YONDELIS
ADIUM
trabectedina
Antineoplásico.
Apresentações.
Pó liofilizado para solução para infusão.
Yondelis® é apresentado em embalagem contendo 1 frasco-ampola com 1 mg de trabectedina para uso único.
USO INTRAVENOSO
USO ADULTO
Composição.
Cada frasco-ampola de Yondelis® contém: trabectedina 1 mg. Excipientes: sacarose, fosfato de potássio monobásico, ácido fosfórico, hidróxido de potássio.
Após reconstituição, 1 mL de solução reconstituída contém 0,05 mg de trabectedina.
Informações técnicas.
1. INDICAÇÕES
Yondelis® (trabectedina) é indicado para o tratamento de pacientes adultos com sarcoma avançado dos tecidos moles, após falha de antraciclinas e ifosfamida, ou pacientes que não são elegíveis para receber esses agentes. Os dados de eficácia baseiam-se principalmente em pacientes com lipossarcoma e leiomiossarcoma.
2. RESULTADOS DE EFICÁCIA
A eficácia e segurança de Yondelis® (trabectedina) no tratamento de sarcoma dos tecidos moles (STM) baseiam-se num estudo randomizado em pacientes com lipossarcoma ou leiomiossarcoma localmente avançados ou metastásicos, cuja doença havia progredido ou sofrido uma recaída após o tratamento com, pelo menos, antraciclinas e ifosfamida. Neste estudo, a trabectedina foi administrada a 1,5 mg/m2 como infusão intravenosa de 24 horas a cada 3 semanas, ou a 0,58 mg/m2 semanalmente como infusão intravenosa de 3 horas durante 3 semanas de um ciclo de 4 semanas. A análise final, especificada pelo protocolo, do tempo até a progressão (TTP) mostrou uma redução de 26,6% no risco relativo de progressão para os pacientes tratados no grupo de 24-h/3sem. [Razão de Risco (RR) = 0,734, Intervalo de Confiança (IC): 0,554-0,974]. Os valores medianos de TTP foram de 3,7 meses (IC: 2,1-5,4 m) no grupo 24-h /3sem e de 2,3 meses (IC: 2,0-3,5 m) no grupo 3-h/sem. (p=0,0302). Não se detectaram diferenças significativas na sobrevida global (SG). A sobrevida mediana com o regime 24-h/3sem. foi de 13,9 meses (IC: 12,5-18,6) e 60,2% dos pacientes estavam vivos ao final de 1 ano (IC: 52,0-68,5%).
Estão disponíveis dados adicionais de eficácia, de 3 estudos de Fase II em populações semelhantes tratadas com o mesmo regime. Estes estudos avaliaram um total de 100 pacientes com lipossarcoma e leiomiossarcoma e 83 pacientes com outros tipos de sarcoma.
Os resultados de um programa ampliado de acesso para pacientes com STM (estudo ET743-SAR-3002) revelam que entre os 903 indivíduos examinados quanto a SG, o tempo médio de sobrevida foi de 11,9 meses (IC 95%: 11,2, 13,8). A sobrevida média por tipo histológico de tumor foi de 16,2 meses [IC 95%: 14,1, 19,5] para indivíduos com leiomiossarcomas e lipossarcomas e 8,4 meses [IC 95%: 7,1, 10,7] para indivíduos com outros tipos de sarcomas. A sobrevida média para indivíduos com lipossarcoma foi de 18,1 meses [IC 95%: 15,0, 26,4] e no caso de indivíduos com leiomiossarcoma foi de 16,2 meses [IC 95%: 11,7, 24,3].
Estão disponíveis dados adicionais sobre eficácia de um estudo randomizado de fase III ativo-controlado de trabectedina versus dacarbazina (Estudo ET743-SAR-3007), em pacientes com lipossarcoma ou leiomiossarcoma não ressecável ou metastático que foram previamente tratados com um regime contendo, pelo menos, uma antraciclina e ifosfamida, ou um regime contendo antraciclina e um regime adicional de quimioterapia citotóxica. Os pacientes no braço da trabectedina receberam uma injeção intravenosa de dexametasona 20 mg antes de cada infusão de trabectedina. Em geral, 384 pacientes foram randomizados para o grupo da trabectedina [1,5 mg/m2 uma vez a cada 3 semanas (24-h/3sem)] e 193 pacientes para o grupo da dacarbazina (1 g/m2 uma vez a cada 3 semanas). A idade mediana dos pacientes era de 56 anos (dos 17 aos 81), 30% eram homens, 77% caucasianos, 12% afro-americanos e 4% asiáticos. Os pacientes dos braços de trabectedina e dacarbazina receberam uma média de 4 e 2 ciclos, respetivamente. O desfecho primário de eficácia do estudo foi SG, que incluiu 381 eventos de morte (66% de todos os pacientes randomizados): 258 (67,2%) mortes no grupo da trabectedina e 123 (63,7%) mortes no grupo da dacarbazina (HR 0,927 [95% CI: 0,748, 1,150; p=0,4920]). A análise final não demonstrou uma diferença significativa com um acompanhamento de sobrevida média de 21,2 meses que resultou numa média de 13,7 meses (95% CI: 12,2, 16,0) para o braço da trabectedina e 13,1 meses [95% CI: 9.1, 16.2] para o braço da dacarbazina. Os principais desfechos secundários são resumidos na tabela abaixo: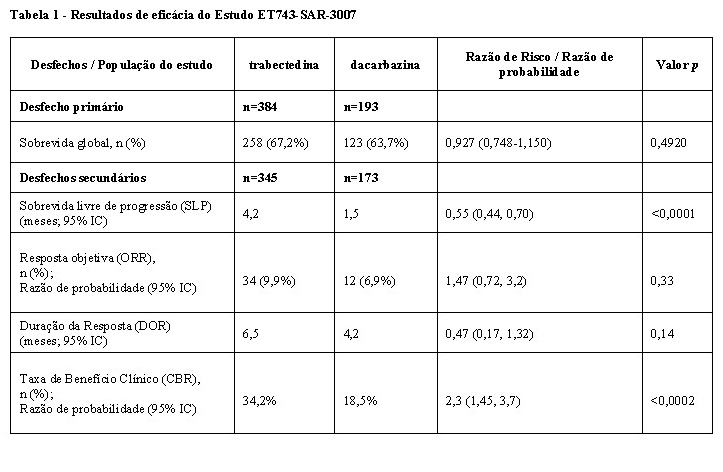
3. CARACTERÍSTICAS FARMACOLÓGICAS
Mecanismo de ação
A trabectedina liga-se ao sulco menor do ácido desoxirribonucleico (DNA), curvando a hélice para o sulco maior. Esta ligação ao DNA desencadeia uma cascata de acontecimentos que afetam diversos fatores de transcrição, proteínas de ligação e vias de reparação do DNA, resultando numa alteração do ciclo celular.
