ZIEXTENZO
SANDOZ
pegfilgrastim
Fator de crescimento hematopoiético.
Apresentações.
Cartucho com 1 seringa preenchida, com protetor de seringa, com 0,6 mL de solução injetável contendo 6 mg de pegfilgrastim.
USO SUBCUTÂNEO
USO ADULTO
Composição.
Cada seringa preenchida contém: 6 mg/0,6 mL
pegfilgrastim 6 mg, excipientes q.s.p. 0,6 mL
(ácido acético, sorbitol, polissorbato 20, hidróxido de sódio, água para injetáveis)
Informações técnicas.
1. INDICAÇÕES
Ziextenzo® é indicado para redução na duração da neutropenia e da incidência de neutropenia febril em pacientes tratados com quimioterapia citotóxica para malignidade (exceto leucemia mieloide crônica e síndromes mielodisplásicas).
2. RESULTADOS DE EFICÁCIA
Ziextenzo® é um medicamento biológico desenvolvido pela via da comparabilidade (biossimilar). O programa de desenvolvimento do medicamento foi projetado para demonstrar a comparabilidade entre Ziextenzo® e o medicamento biológico de referência Neulastim® .
Resultados de eficácia do medicamento biológico de referência
Em dois estudos clínicos pivotais, randomizados, duplo-cegos em pacientes de alto risco com câncer de mama em estágio II-IV, submetidos à quimioterapia mielossupressora composta por doxorrubicina e docetaxel, o uso de pegfilgrastim administrado uma única vez por ciclo reduziu a duração da neutropenia e a incidência de neutropenia febril de forma semelhante ao observado com as administrações diárias de filgrastim (mediana de 11 administrações diárias). Na ausência de terapia de suporte com fator de crescimento, esse esquema quimioterápico resultou em neutropenia de grau 4 com duração média de 5 a 7 dias, e em uma incidência de neutropenia febril de 30 a 40%. Em um destes estudos (n = 157), em que se empregou uma dose fixa de pegfilgrastim de 6 mg, a duração média de neutropenia de grau 4 foi de 1,8 dias para o grupo de pegfilgrastim e de 1,6 dias no grupo do filgrastim (diferença de 0,23 dias, 95% IC de -0,15; 0,63). Durante todo o estudo, a taxa de neutropenia febril foi de 13% nos pacientes tratados com pegfilgrastim e de 20% nos pacientes tratados com filgrastim (diferença 7%, 95% IC de -19%; 5%). Em um segundo estudo (n = 310), em que se empregou uma dose ajustada ao peso (100 mg/kg), a duração média da neutropenia de grau 4 no grupo tratado com pegfilgrastim foi de 1,7 dias e de 1,8 dias no grupo tratado com filgrastim (diferença 0,03 dias, 95% IC de -0,36; 0,30). A taxa global de neutropenia febril foi de 9% nos pacientes tratados com pegfilgrastim e de 18% nos pacientes tratados com filgrastim (diferença de 9%, 95% IC de -16,8%; -1,1%). Em um estudo clínico controlado com placebo, duplo-cego em pacientes com câncer de mama, o efeito de pegfilgrastim sobre a incidência de neutropenia febril foi avaliado após uma administração de um esquema quimioterápico associado a uma taxa de neutropenia febril de 10 a 20% (docetaxel 100 mg/m2, a cada 3 semanas, durante 4 ciclos). Novencentos e vinte e oito pacientes foram randomizados para receber uma dose única de pegfilgrastim ou placebo, aproximadamente 24 horas (Dia 2) após a quimioterapia em cada ciclo. A incidência de neutropenia febril foi inferior nos pacientes randomizados para receber pegfilgrastim, em comparação aos que receberam placebo (1% versus 17%, p ≤ 0,001). A incidência de hospitalizações e de uso de anti-infecciosos IV associados ao diagnóstico clínico de neutropenia febril foi menor no grupo que usou pegfilgrastim comparado ao placebo (1% versus 14%, p < 0,001; e 2% versus 10%, p < 0,001). Um estudo clínico pequeno (n = 83), de Fase II, randomizado, duplo-cego, realizado com pacientes recebendo quimioterapia para leucemia mieloide aguda de novo, comparou pegfilgrastim (dose única de 6 mg) com filgrastim administrado durante a quimioterapia de indução. O tempo mediano para a recuperação de neutropenia grave foi estimado em 22 dias, nos dois grupos de tratamento. O resultado a longo prazo não foi estudado (vide "5. ADVERTÊNCIAS E PRECAUÇÕES").
Resultados obtidos nos estudos comparativos entre o biossimilar e o medicamento biológico de referência Estudos clínicos comparativos Desenho do estudo comparativo e dados demográficos do estudo
Um estudo clínico comparativo em indivíduos saudáveis foi conduzido para apoiar a similaridade entre o Ziextenzo® e o medicamento biológico de referência (Neulastim® licenciado pela UE e pelos EUA). Este foi um estudo randomizado, duplo-cego, cruzado de três vias para comparar a farmacocinética, a farmacodinâmica e a segurança de uma administração subcutânea única de 6 mg de Ziextenzo® , Neulastim® licenciado pela UE e pelos EUA em indivíduos saudáveis (LAEP06-104). Uma visão geral do desenho do estudo e das características demográficas dos indivíduos inscritos no estudo clínico é apresentada na Tabela 1.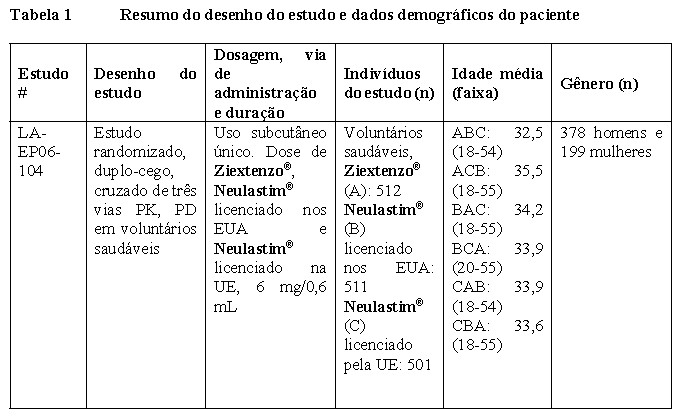
Resultados do estudo comparativo
Estudos Comparativos de Biodisponibilidade
A semelhança PK e PD de Ziextenzo® foi demonstrada em um estudo cruzado de três vias de dose única em indivíduos saudáveis com uma administração s.c. de Ziextenzo® e do medicamento biológico de referência (LA-EP06-104).
