ZEDORA
LIBBS
150 mg
trastuzumabe
Antineoplásico.
Apresentações.
Zedora 150 mg: pó liofilizado para solução injetável.
Cada embalagem contém um frasco-ampola de dose única com 150 mg de pó liofilizado de trastuzumabe para solução injetável para infusão via intravenosa.
VIA INTRAVENOSA
USO ADULTO
Composição.
Princípio ativo: cada frasco-ampola dose única contém 150 mg de pó liofilizado de trastuzumabe para solução injetável. O concentrado de Zedora reconstituído contém 21 mg/mL de trastuzumabe. Excipientes: Frasco-ampola de Zedora 150 mg: L-histidina, cloridrato de histidina monoidratado, macrogol, sorbitol, ácido clorídrico e hidróxido de sódio.
Informações técnicas.
1. INDICAÇÕES
Câncer de mama metastático
Zedora é indicado para o tratamento de pacientes com câncer de mama metastático que apresentam tumores com superexpressão do HER2:
• Em monoterapia para o tratamento de pacientes que já tenham recebido um ou mais tratamentos quimioterápicos para suas doenças metastáticas;
• Em combinação com paclitaxel ou docetaxel para o tratamento de pacientes que ainda não tenham recebido quimioterapia para suas doenças metastáticas.
Câncer de mama inicial
Zedora é indicado para o tratamento de pacientes com câncer de mama inicial HER2-positivo:
• Após cirurgia, quimioterapia (neoadjuvante ou adjuvante) e radioterapia (quando aplicável);
• Após quimioterapia adjuvante com doxorrubicina e ciclofosfamida, em combinação com paclitaxel ou docetaxel;
• Em combinação com quimioterapia adjuvante de docetaxel e carboplatina;
• Em combinação com quimioterapia neoadjuvante seguida por terapia adjuvante com Zedora para câncer de mama localmente avançado (inclusive inflamatório) ou tumores > 2 cm de diâmetro.
Câncer gástrico avançado
Zedora em associação com capecitabina ou 5-fluorouracil (5-FU) intravenoso e um agente de platina é indicado para o tratamento de pacientes com adenocarcinoma inoperável, localmente avançado, recorrente ou metastático do estômago ou da junção gastroesofágica, HER2-positivo, que não receberam tratamento prévio contra o câncer para a doença metastática.
2. RESULTADOS DE EFICÁCIA
O medicamento Zedora (trastuzumabe) é um medicamento biológico desenvolvido pela via de comparabilidade (biossimilar). O programa de desenvolvimento do produto foi projetado para demonstrar a comparabilidade entre Zedora e o medicamento comparador (Herceptin®).
A) Câncer de mama metastático
O trastuzumabe (Herceptin®) como monoterapia foi utilizado em estudo clínico fase II para pacientes com câncer de mama metastático que apresentavam tumores com superexpressão do HER2 tratados sem sucesso com um ou mais esquemas quimioterápicos prévios para essas doenças metastáticas. Trastuzumabe (Herceptin®) como agente único produziu respostas objetivas duradouras em mulheres com câncer de mama metastático HER2-positivo que progrediu após a quimioterapia para doença metastática.1
O trastuzumabe (Herceptin®) foi utilizado em estudos clínicos, fase III em combinação com paclitaxel ou com uma antraciclina (doxorrubicina ou epirrubicina) mais ciclofosfamida (AC), como terapia de primeira linha para pacientes com câncer de mama metastático que apresentavam tumor com superexpressão HER2.2 Pacientes que tinham recebido previamente quimioterapia adjuvante à base de antraciclina foram tratados com paclitaxel (175 mg/m2, com infusão durante três horas) com ou sem trastuzumabe (Herceptin®). Os pacientes poderiam ser tratados com trastuzumabe (Herceptin®) até a progressão da doença.2
A monoterapia com trastuzumabe (Herceptin®), utilizada no tratamento de segunda ou terceira linha de mulheres com câncer de mama metastático com superexpressão do HER2, resultou em taxa de resposta objetiva (completa e parcial) de 15% e sobrevida mediana de 13 meses.
A utilização de trastuzumabe (Herceptin®), em combinação com paclitaxel como tratamento de primeira linha de mulheres com câncer de mama metastático com superexpressão do HER2, prolonga significativamente o tempo mediano até a progressão da doença em comparação com paclitaxel em monoterapia. O aumento no tempo mediano até a progressão da doença para os pacientes tratados com trastuzumabe (Herceptin®) e paclitaxel é de 3,9 meses (6,9 meses versus 3,0 meses). A resposta tumoral e a taxa de sobrevida em um ano também aumentaram com trastuzumabe (Herceptin®) em combinação com paclitaxel versus paclitaxel isolado.2
O trastuzumabe (Herceptin®) também foi avaliado em estudo fase III randomizado, controlado, em combinação com docetaxel, como tratamento de primeira linha de mulheres com câncer de mama metastático. A combinação de trastuzumabe (Herceptin®) com docetaxel aumentou significativamente a taxa de resposta global (61% versus 34%,) e prolongou a mediana de tempo até a progressão da doença (em 5,6 meses), em comparação com pacientes tratados apenas com docetaxel. A sobrevida mediana também aumentou de forma significativa em pacientes tratados com a combinação, em comparação com aqueles que receberam docetaxel isoladamente (31,2 meses versus 22,7 meses).3
B) Câncer de mama inicial
1) Tratamento adjuvante
No tratamento adjuvante, trastuzumabe (Herceptin®), foi investigado em quatro grandes estudos de Fase III, multicêntricos e randomizados:
• O estudo BO16348 (estudo HERA) foi desenhado para comparar um4,5 e dois5 anos de tratamento com trastuzumabe (Herceptin®), a cada três semanas versus observação em pacientes com câncer de mama inicial HER2-positivo após cirurgia, quimioterapia e radioterapia (se aplicável). As pacientes haviam recebido tratamento locoregional e pelo menos 4 ciclos de quimioterapia adjuvante ou neoadjuvente. Pacientes designados para trastuzumabe (Herceptin®) receberam uma dose de ataque inicial de 8 mg/kg, seguida por 6 mg/kg, a cada três semanas, durante um ou dois anos.
• Os estudos NCCTG N9831 e NSABP-B31, que incluem a análise conjunta, foram desenhados para investigar o uso clínico do tratamento combinado de trastuzumabe (Herceptin®), intravenoso (IV) com paclitaxel após quimioterapia AC (adriamicina e ciclofosfamida). Adicionalmente o estudo NCCTG N9831 investigou a adição de trastuzumabe (Herceptin®), após a quimioterapia de AC-paclitaxel em pacientes com câncer de mama inicial HER2-positivo após cirurgia.6
• O estudo BCIRG 006 foi desenhado para investigar o tratamento combinado de trastuzumabe (Herceptin®) IV com docetaxel após a quimioterapia AC ou em combinação com docetaxel e carboplatina em pacientes com câncer de mama inicial HER2-positivo após cirurgia.7
Estudo BO16348 (Hera)
O câncer de mama inicial foi limitado a operável, primário, adenocarcinoma invasivo da mama, com tumores de nódulos axilares positivos ou negativos de, pelo menos, 1 cm de diâmetro.4,5
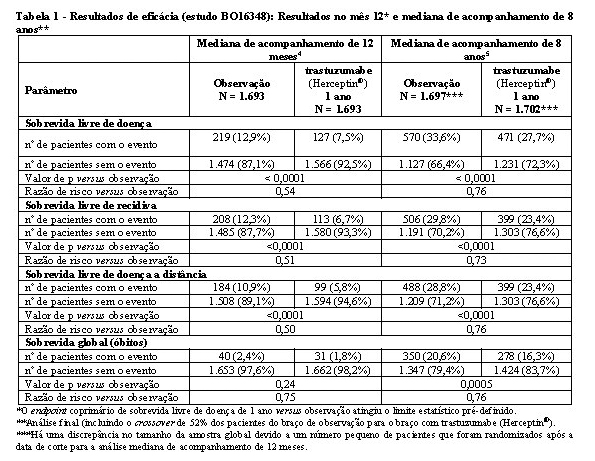
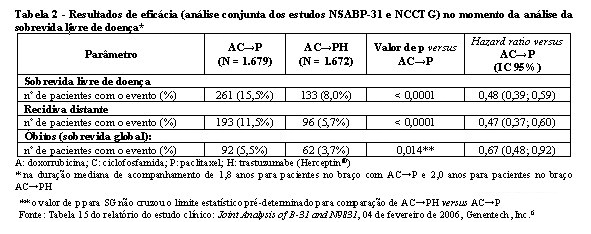
Os resultados de eficácia do estudo BO16348 estão resumidos na tabela 1.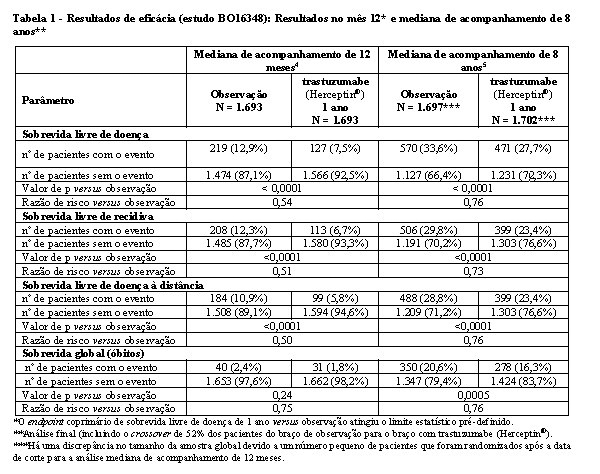
Os resultados de eficácia da análise interina cruzaram o limite estatístico pré-determinado no protocolo para a comparação estatística de um ano de trastuzumabe (Herceptin®) versus observação. Após a mediana de acompanhamento de 12 meses, a razão de risco (RR) para a sobrevida livre de doença (SLD) foi de 0,54 (IC 95% 0,44-0,67), que se traduz em um benefício absoluto, em termos de taxa de SLD durante dois anos de 7,6 pontos percentuais (85,8% versus 78,2%) favoráveis ao braço com trastuzumabe (Herceptin®).4
A análise final foi realizada após a mediana de acompanhamento de 8 anos, que demonstrou que o tratamento com trastuzumabe (Herceptin®) por um ano está associado a uma redução do risco de 24% em relação à observação somente (HR = 0,76, IC 95% 0,67, 0,86). Isso se traduz em um benefício absoluto em termos de taxa de sobrevida livre de doença durante 8 anos, de 6,4 pontos percentuais a favor de um ano de tratamento com trastuzumabe (Herceptin®).5
Nessa análise final, a extensão do tratamento com trastuzumabe (Herceptin®) por um período de dois anos não mostrou benefício adicional sobre o tratamento por um ano [SLD RR na população com intenção de tratamento (ITT) de dois anos versus um ano = 0,99 (IC 95% 0,87, 1,13, valor de p = 0,90) e sobrevida global (SG) RR = 0,98 (IC 95% 0,83-1,15, p = 0,78). A taxa de disfunção cardíaca assintomática foi maior no grupo de tratamento de dois anos (8,1% versus 4,6% no grupo de tratamento de um ano). Mais pacientes tiveram pelo menos um evento adverso de grau 3 ou 4 no grupo de tratamento de dois anos (20,4%) em comparação com o grupo de tratamento de 1 ano (16,3%).5
Estudos NCCTG N9831 e NSABP-B31
Na análise conjunta dos estudos NCCTG N9831 (North Central Cancer Treatment Group) e NSABP-B31 (National Surgical Adjuvant Breast and Bowel Project)6 o câncer de mama inicial foi limitado a mulheres com câncer de mama operável de alto risco, definido como HER2-positivo e linfonodo axilar positivo ou HER2-positivo e linfonodo negativo com características de alto risco (tamanho do tumor > 1 cm e receptor hormonal negativo ou tamanho do tumor > 2 cm, independentemente do status hormonal). Trastuzumabe (Herceptin®) foi administrado em combinação com paclitaxel após quimioterapia AC. O paclitaxel foi administrado conforme segue:
- paclitaxel intravenoso: 80 mg/m2, na forma de infusão intravenosa contínua, administrada toda semana, por um período de 12 semanas;
ou
- paclitaxel intravenoso: 175 mg/m2, na forma de infusão intravenosa contínua, administrada a cada três semanas, por um período de quatro ciclos (dia 1 de cada ciclo).
