ZEJULA
GLAXOSMITHKLINE
niraparibe
Antineoplásico.
Apresentações.
Cápsulas duras, contendo 100 mg de niraparibe, é apresentado em embalagens contendo 28 ou 56 cápsulas.
USO ORAL
USO ADULTO
Composição.
Cada cápsula de Zejula contém: Niraparibe 100 mg (equivalentes a 159,4 mg de tosilato de niraparibe monoidratado), excipientes* q.s.p. 1 cápsula
*lactose monoidratada, estearato de magnésio, cápsula gelatinosa dura (dióxido de titânio, gelatina, FD&C azul, FD&C vermelho e FD&C amarelo), tinta de impressão preta (goma laca, álcool etílico, álcool isopropílico, álcool butílico, propilenoglicol, água purificada, solução de amônia concentrada, hidróxido de potássio, óxido de ferro preto) e tinta de impressão branca (goma laca, álcool etílico, álcool isopropílico, álcool butílico, propilenoglicol, hidróxido de sódio, povidona e dióxido de titânio).
Informações técnicas.
1. INDICAÇÕES
Zejula cápsulas é indicado para:
- terapia de manutenção de pacientes adultas com carcinoma de ovário da trompa de Falópio ou peritoneal primário avançado (Estágios III e IV - FIGO) de alto grau, que responderam completamente ou em parte, após a conclusão da quimioterapia de primeira linha à base de platina.
- terapia de manutenção de pacientes adultas com carcinoma epitelial de ovário, da trompa de Falópio ou peritoneal primário seroso de alto grau, recorrente e sensível à platina. A paciente deve ter respondido completamente ou em parte à quimioterapia à base de platina.
2. RESULTADOS DE EFICÁCIA
Terapia de manutenção de primeira linha para câncer de ovário
O estudo PRIMA foi um estudo duplo-cego, controlado por placebo, no qual as pacientes (n = 733) com carcinoma de ovário, da trompa de Falópio ou peritoneal primário avançado de alto grau com resposta completa ou parcial à quimioterapia de primeira linha à base de platina foram randomizadas na proporção de 2:1 para o tratamento com Zejula ou foram atribuídas a um placebo correspondente.
Após a conclusão da quimioterapia de primeira linha à base de platina, com ou sem cirurgia, as pacientes foram randomizadas. O bevacizumabe foi permitido juntamente com a quimioterapia. Pacientes que tinham recebido bevacizumabe com a quimioterapia, mas não podiam receber bevacizumabe como tratamento manutenção foram permitidas (n=6). As pacientes não poderiam ter recebido outro inibidor da PARP. As pacientes foram randomizadas dentro de 12 semanas do primeiro dia do último ciclo de quimioterapia. As pacientes receberam entre 6 e 9 ciclos de quimioterapia à base de platina. Após a cirurgia de citorredução com intervalo (IDS) as pacientes tiveram 2 ciclos ou mais de quimioterapia à base de platina pós-operatória. As pacientes que receberam quimioterapia neoadjuvante seguida de cirurgia de citorredução com intervalo (IDS) poderiam ter um tumor residual visível ou nenhum tumor residual. As pacientes com doença estágio III que tiveram citorredução completa (isto é, sem doença residual visível) após cirurgia primária de citorredução (PDS) foram excluídas.
As pacientes deveriam ter CA-125 na faixa normal ou CA-125 reduzida em mais de 90% durante a quimioterapia de primeira linha à base de platina que é estável por, pelo menos, 7 dias (por exemplo, nenhum aumento > 15% do nadir).
A randomização foi estratificada com base na melhor resposta durante regime de primeira linha à base de platina (resposta completa vs. parcial), quimioterapia neoadjuvante (NACT; sim vs. não) e status de deficiência de recombinação homóloga (HDR; positivo vs. negativo ou indeterminado). O teste de HDR foi realizado usando um teste de HDR no tecido tumoral, que foi retirado do diagnóstico inicial.
Em alguns casos, outros critérios além do RECIST, como sinais clínicos e sintomas e aumento do CA-125 foram empregados.
O estudo PRIMA foi iniciado com uma dose inicial de 300 mg uma vez ao dia em ciclos contínuos de 28 dias (a seguir denominada dose inicial fixa ou FSD). Com base na análise retrospectiva do estudo NOVA, a dose inicial no estudo PRIMA foi alterada pela Emenda 2 do protocolo. A partir desse momento, as pacientes com peso corporal ≥77 kg e contagem plaquetária ≥150.000/mL receberam Zejula 300 mg (cápsulas 3 × 100 mg) ou placebo (3 cápsulas) diariamente no início do estudo. As pacientes com peso corporal < 77 kg ou contagem de plaquetas < 150.000/mL no início do estudo receberam Zejula 200 mg (cápsulas 2 × 100 mg) ou placebo (2 cápsulas) diariamente (a seguir denominada dose inicial ajustada individualmente ou ISD).
Antes da Emenda 2, os475 pacientes receberam a dose inicial fixa de 300 mg (317- niraparibe, 158-placebo). Após a Emenda, 258 pacientes receberam ISD (44-300mg, 125-200mg, 86-placebo, 3-não dosados após randomização).
O parâmetro decisivo para o resultado da eficácia, PFS (sobrevida livre de progressão), foi determinado com base nos critérios RECIST (versão 1.1.) por revisão central independente cega (BICR). A sobrevida global foi um importante objetivo secundário. Os testes de PFS foram realizados de forma hierárquica: primeiro na população positiva para deficiência da recombinação homóloga e depois na população total. A idade média de 62 anos foi resultado de uma faixa etária de 32 a 85 anos em pacientes randomizadas para Zejula e uma faixa etária de 33 a 88 anos em pacientes randomizadas para placebo. 89% de todas as pacientes eram caucasianas. 69% das pacientes randomizadas para Zejula e 71% daquelas randomizadas para placebo tiveram um ECOG de 0 no início do estudo. Na população total, 65% das pacientes apresentavam doença em estágio III e 35% em estágio IV. 67% das pacientes receberam quimioterapia neoadjuvante. 69% das pacientes apresentaram resposta completa à quimioterapia de primeira linha à base de platina.