Farmacocinética
A avaliação da similaridade de PK foi baseada nos ICs de 90% da proporção das médias geométricas entre Ziextenzo® e o medicamento biológico de referência (licenciado nos EUA) para os três parâmetros de PK primários, AUC0-inf, AUC0-último e Cmáx, que foram todos contidos nas margens de semelhança PK predefinidas de 0,80 a 1,25 (Tabela 2). A biossimilaridade também foi alcançada entre o Ziextenzo® e o medicamento biológico de referência licenciado pela UE e entre os medicamentos biológicos de referência licenciados pelos EUA e pela UE (dados não mostrados), o que suporta análises de segurança comparativas entre o Ziextenzo® e o medicamento biológico de referência licenciado pela UE (vide adiante "Segurança e eficácia comparativas").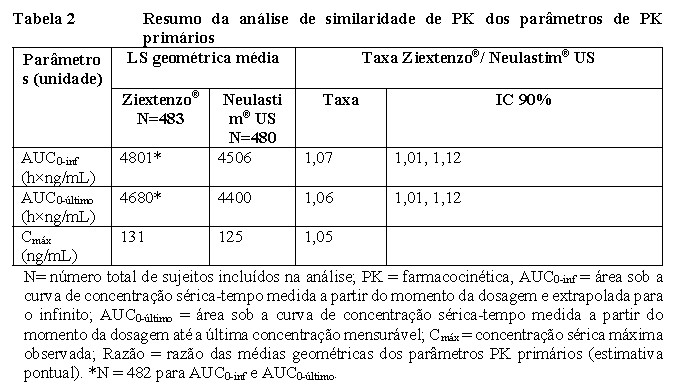
Farmacodinâmica
A farmacodinâmica (PD) foi estudada usando contagem absoluta de neutrófilos (ANC) ao longo do tempo. A contagem absoluta de neutrófilos é um marcador substituto de eficácia estabelecido. A similaridade de PD entre Ziextenzo® e o medicamento biológico de referência (Neulastim® licenciado nos EUA) foi demonstrada com os ICs de 95% (intervalos de confiança) das razões geométricas médias dos endpoints de PD primários AUEC0-último e Emáx estando inteiramente contidos dentro das margens predefinidas de 0,80 a 1,25 (Tabela 3). A biossimilaridade também foi alcançada entre Ziextenzo® e o medicamento biológico de referência licenciado pela UE e entre os medicamentos biológicos de referência licenciados pelos EUA e pela UE (dados não mostrados).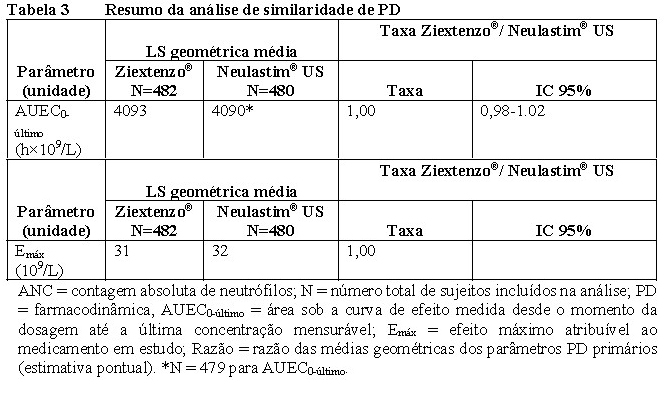
Segurança e eficácia comparativas
Em dois estudos independentes duplo-cegos de Fase 3, pacientes do sexo feminino com câncer de mama recebendo quimioterapia mielossupressora estabelecida foram randomizados 1:1 para Ziextenzo® ou Neulastim® EUA administrado no Dia 2 de cada quimioterapia (docetaxel 75 mg/m2) em combinação com doxorrubicina (50 mg/m2) e ciclo de ciclofosfamida (500 mg/m2) por até 6 ciclos. Em ambos os estudos, Ziextenzo® ou Neulastim® EU foi administrado como uma dose de 6 mg s.c. uma vez em cada ciclo de quimioterapia e a duração do tratamento foi de até 18 semanas. Não foram observadas diferenças na segurança entre Ziextenzo® e Neulastim® EU em pacientes com câncer de mama. Perfis de segurança semelhantes entre Ziextenzo® e o medicamento biológico de referência também foram observados no estudo clínico em voluntários saudáveis (LA-EP06-104).
Imunogenicidade
A imunogenicidade de Ziextenzo® e do medicamento biológico de referência foi comparada em indivíduos saudáveis e pacientes com câncer de mama. A incidência de ADAs (anticorpos anti-drogas) foi semelhante em todos os grupos de tratamento. No estudo LA-EP06-104, três indivíduos relataram neutralizadores de ADAs (NAbs), dois no grupo Neulastim® licenciado pela UE e um no grupo Ziextenzo® (apenas no período 1). Não houve comportamento incomum observado em perfis individuais de PK e contagem absoluta de neutrófilos (ANC), indicando que os ADAs tiveram efeitos mínimos na depuração sistêmica do pegfilgrastim ou na produção e liberação de neutrófilos. Houve também uma baixa taxa de detecção de ADAs em pacientes com câncer de mama nos estudos de Fase III duplo-cegos independentes, o que demonstra um baixo potencial de imunogenicidade de Ziextenzo® , semelhante ao relatado para o medicamento biológico de referência.
Farmacodinâmica Não Clínica Comparativa
Estudos in vitro
As características biológicas de Ziextenzo® , Neulastim® EU e Neulastim® US foram avaliadas usando um ensaio de proliferação celular e um ensaio de ligação ao receptor, conforme descrito na Tabela 4.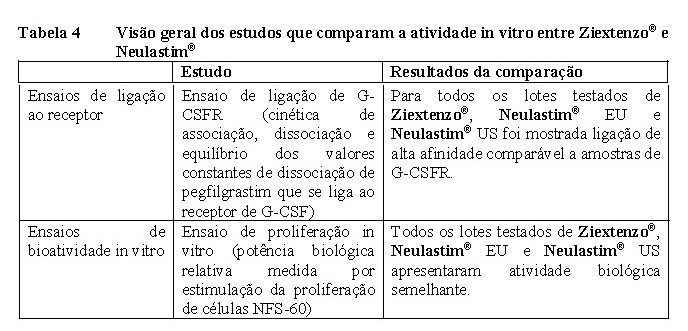
No geral, a bioatividade in vitro e os ensaios de ligação ao receptor demonstraram atividade biológica comparável e afinidade de ligação ao G-CSFR entre os lotes de Ziextenzo® , Neulastim® EU e Neulastim® US.