Pela semelhança do desenho dos estudos, os resultados de eficácia dos estudos NCCTG N9831 e NSABP-B31 foram analisados em conjunto e estão resumidos na tabela 2.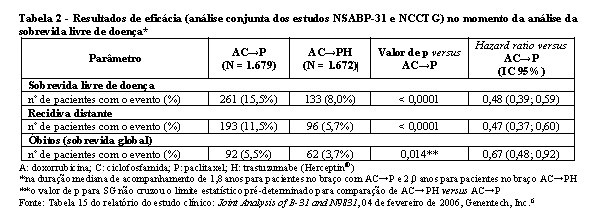
Considerando o objetivo primário SLD, a adição de trastuzumabe (Herceptin®) à quimioterapia com paclitaxel resultou em redução de 52% no risco de recidiva da doença. A RR transforma-se em um benefício absoluto, em termos de taxa de SLD durante três anos, de 11,8 pontos percentuais (87,2% versus 75,4%) favoráveis ao braço de AC→PH (trastuzumabe - Herceptin®).6
A análise final pré-planejada da SG a partir da análise conjunta dos estudos NSABP-B31 e NCCTG N9831 foi realizada quando 707 mortes ocorreram (acompanhamento mediano de 8,3 anos no grupo AC→PH). O tratamento com AC→PH resultou em uma melhora significativa da SG comparada com AC→P (estratificado RR 0,64, IC 95% 0,55-0,74; p < 0,0001). Em 8 anos, a taxa de sobrevida global foi estimada em 86,9% para o braço AC→PH e 79,4% para o braço AC→P, um benefício absoluto de 7,4% (IC 95% 4,9%, 10,0%).6
A análise final de SG a partir da análise conjunta dos estudos NSABP-B31 e NCCTG N9831 foi resumida na tabela 3.
Estudo BCIRG 006
No estudo BCIRG 006, o câncer de mama inicial HER2-positivo foi estudado em pacientes com doença invasiva, linfonodo positivo ou nódulo negativo de alto risco (definido como envolvimento de linfonodo negativo (pN0) e com, pelo menos, um dos seguintes fatores: tamanho do tumor maior que 2 cm, receptor de estrógeno e progesterona negativo, grau histológico e/ou nuclear 2-3 ou idade < 35 anos). Trastuzumabe (Herceptin®) foi administrado em combinação com docetaxel, após quimioterapia AC (AC-DH) ou em combinação com docetaxel e carboplatina (DCarbH).7
O docetaxel foi administrado conforme segue:
- docetaxel intravenoso: 100 mg/m2, na forma de infusão intravenosa, durante uma hora, administrada a cada três semanas, por um período de quatro ciclos (dia 2 do primeiro ciclo de docetaxel e dia 1 de cada ciclo subsequente);
- docetaxel intravenoso: 75 mg/m2, na forma de infusão intravenosa, durante uma hora, administrada a cada três semanas, por um período de seis ciclos (dia 2 do ciclo 1 e dia 1 de cada ciclo subsequente);
Que foi seguido por:
- carboplatina: objetivo de AUC = 6 mg/mL/min administrada por infusão intravenosa durante 30-60 minutos, repetida a cada três semanas para um total de seis ciclos.
Os resultados de eficácia do estudo BCIRG 006 estão resumidos nas tabelas 4 e 5.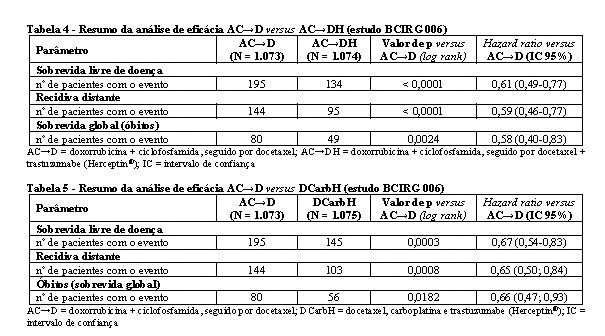
No estudo BCIRG 006, para o endpoint primário, sobrevida livre de doença, o hazard ratio transforma-se em um benefício absoluto, em termos de taxa de sobrevida livre de doença durante três anos, de 5,8 pontos percentuais (86,7% versus 80,9%) favoráveis ao braço de AC→DH (Herceptin®) e 4,6 pontos percentuais (85,5% versus 80,9%) favoráveis ao braço de DCarbH (Herceptin®) comparados a AC→D.7
Para o endpoint secundário, sobrevida global, o tratamento com AC→DH reduziu o risco de óbito em 42% quando comparado a AC→D [hazard ratio 0,58 (IC 95%: 0,40; 0,83); p = 0,0024; teste log-rank], e o risco de óbito foi reduzido em 34% em pacientes tratados com DCarbH quando comparado aos pacientes tratados com AC→D [hazard ratio 0,66 (IC 95%: 0,47; 0,93); p = 0,0182]. Na segunda análise interina do estudo BCIRG 006, 185 pacientes randomizados foram a óbito: 80 pacientes (7,5%) no braço AC→D, 49 (4,6%) no braço AC→DH e 56 pacientes (5,2%) no braço DCarbH. A duração mediana do acompanhamento foi 2,9 anos para o braço AC→D e 3,0 anos para os braços AC→DH e DCarbH.7
2) Tratamento neoadjuvante-adjuvante
No tratamento neoadjuvante-adjuvante, trastuzumabe (Herceptin®), foi avaliado em um estudo Fase III:8
O estudo MO16432 investigou um total de 10 ciclos de quimioterapia neoadjuvante [uma antraciclina e um taxano (AP+H) seguido por P+H, seguido por CMF+H] concomitantemente com terapia neoadjuvante-adjuvante com trastuzumabe (Herceptin®), ou quimioterapia neoadjuvante isolada seguida por tratamento adjuvante com trastuzumabe (Herceptin®), até a duração total de um ano de tratamento em pacientes com diagnóstico recente de câncer de mama HER2-positivo localmente avançado (estágio III) ou inflamatório.
O MO16432 é um estudo de Fase III, aberto e randomizado, de comparação de um ano de tratamento neoadjuvante e adjuvante de trastuzumabe (Herceptin®) com observação em 231 pacientes com câncer de mama HER2-positivo localmente avançado ou inflamatório, tratados com um regime de quimioterapia neoadjuvante sequencial que incluiu doxorrubicina, paclitaxel, ciclofosfamida, metotrexato e 5-fluorouracil. A população alvo para o estudo MO16432 consistia em mulheres ≥ 18 anos que foram recentemente diagnosticadas com câncer de mama localmente avançado e que não haviam recebido qualquer tratamento anterior para uma doença invasiva. O tumor primário deveria ser T3N1 ou T4 (invasão do mamilo ou da pele, peau d'orange, extensão para a parede torácica ou carcinoma inflamatório); qualquer T mais N2 ou N3; ou qualquer T mais envolvimento dos nódulos supraclaviculares ipsilaterais. As pacientes precisavam ter doença HER2-positivo, definida como doença com superexpressão de HER2 por imunohistoquímica IHC 3+ e/ou amplificação de HER2 de acordo com a hibridização fluorescente in situ (FISH), com base na confirmação do laboratório central (entretanto, permitiu-se que as pacientes entrassem no estudo com base em um resultado IHC 3+/FISH central negativo).
Os resultados de eficácia do estudo MO16432 estão resumidos na tabela a seguir. A mediana de duração do acompanhamento no braço de trastuzumabe (Herceptin®) foi 3,8 anos.8
Para o endpoint primário, sobrevida livre de evento, a adição de trastuzumabe (Herceptin®) à quimioterapia neoadjuvante, seguida pelo tratamento adjuvante com trastuzumabe (Herceptin®) para uma duração total de 52 semanas, resultou em redução de 35% no risco de recidiva/progressão da doença. O hazard ratio traduz-se em um benefício absoluto, em termos de taxa de sobrevida livre de evento de três anos, estimada em 13 pontos percentuais (65% versus 52%) favoráveis ao braço com trastuzumabe (Herceptin®).8
C) Câncer gástrico avançado
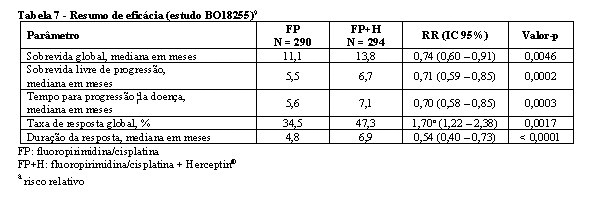
Os resultados de eficácia do estudo fase III BO18255 (Trastuzumab for Gastric Cancer = ToGA) estão resumidos na tabela 7. Os pacientes com adenocarcinoma localmente avançado inoperável ou metastático e/ou recorrente do estômago ou da junção gastroesofágica, HER2-positivo, sem possibilidade de terapia curativa e não tratados previamente, foram recrutados para o estudo. O endpoint primário foi a sobrevida global, a qual foi definida como o tempo a partir da data de randomização até o dia do óbito por qualquer causa. No momento da análise, um total de 349 pacientes randomizados foi a óbito: 182 pacientes (62,8%) no braço controle e 167 pacientes (56,8%) no braço tratamento. A maioria dos óbitos foi devida a eventos relacionados com o câncer subjacente.9
A sobrevida global foi significativamente maior no braço trastuzumabe (Herceptin®) + capecitabina/5-FU e cisplatina comparada ao braço capecitabina/5-FU e cisplatina (p = 0,0046, teste log-rank). O tempo mediano da sobrevida foi de 11,1 meses com capecitabina/5-FU e cisplatina e 13,8 meses com trastuzumabe (Herceptin®) + capecitabina/5-FU e cisplatina. O risco de óbito diminuiu em 26% (RR 0,74, IC 95% 0,60-0,91) para pacientes no braço com trastuzumabe (Herceptin®) comparado ao braço com capecitabina/5-FU.9
Análises de subgrupo post-hoc indicam que ter como alvo tumores com níveis mais elevados da proteína HER2 (IHQ 2+/FISH+ e IHQ 3+/independentemente do status FISH) resulta em melhor efeito terapêutico. A mediana de sobrevida global para o grupo com alta expressão de HER2 foi de 11,8 meses versus 16 meses, HR 0,65 (IC 95% 0,51-0,83), e a mediana de sobrevida livre de progressão foi de 5,5 meses versus 7,6 meses, HR 0,64 (IC 95% 0,51-0,79) para capecitabina/5-FU e cisplatina e trastuzumabe (Herceptin®) + capecitabina/5-FU e cisplatina, respectivamente.9
Em estudo de comparação de método, um alto grau de concordância ( > 95%) foi observado para as técnicas SISH e FISH para a detecção da amplificação do gene HER2 em pacientes com câncer gástrico.10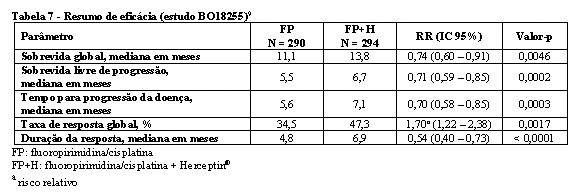
D) Estudos de comparabilidade entre Zedora e o medicamento comparador (Herceptin®).