Farmacodinâmica
A trabectedina mostrou exercer atividade antiproliferativa in vitro e in vivo contra um leque de linhagens celulares tumorais humanas e tumores experimentais, incluindo tumores malignos como sarcoma, câncer de mama, câncer de pulmão de células não pequenas, câncer de ovário e melanoma.
Exames complementares de diagnóstico eletrocardiograma (ECG)
Em um estudo de QT/QTc controlado por placebo, a trabectedina não prolongou o intervalo QTc em pacientes com tumores malignos sólidos em estado avançado.
Farmacocinética
Distribuição
A exposição sistêmica após a administração intravenosa como infusão a velocidade constante é proporcional à dose, em doses até 1,8 mg/m2. O perfil farmacocinético de trabectedina é consistente com um modelo de disposição de compartimentos múltiplos. Após a administração intravenosa, a trabectedina demonstra um volume aparente de distribuição elevado, consistente com uma ligação extensiva aos tecidos e às proteínas plasmáticas (94 a 98% da trabectedina no plasma encontra-se ligada a proteínas). O volume de distribuição da trabectedina no estado estacionário em seres humanos excede 5.000 L.
Biotransformação
O citocromo P450 3A4 é a principal isoenzima do citocromo P450 responsável pelo metabolismo oxidativo da trabectedina em concentrações clinicamente relevantes. Outras enzimas P450 poderão contribuir para o metabolismo. A trabectedina não induz nem inibe as principais enzimas do citocromo P450.
Eliminação
A eliminação renal de trabectedina inalterada em seres humanos é baixa (inferior a 1%). A semi-vida terminal é longa (valor da população da fase de eliminação terminal: 180-h). Após a administração de uma dose de trabectedina com radiomarcação em pacientes oncológicos, a média (DP) da recuperação fecal da radioatividade total é de 58% (17%), e a média (DP) de recuperação urinária é de 5,8% (1,73%). Com base na estimativa populacional para a depuração plasmática da trabectedina (30,9 L/h) e na razão sangue/plasma (0,89), a depuração da trabectedina no sangue completa é de cerca de 35 L/h. Este valor é cerca de metade da taxa do fluxo sanguíneo hepático em seres humanos. Portanto, a razão de extração de trabectedina pode ser considerada como moderada. A variabilidade interpacientes da estimativa populacional para a depuração plasmática da trabectedina foi de 49% e a variabilidade intrapacientes foi de 28%.
Populações especiais
Etnia
Uma análise farmacocinética da população indicou que a depuração plasmática da trabectedina não é influenciada pela idade (intervalo de 19-83 anos), sexo, peso corporal total (intervalo de 36 a 148 kg) ou área de superfície corporal (intervalo de 0,9 a 2,8 m2). Uma análise farmacocinética realizada na população revelou que as concentrações de trabectedina no plasma, observadas na população japonesa na dose de 1,2 mg/m2 eram equivalentes às obtidas na população ocidental não japonesa de 1,5 mg/m².
Comprometimento renal
Não existe influência relevante da função renal, medida pela depuração da creatinina, sobre a farmacocinética da trabectedina dentro do intervalo de valores (≥ 30,3 mL/min) presente nos pacientes incluídos nos estudos clínicos. Não estão disponíveis dados para pacientes com uma depuração da creatinina inferior a 30,3 mL/min. A baixa recuperação ( < 9% em todos os pacientes estudados) da radioatividade total na urina após uma única dose de trabectedina marcada com 14C indica que o comprometimento renal tem pouca influência sobre a eliminação da trabectedina ou dos seus metabólitos.
Comprometimento hepático
O efeito da disfunção hepática na farmacocinética da trabectedina foi avaliado em 15 pacientes com câncer em doses que variavam de 0,58 a 1,3 mg/m2 administradas numa infusão de 3 horas. A dose média geométrica normalizada da exposição à trabectedina (AUC) aumentou 97% (IC 90%: 20%, 222%) em 6 pacientes com insuficiência hepática moderada (aumento dos níveis séricos de bilirrubina de 1,5 a 3 x LSN e um aumento de aminotransferases (AST ou ALT) < 8 x LSN) após a administração de uma dose única de trabectedina de 0,58 mg/m2 (n=3) ou 0,9 mg/m2 (n=3) em comparação com 9 pacientes com função hepática normal após a administração de uma dose única de trabectedina de 1,3 mg/m2 (vide seções 4. CONTRAINDICAÇÕES e 8. POSOLOGIA E MODO DE USAR).