Estudos in vivo
Uma comparação da resposta farmacodinâmica ao Ziextenzo® em comparação com o medicamento biológico de referência licenciado pela UE (Neulastim®) foi realizada em ratos naive e neutropênicos, e também em coelhos e cães naives. Em ratos naive, a administração subcutânea de Ziextenzo® induziu um aumento rápido e dependente da dose de neutrófilos no sangue periférico. O aumento na contagem de neutrófilos foi semelhante em duração e magnitude ao induzido por uma dose igual do medicamento biológico de referência. Em ratos neutropênicos, a administração de Ziextenzo® também demonstrou aumentar a recuperação dos neutrófilos com cinética e magnitudes semelhantes às obtidas com as mesmas doses do medicamento biológico de referência. Em ambos, coelhos e cães ingênuos, a administração subcutânea de Ziextenzo® induziu um rápido aumento de neutrófilos no sangue periférico, que foi semelhante em duração e magnitude ao induzido por uma dose igual do medicamento biológico de referência.
Toxicologia Comparativa
A segurança do Ziextenzo® em comparação com o medicamento biológico de referência licenciado pela UE (Neulastim®) foi avaliada em um estudo de toxicidade geral de dose repetida em ratos. Foi administrada dosagem subcutânea repetida de até 200 mcg/kg a cada dois dias durante 4 semanas ou 1000 mcg/kg uma vez por semana durante 5 semanas, o que cobre adequadamente o regime clínico. As avaliações toxicocinéticas confirmaram que os animais foram expostos a níveis sistêmicos apropriados de rhG-CSF. A avaliação dos anticorpos antidroga demonstrou que o Ziextenzo® tinha uma imunogenicidade semelhante ao fármaco biológico de referência nestes estudos e que a geração de anticorpos não afetou a resposta farmacodinâmica. Ambas as preparações foram bem toleradas, sem nenhuma morte relacionada ao tratamento ou efeitos no peso corporal e no consumo de alimentos. As alterações relacionadas ao tratamento foram limitadas aos efeitos adversos esperados após a administração de rhG-CSF, como aumento da granulocitopoiese na medula óssea e ativação da hematopoiese/granulocitopoiese no baço e fígado. Todas as alterações ocorreram de forma semelhante com o Ziextenzo® e com o medicamento biológico de referência e foram consideradas em linha com uma resposta farmacodinâmica exagerada ao rhG-CSF. Essas alterações foram menos graves nos grupos que receberam 1000 mcg/kg uma vez por semana durante 5 semanas do que aqueles que receberam 200 mcg/kg a cada dois dias durante 4 semanas. Todas as alterações diminuíram no final do período de recuperação de 2 meses sem dosagem, exceto por um leve aumento no peso do baço. Portanto, a toxicologia pré-clínica não mostrou quaisquer diferenças relevantes entre o Ziextenzo® e o medicamento biológico de referência.
A tolerabilidade local foi comparada após administração de dose única e múltipla por via subcutânea. Uma tolerabilidade local boa e semelhante após doses subcutâneas únicas e múltiplas foi demonstrada para Ziextenzo® e o medicamento biológico de referência nos estudos de PK/PD e toxicidade. Em conclusão, os estudos não clínicos farmacodinâmicos, de tolerância local e toxicológicos confirmaram que a eficácia e a toxicidade são semelhantes entre Ziextenzo® e o medicamento biológico de referência.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Farmacodinâmica
Grupo farmacoterapêutico: imunoestimulantes, fator estimulador de colônia, Código ATC: L03AA13.
O fator estimulador de colônias de granulócitos humanos (G-CSF) é uma glicoproteína, que regula a produção e liberação de neutrófilos da medula óssea. O pegfilgrastim é um conjugado covalente do G-CSF humano recombinante (r-metHuG-CSF) com uma molécula única de polietilenoglicol (PEG) de 20 kd. O pegfilgrastim é uma forma de filgrastim de longa duração devido à depuração renal diminuída. Foi demonstrado que pegfilgrastim e filgrastim apresentam o mesmo mecanismo de ação, causando acentuado aumento no número de neutrófilos no sangue periférico dentro de 24 horas, com aumentos mínimos dos monócitos e/ou linfócitos. Da mesma forma que filgrastim, os neutrófilos produzidos em resposta a pegfilgrastim apresentam função normal ou aumentada, conforme demonstrado em ensaios sobre a função quimiotática e fagocítica. Assim como outros fatores de crescimento hematopoiético, o G-CSF demonstrou in vitro possuir propriedades estimuladoras sobre as células endoteliais humanas. O G-CSF pode promover o crescimento de células mieloides in vitro, incluindo células malignas, e efeitos similares podem ser observados em algumas células não mieloides in vitro.
Farmacocinética
Absorção
Após uma administração subcutânea única de pegfilgrastim, o pico da concentração sérica de pegfilgrastim ocorre dentro de 16 a 120 horas.
Distribuição
As concentrações séricas de pegfilgrastim são mantidas durante o período de neutropenia após quimioterapia mielossupressora.