O estudo PRIMA mostrou uma melhora estatisticamente significativa na PFS em pacientes randomizadas para Zejula em comparação com o placebo na população positiva para deficiência da recombinação homóloga e na população total (Tabela 1; Figuras 1 e 2).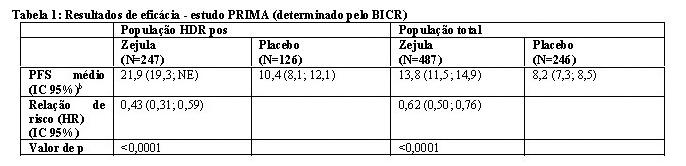
Em uma análise de subgrupo exploratória das pacientes que receberam as doses de 200mg ou 300mg de niraparibe com base no seu peso corporal basal ou contagem plaquetária basal (grupo ISD), eficácia comparável (PFS avaliada pelo investigador) foi observada para o grupo com a dose fixa inicial de 300mg (FSD), com RR de 0,54 (IC 95% 0,33; 0,91) na população HDR pos e RR de 0,68 (IC 95% 0,49; 0,94) na população total. No subgrupo HDR neg, a dose de 200 mg pareceu produzir um efeito de tratamento inferior em comparação com a dose de 300 mg.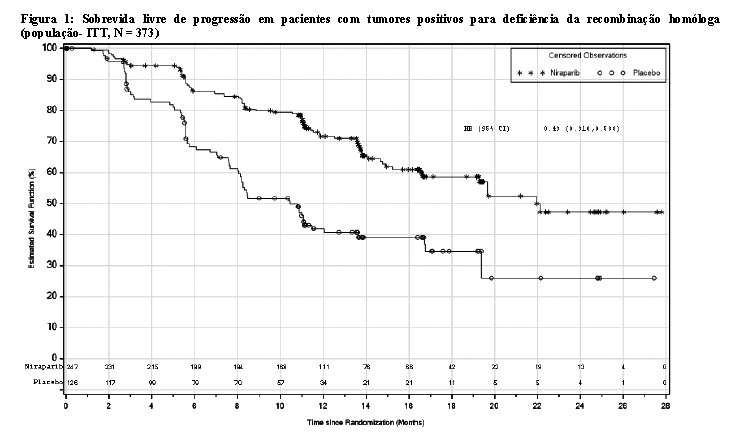
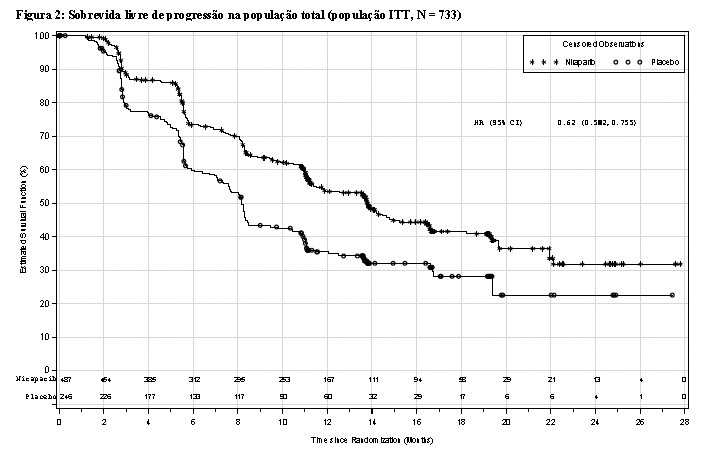
Dentro da população positiva para deficiência da recombinação homóloga, uma taxa de risco de 0,40 (IC 95% [0,27; 0,62]) foi encontrada no subgrupo de pacientes com câncer de ovário mutado BRCA (N = 223). No subgrupo de pacientes sem uma mutação BRCA (N = 150), a taxa de risco foi de 0,50 (IC 95% [0,31; 0,83]).
Para a população negativa para deficiência da recombinação homóloga (HDR negativa) (N = 249), a taxa de risco foi de 0,68 (IC 95% [0,49, 0,94]).
No momento da análise da PFS, os dados de sobrevida global eram limitados com 11% de mortes na população total.
Não houve diferenças estatisticamente significantes entre Zejula e placebo em termos de sintomas relatados pelas pacientes ou QVRS (qualidade de vida relacionada à saúde) devido às taxas de melhora e deterioração medidas com FOSI, EQ-5D-5L e EORTC-QLQ.
O tratamento com Zejula não teve um impacto negativo nos resultados relatados pelas pacientes ou na qualidade de vida relacionada à saúde, conforme avaliado por FOSI e EQ-5D-5L. Exceções foram observadas nos resultados gastrointestinais do EORTC-QLQ-C30 nas pacientes tratadas com niraparibe [constipação, náusea/vômito, perda de apetite, dispneia] e nas pacientes com placebo [diarreia]. Além disso, nenhuma diferença relacionadas à piora abdominal/GI foi observada conforme medido pelo EORTC-QLQ-OV28, entre as pacientes tratadas com niraparibe e placebo.
Terapia de manutenção para câncer de ovário recorrente
A segurança e a eficácia do niraparibe como terapia de manutenção foram investigadas em um estudo internacional de fase 3, randomizado, duplo-cego e controlado por placebo (NOVA) em pacientes com carcinoma de ovário epitelial seroso, das trompas de Falópio ou peritoneal primário, principalmente de alto grau, anteriormente sensíveis à platina. As pacientes deveriam ter uma histologia serosa de alto grau (ou Grau 3), uma histologia predominantemente serosa ou uma gBRCAmut conhecida. No entanto, nos dados clínicos, o grau numérico do câncer de ovário, das trompas de Falópio ou peritoneal primário não foram coletados.