Dados de segurança pré-clínica
Os dados pré-clínicos indicam que a trabectedina tem um efeito limitado sobre os sistemas cardiovascular, respiratório e sistema nervoso central para exposições abaixo do intervalo clínico terapêutico, em termos de AUC.
Os efeitos da trabectedina sobre as funções cardiovascular e respiratória foram investigados in vivo (macacos Cynomolgus (Macaca fascicularis) anestesiados). Selecionou-se um plano de infusão de 1 hora para atingir níveis plasmáticos máximos (valores Cmax) dentro do intervalo dos observados na prática clínica. Os níveis plasmáticos de trabectedina foram de 10,6 ± 5,4 (Cmax), mais elevados do que os atingidos nos pacientes após infusão a 1.500 mg/m2 para 24 horas (Cmax de 1,8 ± 1,1 ng/mL) e semelhantes aos atingidos após administração da mesma dose através de infusão de 3 horas (Cmax de 10,8 ± 3,7 ng/mL).
A mielossupressão e hepatotoxicidade foram identificadas como toxicidade primária para a trabectedina. Os resultados observados incluíram toxicidade hematopoiética (leucopenia grave, anemia e depleção linfoide e da medula óssea), bem como aumentos nos testes da função hepática, degeneração hepatocelular, necrose epitelial intestinal, e reações locais graves no local da injeção. Foram detectados resultados toxicológicos renais em estudos de toxicidade multiciclo conduzidos em macacos. Estes resultados foram secundários a reação local grave no local de administração e, por conseguinte, de atribuição incerta à trabectedina; contudo, deve garantir-se cuidado na interpretação destes resultados renais, e não pode excluir-se toxicidade relacionada com o tratamento.
A trabectedina é genotóxica tanto in vitro como in vivo. Não foram realizados estudos de carcinogenicidade a longo prazo.
Não foram realizados estudos de fertilidade com trabectedina, mas observaram-se alterações histopatológicas limitadas nas gónadas em estudos de toxicidade de dose repetida. Considerando a natureza do composto (citotóxico e mutagênico) é provável que este afete a capacidade reprodutiva.
A transferência placentária de trabectedina e a exposição fetal a trabectedina foram observadas num estudo em camundongos que receberam uma única dose de 14C-trabectedina a 0,061 mg/kg por via intravenosa durante a gravidez. A concentração de radioatividade máxima no tecido fetal foi semelhante à observada no plasma ou sangue materno.
4. CONTRAINDICAÇÕES
Yondelis® (trabectedina) é contraindicado em pacientes com hipersensibilidade conhecida à trabectedina ou a outro componente da fórmula. Yondelis® não deve ser usado por gestantes e lactantes.
Yondelis® não deve ser administrado em pacientes com infecção ativa grave ou não controlada.
Yondelis® não deve ser administrado em combinação com vacina contra a febre-amarela (vide seção 5. ADVERTÊNCIAS E PRECAUÇÕES).
Categoria de risco na gravidez: D
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
5. ADVERTÊNCIAS E PRECAUÇÕES
Comprometimento hepático
Os pacientes devem atender a critérios específicos sobre os parâmetros da função hepática para iniciar o tratamento com Yondelis® (trabectedina). Uma vez que a exposição sistêmica à trabectedina é em média aproximadamente duplicada (vide seção 3. CARACTERÍSTICAS FARMACOLÓGICAS - Farmacocinética) devido a insuficiência hepática e, portanto, o risco de toxicidade pode ser aumentado, os pacientes com doenças hepáticas clinicamente relevantes, tais como hepatite crônica ativa, devem ser monitorados de perto e a dose ajustada se necessário. Os pacientes com níveis séricos elevados de bilirrubina não devem ser tratados com trabectedina (vide seção 8. POSOLOGIA E MODO DE USAR).
Comprometimento renal
A depuração da creatinina deve ser monitorada antes e durante o tratamento. Yondelis® não deve ser utilizado em pacientes com depuração da creatinina < 30 mL/min (vide seção 8. POSOLOGIA E MODO DE USAR).
Neutropenia e trombocitopenia
Foram notificadas com muita frequência neutropenia e trombocitopenia de graus 3 ou 4 associadas ao tratamento com Yondelis®. Deve-se efetuar um hemograma completo, incluindo a contagem diferencial e plaquetária, deve ser realizada no início do tratamento (basal), semanalmente durante os dois primeiros ciclos e, em seguida, uma vez entre os ciclos (vide seção 8. POSOLOGIA E MODO DE USAR). Os pacientes que desenvolvem febre devem procurar atendimento médico imediatamente. Se isso ocorrer, a terapia de suporte ativa deve ser iniciada imediatamente.
Yondelis® não deve ser administrado em pacientes com contagem inicial de neutrófilos inferior a 1.500 células/mm3 e contagem de plaquetas inferior a 100.000 células/mm3. Se ocorrer neutropenia grave (CAN < 500 células/mm3) com duração superior a 5 dias ou associada a febre ou infeção, recomenda-se a redução da dose (vide seção 8. POSOLOGIA E MODO DE USAR).
Náusea e vômito
A profilaxia antiemética com corticosteroides como a dexametasona deve ser administrada a todos os pacientes (vide seção 8. POSOLOGIA E MODO DE USAR).