Eliminação
A eliminação de pegfilgrastim não é linear com relação à dose, e a depuração sérica de pegfilgrastim diminui com o aumento da dose. O pegfilgrastim parece ser eliminado, principalmente, pela depuração mediada pelos neutrófilos que se tornam saturados com doses mais elevadas. Consistente com um mecanismo de depuração autorregulado, a concentração sérica de pegfilgrastim diminui rapidamente após o início da recuperação dos neutrófilos (vide Figura 1).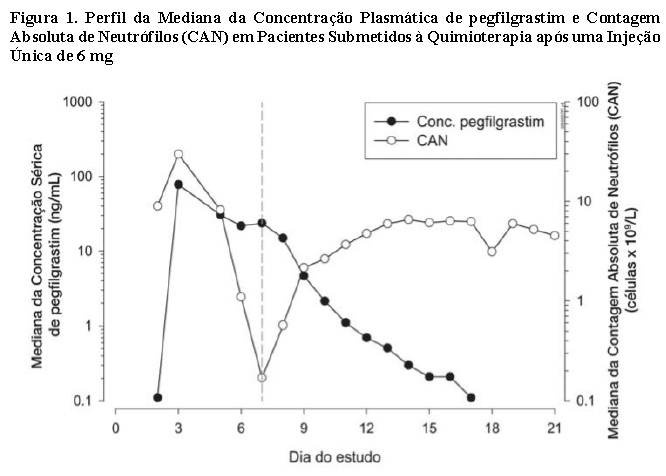
Farmacocinética em populações especiais
Alterações da Função Renal e Hepática
Devido ao mecanismo de depuração mediada por neutrófilos, não se espera que a farmacocinética do pegfilgrastim seja afetada por disfunção renal ou hepática. Em um estudo clínico aberto com dose única (n = 31), vários estágios de comprometimento renal, incluindo insuficiência renal em estágio final, não tiveram impacto na farmacocinética do pegfilgrastim.
População Geriátrica
Dados limitados indicam que a farmacocinética de pegfilgrastim em pacientes idosos ( > 65 anos) é semelhante à de adultos.
Segurança Pré-clínica
Teratogenicidade
Não foram observados efeitos adversos nas ninhadas de ratas prenhas que receberam pegfilgrastim por via subcutânea, mas em coelhos, pegfilgrastim causou toxicidade embriofetal (perda fetal) em doses cumulativas de aproximadamente 4 vezes a dose humana recomendada, o que não ocorreu quando coelhas prenhas foram expostas à dose humana recomendada. Em estudos conduzidos em ratos, observou-se que pegfilgrastim pode atravessar a placenta. Estudos em ratos indicaram que o desempenho reprodutivo, a fertilidade, o ciclo estral, o número de dias entre o acasalamento e a cópula e a sobrevida intrauterina não foram afetados por pegfilgrastim administrado por via subcutânea. A relevância desses achados para os seres humanos é desconhecida.
Outros
Os dados de estudos pré-clínicos convencionais de toxicidade de doses repetidas revelaram os efeitos farmacológicos esperados incluindo o aumento da contagem leucocitária, hiperplasia mieloide da medula óssea, hematopoiese extramedular e esplenomegalia.
4. CONTRAINDICAÇÕES
Hipersensibilidade ao princípio ativo ou a qualquer um dos excipientes.
Este medicamento é contraindicado para menores de 18 anos.
5. ADVERTÊNCIAS E PRECAUÇÕES
Dados clínicos limitados sugerem um efeito comparável entre pegfilgrastim e filgrastim no tempo de recuperação da neutropenia grave em pacientes com leucemia mieloide aguda de novo, (vide"2. RESULTADOS DE EFICÁCIA"). No entanto, os efeitos de pegfilgrastim a longo prazo não foram ainda estabelecidos em leucemia mieloide aguda (LMA), portanto, pegfilgrastim deve ser utilizado com cautela nesta população de pacientes. O G-CSF pode promover o crescimento de células mieloides in vitro e efeitos semelhantes também podem ser observados em algumas células não mieloides in vitro. A segurança e eficácia de pegfilgrastim não foram ainda investigadas em pacientes com síndrome mielodisplásica, leucemia mieloide crônica e em pacientes com Leucemia Mieloide Aguda (LMA) secundária. Portanto, não deve ser usado em tais pacientes. Deve-se ter cuidado especial na distinção de um diagnóstico de transformação blástica de leucemia mieloide aguda em leucemia mieloide crônica. A segurança e a eficácia da administração de pegfilgrastim em pacientes portadores de LMA de novo com < 55 anos de idade com citogenética t(15;17) não foram estabelecidas. A segurança e a eficácia de pegfilgrastim não foram investigadas em pacientes recebendo altas doses de quimioterapia. Este medicamento não deve ser utilizado para aumentar a dose da quimioterapia citotóxica além dos regimes posológicos estabelecidos.
Eventos Adversos Pulmonares
Reações adversas pulmonares incomuns, em particular pneumonia intersticial, foram relatadas após a administração do G-CSF. Pacientes com um histórico recente de infiltrados pulmonares ou pneumonia podem ter um risco maior (vide "9. REAÇÕES ADVERSAS"). O início de sinais pulmonares, como tosse, febre e dispneia, em associação com sinais radiológicos de infiltrados pulmonares e deterioração da função pulmonar concomitantemente com o aumento do número de neutrófilos, podem corresponder a sinais preliminares indicativos da síndrome da angústia respiratória aguda (SARA). Em tais circunstâncias, o uso de pegfilgrastim deve ser descontinuado a critério do médico, e o tratamento apropriado deve ser instituído (vide "9. REAÇÕES ADVERSAS").
Glomerulonefrite
Glomerulonefrite tem sido relatada em pacientes tratados com filgrastim e pegfilgrastim. Geralmente, os eventos de glomerulonefrite são resolvidos após a redução da dose ou a descontinuação de filgrastim e pegfilgrastim. Monitoramento por urinálise é recomendado.
Síndrome do Extravasamento Capilar
Síndrome de extravasamento capilar foi relatada após a administração do G-CSF e é caracterizada por hipotensão, hipoalbuminemia, edema e hemoconcentração. Os pacientes que desenvolverem sintomas de síndrome de extravasamento capilar sistêmico devem ser monitorados rigorosamente, e devem receber tratamento sintomático padrão, o que pode incluir a necessidade de cuidados intensivos (vide "9. REAÇÕES ADVERSAS").
Esplenomegalia e Ruptura Esplênica
Casos geralmente assintomáticos, de esplenomegalia e casos de ruptura esplênica, incluindo alguns casos fatais, foram relatados após a administração de pegfilgrastim (vide "9. REAÇÕES ADVERSAS"). Portanto, as dimensões do baço devem ser cuidadosamente monitoradas (exemplo: exame clínico, ultrassonografia). Um diagnóstico de ruptura esplênica deve ser considerado em pacientes relatando dor abdominal no quadrante superior esquerdo ou dor na extremidade do ombro esquerdo.