Todos os estudos foram conduzidos de acordo com as Boas Práticas Clínicas da International Council for Harmonization, com os princípios da Declaração de Helsinque, com as regras americanas e europeias e com os regulamentos e diretrizes locais/regionais sobre a realização de estudos clínicos.11
Estudos Pivotais
• Estudo MYL-Her-1001: Centro único, de dose única, em 2 períodos, randomizado, duplo-cego, estudo de cruzamento de grupos em homens voluntários sadios. No período I administrou-se MYL-1401O (Zedora) ou Herceptin® aprovado na UE e no período II alternou-se a medicação recebida. O objetivo primário foi confirmar a bioequivalência entre Zedora e Herceptin® na dose única de 8 mg/kg IV durante 90 minutos. Ainda foi avaliada a segurança e a tolerabilidade de Zedora. Além disso, os seguintes parâmetros farmacodinâmicos (PD) foram avaliados: imunomodulação ex vivo em células mononucleares estimuladas de sangue periférico e produção de citocinas séricas; atividade antiproliferativa de amostras de soro em BT-474 (linhagem celular com receptores que superexpressam HER2-positivo; marcadores de apoptose (ativação da caspase-3, fragmentação do ácido desoxirribonucleico) e fosforilação de Akt; análise farmacocinética (PK) não-compartimental; e correlações PK/PD. O estudo foi realizado na Suíça e foi concluído em 17 de dezembro de 2012.11
Em cada período do estudo coletou-se amostras de voluntários sadios para estudos de PK nas horas 0, 45 e 90 minutos antes do final da infusão; nas horas 3, 6, 9, 24, 48 e 96 horas após o início da infusão; e nos dias 8, 11, 15, 22, 29, 43, 57, 71 e 99 após a dose. Os parâmetros de PK foram calculados usando técnicas não compartimentais. A bioequivalência foi declarada se o intervalo de confiança de 90% de dois lados da relação das médias geométricas dos parâmetros primários de PK para Zedora e Herceptin® ficasse entre 80-125%. Os resultados mostraram que Zedora e Herceptin® aprovado na UE são bioequivalentes.11
• Estudo MYL-Her-1002: Centro único, de dose única, randomizado, duplo-cego, paralelo de 3 braços em homens voluntários sadios, para avaliar a bioequivalência entre Zedora e o Herceptin® aprovado na UE e licenciado nos EUA e o Herceptin® aprovado nos EUA e licenciado na UE. Também avaliou a bioequivalência entre o Herceptin® aprovado na UE e o licenciado nos EUA. O objetivo primário foi confirmar a bioequivalência na dose única de 8 mg/kg IV durante 90 minutos sob condições de jejum. Outros parâmetros de PK foram avaliados para confirmação da similaridade com Herceptin®, além de avaliações de segurança (incluindo imunogenicidade) e tolerância local. O desenho paralelo de 3 vias serviu ao propósito de estabelecer uma ponte entre os resultados do Herceptin® licenciado nos EUA e o Herceptin® aprovado pela UE. O estudo foi realizado nos EUA e foi concluído em 27 de fevereiro de 2014.11
Em cada período do estudo coletou-se amostras de voluntários sadios para estudos de PK nas horas 0, 45 e 90 minutos antes do final da infusão; nas horas 3, 6, 9, 24, 48 e 96 horas após o início da infusão; e nos dias 8, 11, 15, 22, 29, 43, 57 e 71 após a dose. Os parâmetros de PK foram calculados usando técnicas não compartimentais. A bioequivalência foi declarada se o intervalo de confiança de 90% de dois lados da relação das médias geométricas dos parâmetros primários de PK para Zedora e Herceptin® ficasse entre 80-125%. Os resultados mostraram que Zedora é bioequivalente tanto ao Herceptin® aprovado na UE quanto ao Herceptin® licenciado nos EUA.11
• Estudo MYL-Her-3001: Multicêntrico, duplo-cego, randomizado, de grupo paralelo, de comparabilidade para a eficácia e segurança de Zedora + taxano (docetaxel ou paclitaxel) versus Herceptin® aprovado na UE + taxano em mulheres com câncer de mama metastático HER2-positivo, sem tratamento anterior, com continuação do tratamento com Zedora versus Herceptin®, ambos em monoterapia, em pacientes com pelo menos doença estável, a fim de avaliar a segurança e a imunogenicidade.
Na parte 1 do estudo (até a semana 24), Zedora + taxano ou Herceptin® + taxano foram administrados por pelo menos 8 ciclos de tratamento (1 ciclo de tratamento = 3 semanas com base na administração de trastuzumabe), a menos que o paciente tivesse eventos adversos inaceitáveis, progressão da doença ou fosse retirado prematuramente do estudo. A escolha do taxano (docetaxel ou paclitaxel) foi feita a critério do pesquisador e aplicada a todos os pacientes deste pesquisador. A avaliação do tumor (com base numa revisão centralizada) foi feita a cada 6 semanas (± 3 dias). Assim termina a parte 1 do estudo. O último paciente da parte 1 do estudo completou sua avaliação em 25 de janeiro de 2016. Na parte 2 do estudo, ainda em andamento, depois de pelo menos 8 ciclos de tratamento, todos os pacientes com pelo menos doença estável continuaram a receber apenas Zedora ou Herceptin® (produto que já haviam sido randomizados no início do tratamento) até a progressão da doença, toxicidade inaceitável ou morte, o que ocorresse primeiro. A avaliação do tumor é feita a cada 12 semanas (± 3 dias).11
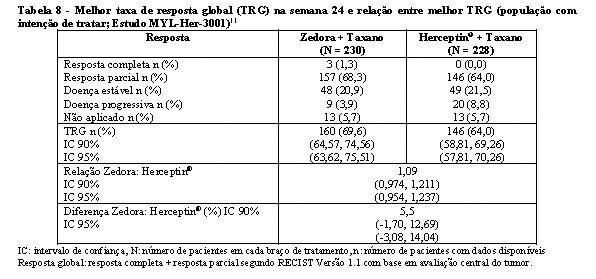
O objetivo primário foi a taxa de resposta global na semana 24, determinada por avaliação radiológica central do tumor. A equivalência seria declarada se o intervalo de confiança estivesse dentro do intervalo de equivalência de 0,81-1,24. Na semana 24, a taxa de resposta global foi de 69,6% para o grupo com Zedora e de 64% para o grupo com Herceptin®, com RR 1.09 (IC 95% 0,974-1,211), comprovando a equivalência terapêutica entre as medicações.11
A equivalência é declarada quando o intervalo de confiança 90% estiver dentro do intervalo de equivalência de 0,81-1,24. Critérios mais rígidos de equivalência podem ser utilizados, considerando-se que para o intervalo de confiança de 95% de dois lados da diferença entre a melhor TRG na semana 24 a variação deve estar entre -15% e 15%. A diferença na melhor TRG entre os dois braços de tratamento foi 5,5%, com IC 95% de -3,08% e 14,04%. Assim, a equivalência entre os dois tratamentos foi comprovada por estes dois critérios.11
Na tabela 8 encontram-se os resultados deste estudo em relação a melhor taxa de resposta global (TRG) e diferença da melhor TRG na semana 24.11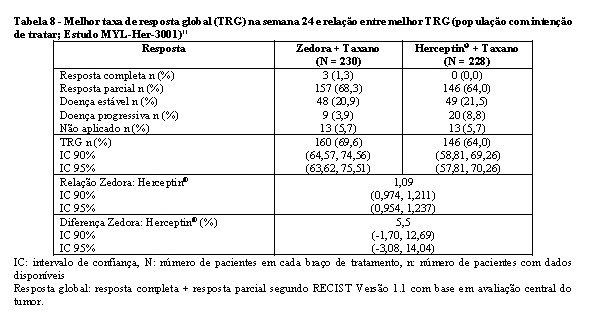
A análise de eficácia secundária mostrou que tempo para progressão, SLP e SG na semana 24 também foram semelhantes entre os grupos. No braço Zedora 35 pacientes (15,2%) tiveram progressão do tumor em comparação com 44 pacientes (19,3%) do braço Herceptin®, p = 0,192. A mediana do tempo para progressão não foi alcançada pela estimativa da curva de Kaplan-Meier devido ao pequeno número de pacientes que teve progressão da doença na semana 24. Quanto a SLP, 189 pacientes (82,2%) que receberam Zedora estavam livres de progressão na semana 24 e 180 pacientes (78,9%) que receberam Herceptin® não tinham doença na semana 24, p=0,303. A SG na semana 24 também não foi estatisticamente diferente entre os dois grupos de estudo, sendo de 97% para Zedora e 96,1% para Herceptin®, p=0,583.11
Referências bibliográficas
1. Cobleigh MA, Vogel CL, Tripathy D, et al. Multinational Study of the Efficacy and Safety of Humanized Anti-HER2 Monoclonal Antibody in Women Who Have HER2-Overexpressing Metastatic Breast Cancer That Has Progressed After Chemotherapy for Metastatic Disease. J Clin Oncol. 1999;17(9):2639-2648.
2. Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresss HER2. N Engl J Med. 2001;344(11):783-792.
3. Marty M, Cognetti F, Maraninchi D, et al. Randomized phase II trial of the efficacy and safety of trastuzumab combined with docetaxel in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer administered as first-line treatment: the M77001 study group. J Clin Oncol. 2005;23(19):4265-4274.
4. Piccart-Gebhart MJ, Procter M, Leyland-Jones B, et al. Trastuzumab after Adjuvant Chemotherapy in HER2-Positive Breast Cancer. N Engl J Med. 2005;353(16):1659-1672.
5. Update Clinical Study Report BO16348 (HERA): A randomized three-arm, multicenter comparison of 1 year and 2 years of Herceptin versus no Herceptin in women with HER2-positive primary breast cancer who have completed adjuvant chemotherapy. Report No. 1044055. March 2013 (dados publicados em Goldhirsch A, Gelber RD, Piccart-Gebhart MJ et al. 2 years versus 1 year of adjuvant trastuzumab for HER2-positive breast cancer (HERA): an open-label, randomised controlled trial. Lancet. 2013;382(9897):1021-1028.)
6. Joint Analysis (B-31 & N9831) Clinical Study Report 2006. Genentech, Inc. (dados publicados em Perez EA, Romond EH, Suman VJ et al. Four-year follow-up of trastuzumab plus adjuvant chemotherapy for operable human epidermal growth factor receptor 2-positive breast cancer: joint analysis of data from NCCTG N9831 and NSABP B-31. J Clin Oncol. 2011;29(25):3366-73 e em Perez EA, Romond EH, Suman VJ et al. Trastuzumab plus adjuvant chemotherapy for human epidermal growth factor receptor 2-positive breast cancer: planned joint analysis of overall survival from NSABP B-31 and NCCTG N9831. J Clin Oncol. 2014;32(33):3744-52.)
7. Slamon D, Eiermann W, Robert N et al. Adjuvant trastuzumab in HER2-positive breast cancer. N Engl J Med. 2011;365(14):1273-83.
8. Gianni L, Eiermann W, Semiglazov V et al. Neoadjuvant chemotherapy with trastuzumab followed by adjuvant trastuzumab versus neoadjuvant chemotherapy alone, in patients with HER2-positive locally advanced breast cancer (the NOAH trial): a randomised controlled superiority trial with a parallel HER2-negative cohort. Lancet 2010, 375:377-384.
9. Bang Y-J, Van Cutsem E, Feyereislova A, et al; for the ToGA Trial Investigators. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687-697.
10. Method Comparison Study of CONFIRM anti-HER2/neu(4B5) Primary Antibody and INFORM HER2 DNA Probe VS Hercep Test and HER2 FISH PharmDx on human gastric cancer. Dated: 27th July 2009.
11. MYL-1401O (a proposed biosimilar to trastuzumab). Clinical Overview, 2016
3. CARACTERÍSTICAS FARMACOLÓGICAS
Zedora foi desenvolvido como um medicamento biológico semelhante a Herceptin® (trastuzumabe) através da tecnologia de ácido desoxirribonucleico recombinante para codificar um anticorpo monoclonal que é 100% idêntico na sequência de aminoácidos à cadeia pesada e cadeia leve da sequência de trastuzumabe. A molécula é uma imunoglobulina G1 monoclonal humanizada produzida em linhagem celular recombinante de ovário de Hamster chinês para obter o anticorpo monoclonal glicosilado. As propriedades físico-químicas e biológicas do Zedora foram avaliadas em detalhe com método analítico ortogonal de última geração.1
Os resultados das extensas caracterizações físico-químicas, estruturais e biológicas e dos estudos pré-clínicos comparativos indicam um alto grau de similaridade entre Zedora e Herceptin®. Além disso, a similaridade também foi demonstrada através de testes funcionais in vitro e estudos em animais in vivo.2,3
Os resultados dos estudos MYL-Her-1001 e MYL-Her-1002 indicaram equivalência farmacocinética entre os produtos (Zedora, Herceptin® aprovado pela UE e Herceptin® licenciado nos EUA).3
Farmacodinâmica
Mecanismo de ação
O trastuzumabe é um anticorpo monoclonal humanizado recombinante que atinge seletivamente o domínio extracelular da proteína do receptor-2 do fator de crescimento epidérmico humano (HER2).
O anticorpo é um isótopo da IgG1 que contém regiões de estrutura humana e regiões que determinam a complementaridade, provenientes de um anticorpo murino anti-p185 HER2 que se liga ao HER2 humano.
O proto-oncogene HER2 ou c-erbB2 codifica uma proteína transmembrana de 185 kDa, semelhante ao receptor, que está estruturalmente relacionada ao receptor do fator de crescimento epidérmico. A superexpressão do HER2 é observada em 15% a 20% dos cânceres de mama primários. A taxa geral de positividade para HER2 em cânceres gástricos avançados observada durante a triagem do estudo BO18255 é 15% para IHC3+ e IHC2+/FISH+ ou 22,1% quando utilizou-se definição mais abrangente de IHC3+ ou FISH+. Uma consequência da amplificação do gene HER2 é o aumento da expressão da proteína HER2 na superfície dessas células tumorais, resultando em uma proteína HER2 constitutivamente ativada.
Os estudos indicam que pacientes com câncer de mama com amplificação ou superexpressão do HER2 apresentam menor sobrevida livre de doença, comparados a pacientes que não apresentam amplificação ou superexpressão do HER2.
Foi demonstrado, tanto nos estudos in vitro quanto em animais, que o trastuzumabe inibe a proliferação das células tumorais humanas com superexpressão HER2. In vitro, demonstrou-se que a citotoxicidade mediada pela célula anticorpo dependente (ADCC), provocada pelo trastuzumabe, é exercida preferencialmente nas células cancerígenas com superexpressão do HER2 em relação às células cancerígenas sem superexpressão do HER2.