Rabdomiólise e elevações graves de CPK ( > 5 x LSN)
A trabectedina não deve ser utilizada em pacientes com CPK > 2,5 x LSN (vide seção 8. POSOLOGIA E MODO DE USAR). A rabdomiólise foi relatada com pouca frequência, geralmente em associação com mielotoxicidade, anormalidades graves nos testes de função hepática e/ou insuficiência renal ou de múltiplos órgãos. Portanto, a CPK deve ser monitorada atentamente sempre que um paciente apresentar qualquer uma dessas toxicidades ou fraqueza muscular ou dor muscular. Se ocorrer rabdomiólise, medidas de suporte como hidratação parenteral, alcalinização da urina e diálise devem ser prontamente estabelecidas, conforme indicado. O tratamento com Yondelis® deve ser descontinuado até que o paciente se recupere totalmente.
Deve-se ter cautela se medicamentos associados à rabdomiólise (por exemplo, estatinas) forem administrados concomitantemente com trabectedina, pois o risco de rabdomiólise pode ser aumentado.
Anormalidades do Teste da Função Hepática (LFT)
Aumentos agudos reversíveis na aspartato aminotransferase (AST) e alanina aminotransferase (ALT) foram relatados na maioria dos pacientes. Yondelis® não deve ser utilizado em pacientes com bilirrubina elevada. Os pacientes com aumentos de AST, ALT e fosfatase alcalina entre os ciclos podem necessitar de ajustes de dose (vide seção 8. POSOLOGIA E MODO DE USAR).
Este medicamento pode causar hepatotoxicidade. Por isso, requer uso cuidadoso, sob vigilância médica estrita e acompanhado por controles periódicos da função hepática, semanalmente durante os dois primeiros ciclos de terapia e pelo menos uma vez entre os tratamentos nos ciclos subsequentes.
Reações no local da injeção
A utilização de acesso venoso central é fortemente recomendada (vide seção 8. POSOLOGIA E MODO DE USAR). Os pacientes podem desenvolver uma reação potencialmente grave no local da injeção quando a trabectedina é administrada através de um cateter venoso periférico.
O extravasamento de trabectedina pode causar necrose tecidual requerendo desbridamento. Não existe antídoto específico para o extravasamento de trabectedina. O extravasamento deve ser gerenciado pela prática padrão local.
Reações alérgicas
Durante a experiência pós-comercialização, foram relatadas reações de hipersensibilidade com ocorrência muito rara de desfecho fatal, associadas à administração de trabectedina (vide seções 4. CONTRAINDICAÇÕES e 9. REAÇÕES ADVERSAS).
Disfunção cardíaca
Os pacientes devem ser monitorados quanto a eventos adversos relacionados ao coração ou disfunção miocárdica.
Uma avaliação cardíaca completa, incluindo a determinação da fração de ejeção do ventrículo esquerdo (FEVE) por ecocardiograma ou varredura de aquisição múltipla (MUGA) deve ser realizada antes do início da trabectedina e em intervalos de 2 a 3 meses até que a trabectedina seja descontinuada.
Pacientes com FEVE inferior ao limite inferior do normal (FEVE < LLN), dose cumulativa anterior de antraciclina > 300mg/m2, idade > 65 anos ou histórico de doença cardiovascular (especialmente naqueles com medicação cardíaca) podem ter risco aumentado de disfunção cardíaca no momento do tratamento com trabectedina.
Para pacientes com eventos adversos cardíacos de grau 3 ou 4 indicativos de cardiomiopatia ou para pacientes com FEVE que diminui abaixo do LIN (avaliado como uma diminuição absoluta da FEVE de ≥ 15% ou < LLN com uma diminuição absoluta de ≥ 5%), a trabectedina deve ser descontinuada.
Síndrome de extravasamento capilar (CLS)
Casos de síndrome de vazamento capilar (CLS) foram relatados com trabectedina (incluindo casos com desfechos fatais). Se surgirem sintomas de possível CLS, como edema inexplicável com ou sem hipotensão, o médico responsável deve reavaliar o nível de albumina sérica. Um rápido declínio no nível de albumina sérica pode ser indicativo de CLS. Se o diagnóstico de CLS for confirmado após exclusão de outras causas, o médico responsável deve descontinuar a trabectedina e iniciar o tratamento para CLS de acordo com as diretrizes institucionais (vide seções 8. POSOLOGIA E MODO DE USAR e 9. REAÇÕES ADVERSAS).
Outros
A administração concomitante de Yondelis® com inibidores potentes da enzima CYP3A4 deve ser evitada (vide seção 6. INTERAÇÕES MEDICAMENTOSAS). Se isso não for possível, é necessário um monitoramento cuidadoso das toxicidades e devem ser consideradas as reduções da dose de trabectedina.
Deve-se ter cautela se medicamentos associados à hepatotoxicidade forem administrados concomitantemente com trabectedina, pois o risco de hepatotoxicidade pode ser aumentado.
O uso concomitante de trabectedina com fenitoína pode reduzir a absorção de fenitoína levando a uma exacerbação das convulsões. A combinação de trabectedina com fenitoína ou vacinas vivas atenuadas não é recomendada e com a vacina contra a febre amarela é especificamente contraindicada (vide seção 4. CONTRAINDICAÇÕES).
A utilização concomitante de trabectedina com álcool deve ser evitada (vide seção 6. INTERAÇÕES MEDICAMENTOSAS).
As mulheres com potencial para engravidar devem utilizar métodos contraceptivos eficazes durante o tratamento e 3 meses depois, e informar imediatamente o médico assistente se ocorrer uma gravidez (vide seção 3. CARACTERÍSTICAS FARMACOLÓGICAS - Dados de segurança pré-clínica).
Os homens em idade fértil devem utilizar métodos contraceptivos eficazes durante o tratamento e 5 meses após o tratamento.
Informe a seu paciente que a doação de sangue é absolutamente contraindicada durante o tratamento com trabectedina e até 6 semanas após seu término, devido ao dano que ele pode causar ao receptor.