A sensibilidade à platina foi definida pela resposta completa (RC) ou resposta parcial (RP) por mais de seis meses ao penúltimo regime terapêutico à base de platina.
Todas as pacientes haviam recebido anteriormente pelo menos dois regimes terapêuticos à base de platina e mostraram uma resposta (completa ou parcial) ao seu último regime terapêutico à base de platina. As concentrações de CA-125 deveriam estar na faixa normal ou terem tido uma redução maior que 90% durante seu último tratamento à base de platina e deveriam estar estáveis por pelo menos 7 dias.. As pacientes não haviam recebido tratamento anterior com um inibidor da PARP, incluindo o niraparibe.
As pacientes elegíveis para o estudo foram designadas para uma das duas coortes, dependendo dos resultados de um teste de mutação da linha germinativa BRCA (coorte gBRCAmut e coorte não gBRCAmut). Em cada coorte, as pacientes foram aleatoriamente designadas para o tratamento com niraparibe ou placebo na proporção de 2:1.
As pacientes foram designadas para a coorte gBRCAmut com base no resultado de teste em amostras de sangue para análise gBRCA realizadas antes da randomização. Nesta coorte, a eficácia foi avaliada na população gBRCAmut global. O estudo atendeu ao objetivo primário de uma melhoria na PFS estatisticamente significativa para o tratamento de manutenção com niraparibe quando comparado ao placebo na coorte gBRCAmut (HR 0,27; IC 95 % 0,173; 0,410; p < 0,0001).
Na coorte não-gBRCAmut, testes exploratórios para tumor com mutação BRCA e HRD realizados antes do estudo foram não-cegos usando tecido obtido no momento do diagnóstico inicial ou no momento da recorrência. A eficácia na coorte não gBRCAmut foi realizada de maneira hierárquica com o teste de subconjunto de HRD positivo (BRCAmut somático e HRD positivo/BRCAwt) realizado inicialmente, seguido por um teste da coorte não-gBRCAmut global caso o primeiro teste fosse estatisticamente significativo.
O estudo atendeu ao desfecho primário na coorte não-gBRCAmut global (RH 0,45; IC95 % 0,338; 0,607; p < 0.0001). O teste experimental HRD avaliou a presença do tumor BRCAmut e três medidas indiretas de instabilidade do genoma do tumor: perda de heterozigosidade, desequilíbrio alélico telomérico (TAI) e transições de forma em grande escala. No grupo positivo para deficiência da recombinação homóloga (HDR positiva), a razão de risco foi de 0,38 (IC 95%, 0,243; 0,586; p < 0,0001). No grupo negativo para deficiência da recombinação homóloga (HDR negativa), a razão de risco foi de 0,58 (IC95% 0,361; 0,922; p = 0,0226). O teste experimental não foi capaz de discriminar quais as pacientes se beneficiariam ou não com a terapia de manutenção com niraparibe.
Dentro de cada coorte, a randomização foi estratificada com base em três critérios: tempo para progressão após o penúltimo tratamento à base de platina antes do recrutamento para o estudo (6 a < 12 meses ou ≥ 12 meses); uso de bevacizumabe com o penúltimo ou último regime terapêutico à base de platina (sim/não); melhor resposta durante o último regime terapêutico à base de platina (resposta total ou parcial).
As pacientes iniciaram o tratamento no ciclo 1/dia 1 com 300 mg de niraparibe ou o placebo correspondente, com administração diária em ciclos contínuos de 28 dias. As visitas à clínica foram realizadas em cada ciclo (4 semanas ± 3 dias).
Durante o estudo NOVA, 48% das pacientes necessitaram de interrupção do tratamento no ciclo 1. Aproximadamente 47% das pacientes retomaram o tratamento com uma dose reduzida no ciclo 2.
A dose mais comum usada em pacientes tratadas com niraparibe no estudo NOVA foi de 200 mg.
A sobrevida livre de progressão como desfecho primário foi determinada por uma avaliação central, independente e cega, de acordo com os critérios RECIST (Critérios de Avaliação de Resposta em Tumores Sólidos, Versão 1.1) ou de acordo com os achados clínicos, os sintomas e o aumento no antígeno CA-125. A sobrevida livre de progressão foi medida entre o tempo de randomização (até 8 semanas após o término do regime de quimioterapia) e a progressão da doença ou óbito.
A análise de eficácia primária para sobrevida livre de progressão foi definida e avaliada prospectivamente e separadamente para a coorte gBRCAmut e a coorte não gBRCAmut. O desfecho primário de eficácia, sobrevida livre de progressão, foi analisado para a coorte não-gBRCAmut usando um esquema de teste hierárquico. Durante a primeira etapa do teste, a PFS foi avaliada no grupo de pacientes com tumores HDR pos; se o resultado fosse significativo, a PFS seria avaliada na coorte não-gBRCAmut completa (BRCAmut somática, HDR pos/BRCA selvagem, HDR neg).
