico prévio de toxicidade muscular com outro inibidor de HMG-CoA redutase ou fibrato,
- abuso de álcool,
- idade > 70 anos,
- situações em que possa ocorrer um aumento nos níveis plasmáticos,
- uso concomitante de fibratos.
Em tais pacientes, o risco do tratamento deve ser considerado em relação ao possível benefício e recomenda-se o monitoramento clínico. Se os níveis de CK estiverem significativamente elevados no período basal ( > 5xLSN), o tratamento não deve ser iniciado.
Durante o tratamento
Os pacientes devem ser solicitados a relatar dor muscular inexplicável, fraqueza ou cãibras imediatamente, particularmente se estiver associada a mal-estar ou febre. Os níveis de CK devem ser medidos nesses pacientes. A terapia deve ser descontinuada se os níveis de CK estiverem acentuadamente elevados ( > 5xLSN) ou se os sintomas musculares forem graves e causarem desconforto diário (mesmo que os níveis de CK sejam < /=5xLSN). Se os sintomas se resolvem e os níveis de CK retornarem ao normal, deve-se considerar a reintrodução da rosuvastatina ou um inibidor alternativo de HMG-CoA redutase na menor dose com monitoramento próximo do paciente. O monitoramento rotineiro dos níveis de CK em pacientes assintomáticos não é garantido. Houve relatos muito raros de uma miopatia necrosante imunomediada (IMNM) durante ou após o tratamento com estatinas, incluindo a rosuvastatina.
A IMNM é clinicamente caracterizada por fraqueza muscular proximal e creatina quinase elevada no sangue, que persistem apesar da interrupção do tratamento com estatina.
Nos estudos clínicos, não houve evidência de aumento dos efeitos do músculo esquelético no pequeno número de pacientes que receberam rosuvastatina e terapia concomitante. No entanto, foi observado um aumento na incidência de miosite e de miopatia em pacientes que receberam outros inibidores de HMG-CoA redutase, juntamente com derivados do ácido fíbrico, incluindo genfibrozila, ciclosporina, ácido nicotínico, antifúngicos azólicos, inibidores de protease e antibióticos macrolídeos.
A genfibrozila aumenta o risco de miopatia quando administrada concomitantemente com alguns inibidores de HMG-CoA redutase. Portanto, a combinação de rosuvastatina e genfibrozila não é recomendada. O benefício de novas alterações nos níveis lipídicos pelo uso combinado de rosuvastatina com fibratos ou niacina deve ser cuidadosamente ponderado com os riscos potenciais de tais combinações. A dose de 40 mg de rosuvastatina é contraindicada com o uso concomitante de fibrato. ZINPASS® EZE não deve ser utilizado em nenhum paciente com uma condição grave e aguda sugestiva de miopatia ou predisposição ao desenvolvimento de insuficiência renal secundária à rabdomiólise (p. ex., sepse, hipotensão, cirurgia de grande porte, traumatismo, distúrbios metabólicos, endócrinos e eletrolíticos graves; ou convulsões descontroladas).
Insuficiência hepática
Devido aos efeitos desconhecidos da exposição aumentada à ezetimiba em pacientes com insuficiência hepática moderada ou grave, ZINPASS® EZE não é recomendado (veja a seção "Propriedades farmacológicas").
Efeitos renais
A proteinúria, detectada pelo teste com tira reagente e principalmente de origem tubular, foi observada em pacientes tratados com doses maiores de rosuvastatina, em particular 40 mg, onde era transitório ou intermitente na maioria dos casos. A proteinúria não mostrou ser preditiva de doença renal aguda ou progressiva (veja a seção "Reações adversas"). A taxa de notificação para os eventos renais graves em uso pós-comercialização é maior na dose de 40 mg. Uma avaliação da função renal deve ser considerada durante o acompanhamento de rotina dos pacientes tratados com uma dose de 40 mg (veja o item "8. Posologia e Modo de Usar").
Diabetes mellitus
Algumas evidências sugerem que as estatinas como uma classe aumentam a glicemia e em alguns pacientes, com alto risco de diabetes no futuro, podem produzir um nível de hiperglicemia onde o tratamento formal do diabetes é apropriado. Este risco, no entanto, é superado pela redução do risco vascular com estatinas e, portanto, não deve ser um motivo para parar o tratamento com estatina. Os pacientes em risco (glicemia de jejum de 5,6 a 6,9 mmol/L, IMC > 30 kg/m2, triglicerídeos aumentados, hipertensão) devem ser monitorados de forma clínica e bioquímica de acordo com as diretrizes nacionais.
No estudo JUPITER, a frequência global relatada de diabetes mellitus foi de 2,8% no grupo da rosuvastatina e 2,3% no grupo do placebo, principalmente em pacientes com glicemia em jejum de 5,6 a 6,9 mmol/L.