Trombocitopenia e Anemia
O tratamento isolado com pegfilgrastim não exclui a possibilidade de trombocitopenia e de anemia, pois a dose completa da quimioterapia mielossupressiva é mantida conforme o regime prescrito. Monitoramento regular da contagem plaquetária e hematócrito é recomendado. Deve-se ter cuidado especial ao administrar agentes quimioterápicos isolados ou em combinação que sejam conhecidos por causar trombocitopenia grave.
Síndrome mielodisplásica e leucemia mieloide aguda em pacientes com câncer de mama e de pulmão
No cenário do estudo observacional pós-comercialização, pegfilgrastim em conjunto com quimioterapia e/ou radioterapia foi associado ao desenvolvimento de síndrome mielodisplásica (SMD) e de leucemia mieloide aguda (LMA) em pacientes com câncer de mama e de pulmão (vide "9. REAÇÕES ADVERSAS"). Pacientes tratados nestes cenários devem ser monitorados para verificação do aparecimento de sinais e sintomas de SMD/LMA.
Anemia Falciforme
Crises falciformes foram associadas com o uso de pegfilgrastim em pacientes com traço falciforme ou doença falciforme (vide "9. REAÇÕES ADVERSAS"). Portanto, os médicos devem ter cautela ao prescreverem pegfilgrastim a pacientes com traço falciforme ou anemia falciforme, devem monitorar apropriadamente os parâmetros clínicos e laboratoriais, e devem estar atentos à possível associação deste medicamento com esplenomegalia e com uma crise vasooclusiva.
Leucocitose
Contagens de leucócitos (WBC) de 100 × 109/L ou superiores foram observadas em menos de 1% dos pacientes tratados com pegfilgrastim. Não foi relatado nenhum evento adverso atribuível a esse grau de leucocitose. Esse aumento de leucócitos é transitório, geralmente observado entre 24 e 48 horas após a administração, e é consistente com o efeito farmacodinâmico desse medicamento. Consistente com os efeitos clínicos e com a possível leucocitose, a contagem de WBC deve ser feita em intervalos regulares durante o tratamento. Se a contagem de leucócitos exceder 50 × 109/L após o nadir esperado, esse medicamento deve ser descontinuado imediatamente.
Hipersensibilidade
Hipersensibilidade, incluindo reações anafiláticas, que ocorrem no tratamento inicial ou subsequente, foi relatada em pacientes tratados com pegfilgrastim. Descontinuar permanentemente pegfilgrastim em pacientes com hipersensibilidade clinicamente significativa. Não administrar pegfilgrastim a pacientes com um histórico de hipersensibilidade a pegfilgrastim ou filgrastim. Se uma reação alérgica séria ocorrer, tratamento apropriado deve ser instituído, com acompanhamento rigoroso do paciente durante vários dias.
Síndrome de Stevens-Johnson
A Síndrome de Stevens-Johnson (SSJ), que pode ameaçar a vida ou ser fatal, foi relatada como rara em associação ao tratamento com pegfilgrastim. Se houver desenvolvimento da SSJ com o uso de pegfilgrastim, então o tratamento com pegfilgrastim não deve ser reiniciado para este paciente.
Imunogenicidade
Assim como todas as proteínas terapêuticas, existe a possibilidade de imunogenicidade. As taxas de geração de anticorpos contra pegfilgrastim geralmente são baixas. Anticorpos de ligação ocorrem conforme esperado com todos os medicamentos biológicos; todavia, não foram associados com atividade neutralizante até o momento.
Aortite
Aortite foi relatada após administração de G-CSF em pacientes saudáveis e com câncer. Os sintomas experimentados incluíram febre, dor abdominal, mal-estar, dor nas costas e aumento de marcadores inflamatórios (exemplo proteína c-reativa e contagem de glóbulos brancos). A maioria dos casos de aortite foi diagnosticada por tomografia computadorizada e geralmente resolvida após suspensão do G-CSF (vide "9. REAÇÕES ADVERSAS").
Outras advertências
A segurança e a eficácia de pegfilgrastim para a mobilização de células progenitoras sanguíneas em pacientes ou doadores saudáveis não foram devidamente avaliadas.
O aumento da atividade hematopoiética da medula óssea em resposta ao tratamento com fator de crescimento foi associada com achados positivos transitórios nos exames de cintilografia óssea. Este fato deve ser considerado ao interpretar os resultados de exames de cintilografia óssea.
Sorbitol -O efeito aditivo de produtos que contêm sorbitol (ou frutose) administrados concomitantemente a ingestão alimentar de sorbitol (ou frutose) deve ser levado em consideração.
Atenção: Contém sorbitol.
Sódio - Este medicamento contém menos de 1 mmol (23 mg) de sódio por 6 mg de dose, isto é, essencialmente "isento de sódio".
Para melhorar a rastreabilidade de fatores estimuladores de colônias de granulócitos (G-CSFs), o nome comercial do produto administrado deve ser registrado claramente no arquivo do paciente.
Gravidez
Há quantidade limitada ou inexistente de dados sobre o uso de pegfilgrastim em mulheres grávidas. Estudos em animais revelaram toxicidade reprodutiva (vide "3. CARACTERÍSTICAS FARMACOLÓGICAS - Segurança Pré-clínica"). Ziextenzo® não é recomendado durante a gravidez e em mulheres férteis que não usam contraceptivos.
Categoria de Risco na Gravidez: C. Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
O uso deste medicamento no período da lactação depende da avaliação e acompanhamento do seu médico ou cirurgião-dentista. Uso criterioso no aleitamento ou na doação de leite humano.
Amamentação
Há poucas informações sobre a excreção de pegfilgrastim /metabólitos no leite materno humano, e não se pode descartar um risco para os recém-nascidos/bebês. Deve-se tomar a decisão de descontinuar a amamentação ou descontinuar/não receber o tratamento com Ziextenzo® levando em conta o benefício da amamentação para a criança e o benefício do tratamento para a mulher.
Fertilidade
O pegfilgrastim não afetou o desempenho reprodutivo ou a fertilidade de ratos machos e fêmeas em doses semanais cumulativas aproximadamente 6 a 9 vezes maiores do que a dose humana recomendada (com base na área de superfície corporal) (vide "3. CARACTERÍSTICAS FARMACOLÓGICAS - Segurança Pré-clínica").