Os dados demográficos, as características basais da doença e a história do tratamento foram geralmente bem equilibrados entre os grupos niraparibe e placebo nas coortes gBRCAmut (n = 203) e não-gBRCAmut (n = 350). A idade mediana, entre tratamentos e coortes, variou de 57 a 63 anos. O tumor primário estava localizado, na maioria das pacientes ( > 80%) em cada coorte, no ovário; na maioria das pacientes ( > 88%), a histologia do tumor mostrou características serosas. Nos dois grupos de tratamento e nas duas coortes, uma alta proporção de pacientes recebeu 3 ou mais linhas de tratamento antes da quimioterapia, incluindo 49% e 34% das pacientes tratadas com niraparibe nas coorte gBRCAmut e não-gBRCAmut, respectivamente. A maioria das pacientes tinha entre 18 e 64 anos (65%), branca (87%) e apresentava um desempenho ECOG de 0 (68%).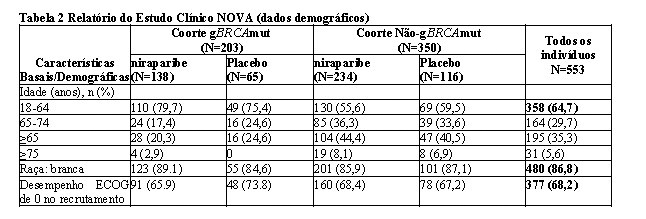
Entre as coortes gBRCAmut e não gBRCAmut, 24% a 27% tinham recebido anteriormente bevacizumabe com o penúltimo ou último regime de platina. O estudo alcançou seu objetivo principal de melhorar estatisticamente a sobrevida livre de progressão com niraparibe como monoterapia de manutenção em comparação com o placebo na coorte gBRCAmut (HR 0,27; IC 95% 0,173-0,410; p < 0,0001); sobrevida média livre de progressão 21,0 meses (IC 95% 12,9 - não alcançado) com niraparibe e sobrevida média livre de progressão 5,5 meses (IC 95% 3,8 - 7,2) com placebo e nas pacientes não-gBRCAmut (HR 0,45; IC 95% 0,338-0,617, p < 0,0001); sobrevida média sem progressão 9,3 meses (IC 95% 7,2 - 11,2) com niraparibe e sobrevida média sem progressão 3,9 meses (IC 95% 3,7 - 5,5) com placebo. A avaliação do pesquisador sobre a sobrevida livre de progressão coincidiu com a do comitê de revisão independente, que realizou a avaliação radiológica central e clínica clínico-oncológica às cegas.
No momento da análise da sobrevida livre de progressão, os dados de sobrevida global disponíveis eram limitados com 17% de mortes entre as duas coortes.
Os desfechos secundários de eficácia, intervalo livre de quimioterapia (CFI), tempo até a primeira terapia subsequente (TFST) e sobrevida livre de progressão 2 (PFS2) mostraram um efeito de tratamento a favor do braço de tratamento com niraparibe na coorte gBRCAmut e em toda a coorte não-gBRCAmut.
Intervalo livre de quimioterapia, definido como o período entre o final do último tratamento à base de platina e o início do tratamento antineoplásico seguinte (com exceção da terapia de manutenção): Na coorte gBRCAmut (HR 0,26, IC95% 0,17 - 0,41); média de 22,8 meses (IC 95% 17,9 - não atingido) para niraparibe e média de 9,4 meses (IC 95% 7,9 - 10,6) para placebo; na coorte não-gBRCAmut (HR 0,50; IC 95% 0,37-0,67); média de 12,7 meses (IC 95% 11,0-14,7) para niraparibe e média de 8,6 meses (IC 95% 6,9-10,0) para placebo.
Duração até o primeiro tratamento subsequente, definido como o período entre a data da randomização e a data do primeiro tratamento antineoplásico subsequente ou morte: Na coorte gBRCAmut (HR 0,31, IC 95% 0,21 - 0,48); média de 21,0 meses (IC 95% 17,5 - não atingido) para niraparibe e média de 8,4 meses (IC 95% 6,6 - 10,6) para placebo; na coorte não-gBRCAmut (HR 0,55; IC 95% 0,41 - 0,72); média de 11,8 meses (IC95% 9,7 - 13,1) para niraparibe e média de 7,2 meses (IC 95% 5,7 - 8,5) para placebo.
Sobrevida livre de progressão 2, definida como o período entre a data da randomização neste estudo e a primeira data da avaliação da progressão no próximo tratamento antineoplásico após o estudo ou a data da morte, independentemente da causa: Na coorte gBRCAmut (HR 0, 48, IC 95% 0,28-0,82); média de 25,8 meses (IC 95% 20,3 - não alcançado) com niraparibe e média de 19,5 meses (IC 95% 13,3 - não alcançado) com placebo; na coorte não-gBRCAmut (HR 0,69; IC 95% 0,49-0,96); média de 18,6 meses (IC 95% 16,2-21,7) com niraparibe e média de 15,6 meses (IC 95% 13,2-20,9) com placebo.
Dados de desfechos reportados pelas pacientes a partir de ferramentas validadas (FOSI -Avaliação Funcional da Terapia para Câncer/ Índice de Sintomas Ovarianos e EQ-5D- escala Europeia de Qualidade de Vida, 5-Dimensões) indicam que as pacientes tratadas com niraparibe não reportaram diferenças em relação aquelas tratadas com placebo em medidas associadas à qualidade de vida.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades Farmacodinâmicas
Código ATC
L01XK02
Mecanismo de ação
O niraparibe é um inibidor das enzimas poli (ADP-ribose) polimerase (PARP), PARP-1 e PARP-2, que desempenham um papel no reparo do DNA. Estudos in vitro mostraram que a citotoxicidade induzida pelo niraparibe pode inibir a atividade enzimática da PARP e aumentar a formação de complexos DNA-PARP, levando a danos no DNA, apoptose e morte celular.
Efeitos Farmacodinâmicos
Foi observado um aumento na citotoxicidade induzida pelo niraparibe em linhagens celulares de tumor com ou sem deficiência na expressão dos genes BRCA1/2. Demonstrou-se que o niraparibe reduz o crescimento do tumor em diferentes modelos experimentais: xenoenxertos implantados em camundongos de tumores humanos de câncer de ovário seroso de alto grau com mutação BRCA 1 e 2; sem mutação BRCA, mas com deficiência da recombinação homóloga; e sem mutação BRCA e sem deficiência da recombinação homóloga detectável.