População pediátrica
Yondelis® não deve ser utilizado em pacientes com menos de 18 anos de idade.
Idosos
Nenhum estudo específico foi realizado em pacientes idosos.
Yondelis® contém potássio
Este medicamento contém potássio, menos de 1 mmol (39 mg) por frasco-ampola, ou seja, é praticamente "livre de potássio".
Gravidez
Não estão disponíveis dados clínicos suficientes sobre a exposição na gravidez. No entanto, com base em seu mecanismo de ação conhecido, a trabectedina pode causar defeitos congênitos graves quando administrada durante a gravidez. A trabectedina atravessou a placenta quando administrada em ratas prenhas. A trabectedina não deve ser utilizada durante a gravidez. Se ocorrer gravidez durante o tratamento, a paciente deve ser informada do potencial risco para o feto (vide seção 3. CARACTERÍSTICAS FARMACOLÓGICAS - Dados de segurança pré-clínica) e monitorada cuidadosamente. Se a trabectedina for usada no final da gravidez, as reações adversas potenciais devem ser monitoradas cuidadosamente nos recém-nascidos.
Categoria de risco na gravidez: D. A trabectedina mostrou evidência positiva de risco fetal humano, no entanto, os benefícios potenciais para a mulher podem eventualmente justificar o risco, por exemplo, em casos de doenças graves ou com risco de vida para as quais não existem outros medicamentos mais seguros.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
Mulheres com potencial para engravidar
As mulheres com potencial para engravidar devem utilizar métodos contraceptivos eficazes durante o tratamento e 3 meses depois, e informar imediatamente o médico responsável se ocorrer uma gravidez (vide seção 3. CARACTERÍSTICAS FARMACOLÓGICAS - Dados de segurança pré-clínica).
Se ocorrer gravidez durante o tratamento, a possibilidade de aconselhamento genético deve ser considerada.
Amamentação
Não se sabe se a trabectedina é excretada no leite humano. A excreção de trabectedina no leite não foi estudada em animais. A amamentação é contraindicada durante o tratamento e 3 meses depois (vide seção 4. CONTRAINDICAÇÕES).
Este medicamento é contraindicado durante o aleitamento ou doação de leite, pois pode ser excretado no leite humano e causar reações indesejáveis no bebê. Seu médico ou cirurgião-dentista deve apresentar alternativas para o seu tratamento ou para a alimentação do bebê.
Fertilidade
Os homens em idade fértil devem utilizar métodos contraceptivos eficazes durante o tratamento e 5 meses após o tratamento (vide seção 5. ADVERTÊNCIAS E PRECAUÇÕES).
A trabectedina pode ter efeitos genotóxicos. Deve-se procurar aconselhamento sobre a conservação de óvulos ou espermatozoides antes do tratamento devido à possibilidade de infertilidade irreversível devido à terapia com Yondelis®.
O aconselhamento genético também é recomendado para pacientes que desejam ter filhos após a terapia.
Efeitos sobre a capacidade de dirigir e operar máquinas
Não foram realizados estudos sobre os efeitos da capacidade de conduzir e utilizar máquinas. No entanto, fadiga e/ou astenia foram relatadas em pacientes recebendo trabectedina. Os pacientes que apresentarem qualquer uma dessas reações adversas durante a terapia não devem dirigir ou operar máquinas.
6. INTERAÇÕES MEDICAMENTOSAS
Efeitos de outras substâncias na trabectedina
Os estudos de interação foram realizados apenas em adultos.
Uma vez que a trabectedina é metabolizada principalmente pelo CYP3A4, é provável que as concentrações de trabectedina no plasma sejam aumentadas em pacientes que recebem concomitantemente medicamentos que inibem potentemente a atividade desta isoenzima. Da mesma forma, a coadministração de trabectedina com indutores potentes de CPY3A4 pode aumentar a depuração metabólica da trabectedina. Dois estudos de fase 1 de interação medicamentosa in vivo confirmaram tendências para exposições aumentadas e diminuídas de trabectedina quando administradas com cetoconazol e rifampicina, respectivamente.
Quando o cetoconazol foi coadministrado com trabectedina, a exposição plasmática da trabectedina aumentou em aproximadamente 21% para Cmax e 66% para AUC, mas não foram identificados novos problemas de segurança. O monitoramento cuidadoso das toxicidades é necessário em pacientes recebendo trabectedina em combinação com inibidores potentes do CYP3A4 (por exemplo, cetoconazol oral, fluconazol, ritonavir, claritromicina ou aprepitanto) e tais combinações devem ser evitadas, se possível. Se tais combinações forem necessárias, ajustes de dose apropriados devem ser realizados em caso de toxicidade (vide seções 5. ADVERTÊNCIAS E PRECAUÇÕES e 8. POSOLOGIA E MODO DE USAR).
Quando a rifampicina foi coadministrada com trabectedina, resultou na redução da exposição plasmática da trabectedina em aproximadamente 22% para Cmax e 31% para AUC. Portanto, o uso concomitante de trabectedina com indutores fortes de CYP3A4 (por exemplo, rifampicina, fenobarbital, Erva de São João) deve ser evitado, se possível (vide seção 5. ADVERTÊNCIAS E PRECAUÇÕES).
O consumo de álcool deve ser evitado durante o tratamento com trabectedina devido à hepatotoxicidade do medicamento (vide seção 5. ADVERTÊNCIAS E PRECAUÇÕES).