Efeitos sobre a Capacidade de Dirigir e Usar Máquinas A influência de Ziextenzo® sobre a capacidade de dirigir e usar máquinas é nula ou insignificante.
População pediátrica
A segurança e a eficácia de Ziextenzo® em crianças não foram estabelecidas. Dados atualmente disponíveis são descritos na seção "Reações Adversas", porém nenhuma recomendação de dose pode ser feita.
Este medicamento não deve ser misturado com outros medicamentos, particularmente com soluções de cloreto de sódio.
6. INTERAÇÕES MEDICAMENTOSAS
Devido a potencial sensibilidade das células mieloides em divisão rápida à quimioterapia citotóxica, Ziextenzo® deve ser administrado pelo menos 24 horas após a administração da quimioterapia citotóxica. Em estudos clínicos, o pegfilgrastim foi administrado com segurança 14 dias antes da quimioterapia. O uso concomitante de Ziextenzo® com qualquer agente quimioterápico não foi avaliado em pacientes. Em modelos animais, a administração concomitante de Ziextenzo® com 5-fluorouracil (5-FU) ou outros antimetabólicos demonstrou potencializar a mielossupressão. Possíveis interações com outros fatores de crescimento hematopoiéticos e citocinas não foram investigadas especificamente em estudos clínicos.
Lítio
O potencial para interação com lítio, que também favorece a liberação de neutrófilos, não foi especificamente investigado. Não há evidências de que tal interação possa ser prejudicial. A segurança e a eficácia Ziextenzo® não foram avaliadas em pacientes recebendo quimioterapia associada com mielossupressão mais tardia, como as nitrosoureias. Não foram realizados estudos específicos de interação ou de metabolismo, mas os estudos clínicos não indicaram interação entre Ziextenzo® e quaisquer outros produtos medicinais.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Armazenar em geladeira (de 2°C a 8°C). Manter nesta embalagem original para proteger da luz. Não congelar.
Ziextenzo® pode ser exposto à temperatura ambiente (não acima dos 35°C) por um único período máximo de 120 horas. Se Ziextenzo® for deixado em temperatura ambiente por mais de 120 horas, deve ser descartado. A exposição acidental a temperaturas de congelamento por um único período inferior a 24 horas não afeta negativamente a estabilidade de Ziextenzo® . Antes da administração, a solução de Ziextenzo® deve ser inspecionada para se assegurar que não contém partículas. Apenas uma solução límpida e incolor a levemente amarelada deve ser administrada. Agitação excessiva pode causar agregação de pegfilgrastim, o que o torna biologicamente inativo. Quando realizar a administração utilizando a seringa preenchida, espere que a seringa preenchida atinja a temperatura ambiente antes de administrar o medicamento.
Prazo de Validade: 36 meses.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Ziextenzo® é uma solução límpida e incolor a levemente amarelada.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
O tratamento com Ziextenzo® deve ser iniciado e supervisionado por médicos com experiência em oncologia e/ou hematologia.
Posologia
Uma dose de 6 mg (uma única seringa preenchida) de Ziextenzo® é recomendada para cada ciclo de quimioterapia, administrada pelo menos 24 horas depois da quimioterapia citotóxica.
Modo de Administração
Ziextenzo® é injetado subcutaneamente. As injeções subcutâneas devem ser aplicadas no abdômen, no braço ou na coxa. Para instruções sobre o manuseio do produto medicinal antes da administração, (vide "Instruções para aplicação da seringa preenchida de Ziextenzo® com protetor de agulha" a seguir).
Populações especiais
Pacientes com Insuficiência Renal
Nenhuma alteração da dose é recomendada para pacientes com insuficiência renal, inclusive aqueles com doença renal em estágio final.
Instruções para aplicação da seringa preenchida de Ziextenzo® com protetor de agulha
Esta seção contém informação sobre como aplicar uma injeção de Ziextenzo® .
Para ajudar a evitar possíveis infecções e garantir o uso correto do medicamento, é importante seguir estas instruções.
Antes de aplicar a injeção, leia as instruções até o fim. É importante não tentar autoaplicar a injeção. Faça isso somente depois de treinado para tal por um médico, enfermeiro ou farmacêutico. O cartucho contém a seringa preenchida lacrada individualmente em uma embalagem de plástico.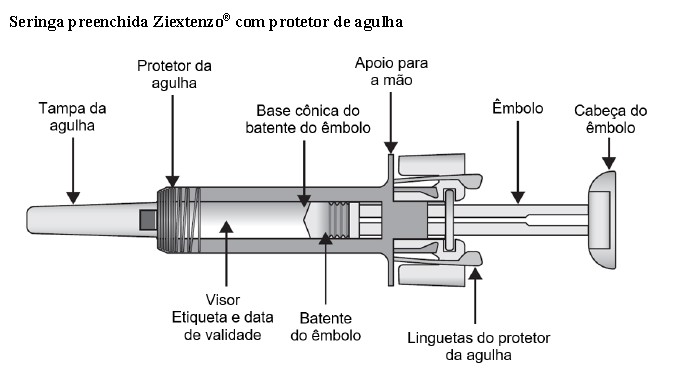
Seringa preenchida Ziextenzo® com protetor de agulha
Após a injeção do medicamento, o protetor da agulha será ativado para cobrir a agulha. O objetivo do protetor da agulha é proteger os profissionais de saúde, cuidadores e pacientes contra picadas de agulha acidentais após a injeção.
Informações importantes sobre segurança Cuidado: mantenha a seringa preenchida fora do campo de visão e do alcance das crianças.
1. Abra o cartucho somente quando estiver pronto para usar a seringa preenchida.
2. Por questão de segurança, não use a seringa preenchida se o lacre da embalagem estiver rompido.
3. Nunca deixe a seringa preenchida sem supervisão onde outras pessoas possam manuseá-la ou violá-la.
4. Não agite a seringa preenchida.
5. Tenha cuidado para não tocar as linguetas do protetor da agulha antes de usá-la. Se tocadas, o protetor da agulha poderá ser ativado antes do momento certo.
6. Remova a tampa da agulha imediatamente antes de aplicar a injeção.
7. A seringa preenchida não pode ser reutilizada. Imediatamente após usar a seringa preenchida, descarte-a em um recipiente para objetos cortantes.