Eletrofisiologia Cardíaca
O niraparibe não demonstrou prolongamento do intervalo QTc clinicamente significativo nos estudos clínicos. O potencial de prolongamento do intervalo QTc com niraparibe foi avaliado em um estudo randomizado e controlado com placebo em pacientes com câncer de ovário (NOVA). A análise QTcF foi conduzida em 58 indivíduos no total (53 com niraparibe, 5 com placebo) derivados do estudo NOVA principal e dois subestudos (Efeito de Alimentos Aberto e QTc Aberto). Nenhuma paciente que foi submetida a monitoramento intensivo de ECG no estudo NOVA principal ou no subestudo QTc apresentou QTcF > 480 ms ou alteração do QTcF do valor basal > 30 ms em qualquer momento pós-administração.
O estudo avaliou os efeitos do niraparibe na repolarização cardíaca após uma dose única de niraparibe (300 mg por via oral) e correlacionou as alterações do valor basal do QTc com as concentrações plasmáticas de niraparibe. Em pacientes que foram submetidas a monitoramento intensivo de ECG no NOVA principal ou subestudo QTc, o maior aumento observado no QTcF desde a linha de base (∆QTcF) foi de 4,3 ± 8,8 ms (média ± DP) 3 horas após administração da dose. O limite superior do IC 95% unilateral do ∆QTcF foi de 6,7 ms 3 horas após a administração da dose. O maior limite superior do IC 95% unilateral da alteração média da linha de base e do placebo no intervalo QTcF (∆∆QTcF) foi de 6,3 ms 4 horas após a administração da dose.
Toxicologia Animal e/ou Farmacologia
In vitro, o niraparibe se ligou ao transportador de dopamina (DAT), transportador de norepinefrina (NET) e transportador de serotonina (SERT) e inibiu a captação de norepinefrina e dopamina em células com valores de IC50 menores do que Cmín no estado de equilíbrio, em pacientes que receberam a dose recomendada. O niraparibe tem o potencial de causar, em pacientes, efeitos relacionados à inibição desses transportadores (por exemplo, cardiovascular ou SNC).
A administração intravenosa de niraparibe a 1, 3 e 10 mg/kg durante 30 minutos a cães vagotomizados resultou em aumento da faixa de pressão arterial de 13-20, 18-27 e 19-25% e aumento da faixa de frequência cardíaca de 2-11, 4-17 e 12-21% acima dos níveis pré-dose, respectivamente. As concentrações plasmáticas de niraparibe não-ligado em cães a estes níveis de dose foram aproximadamente 0,5, 1,5 e 5,8 vezes a Cmáx não ligada no estado de equilíbrio em pacientes recebendo a dose recomendada.
Além disso, o niraparibe atravessou a barreira hematoencefálica em ratos e macacos após administração oral. A razão líquido cefalorraquidiano (LCR): Cmáx plasmático de niraparibe administrado a 10 mg/kg por via oral a dois macacos Rhesus foram de 0,10 e 0,52.
Propriedades Farmacocinéticas
Absorção
Após uma dose única de 300 mg de niraparibe em jejum, o niraparibe foi mensurável no plasma após 30 minutos. A concentração plasmática máxima média (Cmax) de niraparibe [804 ng/mL (coeficiente de variação de 50,2%)] foi atingida após aproximadamente 3 horas. Após doses orais múltiplas de 30 mg a 400 mg de niraparibe uma vez ao dia, o acúmulo de niraparibe foi de aproximadamente 2-3 vezes.
A exposição sistêmica ao niraparibe (Cmax e AUC) aumentou proporcionalmente à dose quando a dose de niraparibe aumentou de 30 mg para 400 mg. A biodisponibilidade absoluta do niraparibe é de aproximadamente 73%.
Uma refeição concomitante com alto teor de gordura não alterou significativamente os parâmetros farmacocinéticos do niraparibe após administração de 300 mg de niraparibe.
Distribuição
Em uma análise farmacocinética populacional de niraparibe, o Vd/F foi de 1311 L em pacientes com câncer, consistente com o volume aparente de distribuição de 1220 L observado no estudo ADME (Absorção, Distribuição, Metabolismo e Eliminação).
Metabolismo
O niraparibe é metabolizado principalmente em M1 por carboxilesterases (CE), esse metabólito não inibe a PARP. Em um estudo de balanço de massa, M1 e M10 (os glicuronídeos de M1 formados posteriormente) foram os principais metabólitos na corrente sanguínea. No plasma, os 3 metabólitos glicuronídeos do M1 juntos representam ~ 55,7% da AUC da radioatividade total, M1 9,3%, niraparibe 2,4% e M1 metilado 2,5%. As isoformas responsáveis da CES e UGT não são totalmente caracterizadas.
Eliminação
Após uma dose oral única de 300 mg de niraparibe, sua meia-vida média terminal (t1/2) variou de 48 horas a 51 horas (aproximadamente 2 dias). Numa análise farmacocinética populacional, a depuração total aparente (CL/F) do niraparibe em pacientes com câncer foi de 16,5 L/h.
O niraparibe é principalmente excretado no fígado e nos rins. Após a administração de uma dose oral única de 300 mg [14C]-niraparibe, uma média de 86,2% (variação de 71% a 91%) da dose foi encontrada na urina e nas fezes novamente durante 21 dias. Da radioatividade detectada, 47,5% da dose (intervalo de 33,4% a 60,2%) estava na urina e 38,8% (intervalo de 28,3% a 47,0%) nas fezes. Nas amostras coletadas durante 6 dias, 40,0% da dose foi encontrada na urina, principalmente como M1, e 31,6% da dose nas fezes, principalmente como niraparibe inalterado.
Carcinogênese
Nenhum estudo de carcinogenicidade foi realizado com niraparibe.