Os dados pré-clínicos demonstraram que a trabectedina é um substrato da P-gp. A administração concomitante de inibidores de P-gp, por exemplo ciclosporina e verapamil, podem alterar a distribuição e/ou a eliminação da trabectedina. A relevância desta interação e a toxicidade do sistema nervoso central (SNC) não foram estabelecidas. Deve-se ter cautela em tais situações.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Yondelis® (trabectedina) apresenta prazo de validade de 36 meses a partir da data de fabricação, devendo ser armazenado sob refrigeração (entre 2°C e 8°C).
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Após reconstituição, Yondelis® demonstrou estabilidade química e física por 30 horas em temperatura/luz ambiente ou sob refrigeração em condições entre 2°C e 8°C.
Do ponto de vista microbiológico, a solução reconstituída deve ser diluída e utilizada imediatamente. Se não for diluída e utilizada imediatamente, os tempos de armazenamento em uso e as condições anteriores ao uso do produto reconstituído são de responsabilidade do usuário e normalmente não devem ser superiores a 24 horas entre 2°C e 8°C, a menos que a reconstituição tenha ocorrido em ambiente com condições assépticas validadas e controladas.
Características físicas e organolépticas
Pó branco ou esbranquiçado, essencialmente livre de partículas visíveis.
A solução reconstituída é clara, sem cor ou uma solução levemente amarelo acastanhada, essencialmente livre de partículas visíveis.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
Yondelis® (trabectedina) deve ser administrado sob a supervisão de um médico com experiência no uso de quimioterapia. Seu uso deve ser restrito a oncologistas qualificados ou outros profissionais de saúde especializados na administração de agentes citotóxicos.
Posologia
A dose recomendada é de 1,5 mg/m2 de área de superfície corporal, administrada por infusão intravenosa durante 24 horas com intervalo de três semanas entre os ciclos.
A dose recomendada em pacientes japoneses é menor do que a dose usual para todas as outras raças e é de 1,2 mg/m2 de área de superfície corporal.
Todos os pacientes devem receber corticosteroides, como por exemplo 20 mg de dexametasona por via intravenosa 30 minutos antes de Yondelis®; não apenas como profilaxia antiemética, mas também porque fornece efeitos hepatoprotetores. Antieméticos adicionais podem ser administrados conforme necessário.
Os seguintes critérios são necessários para permitir o tratamento com Yondelis®:
- Contagem absoluta de neutrófilos (ANC) ≥ 1.500/mm3
- Contagem de plaquetas ≥ 100.000/mm3
- Bilirrubina ≤ limite superior do normal (LSN)
- Fosfatase alcalina ≤ 2,5 x LSN (considerar isoenzimas hepáticas 5-nucleotidase ou gama glutamil transpeptidase (GGT), se a elevação puder ser de origem óssea).
- Albumina ≥ 25 g/L
- Alanina aminotransferase (ALT) e Aspartato aminotransferase (AST) ≤ 2,5 x LSN
- Depuração de creatinina ≥ 30 mL/min e creatinina sérica ≤ 1,5 mg/dL (≤ 132,6 mmol/L)
- Creatina fosfoquinase (CPK) ≤ 2,5 x LSN
- Hemoglobina ≥ 9 g/dL
Os mesmos critérios acima devem ser atendidos antes de um novo tratamento. Caso contrário, o tratamento deve ser adiado por até 3 semanas até que os critérios sejam atendidos.
O monitoramento adicional dos parâmetros hematológicos bilirrubina, fosfatase alcalina, aminotransferases e CPK deve ocorrer semanalmente durante os dois primeiros ciclos de terapia e pelo menos uma vez entre os tratamentos nos ciclos subsequentes.
A mesma dose deve ser administrada para todos os ciclos, desde que não sejam observadas toxicidades de grau 3-4 e que o paciente preencha os critérios de um novo tratamento.
Ajustes de dose durante o tratamento
Antes do novo tratamento, os pacientes devem atender aos critérios de valores basais definidos acima. Se qualquer um dos seguintes eventos ocorrer a qualquer momento entre os ciclos, a dose deve ser reduzida conforme a tabela 2 abaixo, para os ciclos subsequentes:
- Neutropenia < 500/mm3 com duração superior a 5 dias ou associada a febre ou infecção;
- Trombocitopenia < 25.000/mm3;
- Aumento de bilirrubina > LSN e/ou fosfatase alcalina > 2,5 x LSN;
- Aumento de aminotransferases (AST ou ALT) > 2,5 x LSN, que não recuperou até o dia 21;
- Qualquer outra reação adversa de grau 3 ou 4 (como náusea, vômito, fadiga).
Uma vez que uma dose tenha sido reduzida devido à toxicidade, o aumento da dose nos ciclos subsequentes não é recomendado. Se qualquer uma dessas toxicidades reaparecer em ciclos subsequentes em um paciente apresentando benefício clínico, a dose pode ser reduzida novamente (veja abaixo). Fatores estimuladores de colônias podem ser administrados para toxicidade hematológica de acordo com a prática padrão local.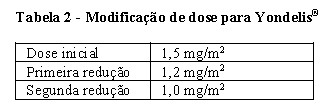
Caso sejam necessárias reduções adicionais da dose, deve ser considerada a descontinuação do tratamento.
Duração do tratamento
Nos ensaios clínicos, não houve limites pré-definidos para o número de ciclos administrados. O tratamento continuou enquanto o benefício clínico foi observado. Yondelis® foi administrado por 6 ou mais ciclos em 29,5% dos pacientes tratados.
Idosos
Não foram realizados estudos específicos em idosos. No geral, 20% dos 1.164 pacientes na análise de segurança integrada dos ensaios clínicos de monoterapia tinham mais de 65 anos. Parece que a depuração plasmática e o volume de distribuição da trabectedina não são influenciados pela idade. Portanto, ajustes de dose com base exclusivamente em critérios de idade não são recomendados rotineiramente.