Farmacocinética
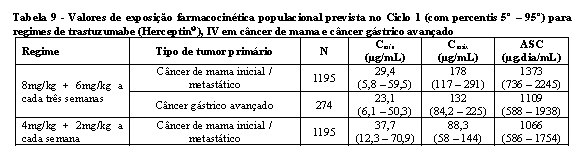
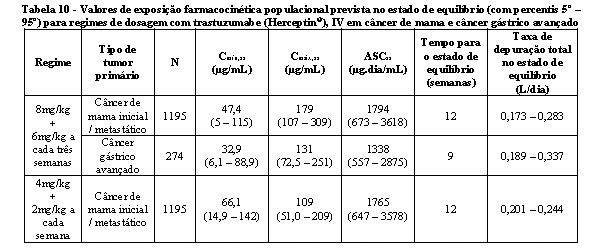
A farmacocinética de trastuzumabe (Herceptin®) foi avaliada em uma análise de modelo de farmacocinética populacional que utilizou um pool de dados de 1.582 pessoas de 18 estudos clínicos de fase I, II e III que estavam recebendo trastuzumabe (Herceptin®) IV. Um modelo de dois compartimentos com eliminação paralela linear e não paralela a partir do compartimento central descreveu o perfil da concentração de trastuzumabe (Herceptin®) ao longo do tempo. Por causa da eliminação não linear, a depuração total aumentou à medida que a concentração diminuiu. A depuração linear foi 0,127 L/dia para o câncer de mama (metastático/inicial) e 0,176 L/dia para câncer gástrico avançado. Os valores do parâmetro de eliminação não linear foram 8,81 mg/dia para a máxima taxa de eliminação (Vmáx) e 8,92 mg/L para a constante de Michaelis-Menten (Km). O volume do compartimento central foi 2,62 L para pacientes com câncer de mama e 3,63 L para pacientes com câncer gástrico avançado.
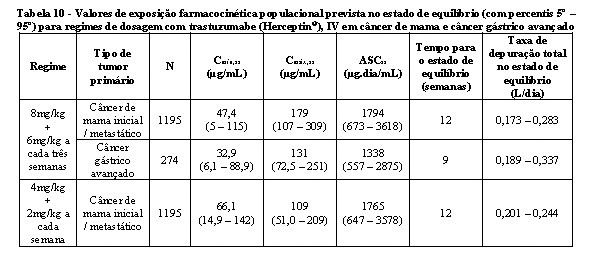
Os valores das exposições de farmacocinética populacional previstos (com percentis 5° - 95°) e do parâmetro farmacocinético em concentrações clinicamente relevantes (Cmáx e Cmín) para câncer de mama e câncer gástrico avançado tratados com os regimes semanal ou a cada três semanas estão descritos nas tabelas 9 e 10.
Washout de trastuzumabe
O tempo de washout de trastuzumabe foi avaliado após a administração de Herceptin® usando modelos farmacocinéticos populacionais. Os resultados dessas simulações indicam que pelo menos 95% dos pacientes alcançarão concentrações séricas de trastuzumabe < 1 mg/mL (aproximadamente 3% de Cmín,ss da população prevista ou em torno de 97% de washout) por 7 meses após a última dose.
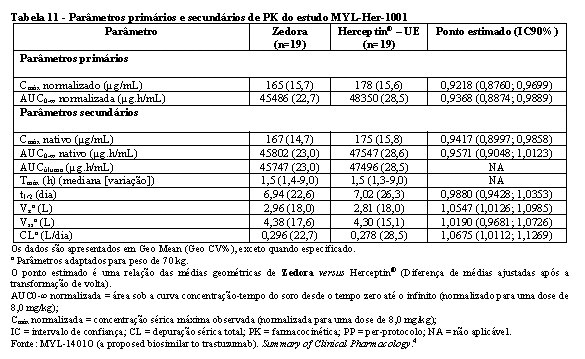
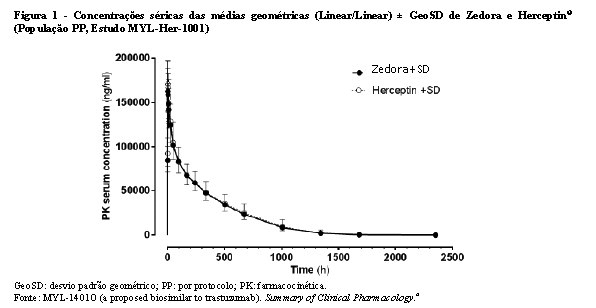
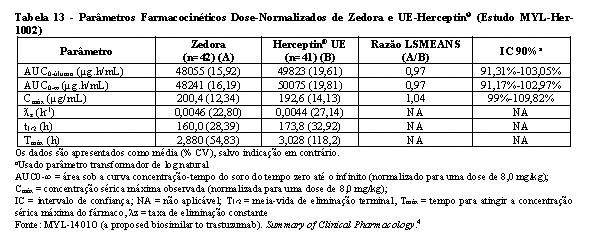
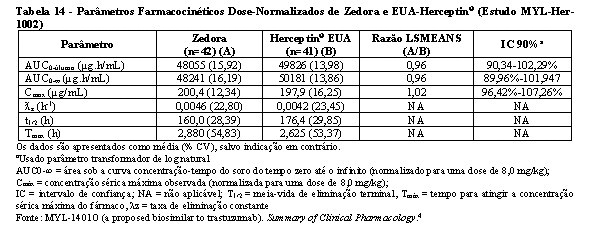
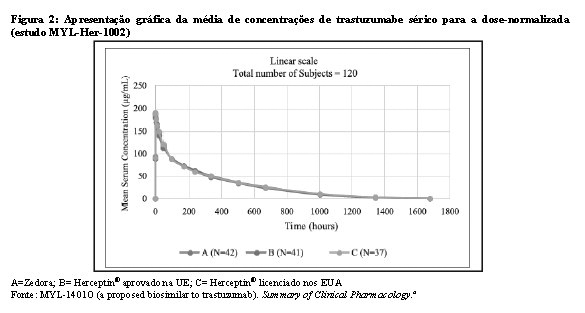
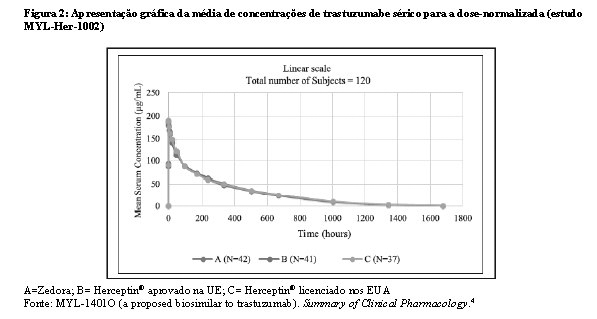
Bioequivalência foi considerada se o IC 90% das razões geométricas médias dos parâmetros primários de PK para Zedora e Herceptin® ficasse entre 80-125%. Os estudos MYL-Her-1001 (Tabelas 11 e 12, Figura 1) e MYL-Her-1002 (Tabelas 13 e 14, Figura 2) demonstraram a bioequivalência entre Zedora e Herceptin®.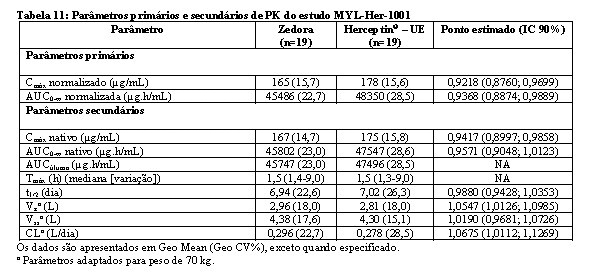
O ponto estimado é uma relação das médias geométricas de Zedora versus Herceptin® (Diferença de médias ajustadas após a transformação de volta).
AUC0-∞ normalizada = área sob a curva concentração-tempo do soro desde o tempo zero até o infinito (normalizado para uma dose de 8,0 mg/kg);
Cmax normalizada = concentração sérica máxima observada (normalizada para uma dose de 8,0 mg/kg);
IC = intervalo de confiança; CL = depuração sérica total; PK = farmacocinética; PP = per-protocolo; NA = não aplicável.
Fonte: MYL-1401O (a proposed biosimilar to trastuzumab). Summary of Clinical Pharmacology.4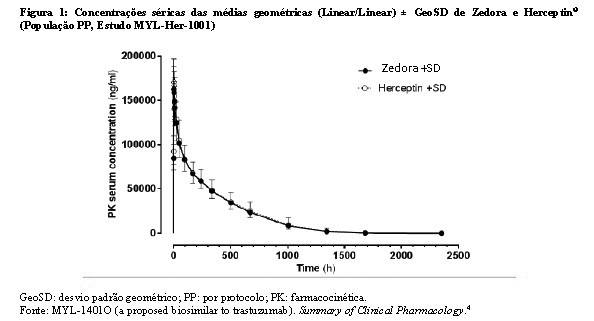

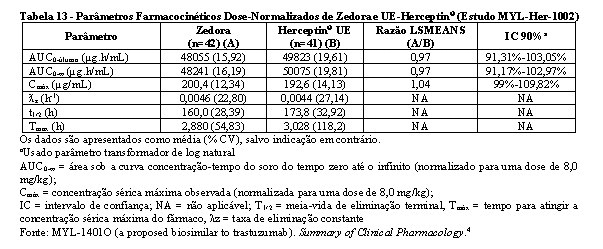
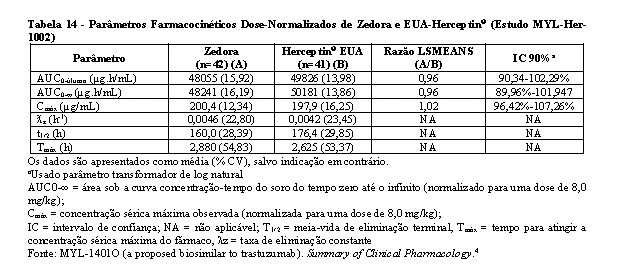
Segurança não clínica
Carcinogenicidade
Não foram realizados estudos de carcinogenicidade para estabelecer o potencial carcinogênico de trastuzumabe.
Diminuição da fertilidade
Os estudos de reprodução foram realizados em macacas Cynomolgus com doses de até 25 vezes a dose semanal humana de manutenção de 2 mg/kg de trastuzumabe IV, e não revelaram evidência de diminuição da fertilidade.
Em um estudo comparativo de 5 semanas de repetição de dose em macacas Cynomolgus, não houve diferenças significativas em segurança, tolerância, exposição ou perfil farmacocinético entre Zedora e Herceptin®. 5
Não foram realizados estudos estendidos em animais no sentido de se estabelecer o potencial carcinogênico do trastuzumabe ou para determinar seus efeitos sobre a fertilidade em indivíduos masculinos.5
Toxicidade reprodutiva
Os estudos de reprodução foram realizados em macacas Cynomolgus com doses de até 25 vezes a dose semanal humana de manutenção de 2 mg/kg de trastuzumabe IV, e não revelaram evidência de danos ao feto. No entanto, em relação à avaliação do risco de toxicidade reprodutiva em humanos, é importante considerar o significado do receptor HER2 dos roedores no desenvolvimento embrionário e na morte de embriões de ratos mutantes que não têm esse receptor. Foi observada transferência placentária de trastuzumabe durante o período de desenvolvimento fetal precoce (dias 20-50 de gestação) e tardio (dias 120-150 de gestação).
Lactação
Um estudo realizado em macacas Cynomolgus lactantes, com doses 25 vezes a dose semanal humana de manutenção de trastuzumabe IV, de 2 mg/kg, demonstrou que trastuzumabe é secretado no leite. A presença de trastuzumabe no soro de macacos recém-nascidos não foi associada com qualquer efeito adverso no seu crescimento ou desenvolvimento desde seu nascimento até 1 mês de idade.
Farmacocinética em populações especiais
Não foram realizados estudos farmacocinéticos detalhados na população geriátrica ou em populações de pacientes com insuficiência renal ou hepática.
População geriátrica
Foi demonstrado que a idade não tem efeito sobre a disponibilidade do trastuzumabe (ver item "5. Advertências e Precauções").
Referências bibliográficas
1. MYL-1401O (a proposed biosimilar to trastuzumab). Summary of Biopharmaceutic Studies and Associated Analytical Methods, 2016
2. MYL-1401O (a proposed biosimilar to trastuzumab). Clinical Overview, 2016
3. MYL-1401O (a proposed biosimilar to trastuzumab). Summary of Clinical Efficacy, 2016
4. MYL-1401O (a proposed biosimilar to trastuzumab). Summary of Clinical Pharmacology, 2016
5. MYL-1401O (a proposed biosimilar to trastuzumab). Non-clinical Overview, 2016
4. CONTRAINDICAÇÕES
Zedora é contraindicado a pacientes com hipersensibilidade conhecida ao trastuzumabe ou a qualquer outro excipiente da fórmula.