Toxicologia Reprodutiva
Não foram realizados estudos em animais sobre toxicidade reprodutiva e de desenvolvimento.
Toxicidade por dose repetida
Estudos de toxicidade de dose repetida, com administração diária por via oral de niraparibe por até 3 meses, foram realizados em ratos e cães.. O principal órgão alvo de ambas as espécies foi a medula óssea, com alterações correspondentes nos parâmetros hematológicos periféricos. Uma diminuição na espermatogênese também foi encontrada em ambas as espécies. Estes resultados foram observados em exposições mais baixas do que as observadas clinicamente e foram amplamente reversíveis nas 4 semanas após a última administração.
Genotoxicidade
O niraparibe não mostrou efeitos mutagênicos no teste de Ames, mas foi clastogênico em um teste in vitro de aberrações cromossômicas em mamíferos e em um teste in vivo de micronúcleos na medula óssea do rato. Essa clastogênese é consistente com a instabilidade genômica resultante da farmacologia primária do niraparibe e indica potencial genotóxico em humanos.
Populações Especiais de Pacientes
Insuficiência Renal
Na análise farmacocinética populacional, as pacientes com insuficiência renal leve (clearance de creatinina 60-90 mL/min) e moderada (30-60 mL/min) tiveram clearance do niraparibe levemente reduzido em comparação com indivíduos com função renal normal (exposição 7-17% maior na insuficiência leve e 17-38% maior na insuficiência renal moderada). A diferença na exposição não é considerada para justificar um ajuste da dose.
Insuficiência Hepática
De acordo com a análise farmacocinética da população, os valores iniciais de albumina sérica, AST, bilirrubina total e ALT não tiveram influência clinicamente significativa na farmacocinética do niraparibe em pacientes com insuficiência hepática leve.
Em um estudo clínico em pacientes com câncer usando os critérios NCI-ODWG para classificar o grau de insuficiência hepática, a AUCinf do niraparibe em pacientes com insuficiência hepática moderada (n = 8) foi 1,56 (IC 90%: 1,06 a 2,30) vezes a AUCinf do niraparibe em pacientes com função hepática normal (n = 9) após administração de uma dose única de 300 mg. O ajuste da dose de niraparibe é recomendado para pacientes com insuficiência hepática moderada (ver Posologia e Modo de Usar). A insuficiência hepática moderada não teve efeito na Cmáx do niraparibe ou na ligação do niraparibe às proteínas.
A farmacocinética de Zejula em pacientes com insuficiência hepática grave é desconhecida.
Idade, peso e raça
Não houve efeito covariável significativo de idade, peso e etnia na farmacocinética do niraparibe; estes parecem não afetar a farmacocinética do niraparibe.
População pediátrica
Nenhum estudo farmacocinético pediátrico foi realizado.
4.CONTRAINDICAÇÕES
Zejula é contraindicado nos seguintes casos de:
- Hipersensibilidade ao ingrediente ativo ou a qualquer um dos excipientes.
- Amamentação durante o tratamento e até 1 mês após a última dose (ver Advertências e Precauções).
Este medicamento é contraindicado para uso por mulheres que estejam amamentando.
5.ADVERTÊNCIAS E PRECAUÇÕES
Efeitos adversos hematológicos
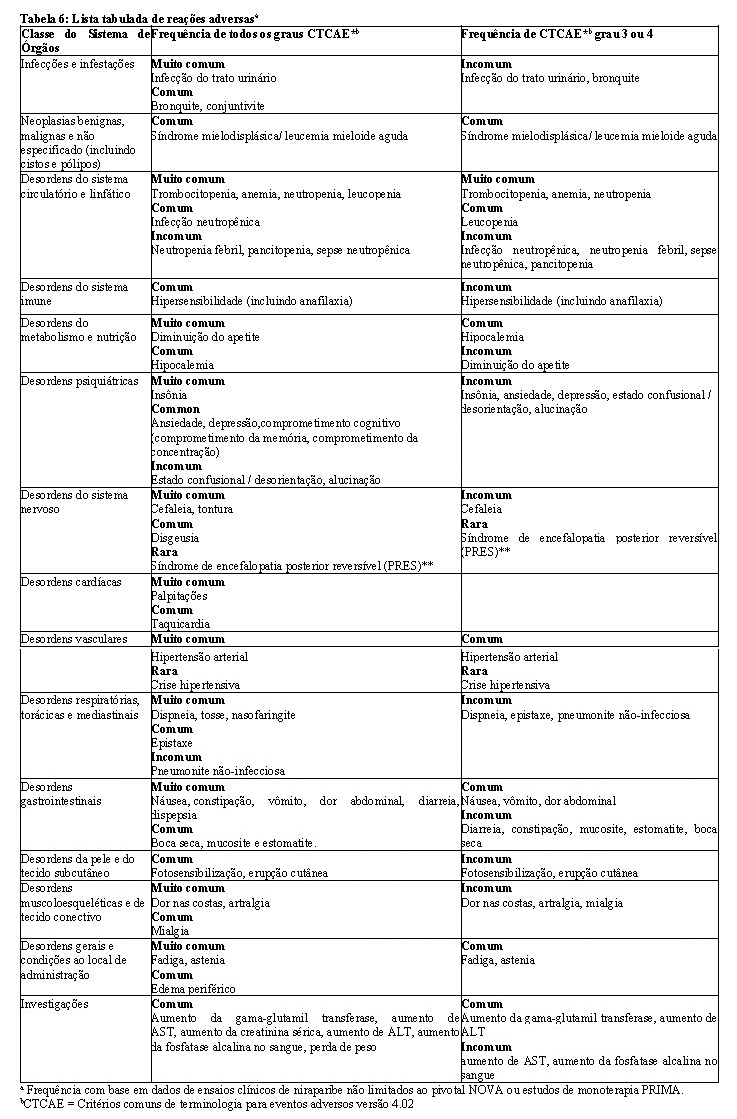
Para descobrir quaisquer alterações clinicamente relevantes nos parâmetros hematológicos durante o tratamento, recomenda-se verificar ohemograma uma vez por semana durante o primeiro mês, depois uma vez por mês durante os próximos 11 meses e, em seguida, em intervalos regulares (ver Posologia e Modo de Usar).Zejula deve ser descontinuado se a paciente desenvolver um hemograma tóxico persistente grave incluindo pancitopenia, que não normalizou no período de 28 dias após a descontinuação. A paciente deve ser encaminhada a um hematologista para investigações adicionais.