Insuficiência hepática
Recomenda-se cautela especial e ajustes de dose podem ser necessários em pacientes com insuficiência hepática, uma vez que a exposição sistêmica à trabectedina é aumentada e o risco de hepatotoxicidade pode ser aumentado. Pacientes com níveis séricos basais elevados de bilirrubina não devem ser tratados com Yondelis®. Os testes da função hepática devem ser monitorados durante o tratamento com Yondelis® uma vez que podem ser indicados ajustes de dose (vide Tabela 2 e seção 5. ADVERTÊNCIAS E PRECAUÇÕES).
Insuficiência renal
Não foram realizados estudos incluindo pacientes com insuficiência renal (depuração da creatinina < 30 mL/min) e, portanto, Yondelis® não deve ser utilizado nesta população de pacientes (vide seção 5. ADVERTÊNCIAS E PRECAUÇÕES). Considerando as características farmacocinéticas da trabectedina (vide seção 3. CARACTERÍSTICAS FARMACOLÓGICAS - Farmacocinética), não são necessários ajustes de dose em pacientes com comprometimento renal leve ou moderado.
Método de administração
A administração intravenosa através de acesso venoso central é fortemente recomendada.
Preparo e administração
Preparação para infusão intravenosa
Yondelis® deve ser reconstituído e diluído antes da infusão intravenosa. Devem ser utilizadas técnicas assépticas apropriadas para preparar a solução para infusão (vide Instruções para reconstituição e diluição).
Instruções para a reconstituição
Cada frasco-ampola de Yondelis® contendo 1 mg de trabectedina é reconstituído com 20 mL de água para injetáveis. A solução obtida tem uma concentração de 0,05 mg/mL e é de uso único.
Uma seringa é usada para injetar 20 mL de água para injetáveis no frasco-ampola. O frasco-ampola deve ser agitado até a dissolução completa. A solução reconstituída resulta numa solução límpida, incolor ou ligeiramente amarelada, essencialmente livre de partículas visíveis.
Esta solução reconstituída contém 0,05 mg/mL de trabectedina. A solução requer diluição adicional e é apenas para uso único.
Instruções para diluição
A solução reconstituída deve ser diluída com solução para infusão de cloreto de sódio a 9 mg/mL (0,9%) ou solução para infusão de glicose a 50 mg/mL (5%). O volume requerido deve ser calculado da seguinte forma: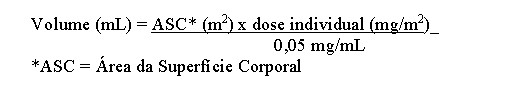
Se a administração for feita através de um acesso venoso central, a quantidade apropriada de solução reconstituída deve ser retirada do frasco-ampola e adicionada a uma bolsa de infusão contendo 500 mL de diluente (cloreto de sódio 9 mg/mL (0,9%) solução para infusão ou glicose 50 mg/mL (5%) solução para infusão), sendo a concentração de trabectedina na solução para infusão ≤ 0,030 mg/mL.
Se o acesso venoso central não for viável e acesso venoso periférico tiver que ser usado, a solução reconstituída deve ser adicionada a uma bolsa de infusão contendo ≥ 1.000 mL de diluente (cloreto de sódio 9 mg/mL (0,9%) solução para infusão ou glicose 50 mg/mL (5%) solução para infusão).
As soluções parenterais devem ser inspecionadas visualmente quanto a partículas antes da administração. Uma vez preparada a infusão, ela deve ser administrada imediatamente.
Instruções para o manuseio e descarte
Yondelis® é um medicamento anticancerígeno citotóxico e, como acontece com outros compostos potencialmente tóxicos, deve-se ter cautela durante o manuseio. Devem ser seguidos os procedimentos para o manuseio e descarte adequados de medicamentos citotóxicos. O pessoal deve ser treinado nas técnicas corretas para reconstituir e diluir o medicamento e deve-se usar roupas de proteção, incluindo máscara, óculos e luvas durante a reconstituição e diluição. As mulheres grávidas devem ser excluídas do trabalho com este medicamento.
O contato acidental com a pele, olhos ou membranas mucosas deve ser tratado imediatamente com água em abundância.
Não foram observadas incompatibilidades entre Yondelis® e frascos de vidro tipo I, bolsas e tubos de cloreto de polivinil (PVC) e polietileno (PE), reservatórios de poli-isopreno e sistemas implantáveis de titânio para acesso vascular.
Qualquer medicamento não utilizado ou resíduos devem ser descartados de acordo com os requisitos locais para medicamentos citotóxicos.
9. REAÇÕES ADVERSAS
Resumo do perfil de segurança
A maioria dos pacientes tratados com Yondelis® (trabectedina) pode apresentar reações adversas de qualquer grau e menos de um terço de reações adversas graves de gravidade grau 3 ou 4. As reações adversas mais frequentes de qualquer grau de gravidade foram neutropenia, náusea, vômito, aumento de AST/ALT, anemia, fadiga, trombocitopenia, anorexia e diarreia.
Reações adversas fatais ocorreram em 1,9% dos pacientes tratados com Yondelis® Elas foram muitas vezes o resultado de uma combinação de eventos, incluindo pancitopenia, neutropenia febril, alguns deles com sepse, envolvimento hepático, renal, insuficiência renal ou de múltiplos órgãos e rabdomiólise.
Resumo tabulado de reações adversas
O seguinte perfil de segurança de Yondelis® baseia-se em reações adversas notificadas em ensaios clínicos, estudos de segurança pós-autorização e notificações espontâneas.