5. ADVERTÊNCIAS E PRECAUÇÕES
A terapia com Zedora deve ser iniciada somente sob a supervisão de um médico experiente no tratamento de pacientes com câncer.
Reações relacionadas à infusão (RRI)
Pode ser difícil diferenciar, clinicamente, as reações relacionadas à infusão das reações de hipersensibilidade.
Pré-medicação pode ser utilizada para reduzir o risco de ocorrência de reações relacionadas à infusão.
Reações graves relacionadas à infusão de trastuzumabe, que incluem dispneia, hipotensão, sibilância, broncoespasmo, taquicardia, redução na saturação de oxigênio e dificuldade respiratória, taquiarritmia supraventricular e urticária foram relatadas (ver item "9. Reações adversas"). O paciente deve ser monitorado em relação às reações relacionadas à infusão.
A interrupção da infusão intravenosa pode ajudar no controle desses sintomas e a poderá ser restituída assim que os sintomas forem controlados. Esses sintomas podem ser tratados com analgésico/antipirético, tais como a meperidina ou paracetamol, ou ainda com anti-histamínico, como a difenidramina. Reações graves têm sido tratadas, com sucesso, com terapias de suporte, tais como oxigenoterapia, beta-agonista e corticoides. Em casos raros, essas reações podem apresentar evolução fatal. Pacientes que apresentam dispneia de repouso decorrente de complicações de doença maligna avançada ou comorbidade podem ter risco aumentado para reação infusional fatal. Portanto, esses pacientes não devem ser tratados com trastuzumabe.
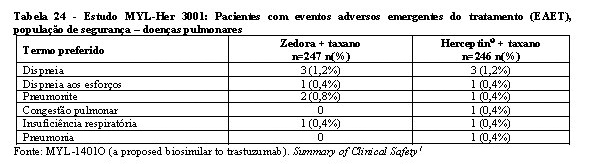
Reações pulmonares
Eventos adversos pulmonares graves com o uso de trastuzumabe foram relatados após sua comercialização. Esses eventos ocasionalmente resultaram em óbito e podem ocorrer como parte da reação relacionada à infusão ou serem de início tardio. Além disso, foram relatados casos de doença pulmonar intersticial, incluindo infiltrado pulmonar, síndrome do desconforto respiratório agudo, pneumonia, pneumonite, derrame pleural, dificuldade respiratória, edema pulmonar agudo e insuficiência respiratória.
Fatores de risco associados com a doença pulmonar intersticial incluem tratamento prévio ou concomitante com outras terapias antineoplásicas conhecidas por serem associadas a essa condição, como taxanos, gencitabina, vinorelbina e radioterapia. Pacientes com dispneia de repouso decorrente de complicações de doença maligna avançada ou comorbidade podem ter risco aumentado para reações pulmonares. Dessa forma, esses pacientes não devem ser tratados com trastuzumabe.
Disfunção cardíaca
Considerações gerais
Pacientes tratados com trastuzumabe apresentam maior risco de desenvolver insuficiência cardíaca congestiva (ICC) (New York Heart Association [NYHA] Classe II-IV) ou disfunção cardíaca assintomática. Esses eventos foram observados em pacientes que receberam trastuzumabe em monoterapia ou em combinação com taxano após regimes quimioterápicos com antraciclina (doxorrubicina ou epirrubicina). A insuficiência cardíaca pode ser de moderada a grave, e já houve casos de óbito (ver item "9. Reações adversas"). Além disso, deve-se ter cautela com pacientes em tratamento que apresentam risco cardíaco aumentado (por exemplo, hipertensão, doença arterial coronariana documentada, insuficiência cardíaca congestiva, disfunção diastólica e idade mais avançada).
Simulações de modelos farmacocinéticos populacionais indicam que o trastuzumabe pode persistir na circulação por até 7 meses após a interrupção do tratamento com trastuzumabe (ver item "3. Características Farmacológicas - Farmacocinética"). Pacientes que utilizam antraciclina após a interrupção do tratamento com trastuzumabe também podem apresentar maior risco de disfunção cardíaca.
Se possível, o médico deve evitar o tratamento com antraciclina por até 7 meses após a interrupção do tratamento com trastuzumabe. Se as antraciclinas forem utilizadas, a função cardíaca do paciente deve ser monitorada cuidadosamente.
Candidatos para o tratamento com trastuzumabe, especialmente aqueles com exposição anterior à antraciclina, devem ser submetidos a uma avaliação cardíaca de base, incluindo histórico e exame físicos, a eletrocardiograma e ecocardiograma ou cintilografia ventricular (MUGA). O monitoramento pode ajudar a identificar os pacientes que podem desenvolver disfunção cardíaca, incluindo sinais e sintomas de ICC. Avaliações cardíacas, como as realizadas inicialmente, devem ser repetidas a cada 3 meses durante o tratamento e a cada 6 meses após a descontinuação do tratamento até 24 meses a partir da última administração de trastuzumabe.
Se a fração de ejeção do ventrículo esquerdo (FEVE) percentual cair dez pontos em relação ao exame basal e abaixo de 50%, trastuzumabe deve ser suspenso, e uma nova avaliação de FEVE deve ser realizada dentro de, aproximadamente, três semanas. Se a FEVE não melhorar, ou diminuir ainda mais, ou se desenvolver uma ICC clinicamente significativa, a descontinuação de trastuzumabe deve ser fortemente considerada, a não ser que os benefícios para o paciente sejam considerados superiores aos riscos.
Os pacientes que desenvolvem disfunção cardíaca assintomática devem ser submetidos a monitoramento mais frequentemente (por exemplo, a cada seis a oito semanas). Se os pacientes continuarem com diminuição da função ventricular esquerda, mas permanecerem assintomáticos, o médico deve considerar a interrupção da terapia, a menos que julgue que os benefícios ao paciente superam os riscos.
A segurança da manutenção ou reintrodução de trastuzumabe em pacientes que apresentam disfunção cardíaca não foram estudadas prospectivamente. Se insuficiência cardíaca sintomática for desenvolvida durante o tratamento com trastuzumabe, deve ser tratada de acordo com a terapia padrão para tal. Em estudos clínicos pivotais, a maioria dos pacientes que desenvolveram insuficiência cardíaca ou disfunção cardíaca assintomática melhorou com a terapia padrão para insuficiência cardíaca, a qual consiste em um inibidor da enzima conversora de angiotensina (ECA) ou um bloqueador do receptor de angiotensina (BRA) e um betabloqueador. A maioria dos pacientes com sintomas cardíacos e com evidências de benefícios clínicos com o tratamento com trastuzumabe continuou o tratamento com trastuzumabe sem apresentar nenhum evento clínico cardíaco adicional.
Câncer de mama metastático
Trastuzumabe e antraciclinas não devem ser administrados concomitantemente para o tratamento do câncer de mama metastático.
Câncer de mama inicial
Para pacientes com câncer de mama inicial, avaliações cardíacas, como as realizadas inicialmente, devem ser repetidas a cada 3 meses durante o tratamento e a cada 6 meses após a descontinuação do tratamento, até 24 meses a partir da última administração de trastuzumabe.1
Para pacientes que utilizam quimioterapia com antraciclina, recomenda-se um monitoramento adicional que deve ser feito anualmente por até 5 anos a partir da última administração de trastuzumabe ou mais, caso seja observada uma diminuição contínua da FEVE.
Pacientes com histórico de infarto do miocárdio, angina pectoris com necessidade de medicação, histórico ou presença de insuficiência cardíaca congestiva (NYHA Classe II-IV), outra cardiomiopatia, arritmia cardíaca com necessidade de medicação, valvulopatia clinicamente significativa, hipertensão mal controlada (hipertensão controlada com medicamentos elegíveis como padrão) e efusão pericárdica hemodinamicamente efetiva foram excluídos dos estudos clínicos para câncer de mama em adjuvância com trastuzumabe.
Tratamento adjuvante
Trastuzumabe e antraciclinas não devem ser administrados concomitantemente para o tratamento adjuvante.
Foi observado em pacientes com câncer de mama inicial, aumento na incidência de eventos cardíacos sintomáticos e assintomáticos quando trastuzumabe foi administrado após quimioterapia com antraciclina, quando comparados com aqueles que receberam tratamento sem antraciclina (a base de docetaxel e carboplatina). A incidência foi mais notável quando trastuzumabe foi administrado concomitantemente com taxanos do que quando administrados sequencialmente a eles. Independentemente do regime de tratamento utilizado, a maioria dos eventos cardíacos sintomáticos ocorreu dentro dos primeiros 18 meses.
Fatores de risco para eventos cardíacos identificados em quatro grandes estudos em adjuvância incluem idade avançada ( > 50 anos), baixo nível basal e diminuição da FEVE ( < 55%), FEVE baixa antes ou após o início do tratamento com paclitaxel, tratamento com trastuzumabe e uso prévio ou concomitante com medicamentos anti-hipertensivos. O risco de disfunção cardíaca em pacientes que receberam trastuzumabe após a conclusão da quimioterapia adjuvante foi associado com alta dose cumulativa de antraciclina administrada antes de iniciar o tratamento com trastuzumabe e com o alto índice de massa corpórea (IMC > 25 kg/m2).
Tratamento neoadjuvante-adjuvante
Em pacientes com câncer de mama inicial elegíveis para o tratamento neoadjuvante-adjuvante, a terapia com trastuzumabe concomitantemente com antraciclinas deve ser usada com cautela e somente em pacientes que nunca receberam quimioterapia. As doses máximas cumulativas dos regimes de baixa dose de antraciclina não devem exceder 180 mg/m2 (doxorrubicina) ou 360 mg/m2 (epirrubicina).
Se os pacientes forem tratados concomitantemente com baixa dose de antraciclinas e trastuzumabe na neoadjuvância, a função cardíaca deve ser monitorada cuidadosamente e nenhuma quimioterapia citotóxica adicional deve ser administrada após cirurgia.
A experiência clínica na neoadjuvância-adjuvância é limitada em pacientes com mais de 65 anos de idade.
Álcool benzílico
A água para injetáveis utilizada para reconstituir os frascos-ampola de dose única de Zedora® 150 mg não contém álcool benzílico.
Gestação e lactação
Categoria de risco na gravidez: D. Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
Trastuzumabe deve ser evitado durante a gravidez, a menos que os potenciais benefícios para a mãe superem os riscos potenciais para o feto. No período de pós-comercialização, foram relatados casos de problemas de crescimento e/ou insuficiência renal em fetos associados ao oligoâmnio em mulheres grávidas que receberam trastuzumabe, alguns associados à hipoplasia pulmonar fatal do feto. As mulheres em idade fértil devem ser instruídas a usar métodos contraceptivos efetivos durante o tratamento com trastuzumabe e por 7 meses após o término do tratamento (ver item "3. Características Farmacológicas - Farmacocinética"). As mulheres que engravidarem devem ser informadas sobre a possibilidade de dano ao feto. Se uma mulher grávida for tratada com trastuzumabe, ou se a paciente engravidar enquanto estiver sendo tratada com trastuzumabe ou dentro do período de 7 meses após a última dose de trastuzumabe, é aconselhável monitoramento cuidadoso por uma equipe multidisciplinar. Se ocorrer gravidez durante o uso ou nos 7 meses seguintes da última dose de Zedora, por favor reporte imediatamente para o Serviço de Atendimento ao Cliente através do telefone 0800 0135044. Informações adicionais serão requeridas durante a gravidez exposta ao Zedora e no primeiro ano de vida do recém-nascido.
Não se sabe se trastuzumabe pode afetar a capacidade de reprodução. Os estudos de reprodução em animais não revelaram evidências de comprometimento na fertilidade ou riscos não aqui relatados para o feto (ver item "3. Características Farmacológicas - Toxicidade reprodutiva").
Lactação
Informe ao seu médico se estiver amamentando.
Não se sabe se o trastuzumabe é excretado no leite humano. Como a imunoglobulina G (IgG) humana é secretada no leite humano e o potencial de danos para os lactentes é desconhecido, a lactação deve ser evitada durante a terapia com trastuzumabe.
Uso geriátrico, pediátrico e em outros grupos de risco
Uso geriátrico
Não foram realizados estudos específicos de farmacocinética na população geriátrica. Os dados existentes sugerem que a disponibilidade de trastuzumabe não se altera com a idade (ver item "3. Características Farmacológicas - Farmacocinética em populações especiais"). Nos estudos clínicos, pacientes com 65 anos de idade ou mais não receberam doses reduzidas de trastuzumabe.
Uso pediátrico
A segurança e a eficácia de trastuzumabe em pacientes menores de 18 anos não foram estabelecidas.