Devido ao risco de trombocitopenia, os anticoagulantes e os medicamentos que diminuem a contagem de plaquetas devem ser usados comcautela (ver Reações Adversas).
Reações adversas hematológicas (trombocitopenia, anemia e neutropenia) foram reportadas em pacientes tratadas com Zejula (ver Reações Adversas).
Pacientes com baixo peso corporal ou baixa contagem plaquetária podem estar sob risco aumentado de apresentar trombocitopenia grau 3 oumaior (ver Posologia e Modo de Usar).
Não iniciar o tratamento com Zejula até que as pacientes tenham se recuperado de toxicidades hematológicas causadas para quimioterapiaprévia.
Síndrome mielodisplásica/leucemia mieloide aguda
Síndrome mielodisplásica/leucemia mieloide aguda (MDS/AML), incluindo casos com desfecho fatal, foi relatada em pacientes que receberam Zejula (ver item Reações Adversas) Casos de síndrome mielodisplásica/leucemia mieloide aguda (MDS/AML) foram observados em pacientes recebendo Zejula em monoterapia ou em combinação e em relatórios pós-comercialização.
Nos estudos clínicos, a duração do tratamento com Zejula antes do aparecimento de uma MDS/AML variou entre 0,5 meses e mais de 4,9 anos. Os casos eram típicos de MDS/AML secundária associada ao tratamento antineoplásico. Todas as pacientes receberam vários esquemas de quimioterapia à base de platina e um grande número delas também recebeu outras substâncias prejudiciais ao DNA e radioterapia. Algumas pacientes tinham histórico de supressão da medula óssea.
Para suspeita de MDS / AML ou toxicidades hematológicas prolongadas, o paciente deve ser encaminhado a um hematologista para avaliação adicional. Se MDS / AML for confirmado, o tratamento com Zejula deve ser interrompido.
Hipertensão, incluindo crise hipertensiva
Foram notificados casos de hipertensão, incluindo crise hipertensiva, durante o tratamento com Zejula (ver Reações Adversas).
A hipertensão arterial pré-existente deve ser efetivamente controlada antes do início do tratamento com Zejula. A pressão sanguínea deve ser verificadas semanalmente nos primeiros dois meses de tratamento com Zejula, depois mensalmente no primeiro ano de tratamento, e posteriormente em intervalos regulares. Pacientes devem ser orientadas a procurar seu médico em caso de aumento da pressão sanguínea. Pacientes com distúrbios cardiovasculares, especialmente insuficiência coronariana, arritmia cardíaca e hipertensão, devem ser cuidadosamente monitoradas. A hipertensão deve ser tratada com medicamentos anti-hipertensivos e a dose de Zejula ajustada, se necessário (ver Posologia e Modo de Usar).
Síndrome de encefalopatia posterior reversível (PRES)
Houve raros relatos (0,09% das pacientes em estudos clínicos) de pacientes tratadas com niraparibe que desenvolveram sinais e sintomas consistentes com Síndrome de Encefalopatia Posterior Reversível (PRES) (ver Reações Adversas). PRES é um distúrbio neurológico raro que pode se apresentar com os seguintes sinais e sintomas, incluindo convulsões, cefaleia, alteração do estado mental, distúrbio visual ou cegueira cortical, com ou sem hipertensão associada. Um diagnóstico de PRES requer confirmação por imagem do cérebro, de preferência imagem de ressonância magnética (MRI). Em pacientes que desenvolvem PRES, é recomendado o tratamento de sintomas específicos, incluindo controle da hipertensão, junto com a descontinuação do niraparibe. A segurança de reiniciar a terapia com niraparibe em pacientes que já experimentaram PRES não é conhecida.
Lactose
As cápsulas de Zejula contêm lactose monohidratada. Pacientes com problemas hereditários raros de intolerância à galactose, deficiência de lactase ou má absorção de glicose-galactose não devem tomar este medicamento.
Excipientes
Este medicamento contém tartrazina (E102), que pode causar reações alérgicas.
Este produto contém o corante amarelo de TARTRAZINA que pode causar reações de natureza alérgica, entre as quais asma brônquica, especialmente em pessoas alérgicas ao ácido acetilsalicílico.
Gravidez e Lactação
Fertilidade
Não existem dados clínicos disponíveis sobre a influência de Zejula na fertilidade. Foi observada uma diminuição reversível da espermatogênese em estudos com animais.
Mulheres com potencial para engravidar/contracepção
Mulheres com potencial para engravidar não devem estar grávidas no início do tratamento e não devem planejar engravidar enquanto estiverem tomando Zejula.
Mulheres com potencial para engravidar devem usar contraceptivos altamente eficazes durante o tratamento e por 6 meses após receber a última dose de Zejula. Para todas as mulheres em idade fértil, um teste de gravidez deve ser realizado antes do tratamento.
Gravidez
Não existem dados disponíveis sobre o uso de Zejula em mulheres grávidas. Não foram realizados estudos em animais sobre toxicidade reprodutiva e de desenvolvimento. No entanto, devido ao seu mecanismo de ação, o niraparibe pode prejudicar o embrião ou o feto quando administrado a uma mulher grávida, incluindo efeitos letais e teratogênicos no embrião. Zejula não deve ser utilizado durante a gravidez.