A tabela abaixo apresenta as reações adversas notificadas em pacientes tratados com trabectedina. Tanto as reações adversas quanto os valores laboratoriais foram usados para fornecer as frequências.
As reações adversas estão listadas por Classe de Sistema de Órgãos e frequência. As frequências são classificadas em muito comuns (≥ 1/10), comuns (≥ 1/100 a < 1/10), incomuns (≥ 1/1.000 a < 1/100) e raras (≥ 1/10.000 a < 1/ 1.000).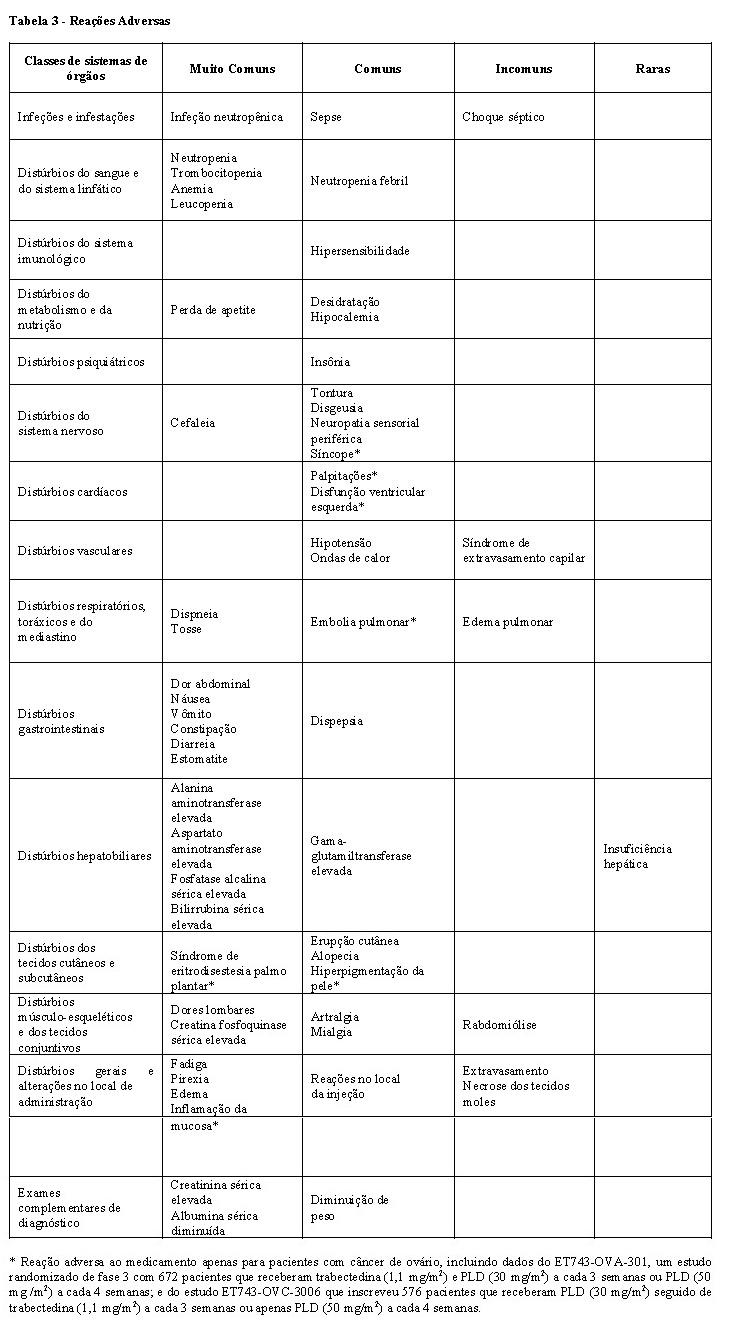
Descrição das reações adversas selecionadas
Reações adversas mais frequentes
Distúrbios do sangue e do sistema linfático
- Neutropenia:
A neutropenia é a toxicidade hematológica mais comum. Seguiu um padrão previsível de início rápido e reversibilidade, e raramente foi associada a febre ou infecção. Os nadirs de neutrófilos ocorreram em uma mediana de 15 dias e se recuperaram em uma semana. A análise por ciclo realizada nos pacientes tratados com Yondelis® mostrou neutropenia de grau 3 e 4 em aproximadamente 19% e 8% dos ciclos, respectivamente. Nesta população, a neutropenia febril ocorreu em 2% dos pacientes e em < 1% dos ciclos.
- Trombocitopenia:
Eventos hemorrágicos associados à trombocitopenia ocorreram em < 1% dos pacientes tratados com Yondelis®. A análise por ciclo realizada nesses pacientes mostrou trombocitopenia de grau 3 e 4 em aproximadamente 3% e < 1% dos ciclos, respectivamente.
- Anemia:
Anemia ocorreu em 93% dos pacientes tratados com Yondelis®. A porcentagem de pacientes anêmicos no início do estudo foi de 46%. A análise por ciclo mostrou anemia de grau 3 e 4 em aproximadamente 3% e 1% dos ciclos, respectivamente.
Distúrbios hepatobiliares
- AST/ALT aumentada:
O tempo médio para atingir os valores de pico foi de 5 dias para AST e ALT. A maioria dos valores tinha diminuído para grau 1 ou resolvido no dia 14-15 (vide seção 5. ADVERTÊNCIAS E PRECAUÇÕES). A análise por ciclo realizada em pacientes tratados com Yondelis® mostrou elevações de grau 3 de AST e ALT em 12% e 20% dos ciclos, respectivamente. Elevações de grau 4 de AST e ALT ocorreram em 1% e 2% dos ciclos, respectivamente