Doença pulmonar intersticial
Foram relatados casos excepcionais de doença pulmonar intersticial com algumas estatinas, especialmente com terapia em longo prazo (veja a seção "Reações adversas"). As características de apresentação podem incluir dispneia, tosse não produtiva e deterioração da saúde geral (fadiga, perda de peso e febre). Caso haja a suspeita de que um paciente desenvolveu doença pulmonar intersticial, a terapia com estatinas deve ser descontinuada.
Reações adversas cutâneas graves
Reação medicamentosa com eosinofilia e sintomas sistêmicos (DRESS) e síndrome de Stevens-Johnson (SJS) foram relatados em associação ao tratamento com ZINPASS® EZE. Os pacientes devem ser informados sobre os sinais e sintomas de manifestações cutâneas graves e monitorados de perto. O tratamento deve ser interrompido ao primeiro aparecimento de erupção cutânea, lesões nas mucosas ou qualquer outro sinal de hipersensibilidade cutânea.
Inibidores de protease
O aumento da exposição sistêmica à rosuvastatina foi observado em sujeitos que receberam rosuvastatina concomitantemente com vários inibidores de protease em combinação com ritonavir. Deve-se considerar o benefício do efeito hipolipemiante pelo uso de rosuvastatina em pacientes com HIV que recebem inibidores de protease e o potencial de aumento das concentrações plasmáticas de rosuvastatina ao iniciar e até a titulação de doses de rosuvastatina em pacientes tratados com inibidores de protease. O uso concomitante com certos inibidores de protease não é recomendado a menos que a dose de rosuvastatina seja ajustada.
Fibratos
A coadministração de ZINPASS® EZE com fibratos não foi estudada, portanto, a coadministração de ZINPASS® EZE e fibratos não é recomendada.
Anticoagulantes
Se ZINPASS® EZE for adicionado à varfarina, outro anticoagulante cumarínico, ou fluindiona, a Razão Normalizada Internacional (INR) deve ser monitorada adequadamente (veja a seção "Interações medicamentosas".
Ácido fusídico
ZINPASS® EZE não deve ser coadministrado com formulações sistêmicas de ácido fusídico ou no prazo de 7 dias após a interrupção do tratamento com ácido fusídico. Em pacientes onde o uso de ácido fusídico sistêmico é considerado essencial, o tratamento com estatina deve ser descontinuado durante todo o período de tratamento com ácido fusídico. Houve relatos de rabdomiólise (incluindo algumas fatalidades) em pacientes que receberam ácido fusídico e estatinas em combinação (veja a seção "Interações medicamentosas"). O paciente deve ser aconselhado a procurar aconselhamento médico imediatamente se apresentarem sintomas de fraqueza, dor ou sensibilidade muscular.
A terapia com estatinas pode ser reintroduzida sete dias após a última dose de ácido fusídico.
Em circunstâncias excepcionais, onde o ácido fusídico sistêmico prolongado é necessário, p. ex., para o tratamento de infecções graves, a necessidade de coadministração de ZINPASS® EZE e ácido fusídico só deve ser considerada caso a caso e sob supervisão médica rigorosa.
Raça
Estudos farmacocinéticos mostram um aumento na exposição da rosuvastatina em sujeitos asiáticos em comparação com os caucasianos.
População pediátrica
ZINPASS® EZE não é recomendado para uso em crianças e adolescentes com menos de 18 anos de idade, devido a dados insuficientes sobre segurança e eficácia (veja a seção "Propriedades farmacológicas"). Pacientes com problemas hereditários raros de intolerância à galactose, deficiência de lactase de Lapp ou mal absorção de glicose-galactose não devem tomar este medicamento.
Este medicamento contém LACTOSE.
Fertilidade, gravidez e aleitamento
ZINPASS® EZE é contraindicado durante a gravidez e a lactação. As mulheres em idade fértil devem usar medidas de contracepção apropriadas.
Gravidez
Não há dados clínicos disponíveis sobre o uso de ezetimiba durante a gravidez. Estudos em animais sobre o uso de ezetimiba em monoterapia não mostraram evidências de efeitos nocivos diretos ou indiretos sobre a gravidez, desenvolvimento embriofetal, nascimento ou desenvolvimento pós-natal.
Uma vez que o colesterol e outros produtos da biossíntese de colesterol são essenciais para o desenvolvimento do feto, o risco potencial da inibição de HMG-CoA redutase supera a vantagem do tratamento durante a gravidez. Estudos em animais fornecem evidências limitadas de toxicidade reprodutiva. Se uma paciente ficar grávida durante o uso de rosuvastatina, o tratamento deve ser descontinuado imediatamente.
Amamentação
Estudos em ratos mostraram que a ezetimiba é secretada no leite materno. Não se sabe se a ezetimiba é secretada no leite materno humano.