Pacientes com insuficiência renal
Em uma análise de farmacocinética populacional, foi demonstrada que a insuficiência renal não afeta a biodisponibilidade de trastuzumabe.
Pacientes com insuficiência hepática
Não foram realizados estudos específicos em populações de pacientes com insuficiência hepática.
Capacidade de dirigir veículos ou operar máquinas
Não foram realizados estudos sobre os efeitos na capacidade de dirigir veículos ou operar máquinas.
Pacientes que apresentam sintomas relacionados com a infusão devem ser orientados a não dirigir veículos ou operar máquinas até que os sintomas sejam resolvidos por completo.
Para aumentar a rastreabilidade dos medicamentos biológicos, o nome comercial e o número de lote do produto administrado devem ser claramente registrados (ou declarados) no prontuário médico do paciente.
Até o momento, não há informações de que Zedora (trastuzumabe) possa causar doping.
Referências bibliográficas
1. MYL-1401O (a proposed biosimilar to trastuzumab). Summary of Clinical Safety, 2016
6. INTERAÇÕES MEDICAMENTOSAS
Não foram realizados estudos formais sobre interações medicamentosas com trastuzumabe em humanos.
Não foram observadas interações clinicamente significativas entre trastuzumabe e a medicação utilizada concomitantemente nos estudos clínicos (ver item "3. Características Farmacológicas - Farmacocinética").
Em estudos nos quais trastuzumabe foi administrado em combinação com docetaxel, carboplatina ou anastrozol, a farmacocinética desses medicamentos não foi alterada, como também a farmacocinética de trastuzumabe não foi alterada.
As concentrações de paclitaxel e doxorrubicina (e os seus principais metabólitos 6-a hidroxipaclitaxel, POH, e doxorrubicinol, DOL) não foram alteradas na presença de trastuzumabe.
No entanto, o trastuzumabe pode aumentar a exposição global de um metabólito da doxorrubicina (7-desoxi-13 di-hidro-doxorrubicinona, D7D). A bioatividade do D7D e o impacto clínico do aumento desse metabólito não são claros. Não foram observadas alterações nas concentrações de trastuzumabe na presença de paclitaxel e doxorrubicina.
Os resultados de um sub-estudo de interação medicamentosa que avaliou a farmacocinética da capecitabina e da cisplatina quando utilizadas com ou sem trastuzumabe, sugerem que a exposição aos metabólitos bioativos da capecitabina (por exemplo, 5-FU) não foi afetada pela utilização concomitante da cisplatina ou pela utilização concomitante da cisplatina mais trastuzumabe. No entanto, a capecitabina por si mesma demonstrou concentrações mais elevadas e uma meia-vida maior quando associada ao trastuzumabe. Os dados também sugerem que a farmacocinética da cisplatina não foi afetada pela utilização concomitante da capecitabina ou pela utilização concomitante da capecitabina mais trastuzumabe.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Antes de aberto, Zedora deve ser conservado sob refrigeração (entre 2°C e 8°C).
Cuidados de conservação da solução reconstituída
O produto reconstituído é física e quimicamente estável durante 48 horas sob temperatura entre 2°C e 8°C após a reconstituição com água para injetáveis. Do ponto de vista microbiológico, a solução reconstituída deve ser adicionalmente diluída em uma solução para infusão imediatamente. Se isso não ocorrer, o tempo e as condições de armazenamento em uso são de responsabilidade do usuário e, normalmente, não devem ultrapassar 24 horas em temperatura entre 2 e 8°C. A solução reconstituída não deve ser congelada.
Cuidados de conservação da solução para infusão com o produto reconstituído
A solução para infusão (solução para infusão de cloreto de sódio a 0,9%) com o produto reconstituído é física e quimicamente estável durante 24 horas (não conservar em temperaturas acima de 30°C).
Do ponto de vista microbiológico, a solução para infusão de Zedora deve ser aplicada imediatamente. Se isso não ocorrer, o tempo e as condições de armazenamento em uso são de responsabilidade do usuário e, normalmente, não devem ultrapassar 24 horas em temperatura entre 2°C e 8°C.
Prazo de validade
Zedora 150 mg possui prazo de validade de 24 meses a partir da data de fabricação.
Após preparo, este medicamento deve ser utilizado conforme as instruções dos cuidados de conservação da solução reconstituída e da solução para infusão com o produto reconstituído.
Zedora (trastuzumabe) em seu frasco-ampola original é um pó liofilizado que apresenta coloração branca a amarela pálida. A solução final é incolor a levemente amarelada.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Descarte de medicamentos não utilizados e/ou com data de validade vencida.
O descarte de medicamentos no meio ambiente deve ser minimizado. Os medicamentos não devem ser descartados no esgoto, e o descarte em lixo doméstico deve ser evitado. Utilize o sistema de coleta local estabelecido, se disponível.
Os seguintes pontos devem ser atendidos rigorosamente em relação ao uso e descarte de seringas e outros materiais médicos perfurocortantes: agulhas e seringas não devem ser reutilizadas; descartar todas as agulhas e seringas utilizadas em recipiente para descarte de material perfurocortante (recipiente descartável à prova de perfuração).
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
É obrigatório avaliar o status HER2 antes de iniciar a terapia com Zedora.
Zedora deve ser administrado por um profissional de saúde qualificado.
É importante conferir a bula e rotulagem do produto para assegurar que o medicamento a ser administrado está consistente com o que foi prescrito para o paciente.
Devem ser usadas técnicas assépticas apropriadas.
Para evitar erros na medicação, é importante verificar os rótulos do frasco-ampola para garantir que a droga que está sendo preparada e administrada é Zedora e não Kadcyla® (trastuzumabe entansina).
Modo de usar
Zedora não deve ser administrado pela via subcutânea.
Este medicamento é de uso hospitalar e, depois de reconstituído, deve ser diluído com soro fisiológico para infusão intravenosa antes de ser administrado. Não administrar rapidamente como injeção intravenosa ou em bolus.
Reconstituição
Zedora deve ser cuidadosamente manuseado durante a reconstituição com água para injetáveis estéril. A formação de espuma excessiva durante a reconstituição ou a agitação da solução de Zedora reconstituído pode resultar em problemas com a quantidade da solução de Zedora que pode ser retirada do frasco-ampola.
- Instruções de reconstituição
1) Usando seringa estéril, injete lentamente 7,2 mL da água para injetáveis estéril no frasco-ampola que contém o pó liofilizado de Zedora, direcionando a corrente para a parte liofilizada.
2) Faça movimentos circulares suaves com o frasco-ampola para auxiliar a reconstituição. NÃO AGITE<33>
A leve formação de espuma do produto durante a reconstituição não é rara. Deixe o frasco-ampola ficar repousando por aproximadamente cinco minutos. Zedora reconstituído resulta em uma solução incolor a levemente amarelada e deve ser essencialmente livre de partículas visíveis.
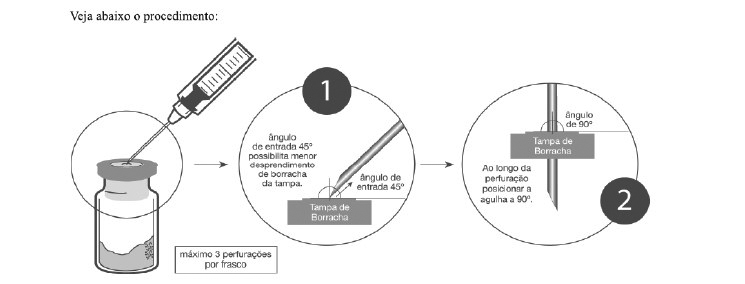
Recomendações de práticas seguras e adequadas para manipulação do medicamento
1. Inserir a agulha de injeção de, no máximo, 1,20x40 mm de calibre;
2. Encher a seringa com o diluente apropriado;
3. Apoiar o frasco-ampola firmemente na posição vertical;
4. Perfurar a tampa de borracha de Zedora dentro do círculo central demarcado, inserindo assepticamente a agulha a 45° com bisel voltado para cima e, ao longo da perfuração, posicioná-la a 90° (figura abaixo);
5. Evitar que as novas perfurações sejam no mesmo local;
6. É recomendado não perfurar mais de 3 vezes a área demarcada (círculo central);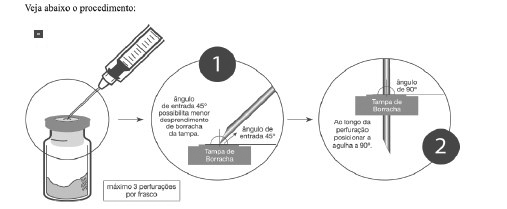
7. A cada 3 perfurações com uma mesma agulha, substituí-la por uma nova.Após a reconstituição, o profissional da saúde deverá inspecionar cuidadosamente, antes de sua utilização, se a solução no interior do frasco-ampola está fluida, livre de fragmentos ou de alguma substância que possa comprometer a eficácia e segurança do medicamento. Não é recomendado a utilização do produto ao verificar qualquer alteração que possa prejudicar a saúde do paciente.
Diluição da solução reconstituída
Determine o volume necessário da solução
- Baseado em uma dose de ataque de 4 mg de trastuzumabe/kg de peso corpóreo ou em uma dose semanal subsequente de 2 mg de trastuzumabe/kg de peso corpóreo:
Volume (mL) = Peso corpóreo (kg) x dose (4 mg/kg de ataque ou 2 mg/kg de manutenção) / 21 (mg/mL, concentração da solução reconstituída)
- Baseado em uma dose de ataque de 8 mg de trastuzumabe/kg de peso corpóreo ou uma dose subsequente, a cada três semanas, de 6 mg de trastuzumabe/kg de peso corpóreo.
Volume (mL) = Peso corpóreo (kg) x dose (8 mg/kg de ataque ou 6 mg/kg de manutenção) / 21 (mg/mL, concentração da solução reconstituída)
A quantidade apropriada da solução deve ser retirada do frasco-ampola e adicionada a uma bolsa de infusão com 250 mL de cloreto de sódio 0,9%. Não deve ser usada solução de dextrose (5%) (ver subitem "Incompatibilidades"). A bolsa deve ser invertida suavemente para misturar a solução e evitar a formação de espuma. Os medicamentos de infusão parenteral devem ser inspecionados visualmente quanto à presença de partículas e alterações da cor antes da administração. Uma vez preparada, a solução para infusão deve ser administrada imediatamente (ver item "7. Cuidados de armazenamento do medicamento").
Posologia
Câncer de mama
Uso semanal
As seguintes doses iniciais (de ataque) e de manutenção são recomendadas em monoterapia e em combinação com paclitaxel ou docetaxel.
Dose de ataque: a dose de ataque inicial recomendada é de 4 mg/kg de peso corpóreo. Zedora deve ser administrado como infusão intravenosa durante 90 minutos.
Doses subsequentes: a dose semanal recomendada de Zedora é de 2 mg/kg de peso corpóreo. Caso a dose anterior tenha sido bem tolerada, a dose pode ser administrada em uma infusão de 30 minutos.
Uso a cada três semanas
Dose de ataque: a dose de ataque inicial recomendada é de 8 mg/kg de peso corpóreo. Zedora deve ser administrado como infusão intravenosa durante 90 minutos.
Doses subsequentes: após 3 semanas da dose de ataque, iniciar Zedora 6 mg/kg de peso corpóreo, mantendo esta dose a cada 3 semanas, em infusões intravenosas de 90 minutos. Caso a dose anterior tenha sido bem tolerada, a dose pode ser administrada em uma infusão de 30 minutos.
Administração em associação com paclitaxel ou docetaxel
Nos estudos clínicos pivotais (H0648g, M77001) o paclitaxel ou o docetaxel foi administrado no dia seguinte à primeira administração de trastuzumabe e imediatamente após as doses subsequentes de trastuzumabe se a dose anterior de trastuzumabe foi bem tolerada.
No estudo MYL-Her-3001 docetaxel foi administrado na dose de 75 mg/m2 de área de superfície corporal IV a cada 3 semanas e paclitaxel na dose de 80 mg/m2 de área de superfície corporal IV semanalmente, ambos durante 1 hora ± 10 minutos. No ciclo 1, o docetaxel foi administrado no Dia 2 do ciclo. Na Semana 1, o paclitaxel foi administrado 24 horas após o término da administração de Zedora ou Herceptin®. A partir do dia 1 do ciclo 2, e em cada ciclo subsequente, os taxanos foram administrados 30 ± 10 minutos após o término da infusão de Zedora ou Herceptin®.1
Câncer gástrico
Uso a cada três semanas
Dose de ataque: a dose de ataque inicial recomendada é de 8 mg/kg de peso corpóreo. Zedora deve ser administrado como infusão intravenosa durante 90 minutos.