Lactação
Não se sabe se o niraparibe ou seus metabólitos são excretados no leite humano. A amamentação é contraindicada durante o tratamento com Zejula e no primeiro mês após a última dose (ver Contraindicações).
Categoria C de risco na gravidez.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Capacidade de realizar tarefas que requerem habilidades motoras, cognitivas ou de julgamento
As pacientes podem sentir fraqueza, cansaço, com dificuldade de concentração e tonturas enquanto estiverem usando Zejula. Pacientes com esses sintomas devem ter cuidado ao conduzir veículos e operar máquinas.
Populações especiais
Ver Populações Especiais de Pacientes, em Características Farmacológicas.
6. INTERAÇÕES MEDICAMENTOSAS
Interações Farmacodinâmicas
A administração simultânea de niraparibe e vacinas ou agentes imunossupressores não foi investigada.
Também existem dados limitados sobre a combinação de niraparibe com medicamentos citotóxicos. Como resultado, deve-se ter cautela quando o niraparibe é coadministrado com vacinas, agentes imunossupressores ou outros medicamentos citotóxicos.
Interações Farmacocinéticas
Nenhum estudo clínico foi realizado sobre interações medicamentosas com Zejula.
O niraparibe inibe o MATE1 (transportador de extrusão de múltiplos medicamentos e toxinas) e o MATE2-K in vitro. O niraparibe pode aumentar a concentração de medicamentos no plasma e nas células tubulares dos rins que são excretados principalmente pelo MATE1 e MATE2-K (por exemplo, metformina).
O niraparibe inibe a proteína de resistência ao câncer de mama (BCRP) em pequena extensão in vitro. A relevância clínica é desconhecida e o niraparibe pode aumentar os níveis plasmáticos de medicamentos cuja distribuição ou eliminação depende do BCRP.
Resumo dos resultados de estudos in vitro
Inibição das UDP-glucuronosiltransferases (UGTs): O niraparibe não apresentou efeito inibitório contra as isoformas da UGT (UGT1A1, UGT1A4, UGT1A9 e UGT2B7) até 200 mM in vitro. Portanto, o potencial de inibição clinicamente relevante das UGTs pelo niraparibe é mínimo.
Inibição do CYP: O niraparibe e seu principal metabólito M1 não são inibidores das enzimas CYPA2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 e CYP3A4.
O niraparibe induz ligeiramente o CYP1A2 in vitro; a relevância clínica não é excluída. Logo, deve-se cautela ao coadministrar niraparibe com substratos da CYP1A2, em especial, aqueles com faixa terapêutica estreita como clozapina, teofilina e ropinirol.
Indução do CYP: O niraparibe e seu principal metabólito M1 não são indutores do CYP3A4.
O niraparibe é um provável substrato da carboxilesterase 1. O papel de outras isoformas da carboxilesterase no metabolismo do niraparibe é desconhecido.
Inibição de sistemas de transporte: O niraparibe é um inibidor do MATE1 e MATE2-K. O metabólito M1 não é um inibidor da P-gp, BCRP ou BSEP. O niraparibe e o M1 não são inibidores dos polipeptídeos de transporte de ânions orgânicos 1B1 (OATP1B1), 1B3 (OATP1B3) ou do transportador de ânions orgânicos 1 (OAT1) e 3 (OAT3) ou do transportador de cátions orgânicos 2 (OCT2).
O niraparibe não é um inibidor de BSEP. In vitro, niraparibe inibe a P-gp muito fracamente e BCRP com um IC50 = 161 mM e 5.8 mM, respectivamente. Portanto, uma interação clinicamente significativa relacionada a uma inibição desses transportadores de efluxo, embora improvável, não pode ser excluída. Deve-se ter cautela quando niraparibe é administrado com substratos do BCRP (irinotecano, rosuvastatina, sinvastatina, atorvastatina e metrotrexato). Substrato do sistema de transporte: O niraparibe é um substrato da glicoproteína P (P-gp) e da proteína de resistência ao câncer de mama (BCRP). O niraparibe não é um substrato da bomba de exportação de sal biliar (BSEP). O metabólito M1 não é um substrato de P-gp, BCRP ou BSEP. Nem niraparibe nem M1 são substratos dos polipeptídeos de transporte de ânions orgânicos 1B1 (OATP1B1), 1B3 (OATP1B3) ou do transportador de cátions orgânicos 1 (OCT1), do transportador de ânions orgânicos 1 (OAT1) e 3 (OAT3) ou o transportador de cátions orgânicos 2 (OCT2). In vitro, niraparibe causou inibição fraca do transportador de cátions orgânicos 1 (OCT1), cuja relevância clínica não foi estudada.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Cuidados de Armazenamento
Mantenha o produto na embalagem original e em temperatura abaixo de 25°C. O prazo de validade é de 36 meses a partir da data de fabricação.
Número do lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Aspectos físicos / Características organolépticas
Cápsulas gelatinosa dura, tamanho 0 com corpo branco gravado '100mg' em preto e tampa roxa gravado 'niraparibe' em branco contendo um pó branco a quase branco.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
Modo de Uso
O tratamento com Zejula deve ser iniciado e monitorado por um médico com experiência no uso de medicamentos antineoplásicos.
Zejula é administrado por via oral. A cápsula deve ser engolida sem necessidade de considerar as refeições.
A administração no momento de dormir pode ser um método em potencial para o manejo da náusea.
Posologia
Terapia de Manutenção de Câncer de Ovário de Primeira Linha
A dose inicial recomendada de Zejula é de 200 mg (duas cápsulas de 100 mg) uma vez ao dia. No entanto, para pacientes com peso ≥ 77 kg e uma contagem plaquetária basal ≥ 150.000/mL, a dose inicial recomendada é de 300 mg (três cápsulas de 1