A rosuvastatina é excretada no leite de ratos. Não há dados relativos à excreção no leite em seres humanos.
Fertilidade
Não há dados de ensaios clínicos disponíveis sobre os efeitos da ezetimiba na fertilidade humana. A ezetimiba não teve efeitos sobre a fertilidade de ratos machos ou fêmeas.
Efeitos sobre a capacidade de dirigir e usar máquinas
Não foram realizados estudos sobre os efeitos sobre a capacidade de dirigir e usar máquinas. No entanto, ao dirigir veículos ou operar máquinas, deve-se levar em consideração que tonturas foram relatadas.
Pacientes idosos
Rosuvastatina: como a idade avançada (≥65 anos) é um fator predisponente para miopatia por estatina, ZINPASS® EZE deve ser prescrito com cautela a idosos. Não é necessário, entretanto, ajuste posológico para pacientes idosos.
Ezetimiba: a concentração plasmática da ezetimiba total é, aproximadamente, 2 vezes mais elevada nos indivíduos idosos ( > 65 anos de idade) em relação aos jovens (18 a 45 anos de idade). A redução de LDL-C e o perfil de segurança são comparáveis em indivíduos idosos e jovens que recebem ezetimiba.
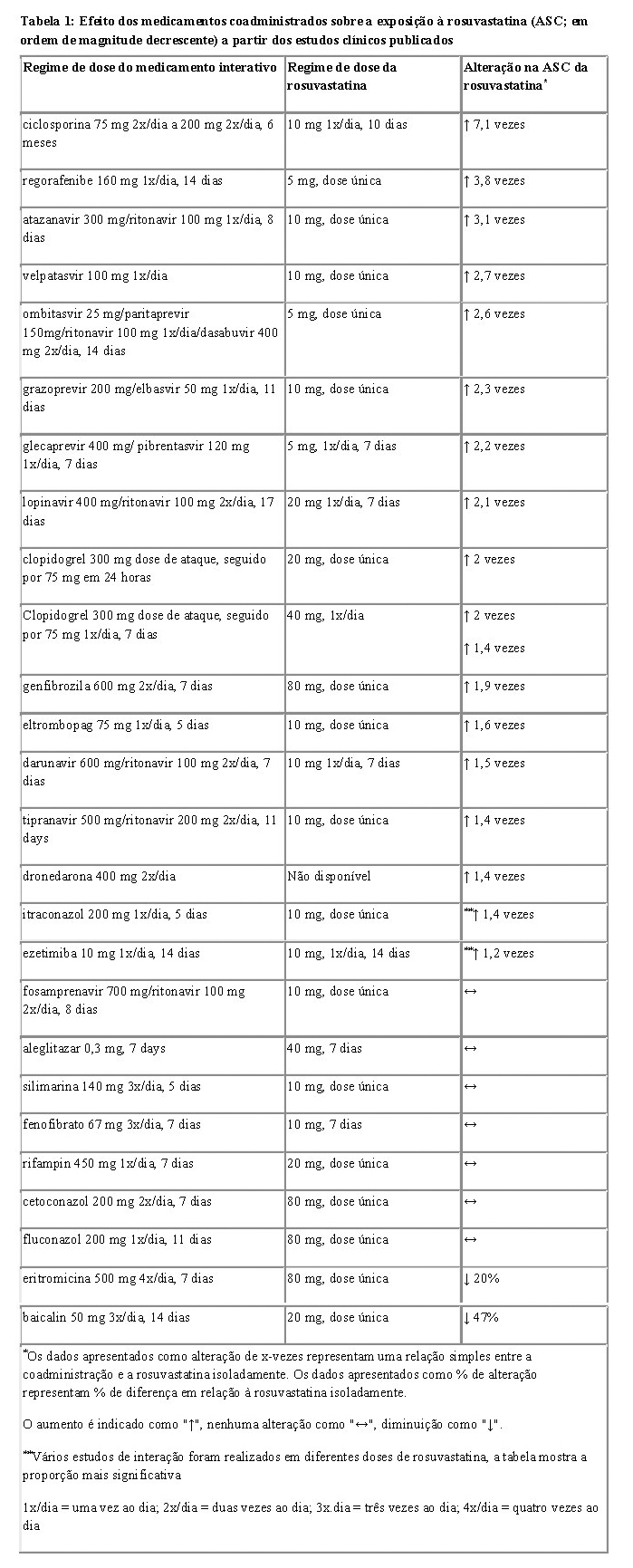
6. INTERAÇÕES MEDICAMENTOSAS
Combinações contraindicadas com ZINPASS® EZE:
Ciclosporina:
-Rosuvastatina: a administração concomitante de ZINPASS® EZE com ciclosporina é contraindicada devido à rosuvastatina (veja a seção "Contraindicações"). Durante o