Doses subsequentes: após 3 semanas da dose de ataque, iniciar Zedora 6 mg/kg de peso corpóreo, mantendo esta dose a cada 3 semanas, em infusões intravenosas de 90 minutos. Caso a dose anterior tenha sido bem tolerada, a dose pode ser administrada em uma infusão de 30 minutos.
Incompatibilidades
Não foram constatadas incompatibilidades entre trastuzumabe e a bolsa de cloreto de polivinila, polietileno ou polipropileno.
Não deve ser usada solução de dextrose (5%), visto que ela causa agregação da proteína.
Trastuzumabe não deve ser misturado ou diluído com outros fármacos.
Duração do tratamento
• Pacientes com câncer de mama metastático devem ser tratados com Zedora até progressão da doença.
• Pacientes com câncer de mama inicial devem ser tratados com Zedora por um ano ou até a recidiva da doença, o que ocorrer primeiro. Estender o tratamento além de um ano para pacientes com câncer de mama inicial não é recomendado (ver item "2. Resultados de Eficácia").
• Pacientes com câncer gástrico avançado devem ser tratados com Zedora até progressão da doença.
Doses não recebidas
Se o paciente deixar de receber uma dose de Zedora no prazo de uma semana ou menos, a dose de manutenção habitual de Zedora (regime semanal: 2 mg/kg; regime a cada três semanas: 6 mg/kg) deve ser administrada o mais rápido possível. Não esperar até o próximo ciclo programado. Doses de manutenção subsequentes de Zedora devem ser administradas 7 dias ou 21 dias mais tarde, conforme regime semanal ou regime a cada três semanas, respectivamente.
Se o paciente deixar de receber uma dose de Zedora durante um prazo superior a uma semana, uma nova dose de ataque (reataque) de Zedora deve ser administrada o mais brevemente possível durante, aproximadamente, 90 minutos (regime semanal: 4 mg/kg; regime a cada três semanas: 8 mg/kg). Doses de manutenção subsequentes de Zedora (regime semanal: 2 mg/kg; regime a cada três semanas: 6 mg/kg, respectivamente) devem ser 7 dias ou 21 dias mais tarde, conforme regime semanal ou regime a cada três semanas, respectivamente.
Redução da dose
Não foram realizadas reduções na dose de Zedora durante os estudos clínicos. Os pacientes podem continuar a terapia com Zedora durante os períodos de mielossupressão reversível induzida pela quimioterapia, mas devem ser monitorados cuidadosamente, durante esse período, quanto a complicações decorrentes da neutropenia. Devem ser seguidas instruções específicas para reduzir ou manter a dose da quimioterapia.
Referências bibliográficas
1. MYL-1401O (a proposed biosimilar to trastuzumab). Summary of Clinical Efficacy, 2016.
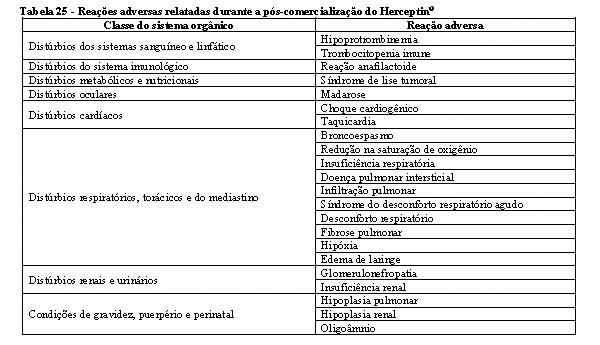
9. REAÇÕES ADVERSAS
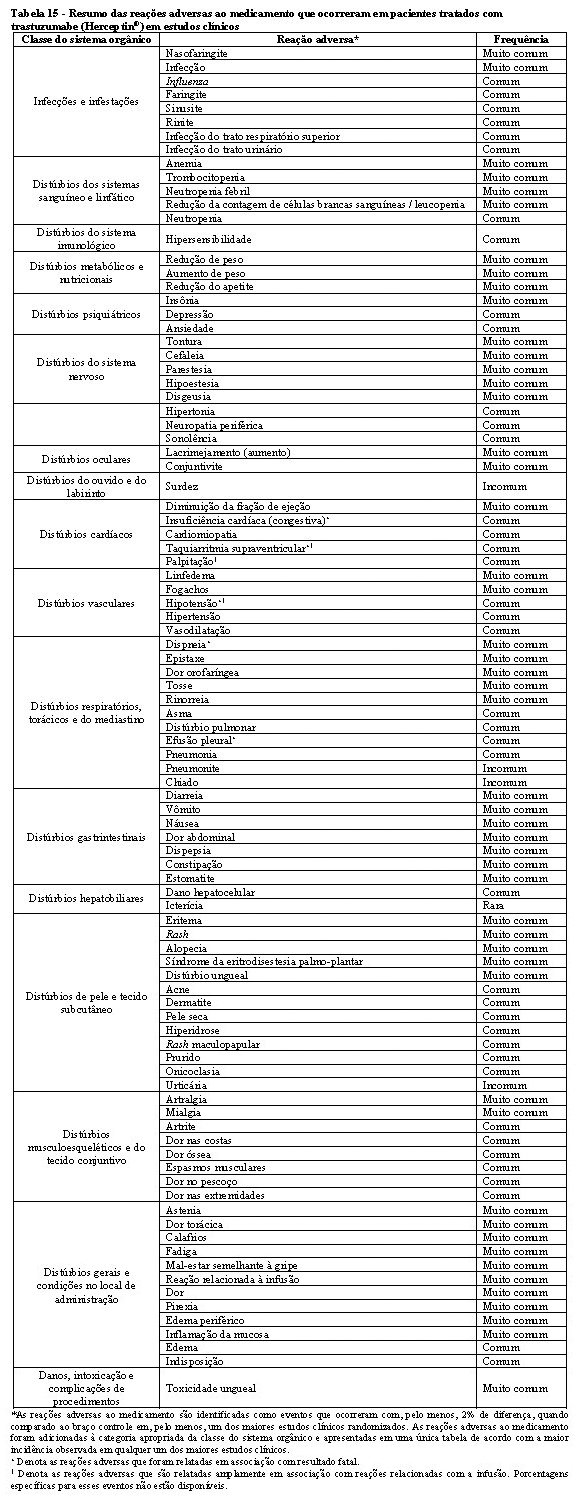
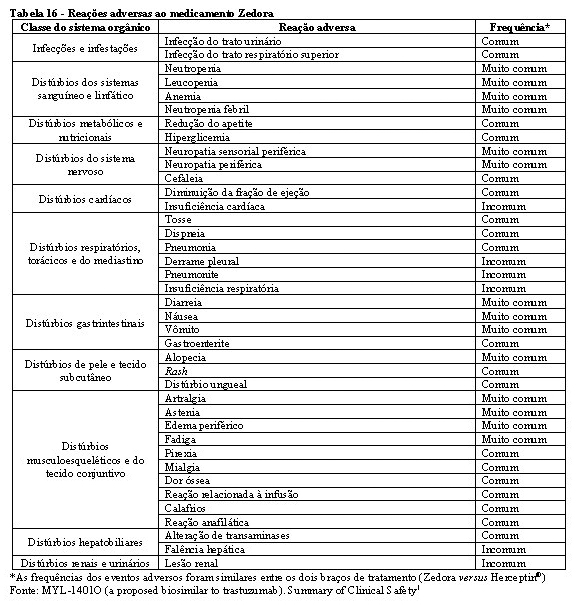
A tabela 15 a seguir apresenta as reações adversas que foram relatadas em associação com o uso de trastuzumabe isolado ou em combinação com quimioterapia em estudos clínicos pivotais do trastuzumabe (Herceptin®). Todos os termos incluídos são baseados na maior porcentagem observada nestes estudos clínicos pivotais.
Tendo em vista que trastuzumabe é comumente utilizado com outros agentes quimioterápicos e radioterapia, geralmente é difícil confirmar a relação causal dos eventos adversos para um fármaco/radioterapia em particular.
A categoria de frequência correspondente para cada reação adversa ao medicamento é baseada na seguinte convenção: muito comum (≥1/10), comum (≥ 1/100 a < 1/10), incomum (≥1/1.000 a < 1/100), rara (≥ 1/10.000 a < 1/1.000), muito rara ( < 1/10.000), não conhecida (não pode ser estimada com base nos dados disponíveis). Dentro de cada grupo de frequência, as reações adversas são apresentadas em ordem decrescente de gravidade.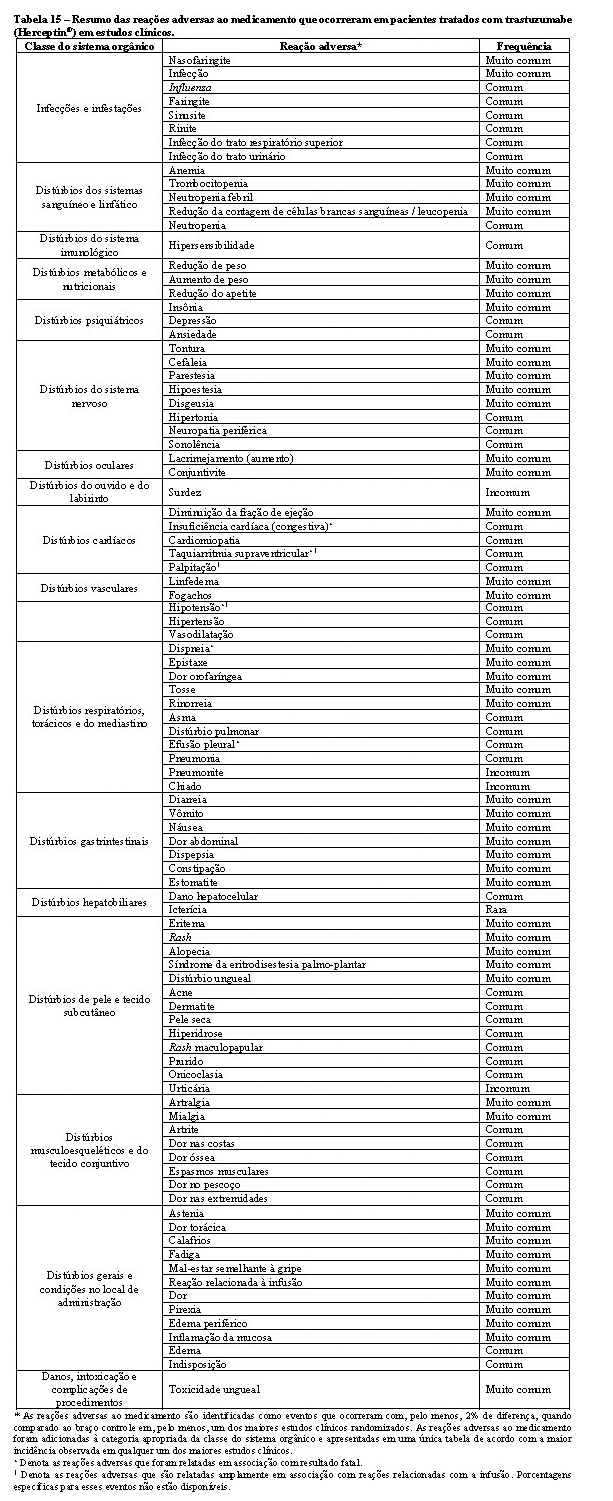
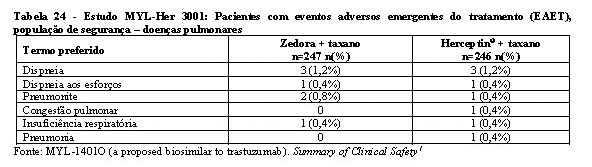
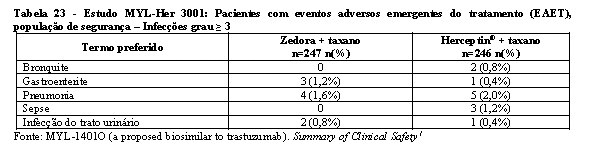
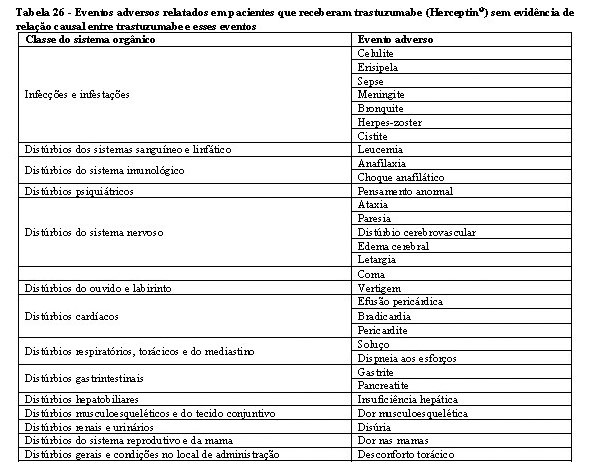
A segurança e a toxicidade de Zedora foram estudados nos 3 estudos pivotais de desenvolvimento da medicação. Os estudos de comparabilidade entre Zedora e o medicamento comparador (Herceptin®) foram MYL-Her-1001, MYL-Her-1002 e MYL-Her-3001. A população estudada de homens saudáveis e o tempo de exposição à medicação (dose única) dos estudos MYL-Her-1001 e MYL-Her-1002 são muito diferentes do estudo MYL-Her-3001 em termos de expectativa de eventos adversos, de modo que não são relevantes para a avaliação de segurança de Zedora. Portanto, os dados de segurança são do estudo MYL-Her-3001.1
Na tabela 16 encontram-se os dados de segurança do estudo MYL-Her-3001.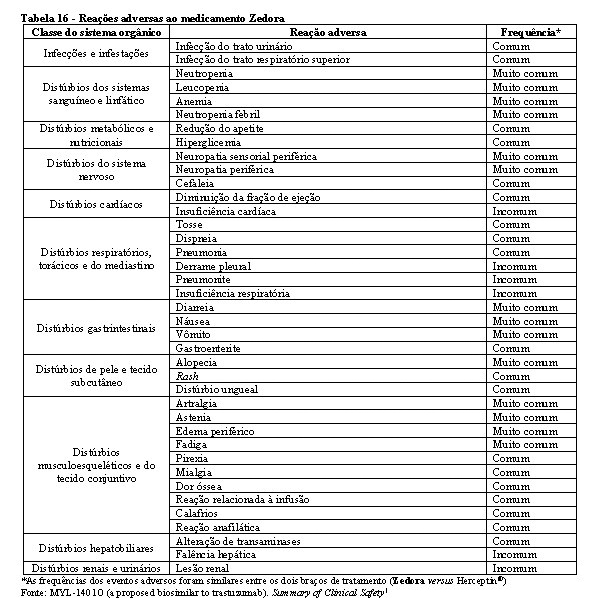
A maioria dos eventos adversos foram graus 1 (77% Zedora x 76% Herceptin®) e 2 (75% Zedora x 76% Herceptin®). No geral, 312 pacientes (63,3%) experimentaram eventos adversos emergentes do tratamento (EAET) de grau 3 ou maior, sendo a incidência destes eventos semelhante entre os grupos de tratamento (63,6% no braço Zedora e 63,0% no braço Herceptin®). Os EAET grau 3 ou maior mais comuns foram neutropenia (44,8%) e leucopenia (14,0%), o que ocorreu em frequências semelhantes entre os braços de tratamento.1
Imunogenicidade
No estudo clínico de câncer de mama inicial na neoadjuvância-adjuvância (BO22227), com mediana de acompanhamento excedendo 70 meses, 10,1% (30/296) dos pacientes do braço tratado com trastuzumabe (Herceptinâ) IV desenvolveram anticorpos contra trastuzumabe. Os anticorpos anti-trastuzumabe neutralizantes foram detectados em amostras pós nível basal em 2 de 30 pacientes do braço tratado com trastuzumabe (Herceptinâ) IV.