8. Não utilize a seringa se esta tiver caído numa superfície dura ou se tiver caído após ter removido a tampa da agulha.
Armazenamento da seringa preenchida Ziextenzo®
1 Armazene a seringa preenchida embalada no cartucho, para protegê-la contra a luz.
2 Armazene no refrigerador entre 2°C e 8°C. Não congele.
3 Antes da utilização, retire a seringa preenchida do refrigerador e deixe Ziextenzo® atingir a temperatura ambiente (até, no máximo, 35°C) por aproximadamente 15 a 30 minutos. Não use a seringa preenchida após a data de validade indicada no cartucho ou na seringa. Se o prazo tiver expirado, devolva a embalagem inteira para a farmácia.
Preparação da seringa preenchida Ziextenzo® para uso
1 Retire o cartucho com a seringa preenchida embalada do refrigerador e deixe-a fechada por aproximadamente 15 a 30 minutos para que atinja a temperatura ambiente.
2 Quando estiver pronto para usar a seringa preenchida, abra a embalagem e lave bem as mãos com água e sabão.
3 Limpe o local da injeção com um swab embebido em álcool.
4 Retire a seringa preenchida da embalagem. Certifique-se de que o protetor da agulha, feito de plástico transparente, esteja situado sobre o cilindro da seringa de vidro. Se o protetor transparente da agulha estiver cobrindo a tampa da agulha (como mostrado abaixoError<33> Reference source not found.), isso indica que a seringa foi ativada. NÃO utilize esta seringa. Utilize uma nova seringa. A figura abaixo mostra uma seringa pronta para uso.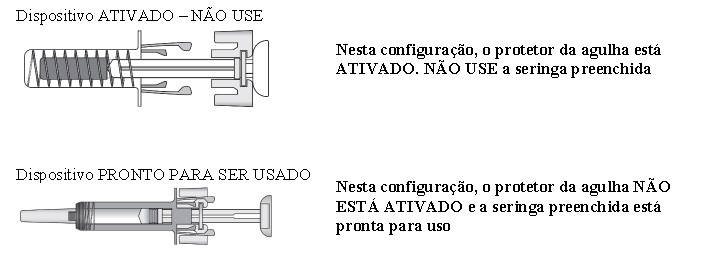
5 Inspecione a seringa preenchida. O líquido deve estar transparente. A cor pode variar deincolor a levemente amarelada. É possível que haja uma pequena bolha de ar no líquido. Isso é normal. Não use a seringa preenchida se forem observadas outras partículas e/ou descoloração.
6 Não use se a seringa estiver quebrada ou ativada. Devolva a seringa preenchida Ziextenzo® e a embalagem para a farmácia.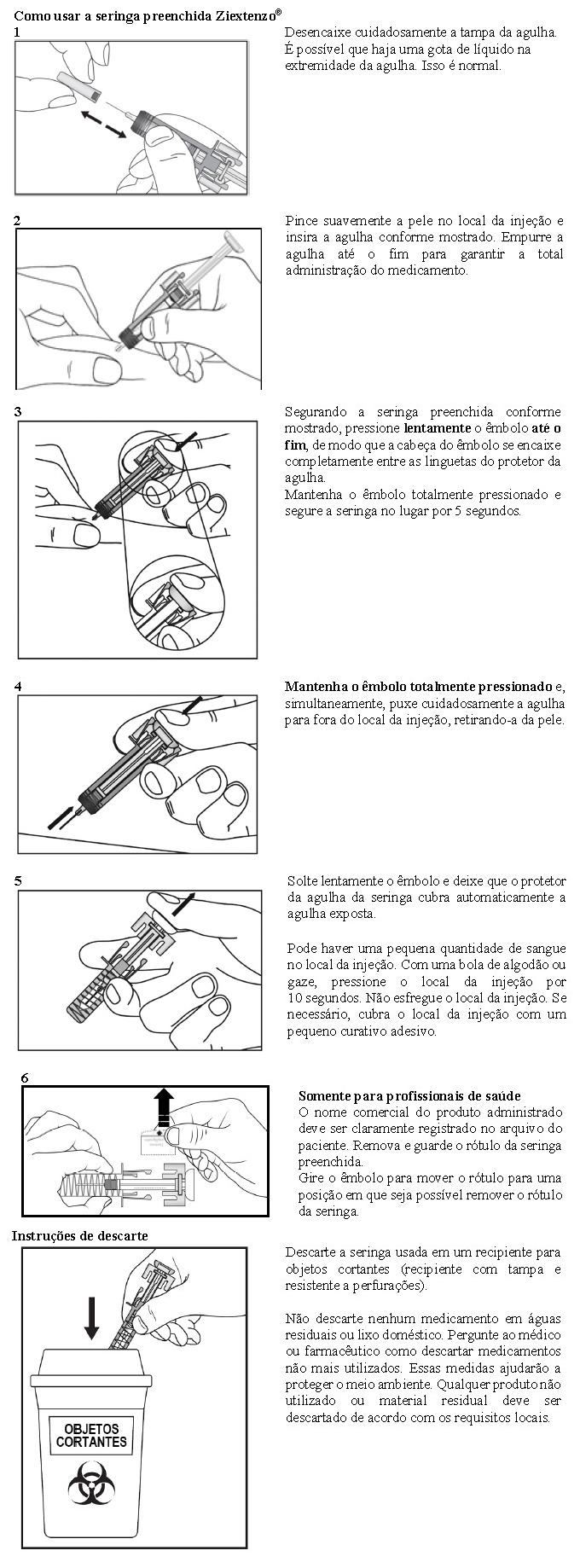
9. REAÇÕES ADVERSAS
Resumo do Perfil de Segurança
As reações adversas mais frequentemente relatadas foram dor óssea (muito comum [≥ 1/10]) e dor musculoesquelética (comum [≥ 1/100 a < 1/10]). A dor óssea geralmente foi de intensidade leve a moderada, transitória e pode ser controlada na maioria dos pacientes com analgésicos convencionais. Reações do tipo hipersensibilidade, incluindo rash cutâneo, urticária, angioedema, dispneia, eritema, rubor e hipotensão, ocorreram no tratamento inicial ou subsequente com Ziextenzo® (incomuns [≥ 1/1.000 a < 1/100]). Reações alérgicas graves, incluindo anafilaxia, podem ocorrer em pacientes recebendo Ziextenzo® (incomuns) (vide "5. ADVERTÊNCIAS E PRECAUÇÕES").