A relevância clínica desses anticorpos é desconhecida. A presença de anticorpos anti-trastuzumabe não teve impacto na farmacocinética, eficácia [determinada pela resposta patológica completa (RpC) e sobrevida livre de doença (SLD)] e segurança (determinada pela ocorrência de reações relacionadas à infusão, RRA) de trastuzumabe IV.Os níveis de anticorpo anti-droga (ADA) foram avaliados no estudo MYL-Her-3001. A taxa global de ADA foi 2,4% (6/247) no braço Zedora e 2,8% (7/246) no braço Herceptin®. O número de doentes positivos diminuiu ao longo do tempo. O potencial imunogênico das duas medicações foi similar e baixo.1
Informações adicionais sobre reações adversas selecionadas
Reações relacionadas à infusão e hipersensibilidade
As reações relacionadas à infusão, tais como calafrios e/ou febre, dispneia, hipotensão, sibilância, broncoespasmo, taquicardia, redução na saturação de oxigênio e insuficiência respiratória, foram observadas em todos os estudos clínicos com trastuzumabe (ver item "5. Advertências e Precauções").
Pode ser difícil diferenciar, clinicamente, as reações relacionadas à infusão de reações de hipersensibilidade.
O índice de todas as reações relacionadas à infusão de todos os graus variou entre os estudos dependendo da indicação, se trastuzumabe foi administrado em concomitante à quimioterapia ou como monoterapia e a metodologia de coleta de dados.
No câncer de mama metastático, o índice das reações relacionadas à infusão variou de 49% a 54% no braço com trastuzumabe, em comparação com 36% a 58% no braço comparador (o qual deve incluir outra quimioterapia). Reações graves (grau 3 ou maior) variaram de 5% a 7% no braço com trastuzumabe em comparação com 5% a 6% no braço comparador.
No câncer de mama inicial, o índice das reações relacionadas à infusão variou de 18% a 54% no braço com trastuzumabe, em comparação com 6% a 50% no braço comparador (o qual deve incluir outra quimioterapia). Reações graves (grau 3 ou maior) variaram de 0,5% a 6% no braço com trastuzumabe, em comparação com 0,3% a 5% no braço comparador.
No tratamento do câncer de mama inicial na neoadjuvância-adjuvância (BO22227), os índices de reações relacionadas à infusão estiveram de acordo com o descrito acima e foi de 37,2% no braço tratado com trastuzumabe (Herceptin®) IV. Reações graves de grau 3 relacionadas à infusão foram de 2% no mesmo braço durante o período de tratamento. Não houve reações relacionadas à infusão de graus 4 ou 5.
Reações anafilactoides foram observadas em casos isolados.
De acordo com o estudo MYL-Her 3001, grande parte das reações relacionadas à infusão ocorreram no primeiro ciclo, e todas resolveram no mesmo dia da interrupção da infusão e/ou tratamento conservativo. Não existem fatores de risco conhecidos.1
Os eventos adversos emergentes do tratamento (EAET) relacionados com a infusão que foram observados no estudo MYL-Her 3001 estão na tabela 17.1 
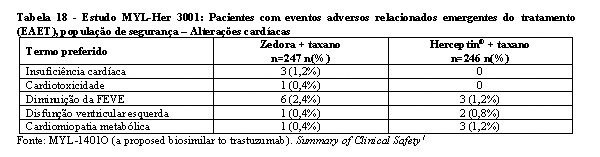
Disfunção cardíaca
Insuficiência cardíaca congestiva (NYHA Classe II-IV) é uma reação adversa comum a trastuzumabe e associada com resultados fatais. Sinais e sintomas de disfunção cardíaca, tais como dispneia, ortopneia, exacerbação da tosse, edema pulmonar, galope S3 ou redução na fração de ejeção do ventrículo esquerdo, foram observados em pacientes tratados com trastuzumabe (ver item "5. Advertências e Precauções").
Trastuzumabe também pode causar cardiomiopatia, diminuição da FEVE, palpitações, arritmias cardíacas, insuficiência cardíaca congestiva, insuficiência cardíaca, alterações cardíacas, disfunção ventricular e hipertensão arterial (ver item "5. Advertências e Precauções").1
Os eventos adversos emergentes do tratamento (EAET) relacionados à disfunção cardíaca que foram observados no estudo MYL-Her 3001 estão na tabela 18.1 
Câncer de mama metastático
Dependendo dos critérios utilizados para definir a insuficiência cardíaca, a incidência de sintomas nos estudos clínicos pivotais realizados em pacientes com doença metastática, variou entre 9% e 12% no grupo de pacientes tratados com trastuzumabe + paclitaxel, comparado com 1% - 4% no grupo de pacientes tratados com paclitaxel isolado. Para a monoterapia com trastuzumabe o índice foi de 6-9%. O índice mais elevado de disfunção cardíaca foi observado em pacientes tratados concomitantemente com trastuzumabe + antraciclina/ciclofosfamida (27%) e foi significativamente mais elevado que o do grupo tratado somente com antraciclina/ciclofosfamida (7-10%). Em outro estudo com monitoramento prospectivo da função cardíaca, a incidência de insuficiência cardíaca sintomática foi de 2,2% em pacientes recebendo trastuzumabe e docetaxel, comparado com 0% nos pacientes recebendo docetaxel isoladamente. A maioria dos pacientes (79%) que desenvolveram disfunção cardíaca nesses estudos apresentou melhora após receber o tratamento padrão para insuficiência cardíaca.
Câncer de mama inicial (adjuvância)
Nos três estudos clínicos pivotais na adjuvância com a administração de trastuzumabe (Herceptin®) em combinação com quimioterapia, a incidência de disfunção cardíaca de grau 3/4 (insuficiência cardíaca congestiva sintomática) foi similar em pacientes que estavam recebendo somente quimioterapia e em pacientes que estavam recebendo trastuzumabe (Herceptin®) sequencialmente após um taxano (0,3 a 0,4%). O índice foi maior em pacientes que estavam recebendo trastuzumabe concomitantemente a um taxano (2,0%). Em 3 anos, o índice de eventos cardíacos em pacientes recebendo AC→P (doxorrubicina mais ciclofosfamida seguidos por paclitaxel) + H (trastuzumabe - Herceptin®) foi estimado em 3,2%, comparado com 0,8% em pacientes tratados com AC→P. Nenhum aumento na incidência cumulativa de eventos cardíacos foi observado em 5 anos de acompanhamento adicional.
Em 5,5 anos, os índices de eventos cardíacos sintomáticos ou eventos relacionados a FEVE foram 1,0%, 2,3% e 1,1%, respectivamente, nos braços de tratamento com AC→D (doxorrubicina mais ciclofosfamida seguidos por docetaxel), AC→DH (doxorrubicina mais ciclofosfamida seguidos por docetaxel mais trastuzumabe - Herceptin®) e DCarbH (docetaxel, carboplatina e trastuzumabe - Herceptin®). Para insuficiência cardíaca congestiva sintomática (NCI-CTC Grau 3-4), os índices de 5 anos foram 0,6%, 1,9% e 0,4%, respectivamente, nos braços de tratamento AC→D, AC→DH e DCarbH. O risco global de desenvolvimento de eventos cardíacos sintomáticos foi baixo e similar para pacientes nos braços de tratamento com AC→D e DCarbH. Com relação aos braços de tratamento AC→D e DCarbH, houve aumento do risco de desenvolvimento de eventos cardíacos sintomáticos para pacientes do braço de tratamento AC→DH, sendo discernível por aumento contínuo no índice cumulativo de eventos cardíacos sintomáticos ou eventos relacionados a FEVE de até 2,3% em comparação com aproximadamente 1% nos dois braços comparadores (AC→D e DCarbH).
Quando trastuzumabe foi administrado após a conclusão da quimioterapia adjuvante, insuficiência cardíaca NYHA Classe III-IV foi observada em 0,6% dos pacientes no braço que receberam trastuzumabe por um ano após mediana de acompanhamento de 12 meses. Após uma mediana de 3,6 anos de acompanhamento, a incidência de insuficiência cardíaca congestiva grave e disfunção ventricular esquerda após a terapia com trastuzumabe permaneceu abaixo de 0,8% e 9,8%, respectivamente.
No estudo BO16348, após uma mediana de acompanhamento de 8 anos, a incidência de insuficiência cardíaca congestiva grave (NYHA Classe III-IV) no braço tratado com trastuzumabe (Herceptin®) por um ano, foi de 0,8%, e o índice de disfunção ventricular esquerda assintomática e sintomática leve foi de 4,6%.
A reversibilidade da insuficiência cardíaca congestiva grave (definida como uma sequência de pelo menos dois valores consecutivos de FEVE ≥ 50% após o evento) foi evidente em 71,4% dos pacientes tratados com trastuzumabe. A reversibilidade da disfunção ventricular esquerda assintomática e sintomática leve foi demonstrada em 79,5% dospacientes. Aproximadamente 17% dos eventos relacionados à disfunção cardíaca ocorreram após a conclusão do tratamento com trastuzumabe.
Na análise conjunta dos estudos NSABP-B31 e NCCTG N9831, com uma mediana de acompanhamento de 8,1 anos para o grupo AC→PH (doxorrubicina mais ciclofosfamida, seguido de paclitaxel mais trastuzumabe - Herceptin®), a incidência por paciente de um novo início de disfunção cardíaca, determinada pela FEVE, permaneceu inalterada em comparação com a análise feita no grupo AC→PH sob mediana de acompanhamento de 2,0 anos: 18,5% dos pacientes no grupo AC→PH com uma redução de FEVE de ≥ 10% a até menos que 50%. A reversibilidade da disfunção ventricular esquerda foi reportada em 64,5% dos pacientes que apresentaram ICC sintomática no grupo AC→PH, sendo assintomática no último acompanhame