Síndrome do Extravasamento Capilar, que pode ser potenciamente fatal se o tratamento for tardio, foi relatada como incomum (≥ 1/1.000 a < 1/100) em pacientes com câncer submetidos à quimioterapia seguidos de uma administração de G-CSF; (vide "5. ADVERTÊNCIAS E PRECAUÇÕES" e seção "Descrição de Reações Adversas Selecionadas" abaixo).
Esplenomegalia, geralmente assintomática, é incomum.
Ruptura esplênica, incluindo alguns casos fatais, foi raramente relatada após a administração de pegfilgrastim (vide "5. ADVERTÊNCIAS E PRECAUÇÕES").
Reações adversas pulmonares incomuns, incluindo pneumonia intersticial, edema pulmonar, infiltrados pulmonares e fibrose pulmonar, foram relatadas. Raramente alguns casos resultaram em insuficiência respiratória ou Síndrome da Angústia Respiratória Aguda (SARA), que pode ser fatal (vide "5. ADVERTÊNCIAS E PRECAUÇÕES"). Casos isolados de crises falciformes foram relatados em pacientes com traço falciforme ou doença falciforme (incomuns em pacientes falciformes) (vide "5. ADVERTÊNCIAS E PRECAUÇÕES").
Lista Tabulada de Reações Adversas
Os dados da tabela abaixo descrevem reações adversas relatadas em estudos clínicos e informadas espontaneamente. Em cada grupo de frequência, efeitos indesejáveis são apresentados em ordem decrescente de gravidade.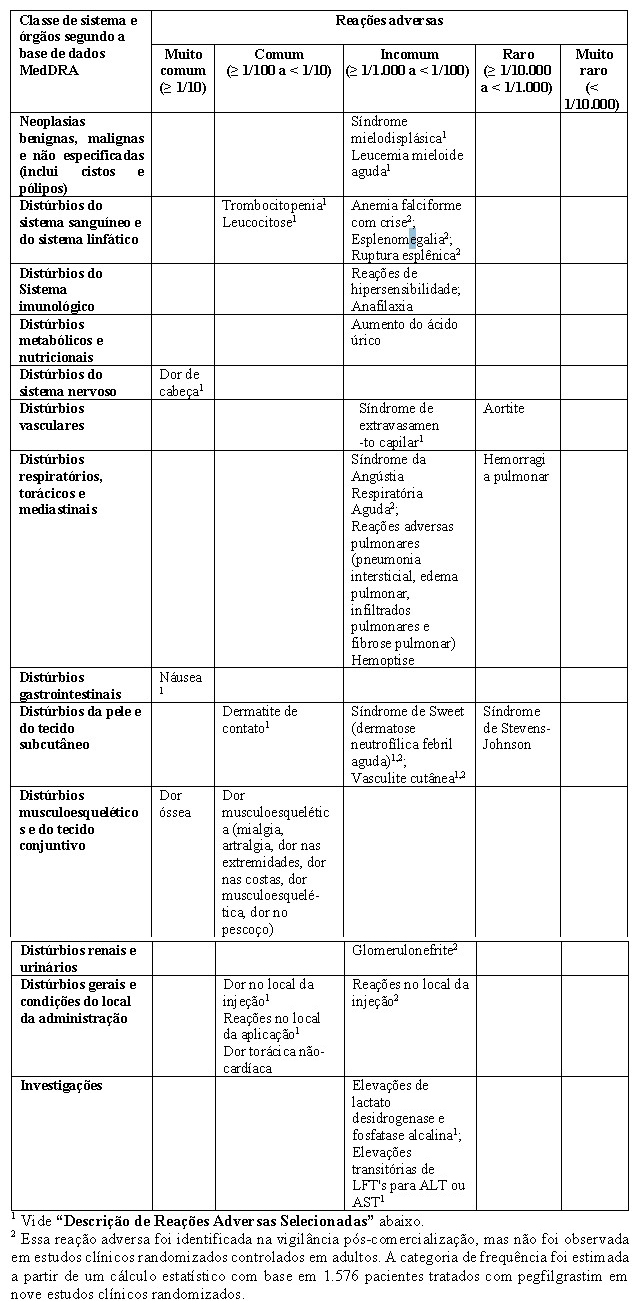
Descrição de Reações Adversas Selecionadas
Casos incomuns de síndrome de Sweet foram relatados, embora em alguns casos malignidades hematológicas de base possam contribuir para a sua ocorrência. Eventos incomuns de vasculite cutânea foram relatados em pacientes tratados com pegfilgrastim. O mecanismo da vasculite em pacientes tratados com pegfilgrastim é desconhecido. Reações no local da injeção, incluindo eritema no local da injeção (incomum (≥ 1/1.000 a < 1/100)), bem como dor no local da injeção (eventos comuns ≥ 1/100 a < 1/10), ocorreram no tratamento inicial ou subsequente com pegfilgrastim.
Casos comuns de leucocitose (Contagem de Leucócitos [WBC] > 100 x 109/L) foram relatados (vide "5. ADVERTÊNCIAS E PRECAUÇÕES"). Aumentos reversíveis leves a moderados de ácido úrico e fosfatase alcalina, sem efeitos clínicos associados, foram incomuns; aumentos reversíveis leves a moderados de lactato desidrogenase, sem efeitos clínicos associados, foram incomuns em pacientes tratados com pegfilgrastim após quimioterapia citotóxica.
Náusea e dores de cabeça foram muito comumente observadas em pacientes tratados com quimioterapia. Aumentos incomuns dos testes de função hepática (LFTs) para ALT (alanina aminotransferase) ou AST (aspartato aminotransferase) foram observados em pacientes depois de receberem pegfilgrastim após a quimioterapia citotóxica. Esses aumentos foram transitórios e retornaram aos valores basais. Risco aumentado de desenvolver SMD/LMA depois do tratamento com pegfilgrastim em conjunto com quimioterapia e/ou radioterapia foi observado em um estudo epidemiológico em pacientes com câncer de mama e de pulmão (vide "5. ADVERTÊNCIAS E PRECAUÇÕES"). Casos comuns de trombocitopenia foram relatados. Casos de síndrome de extravasamento capilar sistêmico foram relatados no período de póscomercialização com o uso de G